

The addition of an N–H bond across a carbon–carbon double or triple bond is potentially one of the most direct and efficient methods of alkylating an amine. Considerable effort has been expended in the search for transition metal complexes that will catalyze this desirable transformation, and while notable successes have been realized, general solutions to the problem have proven elusive.1 As a consequence, the discovery and development of hydroamination reactions remain important goals.
Relatively few early transition metal complexes have been shown to be hydroamination catalyst precursors. Among these are the class of bis(cyclopentadienyl)zirconium and titanium alkyne hydroamination catalysts studied earlier by our group2 and by Doye’s3,4 (eq 1). In both systems it has been assumed that the critical intermediates are the 16-electron imido complexes Cp2M=NR. The purpose of this communication is to report titanium-based imido complexes5,6 that function as catalyst precursors for both allene and alkyne hydroamination reactions.7–9 A detailed mechanistic investigation has revealed an unexpected cyclopentadienide/amide ligand exchange reaction that transforms the biscyclopentadienyltitanium precursors (Cp2TiLn) into a monocyclopentadienyl titanium(amido) complex (Cp(ArNH)TiLn)10 exhibiting substantially higher reactivity than the Cp2TiLn precursor.
(1).
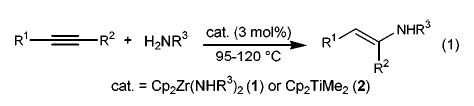
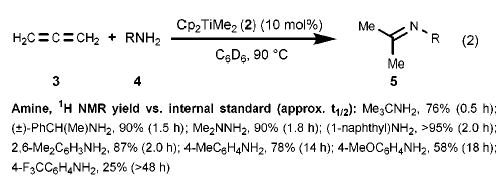
We became interested in catalytic allene hydroamination with the discovery that enantiopure (ebthi)zirconium imido complexes kinetically resolve chiral racemic allenes under stoichiometric conditions (ebthi = ethylene bis(tetrahydroindenyl)).11 Experiments to explore the feasibility of metal-catalyzed allene hydroamination were undertaken with the eventual goal of developing a catalytic variant of this kinetic resolution. A brief screen of catalysts revealed that titanium complexes were more reactive than their zirconium counterparts, and that Cp2TiMe212 was a convenient precursor to an undefined, but competent, catalyst species that hydroaminated 1,2-propadiene in good yields (eq 2). Aryl and alkylamines are both tolerated, as are hydrazines; bulky amines appear to react with greater facility than those with lessened steric demand.
(2).
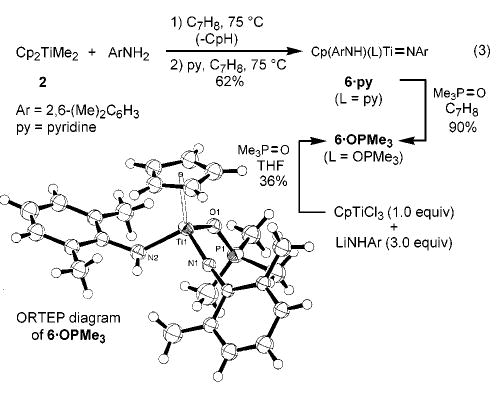
Monitoring these hydroamination reactions by 1H NMR spectroscopy revealed the liberation of cyclopentadiene in amounts that varied with the particular amine being employed.13 In an effort to determine the identity of the true catalyst, stoichiometric reactions of Cp2TiMe2 with 2,6-dimethylaniline (2.0 equiv) were carried out (C6D6, 75 °C, 24 h). This revealed the decomposition of the starting complex, and the formation of four different Cp-containing compounds plus free cyclopentadiene. Thermolysis of this mixture in the presence of added pyridine (1.5 equiv, 75 °C) resulted in formation of the single cyclopentadienyl(amido)-titanium imido complex 6·py in 62% yield versus internal standard (eq 3). We have not established the identity of the products formed prior to pyridine trapping, but it seems likely that pyridine effectively intercepts (or promotes formation of) a monomeric imido species14 from a monomer–dimer equilibrium. Imido complex 6·py, a titanium analogue of a similar imidozirconium complex previously prepared from (η5-C5Me5)ZrCl3, LiNH(2,6-i-Pr2C6H3), and pyridine,15 could be isolated as a brown solid in ~55–60% yield, contaminated with 2,6-dimethylaniline. The difficulties associated with removing amine contaminants from early transition metal imido complexes have been previously noted.16,17 Complex 6·py underwent facile dative ligand exchange with trimethylphosphine oxide to afford complex 6·OPMe3, which was independently prepared from CpTiCl3, LiNH(2,6-Me2C6H3), and Me3PO. An X-ray diffraction study of 6·OPMe3 confirmed the presence of the Ti=N linkage and the bent nature of the amide ligand.
(3).
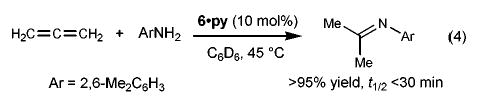
Mono(Cp) complex 6·py catalyzed allene hydroamination at an unusally low temperature (eq 4, 45 °C, t1/2 < 30 min, [allene]0 = 0.40 M, [6·py] = 0.02 M). The mildness of these conditions prompted us to carry out a detailed kinetic analysis of the reaction. Our initial attempts were frustrated by erratic rates presumably caused by the presence of trace amounts of water and oxygen. However, the addition of small quantities of Cp2ZrMe2, a powerful desiccant we have used previously to solve such problems,18 eliminated this difficulty and gave clean pseudo-first-order disappearance of allene in the presence of excess amine.19 Repeating these rate studies in the presence of varying concentrations of catalyst, pyridine, and excess amine revealed that the reaction is zero-order in [amine], inverse first-order in [pyridine], and first-order in [6·py] (eq 5).20 This contrasts the previously studied Cp2Zr(NHR)2 system2 which exhibited an inverse first-order dependence on [amine]. The rate law for the catalysis by 6·py, coupled with the fact that complex 6·py is the only Ti-containing species observed during the catalytic reaction (1H NMR spectroscopy), indicates that addition of ArNH2 to the Ti=N linkage to form CpTi(NHAr)3 is not thermodynamically favored in the titanium system, behavior that also differs from the Cp2-Zr(NHR)2 system.
(4).
(5).
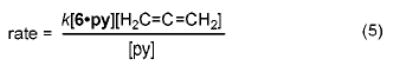
A mechanism consistent with the observed rate law is outlined in Scheme 1. Reversible pyridine dissociation from 6·py generates the Lewis base-free imido complex 7. Formal [2 + 2] cycloaddition with allene21 leads to an azametallacyclobutane (8) that is rapidly protonated by amine to generate tris(amido)Ti complex 9. Elimination expels the enamine product 10 (which tautomerizes to the thermodynamically stable imine 11) and regenerates the catalyst 7.
Scheme 1.
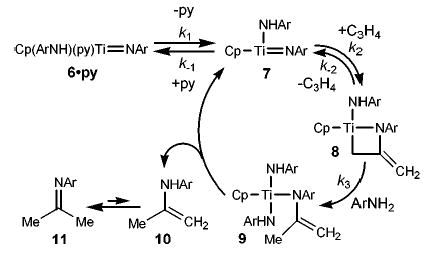
The generally enhanced reactivity of 6·py was apparent when it was compared to that of Cp2TiMe2 (2) in the alkyne hydroamination reaction (eq 6). Complex 6·py (3 mol %) rapidly catalyzed the addition of 2,6-dimethylaniline to diphenylacetylene (75 °C, t1/2 < 15 min, [alkyne]0 = 0.43 M, [6·py] = 0.01 M), while complex 2, under identical conditions, yielded no enamine product after 12 h. In accord with Doye’s observations,3 hydroamination using 2 was achieved only after 3 d at 105 °C; similarly, the use of Cp2Zr(NHAr)2 (1) required 13 d at 120 °C.2
(6).
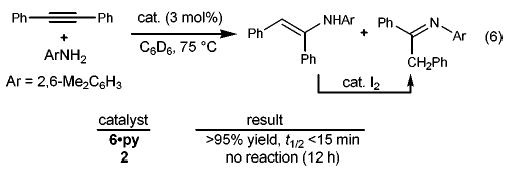
In preliminary experiments we have also utilized imido complex 6·py in the more challenging task of hydroaminating substituted allenes. The catalyzed additions of 2,6-dimethylaniline to 1-phenyl-1,2-propadiene (12), 1,2-nonadiene (13), and 1,2-cyclononadiene (14) proceed to give imines 15–17 in good isolated yields (eq 7).22 Complete regiocontrol is observed in hydroamination of substituted allenes, a direct consequence of the regiochemistry of the [2 + 2] cycloaddition that places the exocyclic olefin β to the metal center in the product metallacycle (8). This regiochemistry is complementary to palladium-catalyzed intermolecular allene hydroamination reactions that afford allylic amine products.7,8
(7).
In summary, we have found that Cp2TiMe2 functions as a catalyst precursor for allene hydroamination. However, mechanistic studies establish that an unusual ligand exchange reaction results in loss of a cyclopentadienyl ligand to generate the highly active monocyclopentadienyltitanium catalyst precursor 6·py. We assume that the same mono-Cp complex is formed in alkyne hydroamination, but its presence in low concentration requires the use of higher temperatures. Accordingly, more rapid alkyne hydroamination is also achieved by directly charging the system with 6·py. Future work will focus on understanding the cause of this Cp-amido ligand exchange and developing highly active, general hydroamination catalysts, as well as asymmetric variants.
Supplementary Material
Acknowledgments
This paper is dedicated to Professor David Evans on the occasion of his 60th birthday. This research was supported by the National Institutes of Health (Grant no. GM-25459 to R.G.B. and a NRSA postdoctoral fellowship to J.S.J.). The structural study of 6·OPMe3 was carried out by Drs. A. G. Oliver and F. J. Hollander of the U.C. Berkeley College of Chemistry X-ray Diffraction Facility (CHEXRAY). We thank Professor R. A. Anderson, Professor J. Arnold, D. M. Tellers, and J. S. Yeston for helpful discussions and experimental assistance. The Center for New Directions in Organic Synthesis is supported by Bristol-Myers Squibb as Sponsoring Member.
Footnotes
Supporting Information Available: Details of kinetic analysis with relevant plots, experimental procedures, characterization data for new compounds, and X-ray diffraction data for 6·OPMe3 (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Müller TE, Beller M. Chem Rev. 1998;98:675. doi: 10.1021/cr960433d. [DOI] [PubMed] [Google Scholar]
- 2.Walsh PJ, Baranger AM, Bergman RG. J Am Chem Soc. 1992;114:1708. [Google Scholar]
- 3.Haak E, Bytschkov I, Doye S. Angew Chem, Int Ed. 1999;38:3389. [PubMed] [Google Scholar]
- 4.Haak E, Siebeneicher H, Doye S. Org Lett. 2000;2:1935. doi: 10.1021/ol006011e. [DOI] [PubMed] [Google Scholar]
- 5.Mountford P. Chem Commun. 1997:2127. [Google Scholar]
- 6.Wigley DE. Prog Inorg Chem. 1994;42:239. [Google Scholar]
- 7.Besson L, Goré J, Cazes B. Tetrahedron Lett. 1995;36:3857. [Google Scholar]
- 8.Al-Masum M, Meguro M, Yamamoto Y. Tetrahedron Lett. 1997;38:6071. [Google Scholar]
- 9.Arredondo VM, McDonald FE, Marks TJ. J Am Chem Soc. 1998;120:4871. [Google Scholar]
- 10.McGrane PL, Jensen M, Livinghouse T. J Am Chem Soc. 1992;114:5459. [Google Scholar]
- 11.Sweeney ZK, Salsman JL, Andersen RA, Bergman RG. Angew Chem, Int Ed. 2000;39:2339. doi: 10.1002/1521-3773(20000703)39:13<2339::aid-anie2339>3.0.co;2-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sieibeneicher H, Doye S. J Prakt Chem. 2000;342:102. [Google Scholar]
- 13.Fuhrhop JH. Tetrahedron Lett. 1969:3205. doi: 10.1016/s0040-4039(01)88388-x. [DOI] [PubMed] [Google Scholar]
- 14.Bennett JL, Wolczanski PT. J Am Chem Soc. 1994;116:2179. [Google Scholar]
- 15.Bai YN, Roesky HW, Noltemeyer M, Witt M. Chem Ber. 1992;125:825. [Google Scholar]
- 16.Arney DJ, Bruck MA, Huber SR, Wigley DE. Inorg Chem. 1992;31:3749. [Google Scholar]
- 17.Blake AJ, Collier PE, Dunn SC, Li WS, Mountford P, Shishkin OV. J Chem Soc, Dalton Trans. 1997:1549. [Google Scholar]
- 18.Proulx G, Bergman RG. Organometallics. 1996;15:684. [Google Scholar]
- 19.Control experiments demonstrate that Cp2ZrMe2 is not catalytically competent under the reaction conditions employed.
- 20.Details of the reaction kinetics may be found in the Supporting Information.
- 21.Bashall A, Collier PE, Gade LH, McPartlin M, Mountford P, Trösch DJM. Chem Commun. 1998:2555. [Google Scholar]
- 22.Attempts to hydroaminate acyclic 1,3-disubstituted allenes, heteroatom-substituted allenes, or unfunctionalized alkenes have to date been unsuccessful.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.