

Abstract
More than 100 point mutations of the superoxide scavenger Cu/Zn superoxide dismutase (SOD; EC 1.15.1.1) have been associated with the neurodegenerative disease amyotrophic lateral sclerosis (ALS). However, these mutations are scattered throughout the protein and provide no clear functional or structural clues to the underlying disease mechanism. Therefore, we undertook to look for folding-related defects by comparing the unfolding behavior of five ALS-associated mutants with distinct structural characteristics: A4V at the interface between the N and C termini, C6F in the hydrophobic core, D90A at the protein surface, and G93A and G93C, which decrease backbone flexibility. With the exception of the disruptive replacements A4V and C6F, the mutations only marginally affect the stability of the native protein, yet all mutants share a pronounced destabilization of the metal-free apo state: the higher the stability loss, the lower the mean survival time for ALS patients carrying the mutation. Thus organism-level pathology may be directly related to the properties of the immature state of a protein rather than to those of the native species.
Amyotrophic lateral sclerosis (ALS) is a motor neuron syndrome where a sub group of 3–6% has been associated with a diverse set of mutations in the superoxide scavenger Cu/Zn superoxide dismutase (SOD; 1.15.1.1) (1, 2). Interestingly, several of these mutations show perfectly native-like in vivo activity (3, 4) and metal coordination (5). As an alternative cause of disease, transgenic mouse models suggest that ALS arises through an adverse, and yet-unidentified, side reaction of the mutated protein causing cytotoxicity. Mice devoid of SOD retain normal motor function (6), as do mice overexpressing wild-type SOD. In contrast, mice expressing high levels of mutant human SOD in addition to endogenous SOD show neural damage. The collective evidence suggests that mutant SOD has gained a cytopathogenic function (7, 8). Models for how the neurodegenerating toxicity arise include the following: (i) enzymatic side-reactions in catalytically promiscuous SOD mutants producing increased levels of oxidative compounds such as hydroxyl radicals (9–11) and peroxynitrite (12), (ii) release of free Cu ions (13), (iii) aberrant binding of SOD to other proteins (14), (iv) binding of mutant SOD to heat shock proteins (15, 16) with the subsequent prevention of their antiapoptotic function (16), (v) altered redox regulation (17), and (vi) formation of toxic SOD aggregates (18–20). Evidence that could potentially distinguish between these models was recently provided from mice lacking the metal-loading chaperone CCS (copper chaperone for SOD; ref. 21). The chaperone is essential for incorporating the copper ion and, hence, to gain the native protein. When the CCS gene was ablated, the ALS-associated mutations were observed to still provoke the disease, indicating that a copper-free precursor state of SOD causes neurotoxicity (21). Along this line, a broad spectrum of mechanistic scenarios is implicated from the folding and assembly pathway of SOD. The protein folds first into a homodimeric apo state (22), and some of the dimers are transported to specific cellular locations, e.g., the mitochondria (23), before the catalytically active Cu ion is loaded by the SOD-specific chaperone CCS (24). The details of the Zn acquisition are yet unknown. Upon extraction of the Cu and Zn ions with chelating agents, SOD reverts spontaneously to the apo state (25). The Cu-free forms of SOD constitute as much as 35% of the SOD content in human lymphoblasts (26). It is thus easy to envisage that even subtle mutational alterations of these precursor species could have significant pathological consequences. Notably, such alterations may not be revealed by the properties of the final metal-loaded protein, which could still appear wild-type-like, as is apparent for several ALS-provoking mutants, both in vitro (27, 28) and in vivo (3, 4, 29).
To investigate this type of indirect folding-related disease mechanism, we have analyzed the stability and unfolding transitions of five ALS-associated SOD mutations that have different structural characteristics and that are all distant from the active site (Fig. 1): the buried mutations SODA4V and SODC6F, which are disruptive, as well as the surface mutations SODD90A, SODG93A, and SODG93C, which display wild-type-like activity and metal coordination (30). As a control we use SODC6A, which is not associated with ALS but is frequently encountered in other organisms (31). The results show that the common denominator of the ALS mutants is a distinct destabilization of the apo state, whereas the properties of the holo protein in several cases are unaffected. This selective effect on the apo protein is not observed in the control, SODC6A.
Fig 1.

Crystal structure of homodimeric Cu/Zn SOD from human (PDB code ), showing the positions of the Cu (green) and the Zn (gray) ions and the side-chain positions mutated in this study (red).
Materials and Methods
All chemicals were of analytic grade and purchased from Sigma, except guanidinium chloride (GdmCl, ultrapure), which was from GIBCO/BRL, and the redox agent tris(2-carboxyethyl)phosphine hydrochloride (TCEP), which was from Pierce. The temperature was 23°C. The experiments were carried out in the presence of 50, 100, or 500 μM TCEP as well as under oxidizing conditions, to account for artifacts from intermolecular crosslinks between the eight thiol side chains in the SOD homodimer. Further, all stability measurements were done at a constant protein concentration of 35 μM (dimer) to cancel contributions from the dimerization equilibrium.
Mutations.
Human Cu/Zn-SOD mutants were constructed by using a QuikChange mutagenesis kit (Stratagene). The identity of the mutations was confirmed by DNA sequencing and mass spectroscopy.
Metal Analysis.
The Cu and Zn contents were determined by graphite furnace atomic absorption spectrometry.
Expression and Purification.
To ensure good metal loading in the bacterial expression system, SOD was coexpressed with the Cu-chaperone CCS from yeast and with a balanced supply of Cu and Zn ions in the growth medium. Cultivation was at 23°C, and CuSO4 (3 mM) and ZnSO4 (30 μM) were added upon induction. Purification was done by heat denaturation (60°C for 20 min) followed by ammonium sulfate precipitation, gel filtration (S100 Sephacryl 100, Amersham Pharmacia), and ion-exchange chromatography (Q-Sepharose). The apoprotein was prepared according to ref. 25, with the addition of a final dialysis against 10 mM EDTA.
GdmCl Denaturation.
The transition midpoints and unfolding m values were obtained by standard GdmCl and urea titrations according to ref. 32,
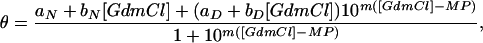 |
where θ is the ellipticity, MP is the transition midpoint, and aN + bN[GdmCl] and aD + bD[GdmCl] are the baselines for the folded (N, native) and denatured state, respectively. In cases where the baseline for the folded species was poorly defined, the lines were replaced with a constant. m is the denaturant dependence of the logarithm of the equilibrium constant, log Kobs = log([D]/[N]), which is assumed to depend linearly on denaturant
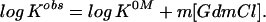 |
CD Measurements.
The data were collected at 216 and 230 nm, with 10-s integration time per point. The buffer in GdmCl experiments was 10 mM potassium phosphate at pH 7.0, and in urea experiments it was 10 mM Mes at pH 6.3. The redox potential was controlled by TCEP, and apo-SOD buffers contained 10 mM EDTA. Protein concentration was 35 μM (dimer). The CD analysis was done at 23°C on a Jobinyvon CD6 Dichrograph and a π*-180 Spectrometer (Applied Photophysics).
Results
SOD Displays Two-State Unfolding in the Presence of TCEP.
The CD monitored GdmCl and urea titrations of holo SODWT and apo-SODWT are shown in Fig. 2. Both proteins produce sigmoidal unfolding transitions that fit nicely to the two-state expression in Eq. 1. The seemingly reversible equilibrium establishes within 20 min and drifts only slowly toward the apo form (days). In the presence of 50 μM TCEP, the transition midpoint is at 3.6 M GdmCl for holo SODWT and at 1.4 M GdmCl for apo-SODWT (Fig. 2). The difference in transition midpoint between the holo- and apoprotein is in overall agreement with earlier results from fluorescence-detected measurements (22, 33) and does not vary significantly with TCEP concentration. Under conditions of fully oxidized cysteines, however, the two-state character is perturbed by a slight stretch of the denatured half of unfolding transition with an accompanying decrease of the m value (data not shown). The perturbation is also revealed by an apparent variation of the transition midpoints (MP) with detection wavelength: at 216 nm MP = 3.9 M but at 230 nm, where the overall CD change is smaller, MP turns out artificially high at 4.2 M if the stretch is not accounted for. The reason for this perturbation is most likely crosslinking of free thiol side chains in the denatured ensemble that would not take place in the reducing environment in the cell. To minimize the complications from such erroneous disulfide bridges, we focus primarily on stability data obtained under reducing conditions. Further, we base our conclusions only on the relative stability changes induced by the mutations (ΔMP), to cancel the background of general stability effects upon addition of TCEP. Thiol reduction generally lowers the transition midpoint by increasing the configurational freedom of the denatured ensemble (32).
Fig 2.

CD-monitored GdmCl titrations of SODWT. θ is in units of 103 deg⋅cm2⋅dmol−1. (Upper) Unfolding transitions of apo-SODWT (▪) and holo-SODWT (•) monitored by CD at 216 nm, under conditions of reduced cysteines. The curves are fits of Eq. 1. The apoprotein shows a significantly lower midpoint and higher m value than the holoprotein (Table 1). (Lower) Unfolding transition of holo-SODWT (•) at 230 nm, and analogous data from a standard protein preparation with heterogeneous metal content (○) under conditions that promote formation of disulfide crosslinks. The latter transition reveals clearly the presence of apo-SOD and other nonnative species that are not discerned in fresh homogenous preparations.
The two-state character of the SOD transition is also affected by the homogeneity of the protein preparation. If the last purification step is excluded (i.e., the Q-Sepharose), the unfolding transition broadens, the ratio between Cu and Zn diverges from unity, and the unfolding transition of apo-SOD becomes apparent in the baseline at low GdmCl (Fig. 2). Thus the sigmoidal transition with linear baselines can be used as a quality control of the protein preparation, indicating a homogenous population of protein with native metal coordination.
Holo-SODWT May Not Completely Unfold at High Denaturant Concentrations.
The m values accompanying the unfolding transitions of holo- and apo-SOD are on average 1.4 and 2.7, respectively (Table 1). The relatively low m value for the holo protein indicates that the unfolding transition is not complete but leads to a partly organized intermediate (34); m is significantly less than expected from full exposure of the molecular surface buried in a 300-residue protein (35). It is possible that this intermediate retains the coordinated metals as has been observed with other Cu proteins (36). It is thus not meaningful to use the m value of holo-SOD to estimate the thermodynamic stability by linear extrapolation of the equilibrium constant to 0 M GdmCl (Eq. 2). Nevertheless, we may use changes of the transition midpoint and of the m values to infer changes in stability and unfolding behavior of holo-SOD upon point mutation.
Table 1.
Stability data from GdmCl and urea titrations on SODWT and the ALS-associated SOD mutations examined in this study
| SOD
|
MPholo, M | ΔMPholo, M |
mholo
|
Cu/Zn ratio
|
MPapo, M | ΔMPapo, M |
mapo
|
Mean survival time, years (ref.)
|
||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 M TCEP GdmCl | 0 M TCEP | 50 μM TCEP | 500 μM TCEP | 0 M TCEP urea | 0 M TCEP | 100 μM TCEP | 500 μM TCEP | |||||
| WT | 3.91 ± 0.03 | 1.42 ± 0.15 | 0.72 | 3.89 ± 0.04 | 2.68 ± 0.36 | — | ||||||
| A4V | 2.65 ± 0.05 | 1.26 | 1.07 | 1.01 | 0.87 ± 0.10 | 0.96 | 1.70 ± 0.05 | 0.86 | 0.76 | 0.92 | 2.71 ± 0.81 | 1.2 ± 1 (58, 59) |
| C6A | 3.77 ± 0.02 | 0.14 | −0.08 | 0.26 | 1.34 ± 0.11 | 0.66 | 3.40 ± 0.08 | 0.19 | 0.1 | 0.14 | 2.27 ± 0.37 | — |
| C6F | 1.76 ± 0.08 | 2.15 | 1.65 | 1.62 | 0.63 ± 0.13 | 1.90 | 1.51 ± 0.19 | 0.93 | 0.88 | 0.78 | 2.26 ± 1.4 | 1 (48) |
| D90A | 3.69 ± 0.03 | 0.22 | 0.04 | 0.08 | 1.22 ± 0.13 | 0.98 | 2.90 ± 0.02 | 0.39 | 0.42 | 0.35 | 2.87 ± 0.47 | 14.2 ± 7 (49) |
| G93A | 3.47 ± 0.02 | 0.44 | 0.29 | 0.11 | 1.14 ± 0.12 | 0.88 | 1.66 ± 0.02 | 0.87 | 0.65 | 0.85 | 3.79 ± 0.43 | 2.3 ± 2 (58, 59) |
| G93C | 3.50 ± 0.03 | 0.41 | 0.32 | 0.16 | 1.18 ± 0.12 | 0.86 | 1.95 ± 0.02 | 0.76 | 0.68 | 0.73 | 3.13 ± 0.46 | 10.1 ± 6 (59) |
ΔMPs are obtained from GdmCl titrations (Eq. 1), except those (
) that are obtained by urea titration and divided by 2.55 to be directly comparable with the GdmCl data (see normalization in Results.
mholo and mapo are the average values from 0, 50, 100, and 500 μM TCEP, and m values from urea data are multiplied by 2.55 to be directly comparable with the GdmCl data. Errors are from the curve-fitting of Eqs. 1 and 2.
The values constitute upper limits because the apo states of A4V and C6F may be partly unfolded even at 0 M denaturant.
Apo-SODWT Is Only Marginally Stable Under Physiological Conditions.
For apo-SOD, where the m value indicates more complete unfolding, we derive a tentative estimate of the fraction of unfolded protein (fD) under physiological conditions according to fD = Kobs/(1 + Kobs), where K0M is obtained by extrapolation according to Eq. 2. The value of log K0M ranges from −3.1 (urea, 500 μM TCEP) to −4.8 (GdmCl, 100 μM TCEP), corresponding to an equilibrium occupancy (fD) of 1 denatured per 1,000 to 60,000 folded apo-SODWT molecules at a protein concentration of 35 μM dimer. The former value is in good agreement with earlier results by Desideri and coworkers (33). At the much lower physiological concentrations of SOD [<2 μM (37)] the value of fD may be even larger, assuming that the denaturation equilibrium for the apo homodimer is N2 ⇄ 2D (38). Notably, these values are indicative of a comparatively low thermodynamic stability of apo-SODWT. For other small proteins fD is typically less than 10−6 at 0 M GdmCl (39), including the homodimeric arc repressor (38).
The low stability of apo-SOD makes GdmCl an unsuitable denaturant for stability measurements: the unfolding transition occurs at denaturant concentrations too low to be accurately resolved. Therefore, we have also determined the stability of apo-SOD by the less potent denaturant urea, which increases the transition midpoint of the SODWT to well above 3 M (Table 1). Normalization of GdmCl and urea data is a standard procedure and normally is accounted for by extrapolation of K0M to 0 M denaturant according to Eq. 2. In this study we opt for a more accurate approach by avoiding the extrapolations and instead deriving the normalization factor directly from the ratio of the GdmCl- and urea-transition midpoints for apo-SODWT at 100 μM TCEP. This yields MPGdmCl = MPurea/2.55, where MPGdmCl = 1.4 M and MPurea = 3.57 M.
ALS-Associated SOD Mutants Show Common Destabilization of the Apo State.
The key result of this study is directly evident from comparison of the GdmCl denaturation curves at 50 μM TCEP in Fig. 3: the ALS-associated mutants show a common and pronounced effect on the apo-state stability, even in cases where the holoprotein is unaffected. A clear example is provided by the most frequently occurring ALS mutation, SODD90A. As the truncated carboxylate group points away from the surface of the protein without being involved in salt bridges or specific hydrogen bonding, the effect on protein folding and stability is expected to be small. Consistently, the denaturation curve of holo SODD90A at 50 μM TCEP is nearly identical to that of the wild-type protein (Fig. 3). Even so, this seemingly benign mutation leads to marked changes of the stability of the apoprotein, indicated by a midpoint shift (ΔMP) of ≈0.4 M GdmCl (Table 1); ΔMP is directly proportional to the stability change upon mutation, according to the linear free-energy relations in Eq. 2. The selective destabilization of apo-SODD90A is apparent at all concentrations of TCEP as well as by the normalized urea data (Fig. 4). The selective destabilization of apo-SOD is even more evident for the ALS-mutant SODG93A and its variant SODG93C (Figs. 3 and 4, Table 1). The only mutants that show substantial effects on the holo stability are SODA4V and SODC6F. SODC6F seems also more plastic than the wild-type protein, manifested in broadened unfolding transitions and decreased values of m (cf. ref. 40). Moreover, structural perturbations of apo-SODC6F are indicated by the altered direction of the urea unfolding transition. The result is not surprising because both Ala-4 and Cys-6 are tightly buried in the hydrophobic core, where the introduction of larger side chains is disruptive. In common with the other ALS mutations, SODA4V and SODC6F show also pronounced effects on the stability of the apoprotein (Figs. 3 and 4, Table 1). Notably, the unfolding transition of all apo-SOD mutants, with the possible exception of D90A, stretches down to 0 M GdmCl, indicating that the apoprotein is partly unfolded (1–25%) under physiological conditions. For comparison, the apo states of SODWT and the control SODC6A are denatured to <0.05% (Eq. 1).
Fig 3.

Unfolding transitions for the ALS-associated mutants (open symbols) and SODWT (filled symbols) measured by CD. The GdmCl data were monitored at 216 nm and the urea data were monitored at 230 nm. The mutants SODD90A, SODG93A, and SODG93C show more pronounced stability effects on the apoprotein (□ and ▪) than on the holoprotein (○ and •), whereas the disruptive mutations SODA4V and SODC6F show radical perturbations of both the apo- and holoprotein. The control mutation SODC6A shows no marked effect on either species.
Fig 4.

Histogram of the stability loss (ΔMP) upon mutation of SOD. The ALS-associated mutations show a common destabilization of the apoprotein (red) even as the effect on holo-SOD varies (blue). The experiments were done at different redox potentials to distinguish possible artifacts from erroneous disulfide bridges. □, 0 M TCEP;  , 50 μM TCEP,
, 50 μM TCEP,  , 100 μM TCEP; and
, 100 μM TCEP; and  , 500 μM TCEP.
, 500 μM TCEP.
As a control, we have engineered the conservative mutation C6A, which is not connected to ALS but is frequently encountered in other organisms (31). SODC6A displays only a small destabilization of the holoprotein without significantly affecting the m value (Fig. 3, Table 1). The result is characteristic for side-chain replacements that solely leave cavities in the protein interior and shows that holo-SOD responds to mutations in a predictable manner (35). It is interesting to note that SODC6A, in contrast to the ALS-associated mutations, shows an equally small effect on the apoprotein (Figs. 3 and 4, Table 1). The reason for the selective destabilization of apo-SOD observed for some of the ALS-associated mutants may be that the structure of the metal-depleted species is stabilized by interactions that are not present in the holoprotein. Alternatively, the targeted side chains are involved in residual structure in the denatured state of the holoprotein.
Spectral Characteristics.
The far-UV CD spectra of the holo, apo, and denatured states of SODWT are shown in Fig. 5, together with the absorbance spectra of holo- and apo-SODWT. Although the CD change for the different SOD species is relatively modest, the holoprotein displays a characteristic absorption band around 680 nm (41) that is missing in the metal-depleted apoprotein (Fig. 5). The mutants with wild-type-like denaturation properties of the holoprotein, i.e., SODD90A, SODG93A, SODG93C, and SODC6A, show wild-type-like CD spectra of the holoprotein, whereas the disruptive mutations SODA4V and SODC6F are different (Fig. 5). The structural perturbation of SODC6F is further evident from its Cu/Zn ratio, which is significantly higher than for the other proteins (Table 1). A similar picture is seen for the apo state: SODD90A, SODG93A, SODG93C, and SODC6A show wild-type-like apo spectra, whereas SODC6F is anomalous in displaying an altogether different apo spectrum with β character (Fig. 5). Notably, the selective effect of mutation on apo-SOD could mean that the metal-depleted species has a structure that is, at least partly, different from that of the holoprotein. Despite this thermodynamic evidence for a structural alteration upon metal binding, the small difference in the far-UV CD spectra between holo- and apo-SOD indicates that their gross structures are still similar (Fig. 5).
Fig 5.

Far-UV CD and absorbance spectra of the various SOD species. θ is in units of 103 deg⋅cm2⋅dmol−1. The only mutations indicative of structural perturbations are SODA4V and SODC6F. (a) CD spectra of holo-SODWT (•), apo-SODWT (○), and holo-SODWT denatured in 6 M GdmCl (▪). (b) Absorbance spectra of holo-SODWT (•) and apo-SODWT (○). (c) CD spectra of the holo-SOD mutants: •, wild type; ♦, A4V; ▪, C6A; ⋄, C6F; ×, D90A; □, G93A; and ▵, G93C. (d) CD spectra of the apo-SOD mutants: ○, wild type; ♦, A4V; ▪, C6A; ⋄, C6F; ×, D90A; □, G93A; and ▵, G93C.
Discussion
Is ALS Provoked by Apo-SOD?
Disease models for ALS have largely been focused on aberrant catalytic properties, or abnormal interactions of native SOD or leftover metal ions (29). The results from this study point instead at the possibility that the disease is triggered by alterations of the metal-free apoprotein. The ALS-associated mutations SODA4V, SODC6F, SODD90A, SODG93A, and SODG93C show a common and distinct destabilization of the apo state, whereas they display variable and often negligible impact on the holoprotein (Table 1). The idea of the apo species playing a critical role in the pathogeneses of SOD was highlighted recently by the finding that a series of ALS-associated SOD mutants (i.e., G37R, G85R, and G93A) cause motor neuron degeneration in transgenic mice independent of the CCS-mediated copper loading (42). Deletion of CCS had no significant effected on the onset or progression of disease, although the production of metal-loaded SOD was substantially diminished (42). The observation suggests that the toxicity is not directly coupled to the final metal-loaded form of SOD, but rather to a species preceding this state. Additional evidence for the involvement of apo-SOD in the disease mechanism is provided by findings that heat-shock proteins selectively bind to partly and fully demetallated SOD mutants, but not to the holoprotein, and block SOD's transfer into the mitochondrion (ref. 16; see also ref. 15).
Two Ways to Disease: Suppressed Formation of Holo-SOD or Alterations of Apo-SOD.
In essence, there are two classes of mutations that are expected to increase the susceptibility of metal-free SOD to provoke disease: (i) mutations that prevent or suppress the formation of the holoprotein and (ii) perturbations of apo-SOD itself that may more directly shift the equilibrium toward the toxic precursor species. Considering the marginal stability of apo-SODWT (33), it is easy to imagine that even subtle energetic perturbations could be critical for populating such partly unfolded or altered conformations. The ALS-associated mutations in Table 1 belong all to the latter category, although SODA4V and SODC6F with their combined effect on the apo- and holoprotein may favor the pathologic side reaction by both mechanisms. Hence, by invoking the apo state as the branch to neurotoxicity, it is easy to explain how such a large number of superficially unrelated mutations can still have a common pathology. At this simplistic level it is assumed that the partly metallated species that may populate under physiological conditions, e.g., Zn-loaded protein, have properties intermediate between those of the apo- and holoprotein.
It was recently observed that introduction of Zn-depleted SOD by liposome fusion caused rapid death of cultured motor neurons. The toxicity was attributed to an anomalous backward conversion of oxygen to superoxide in the Zn-depleted active site, and the superoxide subsequently combined with nitric oxide to form toxic peroxynitrite (43, 44). However, peroxynitrite-mediated toxicity has gained little support from studies in transgenic mice (45), because termination of the neuronal nitric oxide synthase does not attenuate the disease (46). A toxicity pathway of Zn-free SOD based on increased accumulation of the apo species could perhaps reconcile these data. Connection between elevated levels of apo-SOD and cytotoxicity is further implicated from treatment of ALS patients with metal chelators, such as penicillamine, EDTA, and thioctic acid, that sometimes accelerates the disease (47).
Apparent Correlation Between Apo-SOD Stability and ALS Progression.
Notably, SODA4V and SODC6F with their marked effect on both the holo- and apoprotein are also the most disease-provoking mutants in this study, with the victims having life expectancies of <2 years after diagnosis (48) (Table 1). At the other extreme is SODD90A, which has the smallest effect on the apoprotein and a survival time of 14 years (49) (Table 1). Interestingly, the apparent trend between apo-SOD stability and ALS progression is also discerned in a plot of mean survival time after disease onset against ΔMPapo, which produces a linear correlation with R = 0.91 (Fig. 6). In view of the limited number of mutants examined, however, we choose not to draw any detailed conclusions from this apparent correlation.
Fig 6.

Plot of mean survival time after ALS diagnosis versus stability loss of apo-SOD (ΔMP) upon mutation. ΔMP is the average value from the different reducing conditions in Table 1. The fitted line is y = 25.9 years − 28x; R = 0.91. A tentative interpretation would be that the intersect of 25.9 years corresponds to the survival time of (sporadic) ALS cases with no SOD mutation, but with genetic factors that are otherwise identical to those of the patient group in Table 1.
Disease Mechanism Based on Apo-SOD Complies with the Protein-Aggregation Hypothesis.
Reduced stability of the apo species upon ALS-associated mutations fuels the idea that the early events in the pathogenic mechanism of ALS involve protein aggregation. In other systems, such marginally stable and structurally promiscuous states are frequently observed in connection with aggregation or fibrillation processes (50), and may also induce apoptosis in cell cultures (51, 52). Seminal evidence for the aggregation idea was provided from observations that intracellular SOD aggregates form in cultured motor neurons upon microinjection of ALS-associated mutants but not upon injection of the wild-type protein (18). Aggregates resembling inclusion bodies have also been found in human ALS patients (19), and transgenic mice expressing mutant SOD produce inclusion bodies in motor neurons and astrocytes in connection with ALS-like pathology (20). Moreover, mice expressing SODG93A display high molecular weight precursor complexes (IPCs) weeks before the outbreak of disease and the appearance of inclusion bodies (20).
The ability of mutant SODs to undergo ordered aggregation in vitro is nicely demonstrated by their tendency to form solid but yet transparent gels. In this study, such gels were encountered during ultracentrifugation and to some extent during protein preparation. It is yet uncertain whether the SOD gels are composed of fibrillar structures, as commonly observed with other protein gels, because they do not stain with standard amyloid indicators such as Congo red or thioflavin T.
Additional support for the aggregation or misfolding hypothesis is found in the way ALS initiates and progresses. Neurodegeneration starts locally and spreads from one myotome segment to the segment next to it (53–55). This domino-like propagation of the pathology has previously been taken as evidence for a viral infection accompanying the disease (56). However, this is also the disease pattern expected from infectious aggregates or misfolded precursor states. Assemblies of macromolecular aggregates are nucleated events, meaning that they typically appear locally and sometimes after a considerable lag period (57). Once a seeding aggregate is formed, however, it may rapidly catalyze further aggregation in an invasive manner consistent with the pathologic development of ALS.
Acknowledgments
We thank Peter Andersen and Stefan Marklund for stimulating discussions and Katarina Wallgren for technical assistance. The work was supported by the Swedish Research Council.
Abbreviations
ALS, amyotrophic lateral sclerosis
SOD, superoxide dismutase
CCS, copper chaperone for SOD
GdmCl, guanidinium chloride
TCEP, tris(2-carboxyethyl)phosphine hydrochloride
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Rosen D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., Donaldson, D., Goto, J., O'Regan, J. P., Deng, H. X., et al. (1993) Nature 362, 59-62. [DOI] [PubMed] [Google Scholar]
- 2.McCord J. M. & Fridovich, I. (1969) J. Biol. Chem. 244, 6049-6055. [PubMed] [Google Scholar]
- 3.Corson L. B., Strain, J. J., Culotta, V. C. & Cleveland, D. W. (1998) Proc. Natl. Acad. Sci. USA 95, 6361-6366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marklund S. L., Andersen, P. M., Forsgren, L., Nilsson, P., Ohlsson, P. I., Wikander, G. & Oberg, A. (1997) J. Neurochem. 69, 675-681. [DOI] [PubMed] [Google Scholar]
- 5.Nishida C. R., Gralla, E. B. & Valentine, J. S. (1994) Proc. Natl. Acad. Sci. USA 91, 9906-9910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reaume A. G., Elliott, J. L., Hoffman, E. K., Kowall, N. W., Ferrante, R. J., Siwek, D. F., Wilcox, H. M., Flood, D. G., Beal, M. F., Brown, R. H., Jr., et al. (1996) Nat. Genet. 13, 43-47. [DOI] [PubMed] [Google Scholar]
- 7.Gurney M. E., Pu, H., Chiu, A. Y., Dal Canto, M. C., Polchow, C. Y., Alexander, D. D., Caliendo, J., Hentati, A., Kwon, Y. W., Deng, H. X., et al. (1994) Science 264, 1772-1775. [DOI] [PubMed] [Google Scholar]
- 8.Ripps M. E., Huntley, G. W., Hof, P. R., Morrison, J. H. & Gordon, J. W. (1995) Proc. Natl. Acad. Sci. USA 92, 689-693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wiedau-Pazos M., Goto, J. J., Rabizadeh, S., Gralla, E. B., Roe, J. A., Lee, M. K., Valentine, J. S. & Bredesen, D. E. (1996) Science 271, 515-518. [DOI] [PubMed] [Google Scholar]
- 10.Yim M. B., Kang, J. H., Yim, H. S., Kwak, H. S., Chock, P. B. & Stadtman, E. R. (1996) Proc. Natl. Acad. Sci. USA 93, 5709-5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Landeghem G. F., Tabatabaie, P., Beckman, G., Beckman, L. & Andersen, P. M. (1999) Eur. J. Neurol. 6, 639-644. [DOI] [PubMed] [Google Scholar]
- 12.Beckman J. S., Carson, M., Smith, C. D. & Koppenol, W. H. (1993) Nature 364, 584. (lett.). [DOI] [PubMed] [Google Scholar]
- 13.Wong P. C., Pardo, C. A., Borchelt, D. R., Lee, M. K., Copeland, N. G., Jenkins, N. A., Sisodia, S. S., Cleveland, D. W. & Price, D. L. (1995) Neuron 14, 1105-1116. [DOI] [PubMed] [Google Scholar]
- 14.Kunst C. B., Mezey, E., Brownstein, M. J. & Patterson, D. (1997) Nat. Genet. 15, 91-94. [DOI] [PubMed] [Google Scholar]
- 15.Shinder G. A., Lacourse, M. C., Minotti, S. & Durham, H. D. (2001) J. Biol. Chem. 276, 12791-12796. [DOI] [PubMed] [Google Scholar]
- 16.Okado-Matsumoto A. & Fridovich, I. (2002) Proc. Natl. Acad. Sci. USA 99, 9010-9014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferri A., Gabbianelli, R., Casciati, A., Celsi, F., Rotilio, G. & Carri, M. T. (2001) J. Neurochem. 79, 531-538. [DOI] [PubMed] [Google Scholar]
- 18.Durham H. D., Roy, J., Dong, L. & Figlewicz, D. A. (1997) J. Neuropathol. Exp. Neurol. 56, 523-530. [DOI] [PubMed] [Google Scholar]
- 19.Bruijn L. I., Houseweart, M. K., Kato, S., Anderson, K. L., Anderson, S. D., Ohama, E., Reaume, A. G., Scott, R. W. & Cleveland, D. W. (1998) Science 281, 1851-1854. [DOI] [PubMed] [Google Scholar]
- 20.Johnston J. A., Dalton, M. J., Gurney, M. E. & Kopito, R. R. (2000) Proc. Natl. Acad. Sci. USA 97, 12571-12576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong P. C., Waggoner, D., Subramaniam, J. R., Tessarollo, L., Bartnikas, T. B., Culotta, V. C., Price, D. L., Rothstein, J. & Gitlin, J. D. (2000) Proc. Natl. Acad. Sci. USA 97, 2886-2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mei G., Rosato, N., Silva, N., Jr., Rusch, R., Gratton, E., Savini, I. & Finazzi-Agro, A. (1992) Biochemistry 31, 7224-7230. [DOI] [PubMed] [Google Scholar]
- 23.Okado-Matsumoto A. & Fridovich, I. (2001) J. Biol. Chem. 276, 38388-38393. [DOI] [PubMed] [Google Scholar]
- 24.Culotta V. C., Klomp, L. W., Strain, J., Casareno, R. L., Krems, B. & Gitlin, J. D. (1997) J. Biol. Chem. 272, 23469-23472. [DOI] [PubMed] [Google Scholar]
- 25.Sutter B., Bounds, P. L. & Koppenol, W. H. (2000) Protein Expression Purif. 19, 53-56. [DOI] [PubMed] [Google Scholar]
- 26.Petrovic N., Comi, A. & Ettinger, M. J. (1996) J. Biol. Chem. 271, 28331-28334. [DOI] [PubMed] [Google Scholar]
- 27.Rodriguez J. A., Valentine, J. S., Eggers, D. K., Roe, J. A., Tiwari, A., Brown, R. H., Jr. & Hayward, L. J. (2002) J. Biol. Chem. 277, 15932-15937. [DOI] [PubMed] [Google Scholar]
- 28.Hayward L. J., Rodriguez, J. A., Kim, J. W., Tiwari, A., Goto, J. J., Cabelli, D. E., Valentine, J. S. & Brown, R. H., Jr. (2002) J. Biol. Chem. 277, 15923-15931. [DOI] [PubMed] [Google Scholar]
- 29.Julien J. P. (2001) Cell 104, 581-591. [DOI] [PubMed] [Google Scholar]
- 30.Parge H. E., Hallewell, R. A. & Tainer, J. A. (1992) Proc. Natl. Acad. Sci. USA 89, 6109-6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bannister W. H., Bannister, J. V., Barra, D., Bond, J. & Bossa, F. (1991) Free Radical Res. Commun. 12–13, 349-361. [DOI] [PubMed] [Google Scholar]
- 32.Clarke J. & Fersht, A. R. (1993) Biochemistry 32, 4322-4329. [DOI] [PubMed] [Google Scholar]
- 33.Stroppolo M. E., Malvezzi-Campeggi, F., Mei, G., Rosato, N. & Desideri, A. (2000) Arch. Biochem. Biophys. 377, 215-218. [DOI] [PubMed] [Google Scholar]
- 34.Silva N., Gratton, E., Mei, G., Rosato, N., Rusch, R. & Finazzi-Agro, A. (1993) Biophys. Chem. 48, 171-182. [DOI] [PubMed] [Google Scholar]
- 35.Fersht A. R., (1999) Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding (Freeman, New York).
- 36.Leckner J., Bonander, N., Wittung-Stafshede, P., Malmstrom, B. G. & Karlsson, B. G. (1997) Biochim. Biophys. Acta 1342, 19-27. [DOI] [PubMed] [Google Scholar]
- 37.Marklund S. L. (1984) J. Clin. Invest. 74, 1398-1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bowie J. U. & Sauer, R. T. (1989) Biochemistry 28, 7139-7143. [DOI] [PubMed] [Google Scholar]
- 39.Jackson S. E. (1998) Folding Des. 3, R81-R91. [DOI] [PubMed] [Google Scholar]
- 40.Otzen D. E. & Oliveberg, M. (2002) J. Mol. Biol. 317, 639-653. [DOI] [PubMed] [Google Scholar]
- 41.Lyons T. J., Liu, H., Goto, J. J., Nersissian, A., Roe, J. A., Graden, J. A., Cafe, C., Ellerby, L. M., Bredesen, D. E., Gralla, E. B. & Valentine, J. S. (1996) Proc. Natl. Acad. Sci. USA 93, 12240-12244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Subramaniam J. R., Lyons, W. E., Liu, J., Bartnikas, T. B., Rothstein, J., Price, D. L., Cleveland, D. W., Gitlin, J. D. & Wong, P. C. (2002) Nat. Neurosci. 5, 301-307. [DOI] [PubMed] [Google Scholar]
- 43.Estevez A. G., Crow, J. P., Sampson, J. B., Reiter, C., Zhuang, Y., Richardson, G. J., Tarpey, M. M., Barbeito, L. & Beckman, J. S. (1999) Science 286, 2498-2500. [DOI] [PubMed] [Google Scholar]
- 44.Cassina P., Peluffo, H., Pehar, M., Martinez-Palma, L., Ressia, A., Beckman, J. S., Estevez, A. G. & Barbeito, L. (2002) J. Neurosci. Res. 67, 21-29. [DOI] [PubMed] [Google Scholar]
- 45.Williamson T. L., Corson, L. B., Huang, L., Burlingame, A., Liu, J., Bruijn, L. I. & Cleveland, D. W. (2000) Science 288, 399. [DOI] [PubMed] [Google Scholar]
- 46.Facchinetti F., Sasaki, M., Cutting, F. B., Zhai, P., MacDonald, J. E., Reif, D., Beal, M. F., Huang, P. L., Dawson, T. M., Gurney, M. E. & Dawson, V. L. (1999) Neuroscience 90, 1483-1492. [DOI] [PubMed] [Google Scholar]
- 47.Conradi S., Ronnevi, L. O. & Norris, F. H. (1982) Adv. Neurol. 36, 201-231. [PubMed] [Google Scholar]
- 48.Morita M., Aoki, M., Abe, K., Hasegawa, T., Sakuma, R., Onodera, Y., Ichikawa, N., Nishizawa, M. & Itoyama, Y. (1996) Neurosci. Lett. 205, 79-82. [DOI] [PubMed] [Google Scholar]
- 49.Andersen P. M., Nilsson, P., Keranen, M. L., Forsgren, L., Hagglund, J., Karlsborg, M., Ronnevi, L. O., Gredal, O. & Marklund, S. L. (1997) Brain 120, 1723-1737. [DOI] [PubMed] [Google Scholar]
- 50.Dobson C. M. (1999) Trends Biochem. Sci. 24, 329-332. [DOI] [PubMed] [Google Scholar]
- 51.Svensson M., Hakansson, A., Mossberg, A. K., Linse, S. & Svanborg, C. (2000) Proc. Natl. Acad. Sci. USA 97, 4221-4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bucciantini M., Giannoni, E., Chiti, F., Baroni, F., Formigli, L., Zurdo, J., Taddei, N., Ramponi, G., Dobson, C. M. & Stefani, M. (2002) Nature 416, 507-511. [DOI] [PubMed] [Google Scholar]
- 53.Friedman A. P. & Freedman, D. (1950) J. Nerv. Ment. Dis. 111, 1-18. [DOI] [PubMed] [Google Scholar]
- 54.Brooks B. R., Sufit, R. L., DePaul, R., Tan, Y. D., Sanjak, M. & Robbins, J. (1991) Adv. Neurol. 56, 521-546. [PubMed] [Google Scholar]
- 55.Brooks B. R. (1991) Can. J. Neurol. Sci. 18, 435-438. [DOI] [PubMed] [Google Scholar]
- 56.Brahic M., Smith, R. A., Gibbs, C. J., Jr., Garruto, R. M., Tourtellotte, W. W. & Cash, E. (1985) Ann. Neurol. 18, 337-343. [DOI] [PubMed] [Google Scholar]
- 57.Bryngelson J. D., Onuchic, J. N., Socci, N. D. & Wolynes, P. G. (1995) Proteins 21, 167-195. [DOI] [PubMed] [Google Scholar]
- 58.Juneja T., Pericak-Vance, M. A., Laing, N. G., Dave, S. & Siddique, T. (1997) Neurology 48, 55-57. [DOI] [PubMed] [Google Scholar]
- 59.Cudkowicz M. E., McKenna-Yasek, D., Sapp, P. E., Chin, W., Geller, B., Hayden, D. L., Schoenfeld, D. A., Hosler, B. A., Horvitz, H. R. & Brown, R. H. (1997) Ann. Neurol. 41, 210-221. [DOI] [PubMed] [Google Scholar]