

Abstract
Targeted disruption of the Rel/NF-κB family members NF-κB2, encoding p100/p52, and RelB in mice results in anatomical defects of secondary lymphoid tissues. Here, we report that development of Peyer’s patch (PP)-organizing centers is impaired in both NF-κB2- and RelB-deficient animals. IL-7-induced expression of lymphotoxin (LT) in intestinal cells, a crucial step in PP development, is not impaired in RelB-deficient embryos. LTβ receptor (LTβR)-deficient mice also lack PPs, and we demonstrate that LTβR signaling induces p52–RelB and classical p50–RelA heterodimers, while tumor necrosis factor (TNF) activates only RelA. LTβR-induced binding of p52–RelB requires the degradation of the inhibitory p52 precursor, p100, which is mediated by the NF-κB-inducing kinase (NIK) and the IκB kinase (IKK) complex subunit IKKα, but not IKKβ or IKKγ. Activation of RelA requires all three IKK subunits, but is independent of NIK. Finally, we show that TNF increases p100 levels, resulting in the specific inhibition of RelB DNA binding via the C-terminus of p100. Our data indicate an important role of p52–RelB heterodimers in lymphoid organ development downstream of LTβR, NIK and IKKα.
Keywords: aly/IκB kinase/NF-κB/p100 processing/secondary lymphoid organs
Introduction
The transcription factor NF-κB plays a pivotal role in immune responses, inflammation, the regulation of apoptosis and in cancer (Ghosh et al., 1998; Barkett and Gilmore, 1999; Hatada et al., 2000; Karin and Delhase, 2000; Karin and Lin, 2002; Karin et al., 2002). Five members of this family have been identified in vertebrates: NF-κB1 (encoding the precursor molecule p105 and the processed form p50), NF-κB2 (encoding the precursor p100 and the processed form p52), RelA (p65), RelB and c-Rel. In resting cells, Rel/NF-κB proteins associate with IκBs and are retained in the cytoplasm as inactive forms. A wide range of stimuli activate the IκB kinase (IKK) complex, which consists of two catalytic (IKKα and IKKβ) and one regulatory (IKKγ/NEMO) subunit, causing phosphorylation and ubiquitin-dependent degradation of the IκBs. Similarly, the precursor molecules p100 and p105 also sequester NF-κB in the cytoplasm via their C-terminal ankyrin repeats. Degradation of the IκBs and the p100/p105 precursors results in the nuclear translocation of Rel/NF-κB complexes and the regulation of κB target genes (Pahl, 1999; Karin and Ben-Neriah, 2000).
The ‘classical’ NF-κB activity consists of p50–RelA heterodimers, other possible homo- and heterodimeric complexes can occur depending on cell type and activation status. RelB alone does not bind to DNA, but must dimerize with p50 or p52 to form transcriptional activators. In the mouse, high levels of RelB expression are restricted to specific regions of lymphoid organs. Interestingly, the basal κB-binding activity in thymus and spleen largely consists of p50–RelB and p52–RelB heterodimers, suggesting a role of RelB in the constitutive expression of κB-regulated genes in these tissues (Ryseck et al., 1996). Rel/NF-κB proteins have essential and distinct roles in development and function of the immune system (Attar et al., 1997; Gerondakis et al., 1999). RelB-deficient mice display a complex phenotype including multi-organ inflammation, splenomegaly, lack of lymph nodes and multi-focal defects in immune responses (Burkly et al., 1995; Weih et al., 1995, 1997). NF-κB1-deficient mice have specific B cell defects, but do not show major alterations in the architecture of lymphoid organs (Sha et al., 1995; Gerondakis et al., 1999). On the other hand, NF-κB2- and RelB-deficient mice have disorganized B and T cell areas, lack germinal centers (GCs) and splenic marginal zone structures, and show strongly reduced expression of homing chemokines (Caamaño et al., 1998; Franzoso et al., 1998; Poljak et al., 1999; Weih et al., 2001).
The histological organization of Peyer’s patches (PPs) distributed along the intestinal tract resembles other peripheral lymphoid organs, such as lymph nodes and spleen, with specialized structures like GCs, distinct interfollicular T cell areas and high endothelial venules (HEVs). Based on their anatomical location and their contribution to IgA production, PPs form the first front of mucosal immunity and play an important role in gastrointestinal immune defense (Griebel and Hein, 1996; Debard et al., 1999). The earliest marker for developing PPs is the appearance of VCAM-1+ PP organizing centers, which can be detected by whole-mount immunohistochemistry at ∼E15.5 of mouse embryonic development (Adachi et al., 1997).
Recently, it was reported that IL-7Rα signaling through Jak3 in CD45+CD3– cells of the embryonic intestine is required for the production of LTα/β heterotrimers, resulting in VCAM-1 and ICAM-1 expression by LTβR-positive stromal cells (Nishikawa et al., 1998; Yoshida et al., 1999). Mice with targeted disruptions of genes encoding LTα, LTβ or LTβR, which lack PPs and peripheral lymph nodes (Debard et al., 1999; Fu and Chaplin, 1999; Matsumoto, 1999) support this finding. In contrast to LTβR signaling, activation of the p55 TNFR-I is not essential for PP organogenesis, but rather determines the cellular and structural organization of B cell follicles in secondary lymphoid tissues, whereas signaling through the p75 TNFR-II is completely dispensable for lymphoid organ development (Pasparakis et al., 1997; Matsumoto, 1999).
The importance of the LTβR signaling pathway for PP and lymph node development is also shown in mice with a mutant NF-κB-inducing kinase (NIK) (Miyawaki et al., 1994; Shinkura et al., 1999), which is essential for LTβR, but dispensable for TNFR signaling to NF-κB (Matsushima et al., 2001; Yin et al., 2001). NIK was shown to phosphorylate and activate IKKα (Ling et al., 1998) and neither alymphoplasia (aly/aly) mice, which carry a point mutation resulting in an amino acid substitution in the C-terminal interaction domain of NIK (Shinkura et al., 1999), nor IKKα-deficient or IKKαAA knockin animals (NIK phosphorylation sites replaced by alanines) develop VCAM-1+ PP organizing centers (Miyawaki et al., 1994; Matsushima et al., 2001; Senftleben et al., 2001).
In the present study, we analyzed the role of RelB and its heterodimerization partners p50 and p52 during the early steps of PP development, focusing on LT and TNF signaling pathways. We show that activation of LTβR triggers processing of the inhibitory p100 precursor to p52 dependent on NIK and IKKα resulting in the induction of p52–RelB heterodimers, whereas TNF signaling inhibits κB binding of RelB via the increased production of p100. Our observations suggest an important role for p52–RelB heterodimers in PP organogenesis downstream of LTβR, NIK and IKKα.
Results
Lack of PPs in relB–/– and nfkb2–/– mice
Histological examination of adult mice revealed that RelB is required for PP development. Whereas wild-type mice had several easily detectable PPs, serial sections of Swiss roles of the small intestine did not reveal any histological evidence of rudimentary PPs in relB–/– mice. While nfkb1–/– mice had small PPs with a poorly developed microarchitecture, nfkb2–/– mice also lacked PPs and only occasionally had lymphoid aggregates in the small intestine (data not shown). To examine whether PP development is blocked at an early stage in nfkb2–/– and relB–/– mice, we stained whole intestines from newborn mice for VCAM-1. Figure 1 shows that VCAM-1+ PP organizing centers formed normally in wild-type (Figure 1A) and nfkb1–/– mice (Figure 1B), but were absent in nfkb2–/– (Figure 1C) and relB–/– animals (Figure 1D). Thus, both p52/p100 and RelB are essential for the development of PPs, whereas the p50 subunit of NF-κB plays only a minor role in this process.

Fig. 1. Lack of VCAM-1-positive cell clusters in nfkb2–/– and relB–/– mice. Whole-mount anti-VCAM-1 immunohistochemical staining of intestines from newborn wild-type (A), nfkb1–/– (B), nfkb2–/– (C) and relB–/– mice (D). Arrowheads mark VCAM-1-positive spots in wild-type and nfkb1–/– mice. Ce, cecum. Immunohistochemical detection of RelB (brown staining) in developing PPs from wild-type E16.5 (E) and E18.5 (F) embryos. (G) Staining of adult wild-type PP with anti-RelB antibody revealed high expression in the interfollicular region (IFR), the subepithelial dome (SED) and the follicle-associated epithelium (FAE). Reduced numbers of RelB-positive cells were also found in GCs. (H) RelB is expressed in stromal cells of PPs. Immuno histochemical detection of RelB in a PP from a lethally irradiated wild-type mouse reconstituted with relB–/– bone marrow. All sections were counterstained with hematoxylin. Original magnifications: (E and F), 40×; (G), 10×; (H), 20×.
Analysis of RelB expression in PPs
Immunohistochemical analysis of intestine sections from wild-type E16.5 (Figure 1E) and E18.5 embryos (Figure 1F) revealed RelB expression in the developing PP anlage. Fully developed PPs from adult wild-type mice showed strong RelB expression in the interfollicular region (IFR), the subepithelial dome (SED) and the follicle-associated epithelium (FAE). Fewer RelB+ cells were also observed in follicles and GCs (Figure 1G). The analysis of lethally irradiated wild-type mice after adoptive transfer of relB–/– bone marrow revealed that RelB in hematopoietic cells was not required for the maintenance of PP structures and that RelB was also expressed in stromal cells of PPs. While RelB levels were clearly reduced in the IFRs, which predominantly harbor T cells and dendritic cells, RelB+ cells were still detected throughout the chimeric PPs, in particular in the FAE close to the follicle-associated crypt (Figure 1H). Adoptive transfers of wild-type fetal liver or bone marrow cells failed to restore PPs in irradiated newborn or adult relB–/– mice, respectively (data not shown). Together, these results suggest that RelB in non-hematopoietic cells is required for normal PP development.
Normal IL-7-induced LTα expression in relB–/– embryonic intestinal cells
Developing PPs can be detected as early as E15.5 d.p.c. (Adachi et al., 1997) and RT–PCR analysis of intestines revealed RelB mRNA expression in wild-type E14.5 embryos with increasing levels at E16.5 and E18.5, while no expression was detected in relB–/– mice (Figure 2A). NF-κB2 mRNA was also expressed in E14.5 embryonic intestines (data not shown). The induction of LTα/β heterotrimers downstream of the IL-7Rα is crucial for the expression of VCAM-1 and ICAM-1 by LTβR-positive stromal cells in the developing PP (Yoshida et al., 1999). Analysis of various embryonic stages did not reveal significant defects in mRNA levels of IL-7 and the IL-7Rα chain or LTβR and TNFR-I in relB–/– intestine compared with wild-type littermates (data not shown). LTα and LTβ expression was examined in intestinal cell cultures from relB+/+ and relB–/– E16.5 embryos. IL-7 treatment strongly upregulated LTα mRNA levels in these cultures whereas LTβ expression was only slightly induced. However, no significant difference was observed between relB–/– mice and wild-type controls (Figure 2B). These data indicate the presence of normal numbers of CD45+CD3–IL-7Rα+ cells and an intact pathway upstream of LTβR in relB–/– embryonic intestine.
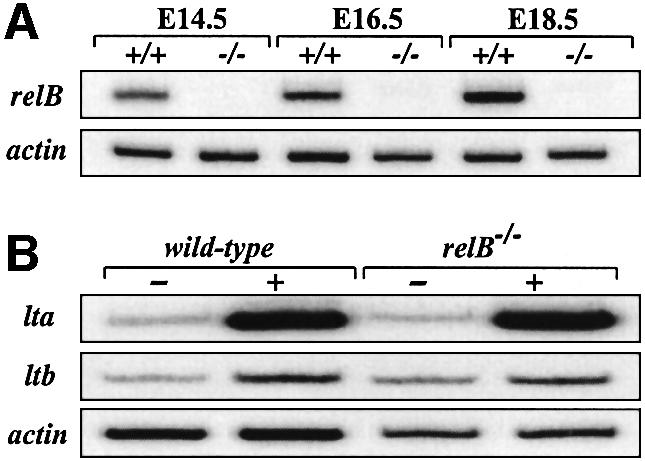
Fig. 2. (A) RT–PCR analysis of relB mRNA levels in intestine from wild-type (+/+) and RelB-deficient (–/–) embryos at E14.5, E16.5 and E18.5 d.p.c. (B) IL-7-induced expression of lta and ltb mRNA in intestine from relB+/+ and relB–/– E16.5 embryos. Intestinal cell cultures were either induced with IL-7 (+) or left untreated (–). After 24 h, total RNA was prepared and analyzed by RT–PCR. Expression of β-actin in (A) and (B) is shown as a control.
LTβR, but not TNFR, signaling induces RelB complexes independent of RelA
Using different primary and established mouse fibroblast lines as a model system for signaling events in stromal cells during early PP development, we examined whether the activation of LTβR, as compared with TNFR, results in the induction of RelB complexes. TNF treatment resulted in strong NF-κB induction after 20 min (complex I), which was maximal at 1 h. In contrast, the kinetics of NF-κB activation after LTβR triggering by the agonistic anti-LTβR monoclonal antibody (mAb) AC.H6 (Rennert et al., 1998) were significantly slower, reaching maximal levels only after 4–8 h (Figure 3A). In addition, LTβR signaling induced a faster migrating heterodimeric complex (II) at 4 and 8 h time points whereas the κB-binding pattern after TNF treatment was rather constant. Dissection of the different Rel/NF-κB complexes in supershift experiments revealed that TNF almost exclusively induced binding of RelA (complex I), whereas LTβR signaling resulted in the activation of both RelA and p52–RelB heterodimers (complex I and II). Activation of LTβR also increased binding of complex III, which consisted of p50–p50 homodimers (Figure 3B). We only observed weak induction of c-Rel complexes in cells treated with anti-LTβR mAb although we cannot rule out the possibility that they did not bind efficiently under our experimental conditions.
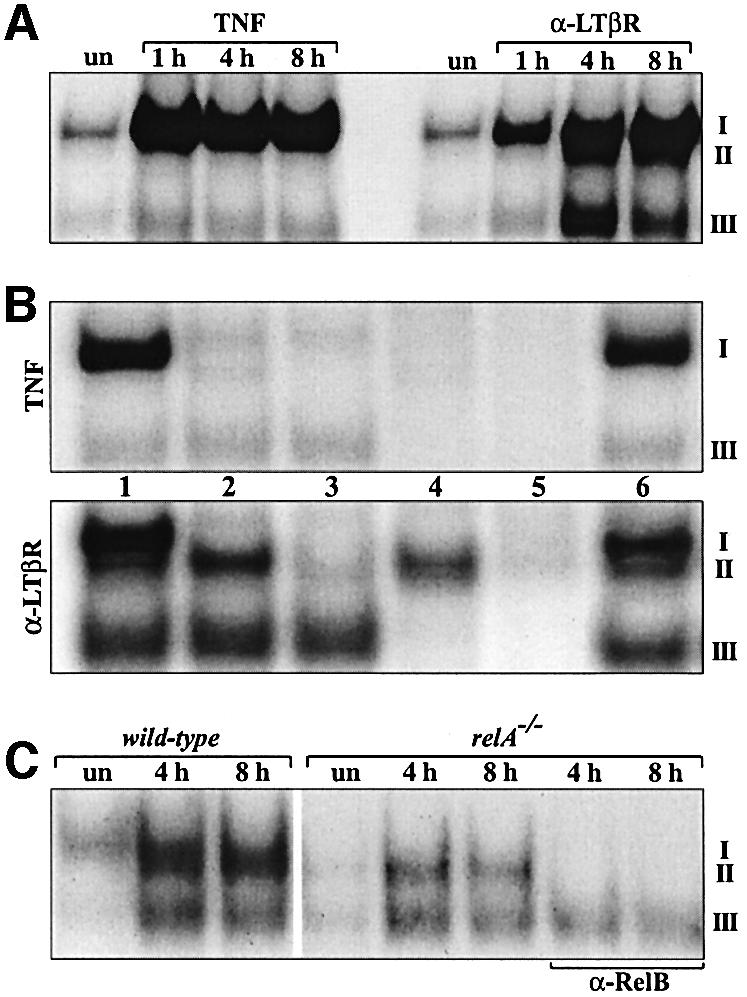
Fig. 3. Induction of RelA complexes and p52–RelB heterodimers by TNFR and LTβR signaling, respectively. (A) Wild-type fibroblasts were either left untreated (un) or treated for 1, 4 and 8 h with TNF or agonistic anti-LTβR mAb and nuclear extracts were analyzed in EMSAs. (B) Extracts from 8 h TNF- or anti-LTβR-treated fibroblasts were used to identify nuclear complexes using following Abs: lane 1, pre-immune serum (p.i.); lane 2, α-RelA; lane 3, α-RelA + α-RelB; lane 4, α-RelA + α-p50; lane 5, α-RelA + α-p50 + α-p52; lane 6, α-cRel. (C) RelA is not essential for the induction of RelB complexes downstream of LTβR. Wild-type and relA–/– fibroblasts were left untreated (un) or were treated for 4 and 8 h with anti-LTβR mAb. RelB-specific Abs were used to confirm the identity of complex II.
To substantiate the results from Figure 3A and B, we performed additional supershift experiments with nuclear extracts from relB–/–, nfkb1–/– and nfkb2–/– mouse embryonic fibroblasts (MEFs) treated with TNF or stimulated with anti-LTβR mAb (see Supplementary figure 1, available at The EMBO Journal Online). In summary, while complex I was not affected, complex II was absent from relB–/– and nfkb2–/– fibroblasts, but still induced by anti-LTβR mAb treatment in nfkb1–/– cells. The data further show that LTβR-induced binding of p50–RelA (complex I) was abolished in nfkb1–/– fibroblasts. Inter estingly, in nfkb1–/– cells, TNF-induced complex I consisted predominantly of p52–RelA heterodimers. Moreover, co-immunoprecipitation experiments provide physical evidence for the formation of p52–RelB heterodimers in response to LTβR signaling (see Supplementary figure 2).
Binding of p52–RelB heterodimers in anti-LTβR-stimulated fibroblasts was preceded by the induction of RelA complexes. Since it was reported that relB gene transcription is regulated by NF-κB (Bren et al., 2001), we analyzed LTβR-mediated activation of NF-κB complexes in relA–/– fibroblasts. Figure 3C shows that while formation of complex I was abolished in relA–/– cells, RelB heterodimers (complex II) were still induced. Together, these data indicate that activation of LTβR, in contrast to TNFR, results in the specific induction of p52–RelB heterodimers and that RelA is not absolutely required for the induction of RelB complexes downstream of LTβR.
LTβR-induced binding of p52–RelB heterodimers requires IKKα and IKKβ, but not IKKγ
To asses which subunits of the IKK complex are involved in LTβR-mediated induction of p52–RelB heterodimers, we compared TNF- and anti-LTβR-stimulated activation of Rel/NF-κB complexes in IKKα-, IKKβ- and IKKγ-deficient fibroblasts. Figure 4A shows that IKKα was absolutely required for LTβR-mediated induction of complex II, but dispensable for TNF-induced binding of NF-κB. The lack of IKKα also resulted in reduced, but still detectable, binding of p50–RelA heterodimers (complex I) after LTβR signaling. IKKβ-deficient fibroblasts also showed an almost complete loss of complex II and strongly reduced binding of complex I. In contrast, LTβR signaling still induced binding of p52–RelB (complex II) and p50–p50 (complex III) in IKKγ-deficient fibroblasts while κB binding was abolished in TNF-treated cells (Figure 4A and B). Quantification of complex II in anti-LTβR-treated IKKγ-deficient cells revealed an ∼3-fold reduction compared with wild-type controls (data not shown).
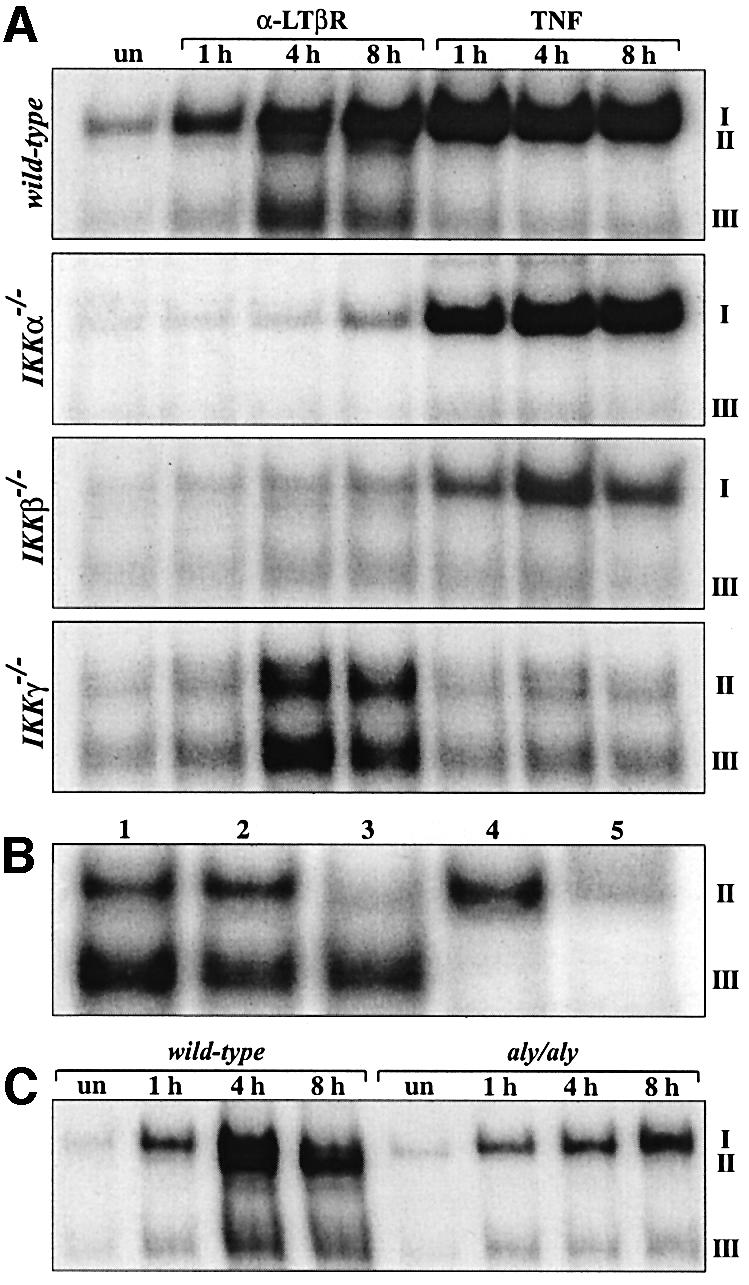
Fig. 4. Analysis of κB binding after LTβR and TNFR signaling in IKK-deficient and aly/aly fibroblasts. (A) IKKα-, IKKβ- and IKKγ-deficient fibroblasts were either left untreated (un) or treated for 1, 4 and 8 h with anti-LTβR mAb or TNF, and nuclear extracts were analyzed in EMSAs. (B) LTβR signaling induces p52–RelB complexes independent of IKKγ. The identity of complexes in nuclear extracts from IKKγ-deficient fibroblasts treated for 8 h with anti-LTβR mAb was determined with the following Abs: lane 1, p.i. serum; lane 2, α-RelA; lane 3, α-RelA + α-RelB; lane 4, α-p50; lane 5, α-p50 + α-p52. (C) NIK is required for the induction of p52–RelB heterodimers. MEFs from wild-type and aly/aly mice were treated for 1, 4 and 8 h with anti-LTβR mAb and nuclear extracts were analyzed for κB binding.
To address the role of NIK in the induction of RelB complexes, we compared MEFs from wild-type and aly/aly mice. Similar to IKKα-deficient cells, aly/aly MEFs completely lacked binding of p52–RelB heterodimers (complex II), whereas the anti-LTβR-induced binding of p50–RelA (complex I) was not reduced (Figure 4C). Thus, binding of p52–RelB heterodimers downstream of LTβR requires NIK, IKKα and IKKβ, but is independent of IKKγ.
IKKα and NIK, but not IKKβ or IKKγ, regulate p100 processing downstream of LTβR
Since LTβR-induced binding of p52–RelB heterodimers may be regulated by the generation of p52 from its precursor molecule p100, we examined p100, p52 and RelB protein levels in wild-type and IKK-deficient fibroblasts that were stimulated with anti-LTβR mAb for different time periods. Whereas IKKα was absolutely required for LTβR-induced processing of p100 to p52, IKKβ was required for normal p100/p52 basal levels, but dispensable for the degradation of the p100 precursor (Figure 5A). Consistent with the results from the electrophoretic mobility shift assays (EMSAs), processing of p100 and accumulation of p52 still occurred in fibroblasts lacking IKKγ although with slower kinetics compared with wild-type cells. Interestingly, the kinetics of p100 processing, the generation of p52 and the increase in RelB levels perfectly coincided with the induced p52–RelB binding (see Figure 3A for 4 and 8 h time points). Similar to p52, basal RelB protein levels depended on IKKα and IKKβ, but were unaffected by the lack of IKKγ (Figure 5A). p100 processing and p52 accumulation downstream of LTβR also required NIK, whereas RelB protein levels were normal in aly/aly MEFs (Figure 5B).

Fig. 5. p100 processing in wild-type, IKK-deficient and aly/aly fibroblasts upon LTβR signaling. Western blot analysis of p100, p52 and RelB protein levels in whole-cell extracts from wild-type, IKKα-, IKKβ- and IKKγ-deficient fibroblasts (A) as well as from wild-type and aly/aly MEFs (B) after stimulation with anti-LTβR mAb for 1, 4 and 8 h. β-actin protein levels are shown as a loading control. *, unspecific band.
TNF induces p100 and RelB levels and promotes the formation of p100–RelB complexes
The C-terminal ankyrin repeat domain of p100, also called IκBδ, can function as a potent inhibitor of RelB complexes (Dobrzanski et al., 1995; Solan et al., 2002). To examine whether the lack of TNF-induced RelB DNA binding (see Figure 3B) is due to the lack of p100 degradation and/or insufficient production of p52 that can heterodimerize with RelB, we analyzed p100 and p52 protein levels in extracts from TNF-treated wild-type fibroblasts. TNF induction resulted in a strong increase of the p100 precursor in both cytoplasm and nucleus. TNF also markedly increased cytoplasmic and nuclear RelB, but had very little effect on p52 levels (Figure 6A). Immunofluorescent staining of fibroblasts with an Ab specific for the C-terminus of p100 confirmed the increase in cytoplasmic and nuclear p100 levels upon TNF stimulation (Figure 6B). To examine whether RelB and p100 interact in vivo immunoprecipitations were performed. Reciprocal experiments with RelB- and p100-specific Abs showed that endogenous RelB was bound to p100 in TNF-stimulated fibroblasts (Figure 6C). This interaction was also observed when nuclear fractions instead of whole-cell extracts were used, but it did not occur in anti-LTβR-treated cells (Supplementary figure 2; data not shown).

Fig. 6. TNF induction of fibroblasts results in increased p100 levels in both cytoplasm and nucleus. (A) Western blot analysis of p100, p52 and RelB levels in cytoplasmic and nuclear extracts from wild-type fibroblasts that were treated with TNF for the indicated time points. Quality of fractions was tested with Abs against cytoplasmic LDH or nuclear c-Jun proteins. XT, extract; *, unspecific band. (B) Immunofluorescent (FITC) staining of untreated (a) and TNF-induced (b) wild-type fibroblasts with an Ab specific for the C-terminus of p100. Staining of p100-deficient fibroblasts is shown as a negative control (c). The inset shows nuclei stained with DAPI. (d) DAPI staining of the TNF-induced fibroblasts from (b). (C) RelB and p100 interact in TNF-simulated fibroblasts. Fibroblasts were induced with TNF and cells were lysed under native conditions. Immunoprecipitations (IP) were carried out with (+) or without (–) lysates using either p100- or RelB-specific Abs. The precipitated material was separated by SDS–PAGE and analyzed in western blots (WB) for p100 and RelB levels. Ig, immunoglobulins.
The C-terminal domain of p100 represses RelB DNA binding downstream of TNFR
RelB requires p50 or p52 as a dimerization partner to bind to DNA. We therefore analyzed whether the limited availability of the p52 subunit in TNF-treated fibroblasts was responsible for the lack of nuclear RelB DNA binding. Fibroblasts that were transfected with a p52 or p50 expression plasmid and stimulated with TNF showed a strong increase of p52 and p50 homodimer binding, respectively. However, no RelB heterodimers were detected, indicating that κB binding of RelB is blocked despite sufficient levels of heterodimerization partners (data not shown).
Another possibility, as suggested by our co-immunoprecipitation experiments, is that the TNF-induced increase of p100 specifically blocks RelB DNA binding. To address this point, we used MEFs that lack the p100 precursor, but still express p52 (Ishikawa et al., 1997). The TNF-induced NF-κB activity was dramatically increased in p100-deficient cells and consisted predominantly of RelB heterodimers, whereas the amount of RelA complexes was similar to wild-type controls. In striking contrast, κB binding in MEFs from mice lacking the C-terminal ankyrin domain of p105, but still expressing p50 (Ishikawa et al., 1998), consisted almost exclusively of RelA complexes (Figure 7A). In both p100–/– and p105–/– MEFs, TNF-induced RelB protein levels were comparable with controls (data not shown). Quantification of TNF-induced κB binding revealed that the ratio of RelB/RelA complexes was increased 10-fold in p100-deficient MEFs and decreased 4-fold in cells lacking p105 (Figure 7B). Together, these results demonstrate that in TNF-induced fibroblasts, the C-terminal domain of p100, but not the corresponding portion of p105, specifically represses RelB DNA binding.

Fig. 7. The C-terminal domain of p100 specifically represses RelB DNA binding in TNF-induced fibroblasts. (A) MEFs from wild-type, p100–/– and p105–/– mice were stimulated for 8 h and nuclear extracts were analyzed for NF-κB activity by EMSAs. Complexes were identified with specific Abs. (B) Quantification of the experiment shown above. The amount of complex retained by the respective Abs in the slot (arrowhead) was quantified, corrected for the signal in samples treated with p.i. serum and the amount of RelB DNA binding in wild-type MEFs was set to one.
Discussion
RelB- and p52-deficient mice lack PPs
Here, we report relB and nfkb2 mRNA expression in embryonic small intestine, RelB protein staining in E16.5 developing PPs and the lack of PPs in both p52- and RelB-deficient mice. The loss of PP structures is reminiscent of mice lacking LTα, LTβ, LTβR or NIK. In contrast, TNF and TNFR-I knockout mice have PPs although their number and size are clearly reduced (Fu and Chaplin, 1999; Matsumoto, 1999). One explanation is that activation of p52–RelB complexes downstream of LTβR is essential for the formation of PP anlagen, whereas p50 complexes and the TNF signaling pathway are not required but rather contribute to the full PP developmental program. The lack of VCAM-1+ PP-organizing centers in RelB- and p52-deficient, but not in p50-deficient, embryonic intestines supports our view.
Paxian et al. also reported that p50-deficient mice have a reduced number and size of PPs and that p52-deficient mice lack PPs. It was suggested that p52 is not required for PP development in principle based on the presence of IL-7Rα mRNA-positive structures (Paxian et al., 2002). IL-7Rα expression may stem from isolated lymphoid follicles in the small intestine, which also contain IL-7Rα+ cells (Hamada et al., 2002). The mere presence of these cells does not indicate normal PP development since they are also found in Jak3-deficient mice, which lack both VCAM-1+-organizing centers and developed PPs (Adachi et al., 1998). To our knowledge, no mutant has yet been described that has VCAM-1+ PP anlagen but lacks PPs.
RelB expression in the embryonic intestine most likely stems from stromal cells since lymphocytes colonize the PP anlage not before E18 (Adachi et al., 1998; Hashi et al., 2001). A crucial step at this stage is the IL-7-induced expression of LTα and LTβ in IL-7Rα+ intestinal cells (Yoshida et al., 1999), which was comparable in wild-type and relB–/– embryonic intestinal cultures. Since IL-7Rα+ cells are the only source of LTα and LTβ at this crucial developmental stage (Yoshida et al., 1999), we conclude that these hematopoietic inducers of PP-organizing centers are present and functional in relB–/– intestine, suggesting a stromal defect downstream of LTβR.
Activation of p52–RelB heterodimers downstream of LTβR regulates PP development
Since LTβR is also expressed in stromal cells of developing PPs (Honda et al., 2001), we used fibroblasts as a model system for LTβR and TNFR signaling. We demonstrated that TNF rapidly induces binding of RelA whereas LTβR signaling stimulates binding of RelA and, with delayed kinetics (2–4 h), also of p52–RelB (Figure 3). RelB heterodimers may have been unnoticed in previous studies (Mackay et al., 1996; VanArsdale et al., 1997; Smith et al., 2001; Yin et al., 2001) due to the undetectable binding of p52–RelB at earlier time points and/or the strong and preceding induction of RelA.
Recently, it was shown that transcription of the relB gene is regulated by NF-κB, possibly via identified RelA- and RelB-binding sites in the promoter region (Bren et al., 2001). A role for RelA in lymphoid organ development has been shown by the analysis of mice deficient for both TNFR-I and RelA (Alcamo et al., 2002). These animals lack PPs, lymph nodes and an organized splenic microarchitecture. However, the precise contribution of RelA to this phenotype is unclear since these organs are already affected by the loss of the TNFR-I pathway. Since LTβR signaling activates RelA complexes with faster kinetics compared with RelB heterodimers, it was important to show that anti-LTβR treatment can also induce binding of p52–RelB independently of RelA. Using relA–/– fibroblasts, we demonstrated that RelB complexes are still induced upon LTβR triggering although at reduced levels. Together, these findings indicate distinct regulatory functions of RelA and RelB in secondary lymphoid organ development, as compared with RelB simply being a transcriptional target of RelA, with a particular role of p52–RelB heterodimers downstream of LTβR in the organization of PP anlagen (see Figure 8). This notion is also supported by mice carrying only one copy of the ltbr and relB genes. Compound heterozygous ltbr–/+relB–/+ animals had fewer and only poorly developed PPs compared with tnfr1–/+relB–/+ compound heterozygous mice, which showed a much milder phenotype with only a slight reduction in PP number and size (data not shown).

Fig. 8. Model of LTβR- and TNFR-I-mediated activation of NF-κB. Signaling through TNFR-I activates the NF-κB pathway involving the β and γ subunits of the IKK complex. Nuclear translocation and DNA binding of p50–RelA heterodimers is accomplished through IκBα phosphorylation and ubiquitin-dependent degradation. This pathway also upregulates the expression of RelB and the p52 precursor, p100, resulting in the specific inhibition of RelB by the C-terminal IκBδ domain of p100. Membrane-bound LTα1β2 heterodimers, on the other hand, activate LTβR, which also results in the induction of RelA complexes, but the requirement for IKK subunits differs from the TNFR pathway. Importantly, LTβR signaling triggers the degradation of p100 in a NIK- and IKKα-dependent manner. As a consequence, p52–RelB heterodimers accumulate in the nucleus and regulate genes that are crucial for the normal development of lymphoid organs.
Differential requirements for NIK and IKK subunits for RelA and RelB activation downstream of LTβR and TNFR
The IKKβ and IKKγ subunits are required for the induction of NF-κB in response to inflammatory signals, such as TNF, whereas NIK and IKKα are dispensable for this function (Matsushima et al., 2001; Smith et al., 2001; Yin et al., 2001). Our data also show that the strong TNF-induced activation of RelA requires only IKKβ and IKKγ, whereas RelB complexes are not, or only very weakly, activated by TNF. However, IKKα is clearly required for LTβR-mediated activation of both RelA and RelB complexes. LTβR-induced binding of p52–RelB heterodimers, in contrast to RelA complexes, also occurs in the absence of IKKγ (for a discussion of the role of IKKβ, see below). This is, to our knowledge, the first report showing that the regulatory IKKγ subunit can be dispensable for the activation of NF-κB in a stimulus- and Rel/NF-κB family member-specific manner.
Similar to IKKα, NIK is not involved in TNF signaling, but plays a crucial role in LTβR-induced binding of p52–RelB heterodimers. In contrast to IKKα, NIK is not required for the activation of the canonical RelA/NF-κB pathway downstream of LTβR, suggesting the assembly of two distinct signaling complexes, which promote the activation of RelA and RelB, respectively (see Figure 8). It is important to note that our results were obtained in untransfected cells and that the requirement for different IKK subunits or NIK was tested in fibroblasts genetically deficient for individual components rather than in overexpression studies using dominant-negative mutants. Thus, related but distinct signals trigger via specific receptors within one cell type the activation of quantitatively and qualitatively distinct NF-κB complexes with very different requirement for upstream kinase components.
LTβR-induced processing of the p100 precursor
In contrast to the classical p50–RelA NF-κB complex, p52–RelB heterodimers are only poorly inhibited by IκBα (Dobrzanski et al., 1994; Lernbecher et al., 1994). Therefore, it is unlikely that LTβR-induced degradation of IκBα accounts for the activation of p52–RelB complexes. Indeed, when LTβR-induced p52–RelB activity was maximal, IκBα levels were unchanged (data not shown). Interestingly, LTβR-induced processing of p100 to p52 is dependent on NIK and IKKα, but not on IKKβ or IKKγ, and perfectly coincides with the induction kinetics of p52–RelB. This finding is in agreement with reports that NIK and IKKα regulate processing of the p100 pre cursor, probably through NIK-mediated activation of IKKα resulting in p100 phosphorylation and degradation (Senftleben et al., 2001; Xiao et al., 2001). Blocking experiments with emetin revealed that LTβR-induced processing of p100 is dependent on protein synthesis. Emetin also blocked nuclear accumulation of RelB without significantly changing overall RelB protein levels, indicating that LTβR-induced nuclear translocation of RelB requires p100 processing (see Supplementary figure 3).
The markedly reduced binding of p52–RelB complexes in IKKβ-deficient fibroblasts most likely stems from the very low steady state expression of p52 and RelB in these cells. The low p52 levels correlate with low expression of the p100 precursor and are not due to impaired LTβR-induced p100 processing. IKKα-deficient fibroblasts, in contrast, show normal p100 levels, but strongly impaired processing of p100 to p52. p52-deficient fibroblasts also have reduced steady state levels of RelB (data not shown), suggesting that RelB expression may be controlled by p52–RelB heterodimers in an autoregulatory feedback loop, resulting in reduced RelB levels in IKKα- and IKKβ-deficient fibroblasts. These data suggest that binding of p52–RelB heterodimers is subject to complex regulation on both the p100 and RelB protein expression level and the processing of p100 to p52. The qualitatively different requirements of IKK subunits for p100 processing and RelB levels further support the notion of selectivity and divergence at the level of the IKK complex (see Figure 8).
The association of the C-terminal portion of p105, also termed IκBγ, with RelA was previously shown to be sufficient for its cytoplasmic retention (Mercurio et al., 1993). We observed that LTβR signaling also induces processing/degradation of the p105 precursor with similar kinetics to p100, but without altering p50 levels. Since binding of RelA, but not RelB complexes is markedly increased in p105-deficient fibroblasts, degradation of p105 most likely functions within the canonical p50–RelA activation pathway. We speculate that there may be a third activation pathway downstream of LTβR, which is IκBα- and p100-independent, but involves p105 processing.
TNF blocks RelB DNA-binding by inducing the accumulation of the p100 inhibitor
The C-terminal domain of the p100 precursor, also called IκBδ, is homologous to IκBα and has been demonstrated to specifically inhibit the transcriptional activity of p52–RelB in Jurkat and COS cells (Dobrzanski et al., 1995). More recently, it was shown in transfected HeLa and 293T cells that RelB preferentially interacts with p100, but not with IκBα, IκBβ, IκBε or p105, resulting in its transcriptional repression (Solan et al., 2002). These studies indicate that p100 is a bona fide inhibitor of RelB.
The marked increase of RelB in nuclear extracts from TNF-induced fibroblasts suggests that it is not impaired nuclear translocation that prevents RelB complexes from binding to DNA. One explanation is that expression of the inhibitory p100 precursor is also strongly upregulated by TNF in both nucleus and cytoplasm of fibroblasts and that RelB is bound by p100 in both compartments, whereas this interaction is not observed in anti-LTβR-treated cells. In vivo evidence for an inhibitory function of the C-terminal domains of p100 and p105 comes from the increased constitutive NF-κB activity and inflammatory phenotype of the respective knockout animals (Ishikawa et al., 1997, 1998). We show here that in fibroblasts, TNF induction results in the specific repression of RelB DNA binding via the C-terminal domain of p100, whereas the corresponding portion of p105 does not participate in the inhibition of RelB complexes. Also, p52 overexpression does not rescue the lack of RelB DNA binding in TNF-induced fibroblasts, suggesting that not the amount of available p52 but rather the release of RelB from its p100 inhibitor is rate limiting. At the present time, it is unclear whether RelB and p100 associate co- or post-translationally, or whether specific modifications of RelB, as proposed for RelA (Naumann and Scheidereit, 1994), are required to form heterodimers with p52.
Collectively, our data indicate that LTβR signaling regulates PP organogenesis via NIK- and IKKα-mediated degradation of the inhibitory p100 precursor, resulting in the accumulation of p52–RelB heterodimers. The identification of target genes that are specifically regulated by RelB complexes should help to better understand how LTβR signaling regulates lymphoid organ development.
Materials and methods
Mice
Generation of nfkb1–/– (Sha et al., 1995), nfkb2–/– (Caamaño et al., 1998) and relB–/– mice (Weih et al., 1995) has been described previously. aly/aly (Miyawaki et al., 1994; Shinkura et al., 1999), ltbr–/– (Fütterer et al., 1998) and tnfr1–/– mice (Pfeffer et al., 1993) were kindly provided by Dr Thomas Böhm and Dr Klaus Pfeffer. Mice were mated overnight and noon on the day that vaginal plugs were observed was accepted as 0.5 d.p.c. Adoptive bone marrow transfers were performed as described previously (Weih et al., 2001). All animals were housed and bred under standardized conditions with water and food ad libitum in the SPF mouse facility of the Forschungszentrum Karlsruhe, Institute of Toxicology and Genetics.
Immunohistochemical analyses
Whole-mount immunohistochemistry was performed as described previously (Yokota et al., 1999). In brief, intestines were fixed in 2% paraformaldehyde overnight, washed with PBS and subjected to serial dehydration with methanol. Following 0.1% H2O2 treatment and rehydration, non-specific binding was blocked with PBSMT [2% skimmed milk, 0.3% Triton X-100 in phosphate-buffered saline (PBS)] and specimens were incubated overnight with anti-VCAM-1 mAb (PharMingen; clone 429, diluted 1:1000). After washing in PBSMT and PBST (0.3% Triton X-100 in PBS) and incubation with anti-rat IgG–horseradish peroxidase (1:500), color reactions were per formed using diaminobenzidine (DAB) and nickel chloride (Vector Laboratories). RelB immunohistochemistry was performed as described previously (Weih et al., 2001).
Preparation of single-cell suspensions from embryonic intestine and of primary fibroblasts
Embryonic intestines and mesentery were collected separately from embryos (with the exception of E14.5), chopped into small pieces and digested at 37°C by type IV-S collagenase (Sigma) for 1 h and 20 min, respectively. Cells were further dissociated using a 21-gauge needle and filtered through a nylon mesh. Following washes with PBS/fetal calf serum (FCS; 2% FCS for mesentery and 5% FCS for intestine cells), cell suspensions were used for either RNA extraction or cell culture. Primary MEFs were prepared from E15.5 embryos according to standard procedures (Hogan et al., 1994). Fibroblasts lacking IKKα, IKKβ, IKKγ or RelA were established from knockout mice and were kind gifts from Dr Michael Karin and Dr Amer Beg, respectively.
Cell culture and induction
Intestine cells collected from E16.5 embryos were cultured in RPMI-1640 supplemented with 10% heat-inactivated FCS, penicillin (100 U/ml), streptomycin (100 µg/ml), l-glutamine (2 mM) and 100 ng/ml stem cell factor (PeproTech). Cultures were either induced with 20 U/ml IL-7 (PeproTech) or left untreated and cells were harvested after 24 h for RNA extraction. Established NIH 3T3 fibroblasts and MEFs were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% heat-inactivated FCS, penicillin (100 U/ml), streptomycin (100 µg/ml) and l-glutamine (2 mM), and induced with recombinant murine TNF (20 ng/ml; PromoCell) or treated with anti-LTβR mAb (1 µg/ml clone AC.H6; a kind gift from Dr Jeffrey Browning and Dr Paul Rennert).
RNA analysis
RNA extraction and semi-quantitative RT–PCR were performed as reported previously (Weih et al., 2001). PCR primers for RelB were CCTCTCTTCCCTGTCACTAACGGTCTC and ACGCTGCTTTGGC TGCTCTGTGATG. Primers for LTα, LTβ, LTβR, TNFR-I and β-actin have been described previously (Weih et al., 2001).
EMSAs, western blots and immunoprecipitations
Preparation of nuclear extracts and EMSAs were essentially performed as described previously (Weih et al., 1994; Vallabhapurapu et al., 2001). The integrity of nuclear extracts was checked using an Oct oligodeoxynucleotide. Whole-cell extracts as well as nuclear and cytoplasmic fractions were prepared according to standard procedures (Schreiber et al., 1989). The quality of nuclear and cytoplasmic fractionation was checked in western blots with Abs against c-Jun (sc-45; Santa Cruz) and lactate dehydrogenase (LDH; AB1222; Chemicon), respectively. Western blots using Abs specific for β-actin (A5441; Sigma), p100/p52 and RelB (sc-7386 and sc-226; Santa Cruz) were essentially performed as described previously (Weih et al., 1994; Vallabhapurapu et al., 2001). Immunoprecipitations under native conditions using Abs specific for RelB (sc-226; Santa Cruz) and the p100 precursor (Dobrzanski et al., 1995) were performed as reported previously (Kovary and Bravo, 1991; Dobrzanski et al., 1994).
Supplementary data
Supplementary data are avaliable at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We gratefully acknowledge Michal Malewicz, Monika Pech, Heike Mondrzak, Miriam Koch and Axel Szabowski for excellent technical assistance and advice. We are indebted to Michael Karin for IKK-deficient fibroblasts and to Amer Beg for RelA-deficient fibroblasts. We thank Jeffrey Browning and Paul Rennert for providing agonistic anti-LTβR mAb and Klaus Pfeffer for LTβR- and TNFR-I-deficient mice. We also thank Peter Angel and Bettina Hartenstein for valuable comments on this manuscript, Peter Herrlich for suggestions and support, as well as Norma Howells, Selma Huber and all the staff in the animal facility at the Institute of Toxicology and Genetics. This work was supported by the Deutsche Forschungsgemeinschaft (grants We2224/1–1 and We2224/2–1).
Note added in proof
Subsequent to the submission of this manuscript, an independent report (Dejardin et al., 2002) has also shown that LTβR signaling activates two NF-κB pathways via the IKK complex/IκBα/p50-RelA and NIK/IKKα/p100/p52-RelB, respectively. In addition, ligation of the BAFF-R (Claudio et al., 2002) or CD40 on B cells (Coope et al., 2002) induces processing of p100, further supporting the existence of an alternative NF-κB activation pathway.
References
- Adachi S., Yoshida,H., Kataoka,H. and Nishikawa,S. (1997) Three distinctive steps in Peyer’s patch formation of murine embryo. Int. Immunol., 9, 507–514. [DOI] [PubMed] [Google Scholar]
- Adachi S., Yoshida,H., Honda,K., Maki,K., Saijo,K., Ikuta,K., Saito,T. and Nishikawa,S.I. (1998) Essential role of IL-7 receptor α in the formation of Peyer’s patch anlage. Int. Immunol., 10, 1–6. [DOI] [PubMed] [Google Scholar]
- Alcamo E., Hacohen,N., Schulte,L.C., Rennert,P.D., Hynes,R.O. and Baltimore,D. (2002) Requirement for the NF-κB family member RelA in the development of secondary lymphoid organs. J. Exp. Med., 195, 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attar R.M., Caamaño,J., Carrasco,D., Iotsova,V., Ishikawa,H., Ryseck, R.-P., Weih,F. and Bravo,R. (1997) Genetic approaches to study Rel/NF-κB/IκB function in mice. Semin. Cancer Biol., 8, 93–101. [DOI] [PubMed] [Google Scholar]
- Barkett M. and Gilmore,T.D. (1999) Control of apoptosis by Rel/NF-κB transcription factors. Oncogene, 18, 6910–6924. [DOI] [PubMed] [Google Scholar]
- Bren G.D., Solan,N.J., Miyoshi,H., Pennington,K.N., Pobst,L.J. and Paya,C.V. (2001) Transcription of the RelB gene is regulated by NF-κB. Oncogene, 20, 7722–7733. [DOI] [PubMed] [Google Scholar]
- Burkly L., Hession,C., Ogata,L., Reilly,C., Marconi,L.A., Olson,D., Tizard,R., Cate,R. and Lo,D. (1995) Expression of relB is required for the development of thymic medulla and dendritic cells. Nature, 373, 531–536. [DOI] [PubMed] [Google Scholar]
- Caamaño J.H., Rizzo,C.A., Durham,S.K., Barton,D.S., Raventos-Suarez,C., Snapper,C.M. and Bravo,R. (1998) Nuclear factor (NF)-κB2 (p100/p52) is required for normal splenic microarchitecture and B cell-mediated immune responses. J. Exp. Med., 187, 185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debard N., Sierro,F. and Kraehenbuhl,J.P. (1999) Development of Peyer’s patches, follicle-associated epithelium and M cell: lessons from immunodeficient and knockout mice. Semin. Immunol., 11, 183–191. [DOI] [PubMed] [Google Scholar]
- Dobrzanski P., Ryseck,R.-P. and Bravo,R. (1994) Differential interaction of Rel/NF-κB complexes with IκBα determine pools of constitutive and inducible NF-κB activity. EMBO J., 13, 4608–4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrzanski P., Ryseck,R.-P. and Bravo,R. (1995) Specific inhibition of RelB/p52 transcriptional activity by the C-terminal domain of p100. Oncogene, 10, 1003–1007. [PubMed] [Google Scholar]
- Franzoso G. et al. (1998) Mice deficient in nuclear factor (NF)-κB/p52 present with defects in humoral responses, germinal center reactions and splenic microarchitecture. J. Exp. Med., 187, 147–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y.-X. and Chaplin,D.D. (1999) Development and maturation of secondary lymphoid tissues. Annu. Rev. Immunol., 17, 399–433. [DOI] [PubMed] [Google Scholar]
- Fütterer A., Mink,K., Luz,A., Kosco-Vilbois,M.H. and Pfeffer,K. (1998) The lymphotoxin β receptor controls organogenesis and affinity maturation in peripheral lymphoid tissues. Immunity, 9, 59–70. [DOI] [PubMed] [Google Scholar]
- Gerondakis S., Grossmann,M., Nakamura,Y., Pohl,T. and Grumont,R. (1999) Genetic approaches in mice to understand Rel/NF-κB and IκB function: transgenics and knockouts. Oncogene, 18, 6888–6895. [DOI] [PubMed] [Google Scholar]
- Ghosh S., May,M.J. and Kopp,E.B. (1998) NF-κB and Rel proteins: evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol., 16, 225–260. [DOI] [PubMed] [Google Scholar]
- Griebel P.J. and Hein,W.R. (1996) Expanding the role of Peyer’s patches in B-cell ontogeny. Immunol. Today, 17, 30–39. [DOI] [PubMed] [Google Scholar]
- Hamada H. et al. (2002) Identification of multiple isolated lymphoid follicles on the antimesenteric wall of the mouse small intestine. J. Immunol., 168, 57–64. [DOI] [PubMed] [Google Scholar]
- Hashi H. et al. (2001) Compartmentalization of Peyer’s patch anlagen before lymphocyte entry. J. Immunol., 166, 3702–3709. [DOI] [PubMed] [Google Scholar]
- Hatada E.N., Krappmann,D. and Scheidereit,C. (2000) NF-κB and the innate immune response. Curr. Opin. Immunol., 12, 52–58. [DOI] [PubMed] [Google Scholar]
- Hogan B., Beddington,R., Costantini,F. and Lacy,E. (1994) Preparing mouse embryo fibroblasts. In Manipulating the Mouse Embryo. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 260–261.
- Honda K. et al. (2001) Molecular basis for hematopoietic/mesenchymal interaction during initiation of Peyer’s patch organogenesis. J. Exp. Med., 193, 621–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H., Carrasco,D., Claudio,E., Ryseck,R.-P. and Bravo,R. (1997) Gastric hyperplasia and increased proliferative responses of lymphocytes in mice lacking the COOH-terminal ankyrin domain of NF-κB2. J. Exp. Med., 186, 999–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H., Claudio,E., Dambach,D., Raventos-Suarez,C., Ryan,C. and Bravo,R. (1998) Chronic inflammation and susceptibility to bacterial infections in mice lacking the polypeptide (p)105 precursor (NF-κB1) but expressing p50. J. Exp. Med., 187, 985–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M. and Ben-Neriah,Y. (2000) Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu. Rev. Immunol., 18, 621–663. [DOI] [PubMed] [Google Scholar]
- Karin M. and Delhase,M. (2000) The IκB kinase (IKK) and NF-κB: key elements of proinflammatory signalling. Semin. Immunol., 12, 85–98. [DOI] [PubMed] [Google Scholar]
- Karin M. and Lin,A. (2002) NF-κB at the crossroads of life and death. Nat. Immunol., 3, 221–227. [DOI] [PubMed] [Google Scholar]
- Karin M., Cao,Y., Greten,F.R. and Li,Z.W. (2002) NF-κB in cancer: from innocent bystander to major culprit. Nat. Rev. Cancer, 2, 301–310. [DOI] [PubMed] [Google Scholar]
- Kovary K. and Bravo,R. (1991) Expression of different Jun and Fos proteins during the G0-to-G1 transition in mouse fibroblasts: in vitro and in vivo associations. Mol. Cell. Biol., 11, 2451–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lernbecher T., Kistler,B. and Wirth,T. (1994) Two distinct mechanisms contribute to the constitutive activation of RelB in lymphoid cells. EMBO J., 13, 4060–4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling L., Cao,Z. and Goeddel,D.V. (1998) NF-κB-inducing kinase activates IKK-α by phosphorylation of Ser-176. Proc. Natl Acad. Sci. USA, 95, 3792–3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay F., Majeau,G.R., Hochman,P.S. and Browning,J.L. (1996) Lymphotoxin β receptor triggering induces activation of the nuclear factor κB transcription factor in some cell types. J. Biol. Chem., 271, 24934–24938. [DOI] [PubMed] [Google Scholar]
- Matsumoto M. (1999) Role of TNF ligand and receptor family in the lymphoid organogenesis defined by gene targeting. J. Med. Invest., 46, 141–150. [PubMed] [Google Scholar]
- Matsushima A., Kaisho,T., Rennert,P.D., Nakano,H., Kurosawa,K., Uchida,D., Takeda,K., Akira,S. and Matsumoto,M. (2001) Essential role of nuclear factor (NF)-κB-inducing kinase and inhibitor of κB (IκB) kinase α in NF-κB activation through lymphotoxin β receptor, but not through tumor necrosis factor receptor I. J. Exp. Med., 193, 631–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercurio F., DiDonato,J.A., Rosette,C. and Karin,M. (1993) p105 and p98 precursor proteins play an active role in NF-κB-mediated signal transduction. Genes Dev., 7, 705–718. [DOI] [PubMed] [Google Scholar]
- Miyawaki S., Nakamura,Y., Suzuka,H., Koba,M., Yasumizu,R., Ikehara,S. and Shibata,Y. (1994) A new mutation, aly, that induces a generalized lack of lymph nodes accompanied by immunodeficiency in mice. Eur. J. Immunol., 24, 429–434. [DOI] [PubMed] [Google Scholar]
- Naumann M. and Scheidereit,C. (1994) Activation of NF-κB in vivo is regulated by multiple phosphorylations. EMBO J., 13, 4597–4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa S., Honda,K., Hashi,H. and Yoshida,H. (1998) Peyer’s patch organogenesis as a programmed inflammation: a hypothetical model. Cytokine Growth Factor Rev., 9, 213–220. [DOI] [PubMed] [Google Scholar]
- Pahl H.L. (1999) Activators and target genes of Rel/NF-κB transcription factors. Oncogene, 18, 6853–6866. [DOI] [PubMed] [Google Scholar]
- Pasparakis M., Alexopoulou,L., Grell,M., Pfizenmaier,K., Bluethmann, H. and Kollias,G. (1997) Peyer’s patch organogenesis is intact yet formation of B lymphocyte follicles is defective in peripheral lymphoid organs of mice deficient for tumor necrosis factor and its 55-kDa receptor. Proc. Natl Acad. Sci. USA, 94, 6319–6323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxian S., Merkle,H., Riemann,M., Wilda,M., Adler,G., Hameister,H., Liptay,S., Pfeffer,K. and Schmid,R.M. (2002) Abnormal organo genesis of Peyer’s patches in mice deficient for NF-κB1, NF-κB2 and Bcl-3. Gastroenterology, 122, 1853–1868. [DOI] [PubMed] [Google Scholar]
- Pfeffer K. et al. (1993) Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell, 73, 457–467. [DOI] [PubMed] [Google Scholar]
- Poljak L., Carlson,L., Cunningham,K., Kosco-Vilbois,M.H. and Siebenlist,U. (1999) Distinct activities of p52/NF-κB required for proper secondary lymphoid organ microarchitecture: functions enhanced by Bcl-3. J. Immunol., 163, 6581–6588. [PubMed] [Google Scholar]
- Rennert P.D., James,D., Mackay,F., Browning,J.L. and Hochman,P.S. (1998) Lymph node genesis is induced by signaling through the lymphotoxin β receptor. Immunity, 9, 71–79. [DOI] [PubMed] [Google Scholar]
- Ryseck R.-P., Weih,F., Carrasco,D. and Bravo,R. (1996) RelB, a member of the Rel/NF-κB family of transcription factors. Braz. J. Med. Biol. Res., 29, 895–903. [PubMed] [Google Scholar]
- Schreiber E., Matthias,P., Müller,M.M. and Schaffner,W. (1989) Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res., 17, 6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senftleben U. et al. (2001) Activation by IKKα of a second, evolutionary conserved, NF-κB signaling pathway. Science, 293, 1495–1499. [DOI] [PubMed] [Google Scholar]
- Sha W.C., Liou,H.-C., Tuomanen,E.I. and Baltimore,D. (1995) Targeted disruption of the p50 subunit of NF-κB leads to multifocal defects in immune responses. Cell, 80, 321–330. [DOI] [PubMed] [Google Scholar]
- Shinkura R., Kitada,K., Matsuda,F., Tashiro,K., Ikuta,K., Suzuki,M., Kogishi,K., Serikawa,T. and Honjo,T. (1999) Alymphoplasia is caused by a point mutation in the mouse gene encoding NF-κB-inducing kinase. Nat. Genet., 22, 74–77. [DOI] [PubMed] [Google Scholar]
- Smith C., Andreakos,E., Crawley,J.B., Brennan,F.M., Feldmann,M. and Foxwell,B.M. (2001) NF-κB-inducing kinase is dispensable for activation of NF-κB in inflammatory settings but essential for lymphotoxin β receptor activation of NF-κB in primary human fibroblasts. J. Immunol., 167, 5895–5903. [DOI] [PubMed] [Google Scholar]
- Solan N.J., Miyoshi,H., Carmona,E.M., Bren,G.D. and Paya,C.V. (2002) RelB cellular regulation and transcriptional activity are regulated by p100. J. Biol. Chem., 277, 1405–1418. [DOI] [PubMed] [Google Scholar]
- Vallabhapurapu S., Ryseck,R.-P., Malewicz,M., Weih,D.S. and Weih,F. (2001) Inhibition of NF-κB in T cells blocks lymphoproliferation and partially rescues autoimmune disease in gld/gld mice. Eur. J. Immunol., 31, 2612–2622. [DOI] [PubMed] [Google Scholar]
- VanArsdale T.L., VanArsdale,S.L., Force,W.R., Walter,B.N., Mosialos, G., Kieff,E., Reed,J.C. and Ware,C.F. (1997) Lymphotoxin-β receptor signaling complex: role of tumor necrosis factor receptor-associated factor 3 recruitment in cell death and activation of nuclear factor κB. Proc. Natl Acad. Sci. USA, 94, 2460–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weih D.S., Yilmaz,Z.B. and Weih,F. (2001) Essential role of RelB in germinal center and marginal zone formation and proper expression of homing chemokines. J. Immunol., 167, 1909–1919. [DOI] [PubMed] [Google Scholar]
- Weih F., Carrasco,D. and Bravo,R. (1994) Constitutive and inducible Rel/NF-κB activities in mouse thymus and spleen. Oncogene, 9, 3289–3297. [PubMed] [Google Scholar]
- Weih F., Carrasco,D., Durham,S.K., Barton,D.S., Rizzo,C.A., Ryseck, R.-P., Lira,S.A. and Bravo,R. (1995) Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-κB/Rel family. Cell, 80, 331–340. [DOI] [PubMed] [Google Scholar]
- Weih F., Warr,G., Yang,H. and Bravo,R. (1997) Multifocal defects in immune responses in RelB-deficient mice. J. Immunol., 158, 5211–5218. [PubMed] [Google Scholar]
- Xiao G., Harhaj,E.W. and Sun,S.-C. (2001) NF-κB-inducing kinase regulates the processing of NF-κB2 p100. Mol. Cell, 7, 401–409. [DOI] [PubMed] [Google Scholar]
- Yin L., Wu,L., Wesche,H., Arthur,C.D., White,J.M., Goeddel,D.V. and Schreiber,R.D. (2001) Defective lymphotoxin-β receptor-induced NF-κB transcriptional activity in NIK-deficient mice. Science, 291, 2162–2165. [DOI] [PubMed] [Google Scholar]
- Yokota Y., Mansouri,A., Mori,S., Sugawara,S., Adachi,S., Nishikawa,S. and Gruss,P. (1999) Development of peripheral lymphoid organs and natural killer cells depends on the helix–loop–helix inhibitor Id2. Nature, 397, 702–706. [DOI] [PubMed] [Google Scholar]
- Yoshida H., Honda,K., Shinkura,R., Adachi,S., Nishikawa,S., Maki,K., Ikuta,K. and Nishikawa,S.I. (1999) IL-7 receptor α+ CD3– cells in the embryonic intestine induce the organizing center of Peyer’s patches. Int. Immunol., 11, 643–655. [DOI] [PubMed] [Google Scholar]
References
- Claudio E., Brown,K., Park,S., Wang,H. and Siebenlist,U. (2002) BAFF-induced NEMO-independent processing of NF-κB2 in maturing B cells. Nat. Immunol., 3, 958–965. [DOI] [PubMed] [Google Scholar]
- Coope H.J., Atkinson,P.G., Huhse,B., Belich,M., Janzen,J., Holman, M.J., Klaus,G.G., Johnston,L.H. and Ley,S.C. (2002) CD40 regulates the processing of NF-κB2 p100 to p52. EMBO J., 21, 5375–5385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejardin E. et al. (2002) The lymphotoxin-β receptor induces different patterns of gene expression via two NF-κB pathways. Immunity, 17, 525–535. [DOI] [PubMed] [Google Scholar]