

Abstract
We report that the multidomain protein POSH (plenty of SH3s) acts as a scaffold for the JNK pathway of neuronal death. This pathway consists of a sequential cascade involving activated Rac1/Cdc42, mixed-lineage kinases (MLKs), MAP kinase kinases (MKKs) 4 and 7, c-Jun N-terminal kinases (JNKs) and c-Jun, and is required for neuronal death induced by various means including nerve growth factor (NGF) deprivation. In addition to binding GTP-Rac1 as described previously, we find that POSH binds MLKs both in vivo and in vitro, and complexes with MKKs 4 and 7 and with JNKs. POSH overexpression promotes apoptotic neuronal death and this is suppressed by dominant-negative forms of MLKs, MKK4/7 and c-Jun, and by an MLK inhibitor. Moreover, a POSH antisense oligonucleotide and a POSH small interfering RNA (siRNA) suppress c-Jun phosphorylation and neuronal apoptosis induced by NGF withdrawal. Thus, POSH appears to function as a scaffold in a multiprotein complex that links activated Rac1 and downstream elements of the JNK apoptotic cascade.
Keywords: apoptosis/JNKs/MLKs/POSH/protein scaffold
Introduction
Apoptotic death of neurons and other cell types often requires activation of specific signaling cascades that culminate in transcriptional regulation of death-associated genes. One such cascade appears to be triggered by the GTP-bound forms of Rac1 and Cdc42 and to lead to phosphorylation and activation of the transcription factor c-Jun (Chuang et al., 1997; Bazenet et al., 1998; Ham et al., 2000). Studies on neuronal and other cell types indicate that the pathway between activated Rac1/Cdc42 and c-Jun includes a sequential kinase cascade consisting of the mixed-lineage kinases (MLKs), mitogen-activated protein (MAP) kinase kinases (MKKs) 4 and 7, and the c-Jun N-terminal kinases (JNKs). Supporting evidence includes observations that overexpression of constitutively active forms of Rac1/Cdc42 or of other members of the pathway promotes apoptotic death and that dominant-negative forms of pathway members block death (Xia et al., 1995; Teramoto et al., 1996; Chuang et al., 1997; Bazenet et al., 1998; Ham et al., 2000; Mota et al., 2001; Xu et al., 2001). Combinatorial co-transfection of such molecules has further permitted their placement in the above sequential scheme (Xu et al., 2001). Additional support comes from findings that CEP-1347, a selective inhibitor of MLK family members (Maroney et al., 2001), protects neurons from death evoked by a variety of apoptotic stimuli (Maroney et al., 1999), that hippocampal neurons of mice null for expression of JNK3 are resistant to kainic acid-induced death (Yang et al., 1997) and that sympathetic neurons cultured from such animals show diminished phosphorylation of c-Jun and apoptotic death in response to nerve growth factor (NGF) deprivation (Bruckner et al., 2001).
Application of various means of interfering with the above pathway (referred to here as the JNK/c-Jun pathway) supports its involvement in death triggered by a wide variety of apoptotic stimuli. In the nervous system in particular, the JNK/c-Jun pathway plays a required role in death evoked by stimuli as diverse as withdrawal of trophic support, DNA damage, oxidative stress, β-amyloid exposure, low potassium and excitotoxic stress (Yang et al., 1997; Maroney et al., 1999; Saporito et al., 2000; Troy et al., 2001a; Whitmarsh et al., 2001; Trotter et al., 2002). Thus, knowledge about this pathway may be relevant not only to insight about the mechanisms of programmed cell death in the developing brain, but also to an understanding and potential amelioration of neuronal death associated with acute and degenerative nervous system disorders.
Despite our extensive knowledge about the JNK/c-Jun pathway, several important issues remain to be resolved. One question involves the role of Cdc42/Rac1 in death. In healthy cells, these small G-proteins act as molecular switches in different signal transduction pathways to control many basic cellular activities such as gene transcription, cytoskeletal rearrangement, cell adhesion, differentiation, the cell cycle and proliferation (reviewed by Tapon and Hall, 1997; Ridley, 2001). How do they become involved in death signaling in response to apoptotic stimuli? A related issue is how Rac1 in particular interfaces with downstream elements of the JNK/c-Jun pathway. There is no evidence for direct interaction of Rac1 with MLKs or with other components of the downstream cascade. A potential solution to these issues would be the presence of a scaffold protein that interacts with Cdc42 and/or Rac1 as well as with MLKs and perhaps other JNK/c-Jun pathway elements, and that directs their activities towards apoptotic signaling. Members of the JIP family are a set of scaffold elements for the JNK/c-Jun pathway that bind both MLKs and JNKs and that appear to play required roles for neuronal death in at least certain apoptotic paradigms (Whitmarsh et al., 2001). However, there is no indication that JIPs and Cdc42/Rac1 can bind one another directly.
POSH (plenty of SH3s) was identified by yeast two-hybrid selection as a Rac1 partner that interacts directly with the GTP form of Rac1 but not the GDP form via a specific binding region (Tapon et al., 1998). Although it does not induce assembly of actin filaments, POSH stimulates the JNK pathway and induces apoptosis when expression constructs are injected into fibroblasts. Expression studies indicated that POSH is present in different tissues, including brain. On the basis of these properties, it was hypothesized that POSH acts as a scaffold protein that contributes to Rac1-dependent signaling functions, including promotion of apoptotic death (Tapon et al., 1998).
We show here that POSH expression induces death of neuronal cells and that it participates as a required element in the death of neurons evoked by withdrawal of trophic support. Our experiments place POSH upstream of MLKs in death signaling and establish that POSH binds members of the MLK family and also interacts with downstream pathway components including MKK4 and 7, and JNKs. It thus appears that POSH acts as a scaffold that links activated Rac1 with MLKs and additional participants in the apoptotic neuronal JNK/c-Jun pathway.
Results
POSH induces apoptosis of naïve and neuronally differentiated PC12 cells and of sympathetic neurons
POSH was cloned into the pCMV.EGFP vector and transiently transfected into both NGF-untreated (naïve) and neuronally differentiated PC12 cells. For both neuronally differentiated (Figure 1A) and naïve cultures (data not shown), there was a large decline in survival by 2 days after transfection with POSH, but not after transfection with an empty vector or with one containing a POSH construct lacking a Kozak start sequence. Loss of POSH-transfected cells was accompanied by a significant increase in the proportion of transfected cells displaying apoptotic nuclei (Figure 1B).

Fig. 1. Induction of apoptosis in neuronal PC12 cells and sympathetic neurons by overexpression of POSH. (A and B) Transient expression of POSH induces cell death in neuronal PC12 cells. Neuronally differentiated PC12 cells were transfected with empty cloning vector (pCMS.EGFP, ‘GFP’ in figures) and full-length POSH with or without a Kozak sequence. (A) Cultures were assessed for cell viability in the presence of NGF at 1, 2 and 3 days after transfection. The percentages of surviving cells was calculated by normalizing to the numbers of cells expressing pCMS.EGFP at 1 day after transfection. Values are the means of counts on three wells ± SEM. Similar results were obtained in three additional independent experiments. (B) Two days after transfection, the percentages of apoptotic nuclei were determined by scoring (per condition) at least 100 nuclei stained with Hoechst 33258 in cells expressing EGFP. Values are the means of four independent experiments ± SEM. (C–E) Expression of POSH induces apoptosis of cultured sympathetic neurons. (C) Representative imunofluorescence photomicrographs (magnification, ×600) of cells transfected with control vector or POSH. Sympathetic neurons were transfected with control vector (pCMS.EGFP) (panels a–c) or POSH (panels d–f). After 48 h, nuclei of the living cells were stained with Hoechst 33258. Normal nuclei (b) and nuclei of cells undergoing apoptosis (e). (D) Sympathetic neurons were transfected with pCMS.EGFP, POSH, or POSH with a deletion or mutation of the ring finger domain (ΔRing or mRing). Numbers of cells with an EGFP signal were counted at both 24 and 48 h after transfection, and values represent the percentages of surviving cells. Values are the means of three wells ± SEM. Similar results were obtained in two additional independent experiments. (E) Sympathetic neurons were transfected as in (D). After 24 h, living cells were stained and the proportions of apoptotic nuclei in transfected cultures were assessed. Values are the means of three independent experiments ± SEM.
Comparable results were obtained with cultured sympathetic neurons. Neurons transfected with POSH showed cell contraction, loss of neurites, surface blebbing and broken, shrunken and condensed (pyknotic) nuclei typical of apoptosis (Figure 1C, compare panels a–c with panels d–f). Apoptotic death of sympathetic neurons evoked by POSH transfection was confirmed and quantified by counting green fluorescent protein (GFP)-positive cells over time (Figure 1D) or nuclei with apoptotic morphology (Figure 1E).
Examination of the POSH sequence (Tapon et al., 1998; Supplementary figure 1 available at The EMBO Journal Online) reveals a putative Zn ring finger domain near the N-terminus. Many such domains possess E3 ubiquitin ligase activity. Constructs in which this domain was either deleted (POSHΔ Ring) or mutated (POSHmRing; C28S/C33S/C36S) to interfere with putative E3 ligase activity also promoted apoptotic death when transfected into PC12 cells (data not shown) or sympathetic neurons (Figure 1E).
POSH acts upstream of the MLK family, MKK4/7 and c-Jun in a neuronal death pathway
Many apoptotic stimuli as well as overexpression of active forms of Rac1 and Cdc42 trigger a death pathway that includes sequential phosphorylation and activation of MLK family members, MKK4/7, JNKs and c-Jun. Because POSH interacts with the activated form of Rac1, stimulates JNK activity and promotes death, we postulated that it acts in the JNK/c-Jun apoptotic pathway. To test this hypothesis, we first used dominant-negative (d/n) forms of MLK family members (MLK1, 2, 3 and DLK) that have been shown to suppress neuronal death evoked by lack of NGF and by UV light-induced DNA damage, as well as by activated forms of Cdc42 and Rac1 (Xu et al., 2001). Co-transfection of the d/n forms significantly reduced POSH-induced death of naïve and neuronally differentiated PC12 cells (Figure 2A; data not shown). These findings place POSH upstream of the MLK family in neuronal death.

Fig. 2. POSH acts upstream of the MLK family, MKK4/7 and c-Jun in the JNK pathway and in a neuronal death pathway. (A) POSH overexpression-induced death is significantly suppressed by dominant-negative forms of MLK family members, MKK4, MKK7 and c-Jun. pCMS.EGFP or POSH in the latter vector were co-transfected with either pCDNA3, d/n MLK family members (MLK1, 2 and 3, and DLK), d/n MKK4 or d/n MKK7 in the pCDNA3 vector. Three days after transfection, the levels of survival and the proportions of apoptotic nuclei were determined. The values are the means of three independent experiments ± SEM. The results from co-transfection of pCMS.EGFP/pCDNA3 and pCMS.EGFP with d/nMLKs and d/n MKK4 or d/n MKK7 were not apparently different from each other, and hence only results with pCMS.EGFP/pCDNA3 are shown. (B) Death induced by POSH expression is suppressed by the JNK pathway inhibitor, CEP-1347, and the pan-caspase inhibitor, BAF. pCMS.EGFP and POSH in pCMS.EGFP vector were transfected into neuronal PC12 cells. Half of the cultures were pre-treated and maintained with either 25 µM BAF or 200 nM CEP-1347. Two days after transfection, cell numbers and percentages of apoptotic nuclei were determined. Numbers of cells transfected with pCMS.EGFP, pCMS.EGFP/BAF or pCMS.EGFP/CEP-1347 were each defined as 100%, and values from the other transfections were normalized to them accordingly. Values are the means of three independent experiments ± SEM. (C) POSH and MLKs are synergetic in induction of the JNK pathway. 293 cells were transfected with pCMS.EGFP or POSH, MLK3, DLK, d/nMLK3 or d/nDLK in the same vector, either alone or in combination as indicated. For the cultures transfected with pCMS.EGFP alone, 3 µg of DNA was used while, for all others, the amount of each construct used was 1.5 µg. CEP-1347 (200 nM) was added where indicated 4 h after transfection. Cell lysates were analyzed by western immunoblotting for levels of phospho-JNK. The membrane was stripped and re-probed with EGFP antibody as a transfection control and JNK as loading control. (D) POSH and MLKs are synergetic in the induction of apoptosis in neuronal PC12 cells. Neuronal PC12 cells were transfected with 0.5 µg of pCMS.EGFP or POSH, MLK3 or DLK in the same vector, with either 0.5 µg of pRK5 or pRK5-POSH in the presence or absence of 200 nM CEP-1347 as indicated. Two days later, the numbers of transfected cells were evaluated. Values are the means of three wells ± SEM. Similar results were obtained in two additional independent experiments. (E) Apoptotic death induced by POSH is suppressed by myristylated AKT (MyrAKT). pCMS.EGFP as well as POSH or DLK in the pCMS.EGFP vector were co-transfected with either pCMV6 (control) vector or pCMV6.MyrAKT (MyrAKT). Three days after transfection, the numbers of surviving transfected cells were counted. Cell numbers for pCMS.EGFP/pCMV6 and pCMS.EGFP/pCMV6.MyrAKT were defined as 100% survival and the cell numbers for other transfections were normalized accordingly. Values are the means from three independent experiments ± SEM.
Because MLKs act upstream of MKK4/MKK7 in neuronal apoptosis (Xu et al., 2001), we next tested whether co-expression of d/n forms of MKK4 and MKK7 suppresses POSH-evoked death. As illustrated in Figure 2A, this was the case. In contrast, d/n forms of MKK3 and 6, which lie upstream of p38, were not protective in this paradigm (data not shown). Finally, we utilized d/n c-Jun, which protects PC12 cells and sympathetic neurons from death evoked by NGF deprivation as well as by overexpression of upstream members of the JNK pathway (Ham et al., 2000). Co-transfection with d/n c-Jun effectively suppressed POSH-evoked death (Figure 2A), thus also placing POSH upstream of c-Jun in the neuronal apoptotic pathway.
POSH induction is suppressed by CEP-1347, BAF and Myr-AKT
We have shown that CEP-1347, a drug that specifically targets MLKs (Maroney et al., 2001), protects neuronal cells from apoptosis evoked by overexpression of Rac1 V12, CDC42 V12 and MLK family members, but not from death evoked by downstream members of the pathway such as MKK4/7 (Xu et al., 2001). As shown in Figure 2B, CEP-1347 effectively rescues neuronal PC12 cells from death caused by POSH overexpression. CEP-1347 also blocked the elevation of JNK phosphorylation induced by POSH overexpression (Figure 2C). Similar results were achieved by co-transfection with d/n DLK and d/n MLK3 (Figure 2C). This supports our above experiments that place POSH upstream of MLKs in the JNK apoptotic pathway.
To assess the role of caspases in POSH-evoked death, cultures of neuronally differentiated PC12 cells were transfected with POSH, treated with the pan-caspase inhibitor Boc-aspartyl-(ome)-fluoromethyl ketone (BAF) (Deshmukh et al., 1996) and scored 2 days later for survival and proportion of apoptotic nuclei. BAF provided significant protection (Figure 2B), thus indicating that POSH, like other components of the JNK pathway, induces apoptosis through caspase activation.
We have reported that co-expression of myr-AKT, a myristylated form of AKT that appears to be constitutively activated in cells, protects neuronal PC12 cells from death evoked by overexpressed MLK family members (Xu et al., 2001). The data in Figure 2E show that myr-AKT also suppresses death promoted by POSH overexpression. These findings are consistent with the interpretation that POSH acts upstream of the MLKs and that AKT can inhibit death by a mechanism that functions either at the level or downstream of MLK activation.
Functional synergism between POSH and MLKs on JNK activation and cell death
Our data indicate that POSH activates the JNK pathway and acts upstream of MLKs in neuronal death. To examine further the functional interaction between POSH and MLKs as well as whether POSH regulates the JNK pathway through MLKs, we transfected 293 cells with POSH, MLK3 and DLK, either alone or in combination. Cells lysates were analyzed for levels of phospho-JNK, an indicator of JNK activation. As shown in Figure 2C, both MLK3 and DLK induce JNK phosphorylation but, when co-transfected with POSH, the phospho-JNK signal is higher even though the number of surviving transfected cells is significantly lower. The latter is indicated by the lower level of enhanced GFP (EGFP) signal in the lysates of co-transfected cells (Figure 2C) and by reduction in survival (Figure 2D; data not shown). Thus, POSH appears to work synergistically with the MLKs in induction of JNK activation and cell death.
POSH binds MLKs
The above data indicate that POSH acts upstream of MLKs and serves as a link between Rac1 and MLK activation. One means by which this might occur is by direct interaction of POSH and MLKs. To evaluate this, PC12 and 293 cells were transfected with expression vectors encoding Myc epitope-tagged POSH and epitope-tagged MLKs, either alone or in combination. Cell lysates were immunoprecipitated with anti-POSH and the immunocomplexes were probed for tagged MLK family members and tagged POSH. MLK2, MLK3 and DLK were detected in the POSH immunocomplexes derived from co-transfected PC12 and 293 cells, but not in cells transfected with MLKs alone (Figure 3A and B; data not shown). In contrast, we were unable to detect co-immunoprecipitation of Myc-tagged POSH with ERK1 and 2 (data not shown)

Fig. 3. POSH interacts with MLK family members in vivo. 293 cells were transfected with expression vectors (pCMS.EGFP) encoding myc epitope-tagged POSH and epitope-tagged members of the MLK family, either alone or in combination as indicated. At 20 h post-transfection, cell extracts were prepared and aliquots were analyzed as described to assess the transfection efficiency of the various constructs. The cell lysates were immunoprecipitated with anti-POSH antibody (A and B), and the immunocomplexes were analyzed for MLK family members. MLK2/d/nMLK2 and DLK were detected with anti-HA antibody and MLK3 was detected with anti-Flag antibody. The membrane was stripped and reprobed with anti-Myc antiserum to detect the presence of POSH and POSH ΔRing (A) or anti-Flag antiserum to detect MLK3 (C). In reciprocal co-expression/immunoprecipitation experiments, cell lysates were subjected to immunoprecipitation with anti-Flag (for MLK3) (C) or anti-HA antiserum (for MLK2 or DLK) (D) and the immunocomplexes were probed for the presence of ΔRingPOSH with anti-Myc antiserum.
When overexpressed, MLK family members become phosphorylated and activated and exhibit a slower electrophoretic mobility (Figure 3B; Leung and Lassam, 1998; Nihalani et al., 2000). As shown in Figure 3B, we found that a kinase-inactive mutant form of MLK2 (MLK2 K125A, or d/n MLK2) also co-immunoprecipitates with POSH. Thus, both active and inactive forms of at least MLK2 interact with POSH.
The data in Figure 3A show that MLK3 co-immunoprecipitates with Myc-ΔRing POSH, a Zn ring finger domain POSH mutant. Similar results were achieved with MLK2 and DLK (data not shown). Thus, POSH’s Zn ring finger domain is not required for interaction with MLKs.
Reciprocal co-expression/immunoprecipitation experiments were carried out with lysates of cells co-transfected with tagged MLKs and POSH/ΔRing POSH. Immuno precipitation of the MLKs again revealed interaction of all three tested family members with POSH and/or ΔRing POSH (Figure 3C and D). Additionally, the reciprocal immunoprecipitation experiments confirmed interaction of POSHΔRing with the kinase-inactive form of MLK2 (Figure 3D).
To confirm further that POSH binds MLKs, we tested the ability of GST–POSH fusion proteins to pull-down MLK family members prepared in an in vitro transcription–translation system. Because of the large size of POSH, we cloned both its N-terminal (amino acids 2–460) and C-terminal (amino acids 437–892) halves into the pGEX2T vector and expressed them as GST fusion proteins. As shown in Figure 4A, MLK1, MLK2, MLK3 and DLK all bind to GST–POSH (437–892), but not to GST alone. Comparable results were achieved with GST–POSH (2–460) (Figure 4B).

Fig. 4. POSH interacts with MLK family members in vitro and with MKK4/7 and JNK1/2 in vivo. (A and B) The POSH C- and N-termini interact with the MLKs in vitro. (A) The C-terminal half of POSH was fused in-frame with GST to form GST–POSH C′ fusion protein, which was expressed and purified as described in Materials and methods. GST–POSH C′ fusion protein was incubated with in vitro translated 35S-labeled full-length MLK family members (MLK1, MLK2, MLK3 and DLK) for the pull-down assays as described in Materials and methods. After extensive washing, radioactive protein that was retained was visualized by SDS–PAGE followed by autoradiography. (B) As in (A), except that the assay used the N-terminal half of POSH. (C and D) POSH co-immunoprecipitates with MKK4/7 and JNK1/2. 293 cells were transfected with expression vectors (pCMS.EGFP) encoding Flag epitope-tagged POSH and Myc epitope-tagged MKK4 and 7, JNK1 and 2, either alone or in combination as indicated. At 20 h post-transfection, cell extract was prepared and analyzed as in (A) for the transfection efficiency of different constructs. The cell lysates were immunoprecipitated with anti-Myc antibody (C), and the immunocomplexes were analyzed for the presence of POSH with anti-Flag antibody. In reciprocal co-expression/immunoprecipitation experiments (D), cell lysates were subjected to immunoprecipitation with anti-Flag antibody and the immunocomplexes were probed for the presence of MKK4, MKK7, JNK1 and JNK2 with anti-Myc antibody.
POSH interacts with MKK4/7 and JNKs
To determine whether components of the JNK pathway downstream of MLKs also interact with POSH, 293 cells were transfected with expression vectors encoding FLAG epitope-tagged POSH and Myc epitope-tagged MKK4, MKK7, JNK1 or JNK2, either alone or in combination. Reciprocal immunoprecipitation experiments (i.e. immunoprecipitation of tagged POSH and probing for MKK4/7 and JNK1/2, and vice versa) revealed specific interaction of all four proteins with POSH (Figure 4C and D). As previously reported, there was also strong interaction with constitutively active Rac1, but not with a d/n form of this protein (data not shown). Thus, POSH appears to act as a scaffold protein in the JNK pathway by interacting with multiple components of the pathway including activated Rac1, MLKs, MKK4, MKK7, JNK1 and JNK2.
POSH plays a required role in c-Jun phosphorylation and neuronal cell death induced by NGF deprivation
Two strategies were used to assess whether POSH plays a required role in neuronal death. The first employed an Antennapedia peptide (penetratin 1)-linked POSH antisense oligonucleotide. This approach has been highly successful in selectively depressing expression of a number of proteins in PC12 cells and sympathetic neurons (Troy et al., 2001b). As shown in Figure 5A and B, this construct, but not a scrambled construct, provided substantial protection of sympathetic neurons and neuronally differentiated PC12 cells from death evoked by NGF deprivation. The POSH antisense construct also significantly reduced the proportion of NGF-deprived sympathetic neurons that stained positive for phospho c-Jun as well as for total c-Jun (Figure 5C and D). The appearance of phospho c-Jun in this death paradigm requires activation of the JNK pathway, and c-Jun elevation itself is a consequence of c-Jun phosphorylation/activation (Eilers et al., 1998). These observations thus indicate that POSH plays a required role in death evoked by NGF deprivation and that depletion of POSH leads, as anticipated, to depression of the JNK/c-Jun signaling pathway. As an alternative approach to deplete endogenous POSH, we also used a POSH small interfering RNA (siRNA) construct. Although endogenous POSH levels are too low to detect with existing antisera, the siRNA effectively decreased expression of co-expressed FLAG-tagged POSH (Figure 5F), thus indicating the efficacy of the construct. The POSH siRNA, but not an irrelevant siRNA against the transcription factor ATF5, also substantially protected neuronally differentiated PC12 cells from NGF deprivation (Figure 5E).

Fig. 5. Penetratin-linked POSH antisense oligonucleotide protects sympathetic neurons and neuronally differentiated PC12 cells from death evoked by NGF deprivation and suppresses c-Jun phosphorylation. (A) Penetratin-linked POSH antisense oligonucleotides protect neuronally differentiated PC12 cells from NGF deprivation. Penetratin-linked antisense oligonucleotides were added to cultures of neuronally differentiated PC12 cells at the same time that NGF deprivation was performed, and the numbers of surviving cells in each culture were compared before and after 24 h of NGF deprivation and are reported as a percentage of survival. (B) Penetratin-linked POSH antisense oligonucleotides protect sympathetic neurons from NGF deprivation. Penetratin-linked antisense oligonucleotides were added to cultures of sympathetic neurons and the percentages of surviving cells were assessed as in (A). (C and D) Penetratin-linked POSH antisense oligonucleotides suppress NGF deprivation-induced c-Jun phosphorylation and c-Jun induction in sympathetic neurons. NGF deprivation and penetratin-linked POSH antisense oligonucleotide treatment of sympathetic neurons was performed as in (A). After 24 h of NGF deprivation, cells were fixed and stained with anti-phospho-c-Jun and anti-c-Jun antisera, and the proportions of neurons that were positive for phospho-c-Jun were determined. Values are the means of triplicate cultures ± SEM. Similar results were obtained in two additional independent experiments. (E) A POSH siRNA protects neuronal PC12 cells from death elicited by NGF deprivation. Neuronally differentiated PC12 cell cultures were co-transfected with pCMS.EGFP and a POSH siRNA or with a control irrelevant siRNA (against rat ATF5), and 2 days later cultured with or without NGF for 1 day. Numbers of surviving cells were monitored and values (±SEM) are reported for NGF-lacking cultures relative to those for NGF-treated cultures (n = 3). Comparable results were achieved in three independent experiments. (F) POSH siRNA blocks expression of Flag-POSH. Neuronal PC12 cells were co-transfected with pCMS. EGFP-Flag-POSH and either an irrelevant siRNA (for mouse caspase 8) or POSH siRNA. One day later, the cultures were monitored by western immunoblotting for expression of Flag-POSH and EGFP.
The Zn ringer finger domain regulates POSH’s expression through the proteasomal pathway
Zn ring finger domains are associated with E3 ubiquitin ligase activity, and their presence permits proteins to target other proteins, and sometimes themselves, for proteasomal degradation (Patterson, 2002). We therefore examined the effect of deleting (POSHΔRing) or mutating (POSHmRing) this domain on expression of POSH protein. As shown in Figure 2, these mutations do not compromise the capacity of POSH to promote neuronal death. At 20 h after transfection of equal levels of constructs into PC12 cells (a time before apparent cell death), the mutant proteins were expressed to at least a 20-fold higher level than wild-type POSH. Moreover, when wild-type POSH was transfected into cells treated with the proteasomal inhibitor lactacystin, its expression reached that of the mutant proteins in the absence of the inhibitor (Figure 6A).
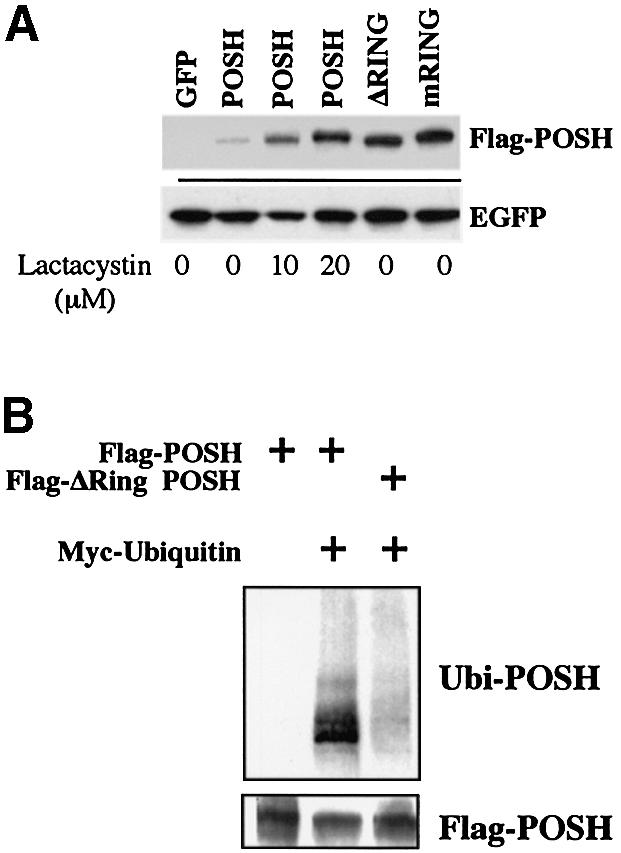
Fig. 6. The POSH zinc ring finger domain regulates POSH expression through the proteasomal pathway. (A) The POSH Zn ring finger domain regulates POSH levels and a proteasome inhibitor elevates POSH expression. PC12 cells were transfected with 2 µg of expression vector (pCMS.EGFP) or expression vectors encoding Flag-tagged wild-type POSH, POSH with a deletion of the RING finger domain (ΔRing POSH) or POSH with a mutation of the RING finger domain (mRing POSH). After 20 h, lactacystin was added to two cultures of cells transfected with wild-type POSH to the indicated final concentrations. At 6 h after lactacystin treatment, all cells were harvested, resolved by SDS–PAGE and immunoblotted with anti-Flag (upper panel) or anti-EGFP antiserum (lower panel). (B) The ring finger domain is required for the ubiquitylation of POSH. 293 cells were transfected with 1 µg of expression vector encoding Flag-POSH or Flag-ΔRing POSH with or without 0.5 µg of expression vector encoding Myc-tagged ubiquitin. After 20 h, lactacystin (10 µM) was added to the medium as indicated, and another 6 h later the cells were lysed, immunoprecipitated with anti-Flag antiserum, subjected to SDS–PAGE and immunoblotted to detect the ubiquitylated POSH with anti-Myc antiserum. The blot was stripped and reprobed with anti-Flag antiserum to detect POSH or ΔRing POSH.
To examine further whether POSH expression is subject to regulation by a ubiquitin-mediated pathway, we co-transfected 293 cells with Myc-tagged ubiquitin and either wild-type POSH or POSHΔRing (both Flag tagged) in the presence of lactacystin. When the cell extracts were subjected to immunoprecipitation with anti-Flag (POSH) and western immunoblotting with anti-Myc (ubiquitin), there was an apparent smear of signal at and above the site of POSH mobility for cells transfected with wild-type POSH. In contrast, there was only a weak signal for cells transfected with POSHΔRing (Figure 6B). Taken together, these findings indicate that POSH is subject to ubiquitylation and proteasomal degradation, and that this requires the ring finger domain.
Discussion
POSH as a required component of the JNK/c-Jun pathway of neuronal apoptotic death
Neuronal apoptotic death induced by NGF deprivation is mediated in part by a sequential pathway that includes GTP-bound Rac1/Cdc42, MLKs, MKK4/7, JNKs and c-Jun. Our findings indicate that the multidomain protein POSH is a required scaffold element in this pathway. We confirmed that POSH binds activated Rac1 and find that it also binds all MLK family members tested and interacts with MKK4/7 as well as JNK1 and JNK2. Expression of exogenous POSH induces apoptotic death of neuronal cells via the JNK pathway, and a POSH antisense oligonucleotide and a POSH siRNA suppress death promoted by NGF deprivation. Prior studies place the JNK/c-Jun pathway upstream of mitochondrial cyto chrome c release and caspase activation (Tournier et al., 2000); our observations that the caspase inhibitor BAF blocks POSH-induced death and that POSH expression evokes mitochondrial cytochrome c release (data not shown) are consistent with such findings. As cited above, various forms of evidence implicate the JNK/c-Jun pathway in death of neuronal and other types of cells evoked by a wide variety of stimuli. Given the apparent required involvement of POSH-interacting MLKs and JNKs in these death paradigms, it seems highly likely that POSH is similarly involved.
POSH is a scaffold for activation of JNK/c-Jun pathway kinases
Propagation of the JNK/c-Jun pathway and induction of neuronal cell death require that MLKs become enzymatically activated via phosphorylation, and the bulk of evidence suggests that this occurs by a mechanism that includes autophosphorylation (Leung and Lassam, 1998; Nihalani et al., 2000; Xu et al., 2001). This raises the question as to why overexpression of MLKs leads to their autophosphorylation/activation whereas endogenous MLKs do not activate spontaneously unless cells are subjected to apoptotic stimuli. A potential explanation is that autophosphorylation requires a degree of MLK–MLK interaction not achieved by the low levels of endogenous MLKs, but that can be reached either by MLK overexpression or by interaction of endogenous material with a scaffold such as POSH in response to apoptotic stimuli. In support of the idea that POSH acts as an activating scaffold for endogenous MLKs, we found that death promoted by POSH overexpression is synergized by co-expression of MLKs and suppressed by CEP-1347 as well as by inactive (d/n) MLK mutants. Similarly, promotion of c-Jun phosphorylation in response to NGF deprivation, which requires MLK activation, was suppressed by a POSH antisense oligonucleotide. In addition, kinetically inactive mutant forms of MLKs interacted with POSH, thus indicating that MLKs need not be phosphorylated/activated to undergo POSH binding. Moreover, the observed association of MLKs with both the N- and C-terminal halves of POSH suggests that a given POSH molecule may interact simultaneously with multiple MLKs. Taken together, these findings support a model in which apoptotic stimuli or POSH overexpression induce direct association between POSH and inactive MLKs. This induction of high MLK proximity in turn permits MLK autophosphorylation and activation, leading to phosphorylation/activation of MKK4/7 and JNKs.
It has been reported that although MLK3 and DLK can be co-immunoprecipitated and although d/n DLK strongly suppresses MLK3-induced JNK activation, MLK3 and DLK do not form heterodimers (Tanaka and Hanafusa, 1998; Nihalani et al., 2000). One explanation as provided by our model is that MLK3 and DLK interact indirectly via POSH with mutual activation when both are wild-type, and suppression of activation when one or the other is a d/n.
This would also explain why d/n forms of all assessed MLK family members block death promoted by POSH overexpression as well as by NGF deprivation or DNA damage (Xu et al., 2001).
Our findings establish that in addition to binding MLKs, POSH interacts with MKK4 and 7 as well as JNK1 and 2. Thus, POSH appears to act as a scaffold for multiple JNK/c-Jun pathway components. This may provide not only induced proximity and activation of MLKs, but also enhanced access of activated MLKs to their MKK targets and of the latter to their JNK targets. Additionally, such a scaffold may serve to deliver activated pathway components to specific intracellular compartments such as the nucleus. Preliminary GST pull-down assays, similar to those described here with MLKs, indicate the lack of in vitro binding between POSH and MKK4/7, JNK1 and JNK2 (N.Kukekov, Z.Xu and L.Greene, unpublished data) and thus that the in vivo interactions between these molecules may be via an intermediate.
As noted above, POSH binds directly to activated (GTP-bound) Rac1. Given the evidence that GTP-Rac1 plays a required role in activation of the apoptotic JNK/c-Jun pathway in neurons and that death promoted by activated Rac1 is blocked by CEP-1347 and d/n forms of MLKs (Xu et al., 2001), it is reasonable to suggest that the GTP-Rac1–POSH interaction plays a role in activation of endogenous MLKs by POSH.
Besides POSH, several other scaffold proteins, including JIP family members and β-arrestin 2, have been shown to induce JNK pathway activation when co-expressed with JNK/c-Jun pathway components and to bind multiple component members (Yasuda et al., 1999; McDonald et al., 2000). Like POSH, JIPs are hypothesized to facilitate activation of the pathway by promoting co-association of MLKs, MKK7 and JNKs (Yasuda et al., 1999). However, in contrast to POSH, JIPs do not appear to bind activated Rac1; nor are they known to bind activated Cdc42. Thus, POSH may serve as a critical link between Rac1 and the JNK/c-Jun cascade and thereby perform a function not presently known for other scaffold proteins. It presently is unclear whether POSH acts independently of JIPs or whether the two sets of scaffold proteins act cooperatively to promote neuronal death.
As in the case of Rac1, d/n Cdc42 overexpression protects sympathetic neurons from death evoked by NGF withdrawal (Bazenet et al., 1998; Mota et al., 2001; Xu et al., 2001), whereas constitutively active forms of Cdc42 promote apoptotic death via MLKs and the JNK/c-Jun pathway (Xu et al., 2001). Such findings have been taken to indicate a role for both Rac1 and Cdc42 in neuronal cell death. However, there is no evidence for interaction of POSH with GTP-Cdc42 (Tapon et al., 1998). This raises the possibility of several alternatives as to how activated Cdc42 might interface with the JNK/c-Jun cascade. One is that yet another presently undefined scaffold molecule links the JNK/c-Jun pathway with GTP-Cdc42. In support of this possibility, it is reported that MLK3 and activated Cdc42 can be co-immunoprecipitated from cellular lysates but that this interaction cannot be recapitulated in an in vitro system (Bock et al., 2000). An alternative, as suggested for NGF deprivation-induced death, is that Cdc42 acts upstream of Rac1 (Bazenet et al., 1998) and thus need not associate with MLKs or other pathway components.
A key question addressed by our study was whether POSH plays a required role in a neuronal cell death paradigm mediated by the JNK/c-Jun pathway. We found that both a POSH antisense construct and a POSH siRNA construct provided substantial protection from death stimulated by NGF deprivation. Although we were unable to monitor levels of endogenous POSH with currently available antisera, the POSH siRNA effectively blocked expression of a co-transfected construct encoding tagged POSH.
Autoregulation of POSH protein expression via the Zn ring finger domain
POSH has a Zn ring finger domain that, by analogy with other such domains, possesses putative E3 ubiquitin ligase activity that could target other proteins for degradation. Many such E3 ligases target themselves for destruction (Hu and Fearon, 1999). Several of our observations indicate that this is the case for POSH. First, deletion of the ring finger domain or introduction of point mutations that should compromise E3 ligase activity permitted cells to accumulate vastly higher levels of the mutant POSH proteins as compared with wild-type POSH. In addition, blockade of proteasomal activity led to accumulation of wild-type POSH to levels similar to those achieved by deletion or mutation of the Zn ring finger domain. Such findings favor a model in which POSH, via the E3 ligase activity of its Zn ring finger domain, directs its own proteasomal degradation, although we cannot exclude the possibility that POSH regulates its own stability indirectly. Autoregulation of POSH stability could account for the very low levels of endogenous POSH. In contrast to its apparent role in regulating POSH expression and degradation, the Zn ring finger domain appears to have no direct role in the pro-apoptotic function of POSH; constructs with deletion/mutation of this domain retained the capacity to promote apoptotic death and to bind components of the JNK/c-Jun cascade.
Given that POSH overexpression promotes MLK-dependent death, an attractive possibility would be that apoptotic stimuli lead to stabilization of endogenous POSH, permitting it to reach levels sufficient to trigger the JNK/c-Jun apoptotic pathway. However, it remains to be seen whether this occurs or whether autoregulation of its own expression will influence POSH’s role in neuronal cell death. An additional scenario worthy of consideration is that because POSH levels appear to be regulated by proteasomal degradation, interference with this pathway in cells might lead to POSH accumulation and consequent JNK/c-Jun-mediated death. It is reported that proteasomal inhibitors promote neuronal death as well as JNK activation (Masaki et al., 2000), and there is increasing evidence for proteasomal dysfunction in neurodegenerative disorders (Chung et al., 2001)
In conclusion, as depicted in Figure 7, our findings indicate that POSH serves as a scaffold for a multiprotein complex that links GTP-Rac1 with a downstream kinase cascade (MLKs, MKK4/7 and JNKs) that culminates in phosphorylation of c-Jun and promotes neuronal cell death. Further studies will be required to determine the mechanism by which activation of Rac1 and its consequent association with POSH influences POSH’s capacity to drive downstream components of the pathway.

Fig. 7. Scheme for the role of POSH as a scaffold for a multiprotein complex that induces phosphorylation of c-Jun and neuronal apoptosis. Apoptotic stimuli such as NGF deprivation promote the formation of a complex of POSH with GTP-Rac1 as well as with downstream members of the JNK cascade. Association leads to autoactivation of MLKs, which in turn phosphorylate and activate MKK4/7, which in turn phosphorylate and activate JNKs. Association of MKK4/7 and JNKs with POSH may be mediated by an additional member of the complex, designated here as ‘X’. The activated JNKs then phosphorylate and activate c-Jun, which triggers subsequent release of cytochrome c, caspase activation and death.
Materials and methods
Materials
Cell growth media RPMI 1640 and Dulbecco’s modified Eagle’s medium (DMEM), and LipofectAMINE 2000 were from Life Technologies, Inc. (Frederick, MD). Human recombinant NGF (hrNGF) was kindly provided by Genentech (South San Francisco, CA). BAF was from Enzyme Systems Products (Dublin, CA). Hoechst dye 33342 and anti-hrNGF antiserum were from Sigma (St Louis, MO). Anti-c-Jun/phospho c-Jun and anti-JNK/phospho JNK (Thr183/Tyr185) antisera were from New England Biolabs (Beverly, MA). CEP-1347 was kindly provided by Cephalon Inc. (West Chester, PA) and applied as described before (Xu et al., 2001).
Cell culture
Cultures of PC12 cells and primary rat sympathetic neurons were generated as described previously (Xu et al., 2001), as was NGF deprivation of PC12 cells. NGF deprivation of sympathetic neurons was performed by washing twice with NGF-free medium and addition of anti-human NGF antibody to 500 ng/ml. Control cells were washed with serum-free medium and maintained in medium supplied with hrNGF. 293 cells were cultured in DMEM with 10% fetal bovine serum.
Transfections and assessment of cell survival
DNA used for transfections was prepared as described previously (Xu et al., 2001). Details for specific constructs are given in the Supplementary data. PC12 cells were transfected using Lipofect AMINE 2000 according to the manufacturer’s protocol. Sympathetic neurons were transfected with the Helios Gene Gun system (Bio-Rad, Hercules, CA) as described previously (Xu et al., 2001). Strip counting and counting of apoptotic nuclei were performed as described previously (Xu et al., 2001).
Co-immunoprecipitation, western immunoblotting and immunofluorescence staining
For co-immunoprecipitation (co-IP) experiments, different full-length cDNAs (2 µg for each plasmid) were transfected into 293 cells as described previously (Angelastro et al., 1998). Cells lysates were prepared 20 h after transfection as described previously (Xu et al., 2001) using lysis buffer (10 mM Tris pH 7.4, 1.0% Triton X-100, 0.5% NP-40, 150 mM NaCl, 20 mM NaF, 0.2 mM sodium orthovanadate, 1 mM EDTA, 1 mM EGTA, 0.2 mM PMSF). For each co-IP, ∼200 µg of protein was incubated for 2–4 h at 4°C with 1 µg of the indicated antibody/antiserum followed by incubation with protein A–Sepharose (Pharmacia) for 1 h. The immunoprecipitates were washed five times with cell lysis buffer and analyzed by western immunoblotting as described previously (Xu et al., 2001). To detect c-Jun/phospho-c-Jun expression, immunofluoresence staining was performed as described previously (Xu et al., 2001). The secondary anti-serum was Alexa Fluor 546 goat anti-mouse IgG (H+L) (1:4000) (Molecular Probe, Eugene, OR).
In vitro binding assays
PGEX plasmids were used to transform E.coli DH5α. After induction of protein expression with 0.4 mM isopropyl-β-d-thiogalactopyranoside (Promega, Madison, WI) for 2–4 h, the bacteria were resuspended in 20 ml of phosphate-buffered saline (PBS) buffer containing 0.5% Tween, 16 µl of NaF (500 mM), 16 µl of Na2VO4 (500 mM), 100 µl PMSF (100 mM), 20 µl of dithiothreitol (DTT; 1 M), 400 µl of lysozyme (50 mg/ml), 30 µl of DNase I (10 mg/ml), and further disrupted by sonification. Following centrifugation at 12 000 g for 20 min, the induced proteins were absorbed to bead-immobilized glutathione according to the manufacturer’s protocol (Amersham, Piscataway, NJ). Full-length cDNAs in pCMS.EGFP vector were transcribed and translated in vitro using the TNT-coupled reticulocyte lysate system (Promega) in the presence of [35S]methionine (New England Nuclear). A 10 µl aliquot of each synthesized polypeptide was incubated with glutathione beads carrying equal amounts of GST or GST–POSH fusion proteins in 300 µl of lysis buffer at 4°C for 4–14 h. The beads were washed six times with 600 µl of the same buffer, boiled in 30 µl of SDS sample buffer, and the solubilized proteins were resolved by electrophoresis on 4–12% Nupage gels (Invitrogen, Carlsbad, CA) and visualized by autoradiography. The amount of protein loaded in control input lanes was approximately one-sixth of the total used in the binding assays.
Preparation and treatment with antisense oligonucleotides and siRNA
Oligonucleotides bearing an SH group at their 5′ end (Qiagen Operon, Alameda, CA) were linked to penetratin 1 (Oncor) as described previously (Troy et al., 2001b). A scrambled penetratin-linked sequence was used as a control. The POSH antisense sequence was 5′-GGCAGACTCATCCATCTTGAG-3′. Penetratin 1-linked antisense oligonucleotides were added at the same time that NGF deprivation was performed. The linked antisense oligonucleotides (2 µl of a 240 nM stock) were first mixed with half of the culture medium (250 µl) containing NGF antibody and then added to the cell cultures in 24-well plates (total volume per well, 500 µl). POSH siRNA was prepared by Xeragon Inc. (Huntsville, AL) based on the sequence 5′-GUCAG ACUUCUGGAUGGCAdTdC-3′. Caspase 8 siRNA used as a control was a gift from Dr C. Troy (Columbia University). siRNAs were transfected using LipofectAMINE 2000 at a final concentration of 25 nM.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank Drs Anna Maroney (Cephalon, Inc.) and James Angelastro (Columbia University) for helpful discussion, and Claudine Bitel for expert technical assistance. This work was supported in part by grants from the NIH-NINDS and the Blanchette Rockefeller Foundation (to L.A.G.). Z.X. was supported in part by NIH Training Grant T32-DK07328-22.
References
- Angelastro J.M., Ho,C.L., Frappier,T., Liem,R.K. and Greene,L.A. (1998) Peripherin is tyrosine-phosphorylated at its carboxyl-terminal tyrosine. J. Neurochem., 70, 540–549. [DOI] [PubMed] [Google Scholar]
- Bazenet C.E., Mota,M.A. and Rubin,L.L. (1998) The small GTP-binding protein Cdc42 is required for nerve growth factor withdrawal-induced neuronal death. Proc. Natl Acad. Sci. USA, 95, 3984–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock B.C., Vacratsis,P.O., Qamirani,E. and Gallo,K.A. (2000) Cdc42-induced activation of the mixed-lineage kinase SPRK in vivo. Requirement of the Cdc42/Rac interactive binding motif and changes in phosphorylation. J. Biol. Chem., 275, 14231–14241. [DOI] [PubMed] [Google Scholar]
- Bruckner S.R., Tammariello,S.P., Kuan,C.Y., Flavell,R.A., Rakic,P. and Estus,S. (2001) JNK3 contributes to c-Jun activation and apoptosis but not oxidative stress in nerve growth factor-deprived sympathetic neurons. J. Neurochem., 78, 298–303. [DOI] [PubMed] [Google Scholar]
- Chuang T.H., Hahn,K.M., Lee,J.D., Danley,D.E. and Bokoch,G.M. (1997) The small GTPase Cdc42 initiates an apoptotic signaling pathway in Jurkat T lymphocytes. Mol. Biol. Cell, 8, 1687–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung K.K., Dawson,V.L. and Dawson,T.M. (2001) The role of the ubiquitin–proteasomal pathway in Parkinson’s disease and other neurodegenerative disorders. Trends Neurosci., 24, S7––S14.. [DOI] [PubMed] [Google Scholar]
- Deshmukh M., Vasilakos,J., Deckwerth,T.L., Lampe,P.A., Shivers,B.D. and Johnson,E.M.,Jr (1996) Genetic and metabolic status of NGF-deprived sympathetic neurons saved by an inhibitor of ICE family proteases. J. Cell Biol., 135, 1341–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers A., Whitfield,J., Babij,C., Rubin,L.L. and Ham,J. (1998) Role of the Jun kinase pathway in the regulation of c-Jun expression and apoptosis in sympathetic neurons. J. Neurosci., 18, 1713–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ham J., Eilers,A., Whitfield,J., Neame,S.J. and Shah,B. (2000) c-Jun and the transcriptional control of neuronal apoptosis. Biochem. Pharmacol., 60, 1015–1102. [DOI] [PubMed] [Google Scholar]
- Hu G. and Fearon,E.R. (1999) Siah-1 N-terminal RING domain is required for proteolysis function and C-terminal sequences regulate oligomerization and binding to target proteins. Mol. Cell. Biol., 19, 724–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung I.W. and Lassam,N. (1998) Dimerization via tandem leucine zippers is essential for the activation of the mitogen-activated protein kinase kinase kinase, MLK-3. J. Biol. Chem., 273, 32408–32415. [DOI] [PubMed] [Google Scholar]
- Maroney A.C. et al. (1999) CEP-1347 (KT7515), an inhibitor of JNK activation, rescues sympathetic neurons and neuronally differentiated PC12 cells from death evoked by three distinct insults. J. Neurochem., 73, 1901–1912. [PubMed] [Google Scholar]
- Maroney A.C. et al. (2001) Cep-1347 (KT7515), a semisynthetic inhibitor of the mixed lineage kinase family. J. Biol. Chem., 276, 25302–25308. [DOI] [PubMed] [Google Scholar]
- Masaki R., Saito,T., Yamada,K. and Ohtani-Kaneko,R. (2000) Accumulation of phosphorylated neurofilaments and increase in apoptosis-specific protein and phosphorylated c-Jun induced by proteasome inhibitors. J. Neurosci. Res., 62, 75–83. [DOI] [PubMed] [Google Scholar]
- McDonald P.H., Chow,C.W., Miller,W.E., Laporte,S.A., Field,M.E., Lin,F.T., Davis,R.J. and Lefkowitz,R.J. (2000) β-Arrestin 2: a receptor-regulated MAPK scaffold for the activation of JNK3. Science, 290, 1574–1577. [DOI] [PubMed] [Google Scholar]
- Mota M., Reeder,M., Chernoff,J. and Bazenet,C.E. (2001) Evidence for a role of mixed lineage kinases in neuronal apoptosis. J. Neurosci., 21, 4949–4957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nihalani D., Merritt,S. and Holzman,L.B. (2000) Identification of structural and functional domains in mixed lineage kinase dual leucine zipper-bearing kinase required for complex formation and stress-activated protein kinase activation. J. Biol. Chem., 275, 7273–7279. [DOI] [PubMed] [Google Scholar]
- Patterson C. (2002) A new gun in town: the U box is a ubiquitin ligase domain. Sci. STKE, 2002, PE4. [DOI] [PubMed] [Google Scholar]
- Ridley A.J. (2001) Cyclin’ round the cell with Rac. Dev. Cell, 1, 160–161. [DOI] [PubMed] [Google Scholar]
- Saporito M.S., Thomas,B.A. and Scott,R.W. (2000) MPTP activates c-Jun NH(2)-terminal kinase (JNK) and its upstream regulatory kinase MKK4 in nigrostriatal neurons in vivo. J. Neurochem., 75, 1200–1208. [DOI] [PubMed] [Google Scholar]
- Tanaka S. and Hanafusa,H. (1998) Guanine-nucleotide exchange protein C3G activates JNK1 by a ras-independent mechanism. JNK1 activation inhibited by kinase negative forms of MLK3 and DLK mixed lineage kinases. J. Biol. Chem., 273, 1281–1284. [DOI] [PubMed] [Google Scholar]
- Tapon N. and Hall,A. (1997) Rho, Rac and Cdc42 GTPases regulate the organization of the actin cytoskeleton. Curr. Opin. Cell Biol., 9, 86–92. [DOI] [PubMed] [Google Scholar]
- Tapon N., Nagata,K., Lamarche,N. and Hall,A. (1998) A new rac target POSH is an SH3-containing scaffold protein involved in the JNK and NF-κB signalling pathways. EMBO J., 17, 1395–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teramoto H., Coso,O.A., Miyata,H., Igishi,T., Miki,T. and Gutkind,J.S. (1996) Signaling from the small GTP-binding proteins Rac1 and Cdc42 to the c-Jun N-terminal kinase/stress-activated protein kinase pathway. A role for mixed lineage kinase 3/protein-tyrosine kinase 1, a novel member of the mixed lineage kinase family. J. Biol. Chem., 271, 27225–27228. [DOI] [PubMed] [Google Scholar]
- Tournier C. et al. (2000) Requirement of JNK for stress-induced activation of the cytochrome c -mediated death pathway. Science, 288, 870–874. [DOI] [PubMed] [Google Scholar]
- Trotter L. et al. (2002) Mitogen-activated protein kinase kinase 7 is activated during low potassium-induced apoptosis in rat cerebellar granule neurons. Neurosci. Lett., 320, 29–32. [DOI] [PubMed] [Google Scholar]
- Troy C.M., Rabacchi,S.A., Xu,Z., Maroney,A.C., Connors,T.J., Shelanski,M.L. and Greene,L.A. (2001a) β Amyloid-induced neuronal apoptosis requires c-Jun N-terminal kinase activation. J. Neurochem., 77, 157–164. [DOI] [PubMed] [Google Scholar]
- Troy C.M., Rabacchi,S.A., Hohl,J.B., Angelastro,J.M., Greene,L.A. and Shelanski,M.L. (2001b) Death in the balance: alternative participation of the caspase-2 and -9 pathways in neuronal death induced by nerve growth factor deprivation. J. Neurosci., 21, 5007–5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmarsh A.J. et al. (2001) Requirement of the JIP1 scaffold protein for stress-induced JNK activation. Genes Dev., 15, 2421–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z., Dickens,M., Raingeaud,J., Davis,R.J. and Greenberg,M.E. (1995) Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science, 270, 1326–1331. [DOI] [PubMed] [Google Scholar]
- Xu Z., Maroney,A.C., Dobrzanski,P., Kukekov,N.V. and Greene,L.A. (2001) The MLK family mediates c-Jun N-terminal kinase activation in neuronal apoptosis. Mol. Cell. Biol., 21, 4713–4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D.D., Kuan,C.Y., Whitmarsh,A.J., Rincon,M., Zheng,T.S., Davis,R.J., Rakic,P. and Flavell,R.A. (1997) Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature, 389, 865–870. [DOI] [PubMed] [Google Scholar]
- Yasuda J., Whitmarsh,A.J., Cavanagh,J., Sharma,M. and Davis,R.J. (1999) The JIP group of mitogen-activated protein kinase scaffold proteins. Mol. Cell. Biol., 19, 7245–7254. [DOI] [PMC free article] [PubMed] [Google Scholar]