

Abstract
A chemical reaction network for the regulation of the quinic acid (qa) gene cluster of Neurospora crassa is proposed. An efficient Monte Carlo method for walking through the parameter space of possible chemical reaction networks is developed to identify an ensemble of deterministic kinetics models with rate constants consistent with RNA and protein profiling data. This method was successful in identifying a model ensemble fitting available RNA profiling data on the qa gene cluster.
With genome sequencing projects supplying an almost complete inventory of the building blocks of life, functional genomics is now facing the challenge of “re-assembling the pieces” (1, 2). Time-dependent mRNA (3) and protein profiling (4), protein–protein (5–8) and protein–DNA (9) interaction mapping, and the in vitro reconstruction of reaction networks (10, 11) are providing insight into the topology and kinetics of a living cell's full biochemical and gene regulatory circuitry. For the first time, it is now possible to place a particular biological circuit like those describing carbon metabolism, transcription, cell cycle, or the biological clock in simple eukaryotes in a larger context, and to examine the coupling of these circuits (12).
New tools in computational biology are needed to identify these reaction networks by using well studied subcircuits like those for carbon metabolism, cell cycle, or the biological clock as a launch point into the entire circuit of a living cell. The qa gene cluster of Neurospora crassa and the GAL gene cluster of Saccharomyces cerevisiae in carbon metabolism have served as early paradigms for eukaryotic gene regulation (13, 14) and are prime candidates for taking a genomic perspective on biological circuits. Mechanisms of regulation in the qa and GAL clusters with their transcriptional activator and repressors are also shared with many other regulatory networks. Because of their relative simplicity, they also provide an opportunity to test new genomic approaches to identifying chemical reaction networks or biological circuits that underlie many fundamental biological processes (15). Three opportunities exist now for identifying and refining biological circuits: the accumulation of transcriptional profiling data (3), a growing number of approaches to modeling gene regulation (11, 15–21), and the ability to carry out the in vitro reconstruction of biological circuits with a diversity of emergent properties including bistable (10) and oscillatory activity (11).
However, initially, the profiling data will be scarce and the unknown parameters plentiful. Identification of the parameters in a reaction network is further complicated by the facts that the data are noisy and that our knowledge of the underlying reaction network's topology and of its participating molecular species is incomplete, even in well studied networks like those for the λ-switch, lac operon, trp operon, or GAL cluster. To circumvent these difficulties, we present a statistical modeling approach called the ensemble method of circuit identification, which bases its predictions not on a single poorly parameterized circuit, but rather on a statistical ensemble (22) of “all” such circuit models consistent with existing profiling data. Our approach thus provides quantitative prediction capabilities, and, most importantly, it permits us to guide the design of new experiments, which further constrain the model ensemble, consistent with the profiling data.
In this report, we develop a simple reaction network for the regulation of the qa cluster and use RNA profiling information to identify the network. As described (14, 23, 24), the qa cluster is composed of five structural genes and two regulatory genes, as shown in Fig. 1. The qa cluster is induced by shift to quinic acid as the sole carbon source. Three of the genes (qa-2, qa-3, and qa-4) in the cluster encode enzymes involved in the catabolism of quinic acid, ultimately for entry into the Krebs cycle. One gene, qa-y, encodes a permease allowing quinic acid into the cell, and another gene, qa-x, encodes an unknown function. All seven genes in the cluster are transcriptionally activated by the product of qa-1F, and the activator is repressed by the product of qa-1S. The cluster is glucose- and sucrose-repressed (23). Our model is based on the gene regulation scheme in Fig. 2, which is taken from ref. 23, in which qa-1F activates expression of all genes in the qa cluster and qa-1S represses the activator.
Fig. 1.

Gene organization in the qa cluster of N. crassa, two regulatory genes, and five structural genes.
Fig. 2.

Regulatory gene control. Arrows indicate RNA transcripts controlled by the protein products (thin lines) of the qa-1F activator and the qa-1S repressor. qa-SG is the five structural genes.
Materials and Methods
Strains and Media.
The N. crassa wild-type strain 74–OR23–1A (#987, Fungal Genetics Stock Center, Kansas City, KS) was used in these experiments. RNA was isolated from conidia germinated as shake cultures for 12 h at 25°C on 1.5% sucrose Fries minimal medium (25) and shifted from 0 to 8 h on 0.3% quinic acid Fries medium. All plasmids used to generate probes for RNA hybridization were grown in Escherichia coli strain JM101.
DNA and RNA Isolation.
Plasmid DNA was prepared as in ref. 26. N. crassa total RNA was isolated as in ref. 27.
RNA Hybridization.
Total RNA was fractionated by electrophoresis on agarose gels containing formaldehyde and transferred to Nytran TM, hybridized in 6× SSPE [standard saline phosphate/EDTA (0.18 M NaCl/10 mM phosphate, pH 7.4/1 mM EDTA)]/2× Denhardt's solution/0.1% SDS/50% formamide at 42°C, and rinsed as recommended (Schleicher & Schuell). Six DNA fragments were labeled with 32P (28) from the qa cluster (29) and the histone gene (H3) of N. crassa as a standard (30). Densitometric analysis of autoradiograms was performed on the Molecular Dynamics 300A. Northern blots were stripped in 50% formamide/2× SSPE at 65°C, as recommended (Schleicher & Schuell), and reprobed.
Biological Circuit Model
Chemical reaction networks have been proposed for the λ phage switch (31), signaling networks (15), the cell cycle in S. cerevisiae (32), and carbon metabolism (21, 33). Our model is a chemical reaction network based on a rate equation framework (15), as follows:
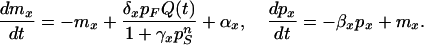 |
Here, mx and px denote the mRNA and protein product concentration of gene x, where x stands for the activator F (≡ qa-1F), the repressor S (≡ qa-1S), and the five structural genes sg (≡ qa-x, qa-y, qa-2, qa-3, and qa-4). All message levels mx are assumed to decay with the same rate constant 1/τ and the model time t, and all mx and px have been rescaled so that all rescaled translation and mRNA decay rate constants are unity; hence, the dimensionless time t in Eq. 1 is related to the physical time t(phys) by t = t(phys)/τ. Possible qa cluster activation by additional, not explicitly included promoter species (14) is modeled by constant basal transcription rates, with rescaled rate coefficients denoted by αx. The rescaled protein decay rates are given by βx. We assume that the concentration of free inducer molecules Q, i.e., quinic acid, decays exponentially according to Q(t) = Q0exp(−κt) or is constant, κ = 0, over time t, where κ is the rescaled decay constant and Q0 is the initial concentration of inducer in the media. The rate of transcription is proportional to the level of inducer and activator protein, with rescaled rate constants denoted by δx. The repressor interacts with the activator, and the effect of the repressor on transcriptional activation is captured in the repressor effects γx. Transcription of the repressor gene is assumed to be unrepressed, i.e., we set γS = 0. The Hill exponent n is a shape parameter controlling the cooperative effect of the repressor on transcription rates. The model does not include posttranscriptional regulation.
Steady-state solutions of Eq. 1 for constant inducer in the media (i.e., κ = 0 and Q = Q0) can be found and local stability analysis performed. For a Hill exponent of n = 1, the steady state is
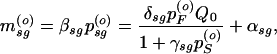 |
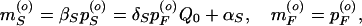 |
where p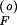 is the unique, positive, stable solution to Ap
is the unique, positive, stable solution to Ap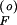 2 + Bp
2 + Bp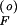 + C = 0, with A = γf1βFδSQ0; B = βF + αSβFγf1 − γf1δSαFQ0 − δFQ0; C = −αF − αFαSγf1; and γf1 = γF/βS.
+ C = 0, with A = γf1βFδSQ0; B = βF + αSβFγf1 − γf1δSαFQ0 − δFQ0; C = −αF − αFαSγf1; and γf1 = γF/βS.
Ensemble Method for Identifying Kinetics Models
As in the study of most biological circuits, for the foreseeable future, biologically realistic models are likely to be parameter rich and data poor, even with the advent of RNA and protein profiling. The approach we take to sidestep this problem is one drawn from statistical mechanics (34) and using Monte Carlo (MC) simulation methods (35–38), which have found increasingly wide application in biology (39). Instead of trying to identify one model, the goal is to identify an ensemble of models consistent with, and constrained by, the available RNA and protein profiling data based on MC simulation techniques. This is termed the ensemble method for circuit identification. We will first give a simplified overview of the method, followed by a full technical description.
Suppose the model and its solution are completely specified by a certain array of parameters that are initially unknown, but are to be constrained by the available experimental data. This array of unknown model parameters is referred to below as the “model parameter vector Θ,” or, for short, the “model Θ.” The basic idea of the ensemble method is to generate an “experimentally constrained” random sample of such Θs, in such a manner that those Θs that yield model predictions “most consistent” with the experimental data are the most likely to be collected into the sample. A model's “degree of consistency” with the experimental data is quantified in terms of a certain figure of merit that measures “how closely” the model's prediction for observed quantities matches the experimental data.
The ensemble simulation starts from an initial Θ that is chosen completely randomly, i.e., without any constraint by experimental data. The simulation then proceeds as a random walk in the “space” of all possible Θ, as follows: From the random walk's current Θ location in the model parameter space, a new Θ is constructed by a certain random “proposal” procedure. If the proposed new Θ improves the figure of merit, it is automatically accepted and becomes the next Θ point visited by the random walk. If the proposed new Θ worsens the figure of merit, the proposal is accepted with a certain probability, Paccept, <1 or rejected with probability 1 − Paccept. If accepted, the proposed new Θ becomes the next point visited by the random walk; if rejected, the next point visited is identical to the current point, i.e., the random walk does not move. Eventually, this random walk settles into a steady state where almost all Θs visited are consistent with the experimental data. A large sample of such Θ vectors, visited by the random walk in steady state, represents the model ensemble.
We now turn to the full technical description of the ensemble method. Let the unknown parameters in the model be denoted by the M-tuple Θ := (Θ1, . . . , ΘM). For the kinetics models explored here, Θ comprises the unknown rate coefficients for all reactions r = 1, 2 , . . . , MR, e.g., in Eq. 1, and all unknown initial concentrations [s]t=0 for all intracellular species s = 1, 2, . . . , MS. Our desired ensemble is then formally described in terms of a probability distribution Q(Θ) on the Θ space of all models.
Next, let Y := (Y1, . . . , YD) denote the D-tuple of all experimental observables, which have been measured in one or a series of ME time-dependent profiling experiments, labeled by e ∈ {1, . . . , ME}, where, in each experiment the concentrations [s] of certain species s are measured at time points t. Different experiments e are distinguished by differing externally controlled and quantitatively known experimental conditions that include, for example, the carbon source and their concentrations, feeding/starvation schedules, choice of measurement time points, and functional presence or absence of certain genes or proteins, as controlled by gene knockout or enzyme inhibition experiments. If, for example, some linear measure of concentration is used, our data vector Y would comprise components
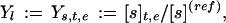 |
with some (known or unknown) reference concentration [s](ref) like, e.g., the maximum [s] level during observation. Alternatively, we may want to use a log-concentration measure (3), Yl := Ys,t,e := ln([s]t,e/[s](ref)). Here l := (s, t, e) and s ∈ S′ labels the MS′ different molecular species, with S′ denoting the subset of all species whose time-dependent concentrations actually have been observed. Typically, S′ is only a subset (generally a small one!) of the set S of all MS participating species in the network. With t ∈ (t1, . . . , tMT) labeling the MT different time points at which concentration measurements have been taken, the dimensionality of our data vector Y is then
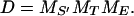 |
Now, let F(Θ) := (F1(Θ), . . . , FD(Θ)) denote the corresponding vector of predicted values for the observables Y in a given model Θ. For the above-described set of observables Ys,t,e, the predicted values Fl(Θ) ≡ Fs,t,e(Θ) are calculated from Θ by solving the circuit's system of rate equations for the rate coefficients and initial conditions comprised in Θ and by then extracting from that solution the linear or log-concentration measure for each observed species s at each observation time point t in each experiment e.
It is reasonable to assume (but not fundamental to our ensemble method!) that the probability distribution P(Y|μ) of the data Y, given their corresponding mean values μ = (μ1, … , μD), is representable as a multivariate Gaussian, without error correlations between different data points Yl. Hence, we will use the following
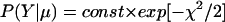 |
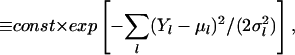 |
with μl and σl denoting the mean and standard deviation of the observable Yl. If multiple realizations of each profiling experiment are performed, then the full variance–covariance matrix can be estimated and used in Eq. 5 in lieu of σ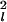 . Based on prior experience with Northern blots, we assume relative standard deviations σl/μl ∼ 0.2 − 0.3 in the simulations reported below. Heteroscedasticity has been reported not to be an issue (40).
. Based on prior experience with Northern blots, we assume relative standard deviations σl/μl ∼ 0.2 − 0.3 in the simulations reported below. Heteroscedasticity has been reported not to be an issue (40).
A given P(Y|μ) does of course not uniquely determine the model ensemble Q(Θ). There is an infinite manifold of Q(Θ) that is consistent with the data distribution P(Y|μ), and we have to make reasonable choices. The simplest choice, which we have adopted here, is to take the likelihood as the posterior (with uniform prior) distribution (41), i.e.,
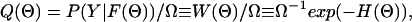 |
with a weight W(Θ) := P(Y|F(Θ)) and normalization factor Ω := ∑ΘW(Θ), where ∑Θ denotes integration over all components of Θ. To emphasize the analogy to the Boltzmann factor in statistical physics, we have also introduced here the analogue of a Hamiltonian or energy function, H(Θ) := −ln W(Θ) (34). More systematic approaches to constructing Q(Θ) can also be used, e.g., a posterior probability derived from Bayesian inference and maximum entropy considerations (41–44). For the present proof-of-principle applications, we will limit ourselves to the choices for P and Q given above.
In standard data-fitting methods, such as maximum likelihood, least-squares fitting, and maximum entropy approaches, one would attempt to construct the correct model by finding a unique Θ that maximizes Q(Θ). Because of the large number of unknown model parameters, the (initial) scarcity of experimental data, and the substantial uncertainties in the data, such approaches are bound to fail in the present context. Our basic philosophy here is that one should not attempt to find a unique Θ, unless it is warranted by the quantity and quality of the underlying data. Rather, one should admit all Θ as possible candidates for the correct model with a probability distribution Q(Θ) that reasonably reflects a Θs degree of consistency with the data. The weight W(Θ) provides a convenient measure of the degree of consistency of the model Θ with the experimental data, and, thus, serves as our figure of merit.
For any ensemble of the general form Q(Θ) := Ω−1W(Θ) with an analytically known or numerically calculable weight function W(Θ) [having a normalization Ω = ∑ΘW(Θ)], we can evaluate the ensemble average of any quantity G(Θ),
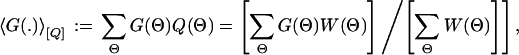 |
as well as, for example, its probability distribution p[G,Q](g): = 〈δ(g − G(.))〉[Q]. This is achieved by a well established MC method from statistical physics (35–38) in which random samples of Θ = (Θ1, . . . ,ΘI), distributed according to Q(Θ) are generated numerically, e.g., by a Metropolis-type (35–38) random walk Markov chain procedure. The desired expectation 〈G(.)〉[Q] is then given, up to controllable statistical sampling errors, by
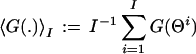 |
over the MC sample, i.e., by the Ergodic Theorem, limI→∞ 〈G(.)〉I = 〈G(.)〉[Q]. Specifically, in our simulations, the basic random updating step in our Markov chain, from a given Θ to the next, Θ+, proceeds as follows: (i) select with equal probability one of the Θ components, Θm, with m ∈ {1, . . . , M}; (ii) propose an update from Θm to Θ′m := Θm + Δm, where Δm is drawn with constant probability from an interval [−Δ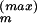 , Δ
, Δ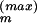 ] with some maximum step width Δ
] with some maximum step width Δ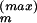 ; (iii) accept the proposed step with the standard Metropolis acceptance probability Paccept(Θ → Θ′) = min[1, Q(Θ′)/Q(Θ)], where Θ′ := (Θ1, . . . , Θ′m, . . . , ΘM). If the proposed step to Θ′ is accepted, set the “next” Θ in the Markov chain Θ+ = Θ′; else, Θ+ = Θ.
; (iii) accept the proposed step with the standard Metropolis acceptance probability Paccept(Θ → Θ′) = min[1, Q(Θ′)/Q(Θ)], where Θ′ := (Θ1, . . . , Θ′m, . . . , ΘM). If the proposed step to Θ′ is accepted, set the “next” Θ in the Markov chain Θ+ = Θ′; else, Θ+ = Θ.
A crucial point here is that only the weight function W(Θ), but not the normalization factor Ω, needs to be evaluated in generating the MC sample, because only ratios Q(Θ′)/Q(Θ) = W(Θ′)/W(Θ) enter into the Metropolis acceptance probability Paccept. Each such updating step does require a completely new solution of the reaction network model to evaluate the new weight W(Θ′) for the proposed new Θ′.
In the following, we will apply the ensemble approach to the kinetics model, Eq. 1, where Θ ≡ (Θ1, . . ., ΘM) comprises (i) the initial concentrations mx,0 and px,0 of the seven mRNA and seven protein species; (ii) the quinic acid initial concentration Q0 and decay rate constant κ; and (iii) the rate coefficients αx, βx, γx, and δx, where the (fixed) γS = 0 is excluded and, for the five structural genes, all five βx are set to the same value βsg, because the sg proteins do not act back on any other species in our model and, hence, βsg does not affect the model predictions for any measured mRNA species. Assuming a fixed Hill exponent n = 1 and a given, fixed mRNA lifetime τ = 60 min, there are, hence, 25 unknown rate constant parameters and 14 initial conditions, i.e., a total of M = 39 Θ-variables, in our model, to be fitted to only D = 42 data points (MS′ = 6 mRNAs × MT = 7 time points × ME = 1 experiment) from Fig. 3.
Fig. 3.

RNA profiles (or Northern blots) for six genes in the qa cluster together with that of the histone (H3) as a control at 30 min, 60 min, 90 min, 2 h, 4 h, 6 h, and 8 h. Sizes of some messages are indicated on the right.
The model Eq. 1 was solved using the fourth-order Runge-Kutta method (45) with time step h = 3.0 min to compute the model solution Fl(Θ) in Eq. 6. For Yl in Eq. 6, we used the linear concentration measures given by the pixel-count data extracted from Fig. 3, with the maximum mRNA level as the reference [s](ref) in Eq. 3 for each measured mRNA species s. At the beginning of the MC random walk, Θ was randomly initialized, with each component Θm drawn from a wide but finite interval Im := [Θ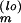 , Θ
, Θ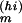 ]. A typical walk in Θ space consisted then of 104 MC “warm-up” steps, to equilibrate the Markov chain, followed by 104-step MC accumulation steps, with all components of Θ and all corresponding solutions for the time-dependent species concentrations sampled after every 102 steps.
]. A typical walk in Θ space consisted then of 104 MC “warm-up” steps, to equilibrate the Markov chain, followed by 104-step MC accumulation steps, with all components of Θ and all corresponding solutions for the time-dependent species concentrations sampled after every 102 steps.
Results
In experimental studies of the mRNA levels of the qa-1F gene, the level of activator mRNA increases for 4 h, decreases slightly by 8 h, and continues to decrease noticeably by 10 h (Fig. 3; ref. 24). At 10 h, the message level is 38% of that at 4 h. The qualitative mRNA dynamics of the qa structural genes is quite similar to the dynamics of qa-1F in Fig. 3. At present, there are no measurements of the mRNA levels of qa-1S. Using the ensemble method described in the previous section, we find that there is a set of models Θ that captures this gene expression dynamic quite well.
The ensemble Q(Θ) represents a complex object in a high-dimensional parameter space. In Fig. 4, we show projections of the ensemble into three arbitrarily chosen 2D Θ planes, displayed as scatter plots of an MC sample. Important interrelations between the parameters can be revealed by such projections. For example, the ensemble appears quite constraining for the rates of protein turnover and induction, as shown in the second and third plane. The basal rates of transcription, shown in the first plane, are much more diffuse and correlated.
Fig. 4.

Projections of an MC sample of Θ, drawn from Q(Θ), into three distinct Θ planes.
In Fig. 5 we show ensemble averages of the numerical solutions of Eq. 1, obtained from the MC sample whose projections are shown in Fig. 4. The dots in Fig. 5 are the experimental data derived from Fig. 3. The shaded areas at each time t are centered around the ensemble averages 〈[s]t,e〉 of the respective species concentrations [s]t,e and comprise 4 ensemble standard deviations of [s]t,e. As Fig. 5 shows, with a few exceptions, these shaded “ensemble” areas cover the experimental data, shown by the dots. Fig. 5 also shows some of the corresponding ensemble predictions for as-yet-unobserved protein time evolutions.
Fig. 5.

Comparison of experimental and ensemble model dynamics of mRNAs of the qa cluster. The dots are the data derived from Fig. 3. The shadowed areas enclose 4 ensemble standard deviations of mx, for x = F, S, sg, centered around the ensemble mean of mx. Also shown are the corresponding ensemble predictions for several protein levels px.
From a microscopic point of view, chemical reactions proceed by a stochastic process involving discrete, random collision events between discrete molecules (or discrete quasimolecular entities such as the gene activator binding sites on a chromosome). A deterministic model like Eq. 1 captures this stochastic dynamics only approximately (46), at the level of statistical averages, thereby neglecting fluctuation effects arising from the discreteness of molecules and molecular collisions. Such fluctuation effects can be important in systems where the total number of molecules of a species is small. For example, it has been shown that fluctuation effects arising in the stochastic dynamics of gene expression (47, 48) may provide an explanation for the phenomenon of phenotypic switching. Binding of a free inducer molecule (i.e., quinic acid in the cell), activator, and repressor to the activator, gene, or repressor, is likely to be a stochastic process subject to substantial fluctuations, because of the small number reactant molecules in the cell (24, 49).
Following ref. 46, we can construct a corresponding stochastic model based on the deterministic model Eq. 1, the circuit in shown Fig. 2, and the parameter estimates provided by the above-described MC sample of model Θ vectors. In this corresponding model, the number of molecules of each species is treated as a stochastically evolving integer, and time is advanced in discrete steps of random lengths, from one collision event to the next, with a time step length distribution determined by the molecular collision rates. The resulting stochastic model has the structure of a discrete-time denumerable Markov chain (50).
Different realizations of the random trajectories of such a stochastic model are shown in Fig. 6. The results in Fig. 6 indicate that the dynamics of the total number of molecules, e.g., for the qa-1F gene product, NmF, behave qualitatively like the experimentally observed and also like the above-described deterministic model dynamics, with maximal expression being reached at ≈4 h. The fact that both the deterministic (Eq. 1) and stochastic models show dynamics in accordance the with experimental data suggests that the gene regulation scheme shown in Fig. 2 is indeed the key molecular mechanism in the gene regulation of the qa cluster.
Fig. 6.

Dynamics of the total number of qa-1F mRNA molecules, obtained by simulations of the stochastic model corresponding to Eq. 1, as described in the text. Different realizations of the stochastic model trajectory generate patterns with maxima located near 4 h in accordance with experimental data in Fig. 3.
Discussion
Simple gene regulation schemes like the one in Fig. 2 are at the core of more complex biological circuits, and they represent a model of how we can think of the functioning of the cell. From such a circuit, a formal model (deterministic or stochastic) can be derived and compared with the temporal dynamics of the RNAs and proteins in the cell. Traditional fitting practices for kinetics models are unlikely to be successful because the models are parameter rich, but the scientist is data poor when the information is derived from profiling experiments. Borrowing from Boltzmann's original ideas in statistical mechanics (34), we are taking the approach of identifying an ensemble of models, rather than trying to find one or a few solutions to an ill conditioned fitting problem. To implement this approach computationally, we initiate a random walk (in particular, the realization of a Markov chain) in the model parameter (Θ) space, guided by some figure of merit that quantifies the deviation of the model from the data. Once this random walk settles into a steady state, typically after a few thousand steps in the parameter space, the ensemble of models is realized by the steady state or stationary walk consistent with the data (Fig. 4). This ensemble (or distribution of fitted models on the parameter space) captures what we know about the biological circuit. This ensemble method of circuit identification allows us to see not only what parameters in the model are well specified (or poorly specified) by providing higher moments (i.e., variances) and (joint) distributions, but it also provides us insights into what are likely to be the most informative new experiments to reduce the uncertainty in our model specification. For example, the basal transcription rates shown in Fig. 4 are poorly constrained by the present experimental data, suggesting the need for additional early-time (<30 min) measurements.
We have successfully used the ensemble method to model profiling data on one of the classic eukaryotic biological circuits, the qa gene cluster. The model Eq. 1 is only the first step for a model of gene regulation in the qa cluster designed to explain existing data. As more profiling data are obtained under a variety of external control conditions, other features could be added to the model, including, for example, coupling to other circuits, such as aromatic amino acid biosynthesis (33, 51). Coupling of such circuits can lead to a richer repertoire of circuit dynamics (15). It has also been argued that translational control may play an important role in the qa gene cluster regulation (14), a feature not included in the current model. The model summarized in Eq. 1 can be extended to study this and other potential complications to yield experimental predictions about possible emergent properties in the biological circuit (15). For example, under some conditions (49, 52), stochastic simulation of this scheme may show oscillatory dynamics or switch-like behavior. This raises the question of whether or not deterministic kinetics models could be used to predict conditions for an oscillatory response of the qa cluster.
Biological circuits such as the qa cluster can be perturbed in a variety of ways. Profiling data will be obtained under varying and/or time-dependent sucrose and QA levels, and in circuits modified by gene knockout and/or enzyme poisoning. By means of a single, joint χ2 function, all such perturbation experiments can be immediately incorporated into the ensemble approach and treated on equal footing, thus leading to systematic refinements and extensions of the circuit model. Extension of the ensemble approach to stochastic reaction kinetics modeling (46) is in principle straightforward. The ensemble method of model identification provides us with a versatile tool, allowing direct inspection of whether a new model “works” in adequately representing the data. A flexible and adaptable fitting tool is key to a detailed understanding of biological circuits because the resulting ensemble models can be used to guide the construction of new costly experiments to extend our understanding of particular real circuits in a genomic context and to maximize the likely information gain from such new experiments.
Abbreviations
MC, Monte Carlo
References
- 1.Venter J. C., Adams, M. D., Myers, E. W., Li, P. W., Mural, R. J., Sutton, G. G., Smith, H. O., Yandell, M., Evans, C. A., Holt, R. A., et al. (2001) Science 291, 1304-1351. [DOI] [PubMed] [Google Scholar]
- 2.International Human Genome Sequencing Consortium (2001) Nature 409, 860-921. [DOI] [PubMed] [Google Scholar]
- 3.DeRisi J. L., Iyer, V. R. & Brown, P. O. (1997) Science 278, 680-686. [DOI] [PubMed] [Google Scholar]
- 4.Gygi S. P., Rist, B., Gerber, S. A., Turecek, F., Gelb, M. H. & Aebersold, R. (1999) Nat. Biotechnol. 17, 994-999. [DOI] [PubMed] [Google Scholar]
- 5.Uetz P. L., Glot, L., Cagney, G., Mansfield, T. A., Judson, R. S., Knight, J. R., Lockshon, D., Narayan, V., Srinivasan, M., Pochart, P., et al. (2000) Nature 403, 623-627. [DOI] [PubMed] [Google Scholar]
- 6.Ito T., Tashiro, K., Muta, S., Ozawa, R., Chiba, T., Nishizawa, M., Yamamoto, K., Kuhara, U. S. & Sakaki, Y. (2000) Proc. Natl. Acad. Sci. USA 97, 1143-1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gavin A.-C., Bosche, M., Krause, R., Grandi, P., Marzioch, M., Bauer, A., Schultz, J., Rick, J. M., Michon, A. M., Cruciat, C. M., et al. (2002) Nature 415, 141-147. [DOI] [PubMed] [Google Scholar]
- 8.Ho Y., Gruhler, A., Heilbut, A., Bader, G. D., Moore, L., Adams, S. L., Millar, A., Taylor, P., Bennett, K., Boutilier, K., et al. (2002) Nature 415, 180-183. [DOI] [PubMed] [Google Scholar]
- 9.Ren B., Robert, F., Wyrick, J. J., Aparicio, O., Jennings, E. G., Simon, I., Zeitlinger, J., Schreiber, J., Hannett, N., Kanin, E., et al. (2000) Science 290, 2306-2309. [DOI] [PubMed] [Google Scholar]
- 10.Gardner T. S., Cantor, C. R. & Collins, J. J. (2000) Nature 403, 339-342. [DOI] [PubMed] [Google Scholar]
- 11.Elowitz M. B. & Leibler, S. (2000) Nature 403, 335-338. [DOI] [PubMed] [Google Scholar]
- 12.Holstege F. C. P., Jennings, E. G., Wyrick, J. J., Lee, T. I., Hengartner, C. J., Green, M. R., Golub, T. R., Lander, E. S. & Young, R. A. (1998) Cell 95, 712-728. [DOI] [PubMed] [Google Scholar]
- 13.Johnston M. (1987) Microbiol. Rev. 51, 458-476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geever R. F., Huiet, L., Baum, J. A., Tyler, B. M., Patel, V. B., Rutledge, B. J., Case, M. E. & Giles, N. H. (1989) J. Mol. Biol. 207, 15-37. [DOI] [PubMed] [Google Scholar]
- 15.Bhalla V. S. & Iyengar, R. (1999) Science 283, 381-387. [DOI] [PubMed] [Google Scholar]
- 16.Wolf D. M. & Eckman, F. H. (1998) J. Theor. Biol. 195, 167-186. [DOI] [PubMed] [Google Scholar]
- 17.Griffith J. S. (1968) J. Theor. Biol. 20, 209-216. [DOI] [PubMed] [Google Scholar]
- 18.McAdams H. M. & Arkin, A. (1998) Annu. Rev. Biophys. Biomol. Struct. 27, 199-224. [DOI] [PubMed] [Google Scholar]
- 19.Hasty J., Pradines, J., Dolnik, M. & Collins, J. J. (2000) Proc. Natl. Acad. Sci. USA 97, 2075-2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holter N. S., Maritan, A., Ciaplak, M., Fedoroff, N. V. & Banavar, R. J. (2001) Proc. Natl. Acad. Sci. USA 98, 1693-1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edwards J. S., Ramakrishna, R. & Palsson, B. O. (2002) Biotechnol. Bioeng. 77, 27-36. [DOI] [PubMed] [Google Scholar]
- 22.Alves R. & Savageau, M. A. (2000) Bioinformatics 16, 534-547. [DOI] [PubMed] [Google Scholar]
- 23.Geever R. F., Baum, J. A., Tyler, B., Case, M. E. & Giles, N. H. (1987) Antonie Leeuwenhoek 53, 343-348. [DOI] [PubMed] [Google Scholar]
- 24.Patel V. P. & Giles, N. H. (1985) Mol. Cell. Biol. 5, 3593-3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davis R. H. & de Serres, F. J. (1970) Methods Enzymol. 17, 79-143. [Google Scholar]
- 26.Ish-Horowicz D. & Burke, J. F. (1981) Nucleic Acids Res. 9, 2989-2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lindegren E. M., Lichens-Park, A., Loros, J. J. & Dunlap, J. C. (1990) Fungal Genet. Newsletter 37, 21. [Google Scholar]
- 28.Feinberg A. & Vogelstein, B. (1984) Anal. Biochem. 137, 266-267. [DOI] [PubMed] [Google Scholar]
- 29.Kelkar H. S., Griffith, J., Case, M. E., Covert, S. F., Hall, R. D., Keith, C. H., Oliver, J. S., Orbach, M. J., Sachs, M. S., Wagner, J. R., et al. (2001) Genetics 157, 979-990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Woudt L. R., Pastink, A., Kempers-Veenstra, A. E., Jansen, A. E. M., Mager, W. H. & Planta, R. J. (1983) Nucleic Acids Res. 11, 5347-5360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gibson M. A. & Bruck, J. (2000) in Computational Modeling of Genetic and Biochemical Networks, eds. Bower, J. M. & Bolouri, H. (MIT Press, Cambridge, MA), pp. 49–71.
- 32.Sveiczer A., Csikasz-Nagy, A., Gyorffy, B., Tyson, J. J. & Novak, B. (2000) Proc. Natl. Acad. Sci. USA 97, 7865-7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Covert M. W., Schilling, C. H. & Palsson, B. O. (2001) J. Theor. Biol. 213, 73-88. [DOI] [PubMed] [Google Scholar]
- 34.Landau L. D. & Lifshitz, E. M., (1980) Statistical Physics (Pergamon, Oxford).
- 35.Landau D. P. & Binder, K., (2000) A Guide to Monte Carlo Simulations in Statistical Physics (Cambridge Univ. Press, Cambridge, U.K.).
- 36.Binder K., (1987) Topics in Current Physics (Springer, Berlin).
- 37.Kalos M. H. & Whitlock, P. A., (1986) Monte Carlo Methods (Wiley, New York).
- 38.Hammersley J. M. & Handscomb, D. C., (1964) Monte Carlo Methods (Methuen, London).
- 39.Thorne J. L., Kishino, H. & Painter, I. S. (1998) Mol. Biol. Evol. 15, 1647-1657. [DOI] [PubMed] [Google Scholar]
- 40.Kerr M. K. & Churchill, G. A. (2001) Proc. Natl. Acad. Sci. USA 98, 8961-8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen M.-H., Shao, Q.-M. & Ibrahim, J. G., (2000) Monte Carlo Methods in Bayesian Computation (Springer, Berlin).
- 42.Jarrell M. & Gubernatis, J. E. (1996) Phys. Rep. 269, 133-195. [Google Scholar]
- 43.Skilling J., (1989) Maximum Entropy and Bayesian Methods (Kluwer Academic, Dordrecht, The Netherlands).
- 44.Erickson G. J. & Smith, C. R., (1988) Maximum Entropy and Bayesian Methods in Science and Engineering (Kluwer Academic, Dordrecht, The Netherlands).
- 45.Press W. H., Teukolsky, S. A., Vetterling, W. T. & Flannery, B. P., (1992) Numerical Recipes (Cambridge Univ. Press, Cambridge, U.K.).
- 46.Gillespie D. T. (1977) J. Phys. Chem. 81, 2340-2361. [Google Scholar]
- 47.McAdams H. M. & Arkin, A. (1997) Proc. Natl. Acad. Sci. USA 94, 814-819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arkin A., Ross, J. & McAdams, H. H. (1998) Genetics 149, 1633-1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kepler T. B. & Elston, T. C. (2001) Biophys. J. 81, 3116-3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kemeny J. G., Snell, J. L. & Knapp, A. W., (1976) Denumerable Markov Chains (Springer, New York).
- 51.Case M. E., Giles, N. H. & Doy, C. H. (1975) Genetics 71, 337-348. [DOI] [PubMed] [Google Scholar]
- 52.Barkai N. & Leibler, S. (2000) Nature 403, 267-268. [DOI] [PubMed] [Google Scholar]