

Abstract
Carotenoids are, along with chlorophylls, crucial pigments involved in light-harvesting processes in photosynthetic organisms. Details of carotenoid to chlorophyll energy transfer mechanisms and their dependence on structural variability of carotenoids are as yet poorly understood. Here, we employ femtosecond transient absorption spectroscopy to reveal energy transfer pathways in the peridinin–chlorophyll-a–protein (PCP) complex containing the highly substituted carotenoid peridinin, which includes an intramolecular charge transfer (ICT) state in its excited state manifold. Extending the transient absorption spectra toward near-infrared region (600–1800 nm) allowed us to separate contributions from different low-lying excited states of peridinin. The results demonstrate a special light-harvesting strategy in the PCP complex that uses the ICT state of peridinin to enhance energy transfer efficiency.
Carotenoids are among the most abundant pigments in nature, having a wide variety of functions in living organisms. In photosynthetic organisms, besides their important regulatory role in the flow of the absorbed energy, carotenoids serve predominantly as light-harvesting pigments, efficiently covering the spectral region 450–550 nm (1). They transfer absorbed light energy to chlorophyll (Chl) or bacteriochlorophyll (BChl) molecules that funnel energy toward the reaction center where a charge separation occurs (1). The ability of the carotenoids to act both as photoprotection agents and light-harvesting pigments is a consequence of their unique photophysical properties (2). A typical carotenoid belongs to an idealized C2h point symmetry group. In the manifold of singlet states of a carotenoid, the one-photon optical transition S0→S1 (1A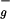 →2A
→2A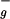 in C2h group notation) is symmetry forbidden, resulting in the absence of an S0→S1 absorption and in very weak fluorescence from the S1 state (2). The well-known absorption of carotenoids in the blue-green region of the visible spectrum is caused by a strongly allowed transition from the S0 (1A
in C2h group notation) is symmetry forbidden, resulting in the absence of an S0→S1 absorption and in very weak fluorescence from the S1 state (2). The well-known absorption of carotenoids in the blue-green region of the visible spectrum is caused by a strongly allowed transition from the S0 (1A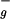 ) state to the S2 (1B
) state to the S2 (1B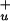 ) state. Usually, fluorescence from the S1 and S2 states of carotenoids has a good spectral overlap with the Qy and Qx bands of Chls/BChls, which favors energy transfer. On the other hand, the forbidden nature of the S1 state results in a very weak transition dipole and puts substantial limits on efficient Förster mechanism of energy transfer (3). Furthermore, although the S2 state has a strong transition dipole moment, there is a rapid internal conversion to the S1 state in 50–300 fs (4), so that energy transfer from the S2 state has to be extremely fast to compete with the S2 to S1 relaxation. Despite these apparent limitations, carotenoids are indeed efficient energy donors in various light-harvesting complexes of bacteria, algae, and higher plants, where efficiency of carotenoid to Chl/BChl energy transfer approaches 100% (1, 5, 6). Such high efficiencies are achieved by tight packing at van der Waals radius of pigments in light-harvesting complexes, minimizing the distances between donor (carotenoids) and acceptor (Chl, BChl) molecules. In most membrane-bound photosynthetic antennae, the principal light-harvesting pigments are either Chl or BChl molecules (6, 7). In contrast, in the dinoflagellates, the main light-harvesting pigment is a carotenoid, peridinin. The 2-Å crystal structure of the peridinin–chlorophyll-a–protein (PCP), a light-harvesting complex from dinoflagellate Amphidinium carterae (8), revealed a trimer of protein subunits with densely packed pigments having a stoichiometry of eight peridinins and two Chl-a molecules in each protein subunit. The pigments are arranged as two essentially identical domains of four peridinins and one Chl-a molecule. Structural information of the PCP complex has provided a good basis for both experimental and theoretical studies of energy transfer pathways within PCP (9–14). These studies demonstrated a high efficiency of peridinin to Chl-a energy transfer, with a characteristic time constant of 2.3–3.2 ps. It was suggested that energy transfer takes place predominantly from the peridinin S1 state to the Qy state of Chl-a (9, 11), although more recent results are interpreted in terms of energy transfer from both S1 and S2 states (14). Peridinin is a highly substituted carotenoid containing a lactone ring and an allene moiety in its structure, breaking the symmetry of the idealized conjugated chain. These substitutions result in observable fluorescence from the S1 state, which could be partly because of a mixing of the S1 and S2 states of peridinin (15), although no absorption due to the S0→S1 transition has been observed. It was also recently shown that the lowest excited state of peridinin exhibits rather unusual behavior, in that its lifetime strongly depends on solvent polarity, the lifetime decreasing with an increase of the medium polarity. Polarity dependence was attributed to an intramolecular charge transfer (ICT) state in the excited state manifold of peridinin (16, 17). Dynamics of the peridinin ICT state in polar solvent were recently studied by near-infrared femtosecond spectroscopy where it was shown that the ICT state can be detected via its pronounced stimulated emission band at ≈950 nm (17). Despite the better understanding of the peridinin ICT state dynamics in solution, its role in energy transfer between peridinin and Chl-a in the PCP complex remained unclear (13, 14). In this study, we applied near-infrared pump-probe femtosecond spectroscopy to the dynamics of the lowest excited states of peridinin in the PCP complex to determine energy transfer pathways between peridinin and Chl-a. We show that the main energy transfer channel from peridinin to Chls employs both the S1 and ICT states of peridinin, resulting in a (3 ps)−1 energy transfer rate. We also confirm that excitation is directly transferred to Chl-a from the peridinin S2 state, as recently reported by Krueger et al. (14), and show that the yield of this channel is ≈25%.
) state. Usually, fluorescence from the S1 and S2 states of carotenoids has a good spectral overlap with the Qy and Qx bands of Chls/BChls, which favors energy transfer. On the other hand, the forbidden nature of the S1 state results in a very weak transition dipole and puts substantial limits on efficient Förster mechanism of energy transfer (3). Furthermore, although the S2 state has a strong transition dipole moment, there is a rapid internal conversion to the S1 state in 50–300 fs (4), so that energy transfer from the S2 state has to be extremely fast to compete with the S2 to S1 relaxation. Despite these apparent limitations, carotenoids are indeed efficient energy donors in various light-harvesting complexes of bacteria, algae, and higher plants, where efficiency of carotenoid to Chl/BChl energy transfer approaches 100% (1, 5, 6). Such high efficiencies are achieved by tight packing at van der Waals radius of pigments in light-harvesting complexes, minimizing the distances between donor (carotenoids) and acceptor (Chl, BChl) molecules. In most membrane-bound photosynthetic antennae, the principal light-harvesting pigments are either Chl or BChl molecules (6, 7). In contrast, in the dinoflagellates, the main light-harvesting pigment is a carotenoid, peridinin. The 2-Å crystal structure of the peridinin–chlorophyll-a–protein (PCP), a light-harvesting complex from dinoflagellate Amphidinium carterae (8), revealed a trimer of protein subunits with densely packed pigments having a stoichiometry of eight peridinins and two Chl-a molecules in each protein subunit. The pigments are arranged as two essentially identical domains of four peridinins and one Chl-a molecule. Structural information of the PCP complex has provided a good basis for both experimental and theoretical studies of energy transfer pathways within PCP (9–14). These studies demonstrated a high efficiency of peridinin to Chl-a energy transfer, with a characteristic time constant of 2.3–3.2 ps. It was suggested that energy transfer takes place predominantly from the peridinin S1 state to the Qy state of Chl-a (9, 11), although more recent results are interpreted in terms of energy transfer from both S1 and S2 states (14). Peridinin is a highly substituted carotenoid containing a lactone ring and an allene moiety in its structure, breaking the symmetry of the idealized conjugated chain. These substitutions result in observable fluorescence from the S1 state, which could be partly because of a mixing of the S1 and S2 states of peridinin (15), although no absorption due to the S0→S1 transition has been observed. It was also recently shown that the lowest excited state of peridinin exhibits rather unusual behavior, in that its lifetime strongly depends on solvent polarity, the lifetime decreasing with an increase of the medium polarity. Polarity dependence was attributed to an intramolecular charge transfer (ICT) state in the excited state manifold of peridinin (16, 17). Dynamics of the peridinin ICT state in polar solvent were recently studied by near-infrared femtosecond spectroscopy where it was shown that the ICT state can be detected via its pronounced stimulated emission band at ≈950 nm (17). Despite the better understanding of the peridinin ICT state dynamics in solution, its role in energy transfer between peridinin and Chl-a in the PCP complex remained unclear (13, 14). In this study, we applied near-infrared pump-probe femtosecond spectroscopy to the dynamics of the lowest excited states of peridinin in the PCP complex to determine energy transfer pathways between peridinin and Chl-a. We show that the main energy transfer channel from peridinin to Chls employs both the S1 and ICT states of peridinin, resulting in a (3 ps)−1 energy transfer rate. We also confirm that excitation is directly transferred to Chl-a from the peridinin S2 state, as recently reported by Krueger et al. (14), and show that the yield of this channel is ≈25%.
Materials and Methods
Monomeric PCP complexes extracted from A. carterae were purified as described (8) and stored in the dark at −40°C. Before the experiments, the samples were dissolved in a buffer (25 mM Tris, pH 7.5/2 mM KCl), in which H2O was replaced by D2O to avoid H2O absorption in the near-infrared region. All transient absorption measurements were done in a 2-mm quartz rotational cuvette. Sample concentrations were adjusted to achieve ≈0.3 OD at 535 nm. Absorption spectra were measured before and after the pump-probe measurements to monitor possible sample degradation. We have not observed any changes in the shape of the absorption spectrum, although in some cases a small (<10%) overall decrease of the sample optical density was noted.
For the near-infrared transient absorption measurements, we used an amplified Ti:sapphire femtosecond laser system operating at a 5-kHz repetition rate. Two optical parametric amplifiers were used for generation of pump pulses in the visible region and probe pulses in near-infrared region (for a detailed description, see ref. 17). The wavelength of the pump pulses was fixed at 535 nm, whereas the wavelength of the probe pulses was scanned over the spectral region 630–1,840 nm. Energy of the excitation pulses was kept below 80 nJ per pulse, corresponding to an excitation density of <1014 photons⋅pulse−1⋅cm−2. The response function of the system was measured by a cross-correlation between probe and pump pulses, yielding a full width half maximum of 160–180 fs. In all measurements, the mutual polarization of the pump and probe beams was set to the magic angle (54.7°).
Results and Discussion
Steady-state absorption spectra of the PCP complex and its principal pigments peridinin and Chl-a are shown in Fig. 1. The absorption spectrum of Chl-a in diethyl ether exhibits sharp Soret and Qy bands centered at 428 and 660 nm, respectively. In solution, the Qy transition has an expressed progression of vibrational bands. In the PCP complex, the Chl-a Soret and Qy bands are shifted to 438 and 671 nm, respectively, due to the interaction with the protein. The weak absorption band at 622 nm originates from the 0–1 vibrational transition of the Qy band and/or from the Qx transition of Chl-a. The peridinin absorption spectrum, which represents the S0→S2 transition, depends strongly on the solvent used. In nonpolar solvents, the absorption spectrum exhibits the vibronic structure of the S2 state, but, in polar solvent, vibronic structure is lost and the absorption spectrum is broadened toward the low energy edge (16). The absorption spectrum of peridinin in the polar and viscous solvent ethylene glycol, which most closely mimics the shape of the peridinin absorption spectrum in PCP, is also shown in Fig. 1. The absorption spectra of peridinin both in ethylene glycol and in the PCP complex are rather featureless, with a maximum centered at ≈480 nm together with a shoulder at ≈520 nm.
Fig 1.

Absorption spectra of the PCP complex (solid line), Chl-a in diethyl ether (dashed line), and peridinin in ethylene glycol (dotted line).
In the present study, we have focused on the excitation dynamics of the lowest singlet excited states of peridinin in the PCP complex and their involvement in energy transfer between peridinin and Chl-a molecules. Monomeric PCP complexes were excited at 535 nm, a wavelength corresponding to the low energy edge of the peridinin S0→S2 absorption (Fig. 1). Excitation at this wavelength minimizes any contribution due to vibrational relaxation within the S2 state, but allows a possible energy transfer from the peridinin S2 state. To characterize transient spectroscopic features of peridinin in the near-infrared region, we measured transient absorption spectra of the PCP complex at delays of 0.5 and 30 ps after the excitation (Fig. 2). Given the known peridinin S1 state lifetime of ≈3 ps in the PCP complex (11, 14), these transient absorption spectra allow us to separate contributions from peridinin and Chl-a molecules. At 30 ps, practically all excitations are transferred to Chl-a, so the 30-ps transient absorption spectrum reflects the Chl-a signal. On the other hand, the transient absorption spectrum 0.5 ps after excitation represents mostly contributions from peridinin. The 30-ps transient spectrum is dominated by a bleaching and stimulated emission (BL/SE) of the Chl-a Qy band at 674 nm. A weaker negative band at 740 nm is due to a stimulated emission from the Qy state to higher vibrational levels of the Chl-a ground state. At wavelengths above 800 nm, a somewhat featureless Chl-a excited state absorption (ESA) is present, apart from a weak ESA band located at 1,290 nm due to a transition from the Chl-a Qy to some state located higher in energy. The energy of this band (7,750 cm−1) fits well to the energy difference between the Qy and Soret bands of Chl-a. For porphyrins, which belong to the D4h symmetry point group, the Soret band is assigned to doubly degenerate B states (18). Because the Qy and B states have the same parity, a one-photon transition between them is forbidden. However, because the Chl-a molecule deviates from a perfect D4h symmetry and the planarity of Chl-a molecules in the PCP complex can be slightly deformed by the protein environment, assignment of the weak 1,290-nm band to the Qy→B transition of Chl-a is feasible. An important observation is a gradual decrease of the Chl-a ESA toward lower energies: it is nearly absent in the near-infrared spectral region above 1,400 nm, allowing us to probe almost exclusively peridinin dynamics in the spectral region 1,500–1800 nm.
Fig 2.

Transient absorption spectra of the PCP complex at 0.5 ps (filled squares) and 30 ps (open circles) after the excitation at 535 nm. (Inset) Enlargement of the spectral region 800–1,800 nm.
The transient absorption spectrum recorded at 0.5 ps, representing predominantly peridinin, exhibits some additional features compared with the transient spectrum recorded at 30 ps. First, a strong positive signal at wavelengths below 750 nm is due to an ESA signal corresponding to the S1/ICT→Sn transition of peridinin, which occurs in the spectral region 500–750 nm (11, 14, 17). Here, due to peridinin to Chl-a energy transfer, the peridinin ESA overlaps with the Chl-a BL/SE band at 674 nm. It is important to note that, even at 0.5 ps after excitation, a distinct Chl-a BL/SE band at 674 nm can be seen on the top of the peridinin ESA, suggesting that some peridinin to Chl-a energy transfer occurs on a subpicosecond time scale. However, the most interesting feature of the 0.5-ps transient spectrum of the PCP complex is a negative band centered at around 930 nm, which is clearly due to the peridinin, because this band is not observed at the 30-ps time delay. A similar band was observed for peridinin in polar solvents and was ascribed to stimulated emission from an ICT state at energy close to that of the peridinin S1 state (17). The presence of the ICT SE band is a clear indication of a polar environment surrounding the peridinin molecules in the PCP complex, because the ICT state was shown to be populated only in polar solvents (11, 17). Further inspection of the 0.5-ps transient absorption spectrum of the PCP complex also reveals an ESA in the spectral region 1,200–1,800 nm, which is the ESA corresponding to the S1→S2 transition of carotenoids (19). As previously discussed for peridinin in solution (17), this ESA is predominantly due to the 0–1 and 0–2 vibrational bands of the S1→S2 transition, because the energy gap between the 0–0 vibronic bands of the S1→S2 transition lies at ≈4,000 cm−1, beyond the sensitivity range of our detection system. Exact assignment of the vibronic bands of peridinin below 1,400 nm in the PCP complex is rather complicated due in part to the overlap with the Chl-a ESA, which, as mentioned above, has a pronounced band at 1,290 nm.
To determine dynamics of different states involved in energy transfer pathways within PCP, the kinetics were followed at various wavelengths spanning the 600- to 1,800-nm region (see Table 1). The kinetics crucial for the following discussion are shown in Fig. 3. The 670-nm kinetic trace is close to the maximum of the Qy Chl-a BL/SE band, and thus reflects the rise of Chl-a signal due to the energy transfer from peridinin. The trace at the low-energy edge of the transient spectrum at 1,800 nm gives predominantly information about the peridinin S1 state (17). Importantly, the trace recorded at 930 nm monitors exclusively the dynamics of the peridinin ICT state. At 1,200 nm, contributions from S2, S1, and ICT states of peridinin overlap at the early times (17), whereas a Chl-a ESA becomes the dominating contribution to the signal at longer times. To obtain characteristic time constants for the relaxation and energy transfer processes involved, all of the kinetics for the different wavelengths spanning the 600- to 1,800-nm spectral region were fitted simultaneously. To obtain satisfactory fits, as much as five time components are needed by the global fitting procedure. Two of the time constants were kept fixed during the fitting procedure, and three others were fitted as global parameters. One fixed time constant was a long-lived component seen in all traces. This component has a time constant of ≈3.7 ± 1 ns, which is clearly the Chl-a lifetime (20). The other fixed component, with a time constant of <100 fs, is due to an ultrafast S2 decay manifested as the S2→SN ESA in the near-infrared region (21), which was also observed for peridinin in solution (17). Another possible origin of the ESA decaying with the time constant <100 fs would be the absorption from a collective excitonic state resulting from the strong interaction between S2 states of closely situated peridinins to some higher excitonic state. A precise resolution of this time constant is beyond the limits of our apparatus, because the S2 lifetime of peridinin in solution is ≈50 fs (17). In PCP, this lifetime is expected to be even shorter because energy transfer also occurs from the peridinin S2 state, as reported by Krueger et al. (14) and as confirmed by our measurements (Fig. 2). In addition to the 3.7-ns and <100-fs components, our global fitting procedure revealed three other components, as follows: 0.7 ± 0.1 ps, 2.5 ± 0.2 ps, and 35 ± 5 ps. The results of the global fitting analysis are summarized in Table 1.
Table 1.
Relative amplitudes of different exponential components
| λ, nm | <0.1 ps amp., % | 0.7 ps amp., % | 2.5 ps amp., % | 35 ps amp., % | 3.7 ns amp., % |
|---|---|---|---|---|---|
| 630 | 18.5 | 62 | 7.5 | 12 | |
| 670 | −19 | −81 | 14 | 86 | |
| 800 | 91 | 5 | −5 | 1 | 3 |
| 930 | −9 | −91 | 0 | 100 | |
| 1,200 | 84 | 3 | −5 | 2 | 11 |
| 1,400 | 69 | 11 | 13 | 4 | 3 |
| 1,800 | 60 | 10 | 24 | 4 | 2 |
Relative amplitudes of five exponential functions with corresponding time constants are extracted from a multiexponential global fitting of kinetics measured at various probing wavelengths for the PCP complex after 535-nm excitation. Negative amplitudes correspond to the rise, and positive to the decay, of the signal. For the 670- and 930-nm kinetics, negative amplitudes correspond to percentage from the total negative amplitude, and, for the 800- and 1,200-nm kinetics, correspond to the percentage from the total positive amplitude. In all cases, uncertainties of the lifetimes and amplitudes are within a limit of ±10%.
Fig 3.

Kinetic traces of the PCP complex measured after the excitation at 535 nm. Probing wavelengths are indicated for each kinetic. Solid lines represent best fits obtained from multiexponential global fitting procedure.
The ubiquitous 2.5-ps component can be firmly attributed to the energy transfer process between peridinin and Chl-a, because it appears as decay at the wavelength characteristic of peridinin (1,800 nm) and as a rise at the wavelength probing the Chl-a (670 nm). This assignment is further supported by good agreement with the peridinin to Chl-a transfer rate observed earlier for energy transfer within the PCP complex (11, 14). However, the most interesting observation here is that the 2.5-ps time constant also appears as a decay of the negative signal at 930 nm, which monitors the peridinin ICT state (17), indicating a direct involvement of the ICT state in energy transfer between peridinin and Chl-a in the PCP complex. The similarity of the transient absorption spectrum of PCP and that of peridinin in the polar solvent methanol (17) leads to the conclusion that peridinin molecules in the PCP complex reside in a polar environment. This finding is in agreement with the available structural data showing that part of each peridinin molecule is surrounded by polar hydrogen-bonding residues together with a number of water molecules. Consequently, the intrinsic peridinin lifetime in PCP is expected to be close to that observed in highly polar solvents such as methanol.
The presence of the Chl-a BL/SE band in the 0.5-ps transient absorption spectrum of the PCP complex suggests an additional energy transfer channel via the S2 state of peridinin. Applying a simple exponential modeling of the kinetic trace measured at 670 nm and assuming that direct Chl-a excitation at 535 nm is negligible, we calculate the energy transfer via the S2 state to be ≈25% of the total. This result is at the lower limit of the 25–50% range previously estimated (14). The rest of the energy transfer must occur via the S1/ICT state. From fluorescence excitation measurements, the overall energy transfer efficiency is 88 ± 2% (11), so ≈63% of the total energy transfer must proceed via the S1/ICT channel, working at ≈84% efficiency. From the energy transfer efficiency and the 2.5-ps S1/ICT state depopulation time in the PCP complex, we calculate that the intrinsic lifetime of the peridinin S1/ICT state in PCP must be ≈16 ps and the energy transfer rate ≈(3 ps)−1. The 16-ps lifetime corresponds to a polar environment surrounding the peridinin molecules in the PCP complex as can be seen from the peridinin lifetime dependence on the solvent polarity (16, 17). This 16-ps intrinsic peridinin lifetime also suggests that the 35-ps time constant obtained from the global fitting procedure is not due to peridinins that do not transfer energy as proposed earlier (14). Rather, the 35-ps component should be attributed to relaxation processes among Chl-a molecules. To clarify the origin of this component, we investigated the excitation intensity dependence of the 670-nm kinetics after excitation at 535 nm (data not shown). An increase of the excitation energy resulted in a shortening of the lifetime of the 35-ps component; a decrease in excitation intensity resulted in a reduction of the signal, which vanished almost completely at an intensity of 6 nJ per pulse. These results confirm that the 35-ps component is due to exciton annihilation between Chl-a molecules.
The origin of the 0.7-ps component revealed by the global fitting is less clear. It represents only a minor contribution except at 800 and 1,200 nm, where it has about the same amplitude as the 2.5-ps component (see Table 1). Because these wavelengths are close to the edges of the ICT emission band (Fig. 2), the 0.7-ps process is probably associated with a shift and/or narrowing of the ICT emission band and thus with a vibrational relaxation and/or cooling in the ICT state. Recent experiments, showing that vibrational relaxation in the lowest singlet excited states of carotenoids occurs on a time scale of 0.5–0.8 ps (22, 23), support this assignment. The 0.7-ps component is also observed as a rise of the Chl-a BL/SE band at 670 nm. This result suggests that it could represent a minor energy transfer channel via a vibrationally hot ICT state, as occurs in higher plant LHCII complexes (24) and LH2 complexes from purple bacteria (25). It is quite possible that the 0.7-ps process corresponds to both relaxation in the ICT state and minor energy transfer via higher vibrational levels of the ICT state.
Having assigned the 2.5-ps component to the main energy transfer channel, we will discuss possible energy transfer mechanisms via the lowest singlet excited states of peridinin. Because the decay of the peridinin ICT SE band matches the Chl-a BL/SE rise, the ICT state must be directly involved in energy transfer between peridinin and Chl-a. In agreement with calculations of energy transfer rates in PCP (13), we can assume that the energy transfer between peridinin and Chl-a occurs predominantly via a Förster mechanism. In such a case, the rate of energy transfer between donor D and acceptor A molecules kDA is given as follows:
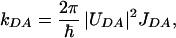 |
where UDA is the electronic coupling between donor and acceptor and JDA is their spectral overlap, the integral of donor emission and acceptor absorption spectra over energy. Although we cannot unequivocally assign the energy of the ICT state from the data presented here, we can make use of fluorescence and transient absorption data for peridinin in methanol. We assume, as has been shown for LHCII complex from higher plants (26) and LH2 complex from purple bacteria (27), that there is no significant change of the carotenoid S1 energy when going from solvent to a protein environment. It is worth underlining that, although we detect the ICT state emission at 930 nm (10,750 cm−1), which is considerably lower than the energy of the Chl-a Qy state (≈14,900 cm−1), the energy transfer via the ICT state does not require an uphill energy transfer, because the spectral origin of the S0-ICT transition is located no more than 1,300 cm−1 below the peridinin S1 state, which is located at 16,400 ± 500 cm−1 in methanol (17). Thus, the 0–0 origin of the S0-ICT transition is still above the Chl-a Qy state, and the extension of the ICT emission up to ≈1,100 nm is due to a substantial shift between the ICT and S0 potential surfaces (17). However, because the ICT state is located slightly below the S1 state, overlap of the ICT emission with the absorption of the Chl-a Qy state may be somewhat poorer than that with the S1 state emission. However, the relative magnitudes of the ICT SE band and the BL/SE band of the Chl-a (Fig. 2) demonstrate that the ICT state has an appreciable transition dipole moment, resulting in a stronger electronic coupling to the Chl-a Qy transition. Because the electronic coupling factor is a squared term whereas the overlap integral is linear in the Förster equation, any reduction in spectral overlap is compensated by the large transition dipole of the ICT state, leading to the highly efficient peridinin to Chl-a energy transfer via the S1/ICT states.
Although the results presented here clearly show that the ICT state is involved in the peridinin to Chl-a energy transfer, it raises the question as to the nature of the interaction between the S1 and ICT states. Do these states represent two separate energy donors or is there a strong coupling between S1 and ICT states, which effectively function as a collective S1/ICT state? The observation of the dominant 2.5-ps energy transfer component at all wavelengths points to the latter possibility. A strong polarity-driven coupling between peridinin S1 and ICT states was previously proposed to explain the uniform lifetime of peridinin in solution over a broad range of detection wavelengths (17, 28). As mentioned above, the intrinsic lifetime of peridinin in the PCP complex (≈16 ps) corresponds to a somewhat polar environment, leading to a strong S1–ICT coupling. Thus, the 2.5-ps process should be seen as an energy transfer between a collective S1/ICT state and the Chl-a Qy state. The importance of the ICT state lies in its capability to enhance the dipole moment of the S1 state, resulting in a more efficient energy transfer between peridinin and Chl-a.
The observed energy transfer rates are those of a net energy transfer. There are four peridinins and one Chl-a molecule in the smallest PCP subunit (8), each of which resides in a local environment with most likely a slightly different polarity. Each peridinin also has a different orientation in respect to the Chl-a that is the acceptor of energy from all four peridinins. Consequently, the (2.5 ps)−1 energy transfer rate is an average. Because it is impossible to resolve spectrally the four peridinins in the domain, their energy transfer rates could be resolved only if one or more were radically different. Here, it is tempting to assign the 0.7-ps component to energy transfer from one of the peridinins having a favorable orientation respect to Chl-a. However, the PCP structure provides no evidence for this. Moreover, calculations of a Coulomb coupling between molecules mimicking peridinin and Chl-a in PCP (13) as well as calculations by Carbonera et al. (10) showed that, in the smallest subunit consisting of four peridinins and one Chl-a molecule, three peridinin molecules have a comparable coupling with Chl-a. Each of these is much stronger than that of the fourth peridinin. Therefore, recalling that the 0.7-ps component in all kinetics has smaller amplitude than the 2.5-ps component, the 0.7-ps cannot be assigned to energy transfer from a single best-placed peridinin molecule within a subunit.
The energy transfer pathways within the PCP complex revealed in this work are depicted schematically in Fig. 4. After excitation into the peridinin S2 state, energy transfer to the Chl-a Qx state is observed with an efficiency of ≈25%, suggesting the rate of energy transfer from the S2 state must be of the order of ≈(200 fs)−1 to compete with the intrinsic S2→S1 relaxation rate in peridinin of ≈(50 fs)−1. The remaining 75% of the peridinin S2 population undergoes a relaxation to populate both the S1 and ICT states. Due to a strong S1–ICT coupling, these two states cannot be separated, and they act as a collective S1/ICT state. The S1/ICT state then decays with a (2.5 ps)−1 rate that represents a sum of the rates of an intrinsic (S1/ICT→S0) peridinin relaxation rate and of the S1/ICT→Chl energy transfer. On the basis of known overall efficiency of peridinin to Chl-a energy transfer in the PCP complex (88%), an 84% yield of the S1/ICT energy transfer is calculated, resulting in an energy transfer rate of ≈(3 ps)−1. The remaining 12% of the total peridinin S2 population (16% of the S1 population) undergoes the S1/ICT→S0 relaxation with a rate of ≈(16 ps)−1. In addition, the results presented here revealed a minor 0.7-ps component that can be ascribed to the peridinin to Chl-a energy transfer via higher vibrational levels of the S1/ICT state. Although the origin of the 0.7-ps component cannot be unequivocally assigned, the spectral profile of this component and its presence in the rise of the Chl-a signal at 670 nm makes the assignment to the energy transfer via the S1/ICT vibrational states feasible.
Fig 4.

Scheme of energy levels and energy transfer pathways between peridinin and Chl-a in the PCP complex. Intramolecular relaxation processes are denoted by wavy arrows, whereas the dashed arrow represents the long-lived Chl-a fluorescence. Solid arrows represent energy transfer channels: (S2→Qx channel, 25%; S1/ICT→Qy channel, 63%). The dotted line represents a possible minor energy transfer channel involving higher vibrational levels of the S1/ICT state. Excitation at 535 nm is shown as a double arrow. All processes are labeled by a corresponding time constant. See text for details.
Using near-infrared transient absorption spectroscopy, we showed that a special light-harvesting strategy is used in the PCP complex from the dinoflagellate A. carterae. In other light-harvesting complexes, such as that from higher plants, the S2 carotenoid-Chl energy pathway clearly dominates (6, 29), although a fractional S1 channel using hot vibrational states was recently proposed (24, 25). On the contrary, the energy transfer between peridinin and Chl-a in the PCP complex uses predominantly the lowest excited state of peridinin. This situation is allowed by the higher energy of the peridinin S1 state (≈16,400 cm−1), which enables a substantial overlap of peridinin emission with the Qy absorption band of Chl-a. In addition, the energy transfer efficiency via the lowest excited state of peridinin is enhanced by presence of the ICT state, which has, in contrast to the S1 state, an appreciable dipole moment. Thus, we have identified a new energy transfer pathway involving an ICT state, whose presence in the peridinin excited state manifold and its coupling to the S1 state is controlled by polarity. The PCP complex therefore represents an example of a system where energy transfer pathways and their efficiencies are finely tuned, not only by the protein structure ensuring a proper orientation of the donor and acceptor molecules, but also by adjusting the polarity of the environment. Too low polarity would lead to a long intrinsic S1/ICT peridinin lifetime, resulting in a better competition of energy transfer with the S1/ICT→S0 relaxation, but at a price of a negligible contribution of the ICT state to energy transfer. On the other hand, too high polarity could enhance the contribution of the ICT state, providing a stronger dipole moment of the donor state, but the resulting short intrinsic S1/ICT lifetime (16) would make the energy transfer less competitive with the S1/ICT→S0 relaxation. Thus, to achieve an efficient S1/ICT energy transfer, the polarity of the environment of peridinin molecules in the PCP complex must be perfectly balanced. It is worth noting that such a light-harvesting strategy using an ICT state is likely to play a significant role in the total photosynthetic production on the Earth. A polarity-controlled ICT state in the excited state manifold of carotenoids was found to be characteristic of the presence of a carbonyl group in the carotenoid-conjugated backbone (28). Carbonyl carotenoids, such as peridinin, fucoxanthin, or siphonaxanthin, occur in various taxonomic groups of oceanic photosynthetic organisms (30), which contribute a substantial part of Earth's photosynthetic CO2 fixation. Consequently, the carotenoid to Chl-a energy transfer mechanism involving an ICT state represents an important light-harvesting strategy. This strategy may be an inevitable correlate of using oxygenated carotenoids and the local protein environment to extend absorption to longer wavelengths in the green region of the visible spectrum.
Acknowledgments
We thank Ivo van Stokkum for useful discussions and Frank Sharples for purifying the PCP complex. The work in Lund was supported by grants from the Swedish Research Council, the Knut and Alice Wallenberg Foundation, and the Crafoord Foundation. The work at Macquarie University was supported by Australian Research Council Project Grant A00000264.
Abbreviations
PCP, peridinin–chlorophyll-a–protein
ICT, intramolecular charge transfer
Chl, chlorophyll
BChl, bacteriochlorophyll
BL, bleaching
SE, stimulated emission
ESA, excited state absorption
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Frank H. A. & Cogdell, R. J. (1996) Photochem. Photobiol. 63, 257-264. [DOI] [PubMed] [Google Scholar]
- 2.Christensen R. L. (1999) in Photochemistry of Carotenoids, eds. Frank, H. A., Young, A. J., Britton, G. & Cogdell, R. J. (Kluwer, Dordrecht, The Netherlands), pp. 137–159.
- 3.Förster T. (1965) in Modern Quantum Chemistry, ed. Sinanoglu, O. (Academic, New York), pp. 93–137.
- 4.Macpherson A. & Gillbro, T. (1998) J. Phys. Chem. A 102, 5049-5058. [Google Scholar]
- 5.Ritz T., Damjanović, A., Schulten, K., Zhang, J.-P. & Koyama, Y. (2000) Photosynth. Res. 66, 125-144. [DOI] [PubMed] [Google Scholar]
- 6.van Amerongen H. & van Grondelle, R. (2001) J. Phys. Chem. B 105, 604-617. [Google Scholar]
- 7.Sundström V., Pullerits, T. & van Grondelle, R. (1999) J. Phys. Chem. B 103, 2327-2346. [Google Scholar]
- 8.Hofmann E., Wrench, P., Sharples, F. P., Hiller, R. G., Welte, W. & Diedrichs, K. (1996) Science 272, 1788-1791. [DOI] [PubMed] [Google Scholar]
- 9.Akimoto S., Takaichi, S., Ogata, T., Nishimura, Y., Yamazaki, I. & Mimuro, M. (1996) Chem. Phys. Lett. 260, 147-152. [Google Scholar]
- 10.Carbonera D., Giacometti, G., Segre, U., Hofmann, E. & Hiller, R. G. (1999) J. Phys. Chem. B 103, 6349-6356. [Google Scholar]
- 11.Bautista J. A., Hiller, R. G., Sharples, F. P., Gosztola, D., Wasielewski, M. R. & Frank, H. A. (1999) J. Phys. Chem. A 103, 2267-2273. [Google Scholar]
- 12.Kleima F. J., Hofmann, E., Gobets, B., van Stokkum, I. H. M., van Grondelle, R., Diederichs, K. & van Amerongen, H. (2000) Biophys. J. 78, 344-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Damjanović A., Ritz, T. & Schulten, K. (2000) Biophys. J. 79, 1695-1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krueger B. P., Lampoura, S. S., van Stokkum, I. H. M., Papagiannakis, E., Salverda, J. M., Gradinaru, C. C., Rutkauskas, D., Hiller, R. G. & van Grondelle, R. (2001) Biophys. J. 80, 2843-2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zimmermann J., Linden, P. A., Vaswani, H. M., Hiller, R. G. & Fleming, G. R. (2002) J. Phys. Chem. B 106, 9418-9423. [Google Scholar]
- 16.Bautista J. A., Connors, R. E., Raju, B. B., Hiller, R. G., Sharples, F. P., Gosztola, D., Wasielewski, M. R. & Frank, H. A. (1999) J. Phys. Chem. B 103, 8751-8758. [Google Scholar]
- 17.Zigmantas D., Polívka, T., Yartsev, A., Hiller, R. G. & Sundström, V. (2001) J. Phys. Chem. A 105, 10296-10306. [Google Scholar]
- 18.Weiss C. J. (1972) J. Mol. Spectrosc. 44, 44-80. [Google Scholar]
- 19.Polívka T., Herek, J. L., Zigmantas, D., Åkerlund, H.-E. & Sundström, V. (1999) Proc. Natl. Acad. Sci. USA 96, 4914-4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koka P. & Song, P.-S. (1977) Biochim. Biophys. Acta 495, 220-231. [DOI] [PubMed] [Google Scholar]
- 21.Polívka T., Zigmantas, D., Frank, H. A., Bautista, J. A., Herek, J. L., Koyama, Y., Fujii, R. & Sundström, V. (2001) J. Phys. Chem. B 105, 1072-1080. [Google Scholar]
- 22.Billsten H. H., Zigmantas, D., Sundström, V. & Polívka, T. (2002) Chem. Phys. Lett. 355, 465-470. [Google Scholar]
- 23.de Weerd F. L., van Stokkum, I. H. M. & van Grondelle, R. (2002) Chem. Phys. Lett. 354, 38-43. [Google Scholar]
- 24.Walla P. J., Linden, P. A., Ohta, K. & Fleming, G. R. (2002) J. Phys. Chem. A 106, 1909-1916. [Google Scholar]
- 25.Papagiannakis E., Kennis, J. T. M., van Stokkum, I. H. M., Cogdell, R. J. & van Grondelle, R. (2002) Proc. Natl. Acad. Sci. USA 99, 6017-6022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Polívka T., Zigmantas, D., Sundström, V., Formaggio, E., Cinque, G. & Bassi, R. (2002) Biochemistry 41, 439-450. [DOI] [PubMed] [Google Scholar]
- 27.Polívka T., Zigmantas, D., Herek, J. L., He, Z., Pascher, T., Pullerits, T., Cogdell, R. J., Frank, H. A. & Sundström, V. (2002) J. Phys. Chem. B 106, 11016-11025. [Google Scholar]
- 28.Frank H. A., Bautista, J. A., Josue, J., Pendon, Z., Hiller, R. G., Sharples, F. P., Gosztola, D. & Wasielewski, M. R. (2000) J. Phys. Chem B 104, 4569-4577. [Google Scholar]
- 29.Croce R., Müller, M. G., Bassi, R. & Holzwarth, A. R. (2001) Biophys. J. 80, 901-915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hiller R. G. (1999) in Photochemistry of Carotenoids, eds. Frank, H. A., Young, A. J., Britton, G. & Cogdell, R. J. (Kluwer, Dordrecht, The Netherlands), pp. 81–96.