

Abstract
Arabidopsis TOM1 (AtTOM1) and TOM2A (AtTOM2A) are integral membrane proteins genetically identified to be necessary for efficient intracellular multiplication of tobamoviruses. AtTOM1 interacts with the helicase domain polypeptide of tobamovirus-encoded replication proteins and with AtTOM2A, suggesting that both AtTOM1 and AtTOM2A are integral components of the tobamovirus replication complex. We show here that AtTOM1 and AtTOM2A proteins tagged with green fluorescent protein (GFP) are targeted to the vacuolar membrane (tonoplast)-like structures in plant cells. In subcellular fractionation analyses, GFP–AtTOM2A, AtTOM2A and its tobacco homolog NtTOM2A were predominantly fractionated to low-density tonoplast-rich fractions, whereas AtTOM1–GFP, AtTOM1 and its tobacco homolog NtTOM1 were distributed mainly into the tonoplast-rich fractions and partially into higher-buoyant-density fractions containing membranes from several other organelles. The tobamovirus-encoded replication proteins were co-fractionated with both NtTOM1 and viral RNA-dependent RNA polymerase activity. The replication proteins were also found in the fractions containing non-membrane-bound proteins, but neither NtTOM1 nor the polymerase activity was detected there. These observations suggest that the formation of tobamoviral RNA replication complex occurs on TOM1-containing membranes and is facilitated by TOM2A.
Keywords: host factor/replication complex/tobamovirus/vacuolar membrane
Introduction
The replication of positive-strand RNA viruses occurs via the negative-strand RNAs on the cytoplasmic surface of intracellular membranes. Brome mosaic virus (BMV) forms replication complexes on the endoplasmic reticulum (ER) membranes (Restrepo-Hartwig and Ahlquist, 1996; Restrepo-Hartwig and Ahlquist, 1999; Schwartz et al., 2002). Tobacco etch virus, cowpea mosaic virus and poliovirus induce proliferation or rearrangement of the ER membranes on which replication complexes are localized (Schaad et al., 1997; Carette et al., 2000; Egger et al., 2000; Rust et al., 2001). The replication complex of Semliki Forest virus is associated with the lysosomal or the endosomal membranes (Froshauer et al., 1988; Kujala et al., 2001). The replication of flock house virus, turnip yellow mosaic virus and alfalfa mosaic virus (AMV) occurs in association with the mitochondrial outer membranes, the chloroplast outer membranes and the vacuolar membranes, respectively (Miller et al., 2001; Prod’homme et al., 2001; van der Heijden et al., 2001). Thus the sites of viral RNA synthesis vary depending on the virus, and the replication complexes are tethered on the membranes in their specific manners. At present, the mechanisms by which each viral replication complex is associated with specific membranes or the precise structures of the replication complexes have not been revealed.
The tobamovirus group belongs to the alphavirus-like superfamily of positive-strand RNA viruses (Buck, 1996). The genome of a tobamovirus encodes non-structural proteins with an approximate molecular mass of 130 kDa (130K protein), its read-through product with an approximate molecular mass of 180 kDa (180K protein), a 30 kDa non-structural protein known as movement protein (MP), which is required for the cell-to-cell movement of the virus, and the coat protein (CP). The 130K and 180K proteins (the 130K/180K proteins) are both involved in viral RNA replication (Ishikawa et al., 1986), whereas MP and CP are dispensable for replication (Meshi et al., 1987; Takamatsu et al., 1987). The 130K protein harbors domains implicated in RNA capping and helicase functions, and the read-through region of the 180K protein contains an RNA polymerase motif. These domains are conserved among the α-like viruses, implying that these viruses replicate through similar mechanisms. The 130K/180K proteins are known to be contained in a membrane-bound RNA-dependent RNA polymerase (RdRp) complex (Osman and Buck, 1996). Immunofluorescent microscopy of tobamovirus-infected plant cells suggested that these replication proteins are localized on the ER membranes (Heinlein et al., 1998; Más and Beachy, 1999), whereas immunoelectron microscopy suggested that they are localized in the cytoplasmic inclusion bodies induced by tobamovirus infection (Hills et al., 1987; Saito et al., 1987).
Arabidopsis thaliana TOM1 (AtTOM1) and TOM2A (AtTOM2A) have been suggested as host factors involved in intracellular multiplication of tobamoviruses, since inactivation of either the AtTOM1 or the AtTOM2A gene results in a decreased multiplication of tobamoviruses (Ishikawa et al., 1993; Ohshima et al., 1998). AtTOM1 and AtTOM2A are predicted to be seven-pass and four-pass transmembrane proteins, respectively, but neither of them possesses well-known sorting signals to specific organelles (Yamanaka et al., 2000; Tsujimoto et al., 2003). AtTOM1 has been shown to interact with the tobamovirus-encoded 130K/180K proteins (Yamanaka et al., 2000). Recent studies have suggested that AtTOM2A interacts both with itself and with AtTOM1 (Tsujimoto et al., 2003). Taking these observations together, we propose that AtTOM1 and AtTOM2A are constituents of the replication complex of tobamoviruses and play important roles in the formation of the complex on the membranes where they co-localize. Furthermore, we have identified the homologs of AtTOM1 and AtTOM2A in Nicotiana tabacum (NtTOM1 and NtTOM2A, respectively) and found that NtTOM1 also interacts with the tobamovirus-encoded 130K/180K proteins (unpublished data).
In this study, the subcellular localizations of At- and NtTOM1 and At- and NtTOM2A were determined by combined analyses of confocal laser scanning microscopy and iodixanol density-gradient centrifugation. These analyses revealed that both TOM1 and TOM2A are mainly localized to the vacuolar membranes (i.e. the tonoplasts). Tobamovirus-encoded membrane-bound 130K/180K proteins were co-fractionated with the activity to synthesize the viral RNA as well as with NtTOM1. Our findings suggest that the replication complex of tobamoviruses is localized at least in part on the tonoplasts and that TOM1 plays a critical role in the formation of the complex on specific membranes.
Results
Subcellular localization of GFP–tagged TOM proteins
To study the intracellular localization of TOM1 and TOM2A in living cells, we used green fluorescent protein (GFP) as a reporter. GFP was fused in frame to either the N- or C-termini of AtTOM1 and AtTOM2A. The resulting constructs, GFP–AtTOM1, AtTOM1–GFP, GFP–AtTOM2A, and AtTOM2A–GFP, were expressed transiently in epidermal cells of A.thaliana leaves by microprojectile bombardment. The GFP fluorescence in the cells was analyzed under a confocal laser scanning microscope. Figure 1A shows a representative image of an epidermal cell transiently expressing free GFP. In addition to its preferential accumulation inside the nucleus, GFP fluorescence was observed in the area lining the plasma membrane. The vacuoles of mature leaf epidermal cells generally occupy most of the intracellular space and press the cytoplasm against the plasma membrane. Therefore the observed fluorescence lining the plasma membrane is likely to represent cytoplasmic localization. Of the two GFP-fused AtTOM1 constructs, only AtTOM1–GFP successfully showed the GFP fluorescence (Figure 1B; data not shown). The fluorescence was found in the area lining the plasma membrane but not in the nucleus. However, it is difficult to specify whether the observed fluorescence is derived from the cytoplasm, tonoplast, plasma membrane or other organelles. Essentially the same fluorescent pattern was observed in cells transiently expressing either GFP–AtTOM2A or AtTOM2A–GFP (Figure 1C; data not shown). AtTOM2A is predicted to possess a putative farnesylation signal (Tsujimoto et al., 2003). This signal functions only at the C-terminus of a protein, so that GFP– AtTOM2A but not AtTOM2A–GFP should preserve the functional signal. Considering the possible importance of AtTOM2A farnesylation, GFP–AtTOM2A was used for further analyses. The distribution patterns of AtTOM1– GFP and GFP–AtTOM2A in epidermal cells of Nicotiana benthamiana and N.tabacum were the same as in A.thaliana cells when transiently expressed (data not shown).
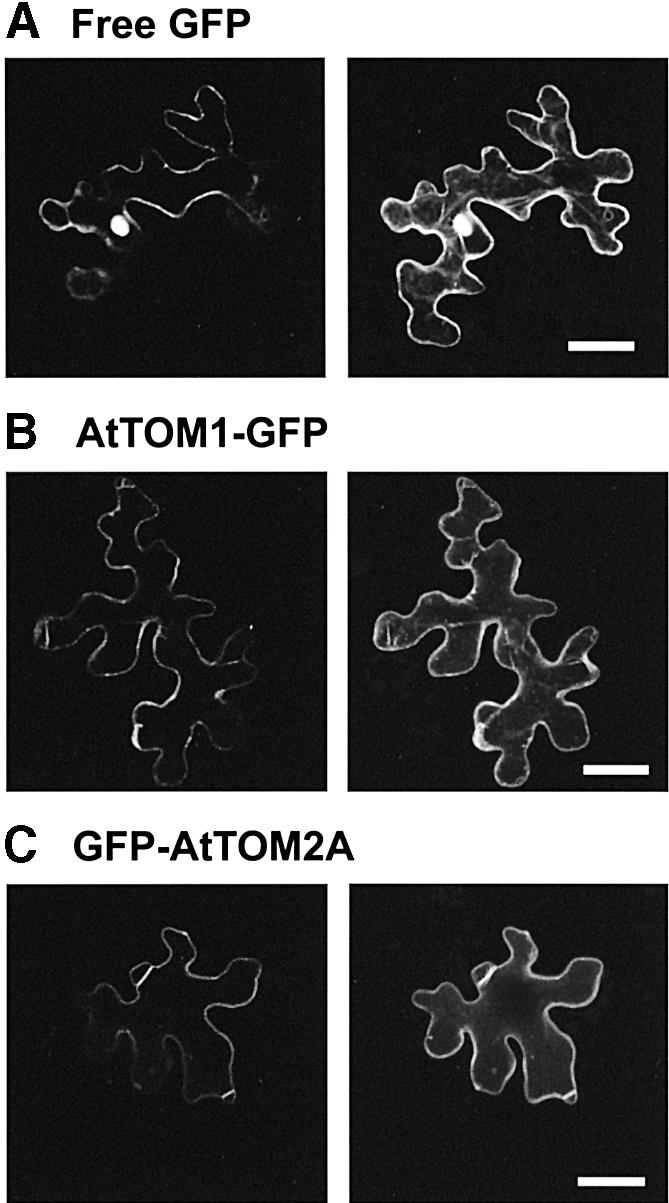
Fig. 1. Confocal laser scanning micrographs of A.thaliana epidermal cells transiently expressing (A) GFP, (B) AtTOM1–GFP and (C) GFP–AtTOM2A. A single optical section of the cell is shown on the left, and a projection of 10 × 1 µm confocal optical sections through the cell is shown on the right. Note that the leaf epidermal cells have a ‘jigsaw-puzzle-like’ appearance and possess huge vacuoles that occupy most of the intracellular space. Scale bars, 25 µm.
Nicotiana tabacum BY-2 suspension-cultured cells are known to have relatively undeveloped vacuoles compared with mature leaf epidermal cells. Owing to the difficulty in determining the subcellular localization of AtTOM1 and AtTOM2A in epidermal cells, we used BY-2 cells for further investigation. When BY-2 cells stably expressing AtTOM1–GFP and GFP–AtTOM2A were analyzed by confocal laser scanning microscopy, strikingly clear images were obtained compared with those of leaf epidermal cells. In cells expressing AtTOM1–GFP (Figure 2A) and GFP–AtTOM2A (Figure 2B), the GFP fluorescence was targeted to the intracellular membranes of round-shaped structures. These structures were indistinguishable from the fluorescent membrane structures observed in BY-2 cells transiently expressing the GFP-fused AtTOM proteins obtained by microprojectile bombardment (data not shown).

Fig. 2. Confocal laser scanning micrographs of transgenic BY-2 cells expressing (A) AtTOM1–GFP, (B) GFP–AtTOM2A, (C) GFP–VM23, (D) GmMan1–GFP, (E) GFP–PAQ1 and (F) mGFP5ER. A single optical section through several cells is displayed on the lower left, and its Nomarski image is shown on the upper left. A high-magnification image is shown on the right. Scale bars, 10 µm.
To identify the observed membrane structures, we analyzed transgenic BY-2 cells stably expressing GFP-tagged in vivo markers for several major organelles, including a tonoplast protein GFP–VM23 (Maeshima, 1992; T.Tsuchiya, S.Suga and M.Maeshima, unpublished data), a Golgi membrane protein GmMan1–GFP (Nebenführ et al., 1999), a plasma membrane protein GFP–PAQ1 (Suga et al., 2001; T.Tsuchiya, S.Suga and M.Maeshima, unpublished data) and an ER-localized GFP marker protein mGFP5ER (Haseloff and Siemering, 1998). It was found that the round-shaped membrane structures shown in the cells expressing AtTOM1–GFP and GFP–AtTOM2A have a strong resemblance to those observed for GFP–VM23 (Figure 2C). Such structures did not appear in cells expressing GmMan1–GFP (Figure 2D), GFP–PAQ1 (Figure 2E) or mGFP5ER (Figure 2F). Furthermore, cells expressing the GFP-tagged AtTOM proteins did not show the ER-like reticulate structures, the Golgi-like fluorescent spots or a fluorescent line observed between two neighboring GFP–PAQ1-expressing cells.
The vital membrane stain FM4-64, which is known as the endocytic tracer, visualizes the vacuolar membranes in yeast (Vida and Emr, 1995) and plant cells (Ueda et al., 2001; Kutsuna and Hasezawa, 2002). In order to verify that AtTOM1–GFP and GFP–AtTOM2A reside on the tonoplast, the transgenic BY-2 cells were pulse-labeled with FM4-64. As shown in Figure 3A, E and I, FM4-64 first labeled the plasma membranes (red signals), which did not overlap the membrane structures visualized by the GFP-fused TOM proteins or the tonoplast marker GFP–VM23 (green signals). Approximately 10 h after labeling, the fluorescence of FM4-64 transported to the tonoplasts by endocytosis overlapped perfectly with the GFP fluorescence in cells expressing AtTOM1–GFP (Figure 3B–D) and GFP–AtTOM2A (Figure 3F–H). The complete overlap of the signals of FM4-64 and GFP–VM23 (Figure 3J–L) confirmed the tonoplast localization of FM4-64. These observations clearly demonstrated that AtTOM1–GFP and GFP–AtTOM2A are both localized mainly to the tonoplasts. This also implies that the fluorescent structures observed in leaf epidermal cells expressing the GFP-fused TOM proteins (see Figure 1B and C) represent the tonoplasts rather than the plasma membranes or appressed cytoplasm.

Fig. 3. Confocal laser scanning micrographs of FM4-64-treated transgenic BY-2 cells expressing (A–D) AtTOM1–GFP, (E–H) GFP–AtTOM2A and (I–L) GFP–VM23. Cells are shown at ∼20 min (A, E, I) and 10 h (B–D, F–H, J–L) after labeling. The GFP and FM4-64 signals are shown as green and red, respectively. In the merged images (A, D, E, H, I, L), the overlapping signals of GFP and FM4-64 appear as yellow. Scale bars, 10 µm.
Subcellular fractionation analysis of TOM proteins
For further investigation of the localization of the TOM proteins, we employed membrane flotation analysis using equilibrium iodixanol density-gradient centrifugation. Through iodixanol gradients, membranes and membrane-associated proteins of different intracellular compartments are finely separated according to their buoyant density, as previously reported (see, for example, Tritel and Resh 2000; Kim et al. 2002). First, we analyzed the distribution of the GFP-fused AtTOM proteins and tobacco TOM homologs, NtTOM1 and NtTOM2A, in the gradients to confirm that the localization of the GFP fusions shown by confocal microscopy reflects that of the non-tagged authentic TOM proteins. The transgenic BY-2 cells expressing AtTOM1–GFP or GFP–AtTOM2A and the non-transgenic BY-2 cells were disrupted and centrifuged at a low speed to pellet and remove cell debris. The lysates were loaded into the bottom of the gradients and centrifuged at 114 000 g for 16 h. The gradients were then fractionated and analyzed by immunoblotting. An antibody against GFP was used to detect AtTOM1–GFP and GFP–AtTOM2A. NtTOM1 and NtTOM2A were detected using antisera against AtTOM1 and AtTOM2A, respectively, and the fractionation patterns were compared.
Figure 4A and B illustrates representative fractionation patterns of AtTOM1–GFP and NtTOM1, respectively. The anti-GFP antibody detected bands migrating around a position corresponding to a molecular mass of 43 kDa, which was smaller than the expected value (∼50 kDa). These bands were also detected by anti-AtTOM1 antiserum (data not shown). Both AtTOM1–GFP and NtTOM1 were detected as double bands, suggesting that TOM1 may undergo modifications. The fractionation pattern of AtTOM1–GFP was similar to that of NtTOM1. Both proteins predominantly floated to the top fraction of the gradient (fraction 1, referred to as Fraction T), while they were also detected in fractions 2–15 with relatively strong signals in the middle part of the gradient (fractions 9–11, referred to as Fraction M). The bands were almost undetectable in the bottom part where the cell lysates were originally loaded (fractions 16–18, referred to as Fraction L). Fraction L contains most of the cytoplasmic non-membrane-bound proteins (data not shown), indicating that TOM1 is exclusively a membrane-bound protein.
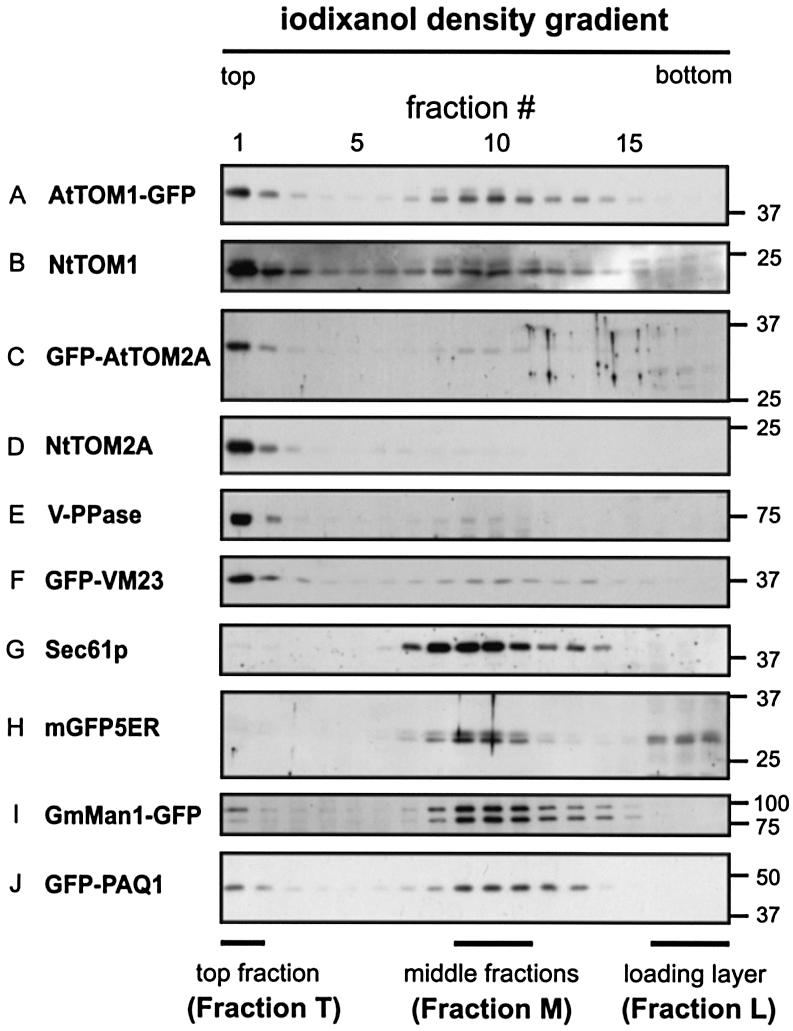
Fig. 4. Equilibrium iodixanol density-gradient analysis of the TOM proteins in BY-2 cells. Lysates from non-transgenic BY-2 cells (B, D, E and G) and transgenic BY-2 cells expressing (A) AtTOM1–GFP, (C) GFP–AtTOM2A, (F) GFP–VM23, (H) mGFP5ER, (I) GmMan1– GFP and (J) GFP–PAQ1 were subjected to density gradient centrifugation. Fractions (1–18, from top to bottom of a gradient) were analyzed by SDS–PAGE and immunoblotting. An anti-GFP antibody was used to detect the GFP-fused proteins. NtTOM1 (B), NtTOM2A (D), V-PPase (E) and Sec61p (G) were detected with the corresponding antibodies. The top (fraction 1), middle (fractions 9–11) and sample-loaded (fractions 16–18) parts are referred to as Fraction T, Fraction M and Fraction L, respectively, as shown below the panels. The positions of molecular mass markers (kDa) are indicated on the right.
The fractionation patterns of GFP–AtTOM2A and NtTOM2A are shown in Figure 4C and D, respectively. Both proteins showed strong signals in Fraction T. They were weakly detected in all other membrane fractions (fractions 2–15) when overexposed (data not shown) but were absent in Fraction L, again indicating that they were completely membrane-bound. In GFP–AtTOM2A, the size of the fusion protein detected by the anti-GFP antibody (∼34 kDa) was smaller than the expected value (∼50 kDa). Nevertheless, the fractionation pattern resembled that of NtTOM2A, indicating that cleavage of GFP–AtTOM2A, if it occurred, did not affect its localization. Similar distribution profiles between AtTOM1– GFP and NtTOM1 and between GFP–AtTOM2A and NtTOM2A suggest that tagging with the GFP reporter did not alter the subcellular localization of AtTOM1 and AtTOM2A. In addition, these results indicate that TOM1 and TOM2A are mainly associated with the membranes that floated to the top part of the iodixanol gradients, while TOM1 is also localized to the membranes contained in Fraction M.
Next, fractionation patterns of marker proteins for the tonoplasts, the ER, and the Golgi and plasma membranes were analyzed. As illustrated in Figure 4E and F, two typical tonoplast proteins, H+-translocating inorganic pyrophosphatase (V-PPase) and GFP-tagged VM23 (see Figure 2C), were mainly detected in Fraction T. Thus, both TOM1 and TOM2A are predominantly distributed to the tonoplast-rich fractions. On the other hand, an ER membrane protein Sec61p was found to be fractionated mainly to Fraction M (Figure 4G), although it was faintly detected in Fraction T when overexposed (data not shown). Similarly, the ER marker protein mGFP5ER (see Figure 2F) was also predominantly fractionated into Fraction M (Figure 4H). This lumenal soluble protein was also detected in Fraction L, probably due to a leak upon disruption of the ER membranes. The fractionation pattern of the Golgi marker GmMan1–GFP (see Figure 2D) is shown in Figure 4I. The anti-GFP antibody detected two bands around the position of the expected molecular mass, but both bands showed a similar fractionation profile. They were predominantly found in Fraction M, although they were also weakly detected in Fraction T. The overall fractionation pattern of the Golgi membranes was found to be similar to that of the ER membranes. The plasma membrane marker GFP–PAQ1 (see Figure 2E) was distributed in a wide range of the membrane fractions and showed relatively strong signals in Fractions T and M (Figure 4J). Taken together, Fraction T, where both TOM1 and TOM2A were mainly detected, was abundant in the tonoplasts, while Fraction M, in which TOM1 but not TOM2A was detected at a significant level, contained membranes from several different organelles.
To rule out the possibility that the buoyant densities of intracellular membranes in BY-2 cells differ from those in A.thaliana cells, we examined the fractionation patterns of AtTOM1, AtTOM2A, V-PPase and Sec61p using A.thaliana Col-0 suspension-cultured cells. The fractionation patterns of these proteins were identical with their respective patterns obtained using BY-2 cells (data not shown). All these findings suggest that both NtTOM1 (AtTOM1) and NtTOM2A (AtTOM2A) are localized predominantly to the tonoplasts. This is consistent with the confocal images illustrating the subcellular localization of AtTOM1–GFP and GFP–AtTOM2A (see Figure 2A and B). The TOM1-bearing membrane(s) in Fraction M remains to be determined.
Co-fractionation of NtTOM proteins and tobamovirus-encoded replication proteins
Previously, we have detected the interaction between membrane-associated AtTOM1 and the helicase domain polypeptides of the 130K/180K proteins of two tobamoviruses, TMV-Cg and TMV-L, using the yeast Sos recruitment system (Yamanaka et al., 2000, 2002). Recently, we have found that AtTOM2A interacts with AtTOM1 in yeast split ubiquitin system (Tsujimoto et al., 2003). Based on these observations, we have proposed that the TOM proteins and tobamovirus 130K/180K proteins are integral parts of the tobamoviral RNA replication complex on membranes. To test this possibility, we investigated subcellular distribution of the 130K/180K proteins of TMV-L and compared their localization with that of the TOM proteins. Protoplasts isolated from BY-2 cells were inoculated with TMV-L RNA. Cells at 6 h post-inoculation (h.p.i.) were disrupted and subjected to iodixanol gradient flotation analysis. The antibody against the helicase domain polypeptide of TMV-L was used to detect both 130K and 180K proteins.
In mock-inoculated protoplasts (Figure 5A), Sec61p, V-PPase, NtTOM1 and NtTOM2A showed similar fractionation patterns to those described for BY-2 cells (see Figure 4). In TMV-L-inoculated protoplasts, two different types of fractionation patterns were obtained. Representative patterns of four out of seven independent inoculations are shown in Figure 5B (‘Infect 1’), while those of the other three inoculations are shown in Figure 5C (‘Infect 2’).
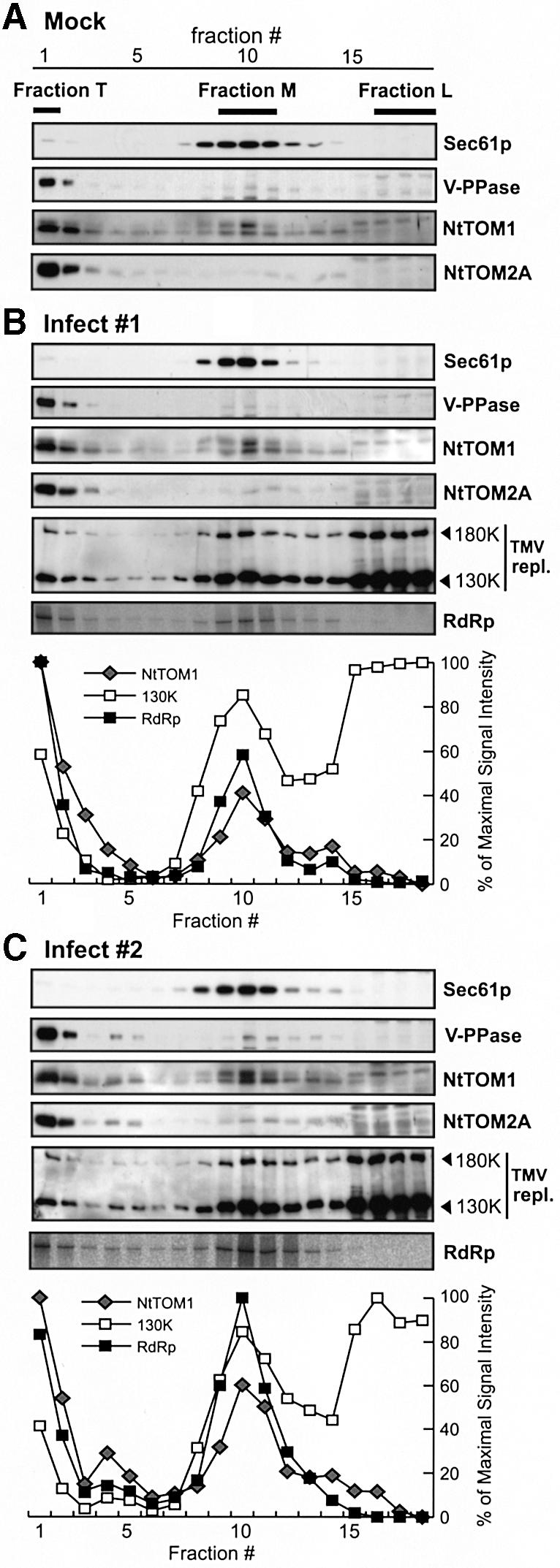
Fig. 5. Equilibrium iodixanol density-gradient analysis of the 130K/180K proteins and the viral RdRp activity of TMV-L-inoculated BY-2 protoplasts. Lysates from mock- or TMV-L-inoculated protoplasts harvested at 6 h.p.i. were subjected to density-gradient centrifugation. Fractions were collected and analyzed by SDS–PAGE and immunoblotting. (A) Immunoblot analysis of Sec61p, V-PPase and the TOM proteins in mock-inoculated protoplasts. (B and C) Immunoblot analysis of Sec61p, V-PPase, TOM proteins, and the 180K and 130K replication proteins in TMV-L-inoculated protoplasts (see text for definition of ‘Infect 1’ and ‘Infect 2’). The RF RNA synthesized in the RdRp assay of each fraction is shown in the bottom panel of (B) and (C). The band intensity for NtTOM1, the 130K protein and the RF RNA was quantitated and represented graphically.
In ‘Infect 1’, the patterns of Sec61p, V-PPase and the TOM proteins were similar to those in mock-inoculated protoplasts. While most of the 130K/180K proteins of TMV-L remained in Fraction L, they also floated up to the membrane fractions (fractions 1–15). Among these fractions, strong signals of the 130K/180K proteins were detected in Fractions T and M, resembling the fractionation pattern of NtTOM1.
In ‘Infect 2’, distinctive fractionation patterns of NtTOM1, NtTOM2A, V-PPase and the 130K/180K proteins were observed (Figure 5C). In addition to the signals observed in ‘Infect 1’, strong signals for NtTOM1, NtTOM2A and V-PPase, but not Sec61p, were detected in fractions 4 and 5. Since these proteins were not detected, or detected at only a very low level, in the corresponding fractions of mock-inoculated protoplasts, it appeared that the buoyant density of part of the tonoplasts was altered upon infection. Remarkably, relatively large amounts of the 130K/180K proteins were also detected in fractions 4 and 5, indicating that the 130K/180K proteins are tethered to the tonoplast-derived membranes found in these fractions.
Subcellular distribution of RNA-dependent RNA polymerase activity
We found that the membrane-associated 130K/180K proteins of TMV-L were abundant in Fractions T and M. In addition, large amounts of the 130K/180K proteins also existed in Fraction L, where non-membrane-bound proteins remained. To investigate whether each fraction contained the functional TMV-L replication complex to synthesize the viral RNA, the fractions shown in Figure 5B (‘Infect 1’) and C (‘Infect 2’) were subjected to an RdRp assay to detect the incorporation of ribonucleotides into the double-stranded replicative form (RF) RNA of TMV-L.
The bottom panels (marked ‘RdRp’) of Figure 5B and C show the 32P-labeled RF RNA produced in each fraction. No RF RNA bands were detected in mock-inoculated protoplast samples (data not shown), indicating that the bands were TMV-L infection-specific. In both ‘Infect 1’ and ‘Infect 2’, the viral RdRp activity (represented by the amount of [32P]RF RNA) was mainly present in Fractions T and M. Furthermore, fractions 4 and 5 in ‘Infect 2’ showed higher RdRp activity than those in ‘Infect 1’ (Figure 5B and C). The RdRp activity was not detected in Fraction L in either case despite the abundance of the 130K/180K proteins. The quantification of the signal intensity of the RF RNA and the protein bands clearly demonstrated that the peaks for the viral RdRp activity, NtTOM1 and the membrane-associated 130K replication protein were located in the same fractions (see the quantification graphs in Figure 5B and C). These observations suggest that the active tobamovirus replication complex is localized on the TOM1-bearing membranes.
Discussion
AtTOM1 and AtTOM2A, encoding putative integral membrane proteins, are required for efficient multiplication of tobamoviruses (Yamanaka et al., 2000; Tsujimoto et al., 2003). In this study, we performed confocal microscopic analyses of the GFP-tagged TOM proteins and subcellular fractionation analyses of the TOM proteins to investigate the subcellular localization of these host factors and their relevance to the localization of the tobamovirus 130K/180K proteins and the viral RdRp activity. The combined results demonstrated that TOM1 and TOM2A were pre dominantly targeted to the vacuolar membranes, although TOM1 was also localized on other uncharacterized membranes (Figure 4A and B, Fraction M). Importantly, the subcellular fractionation pattern of the viral RdRp activity coincided well with that of both NtTOM1 and the membrane-bound 130K/180K proteins. These findings support the hypothesis that the replication complex of tobamoviruses is tethered to the membranes in association with TOM1.
TOM proteins are localized to the tonoplasts
The GFP-tagged AtTOM proteins were localized to the tonoplast-like structures in tobacco BY-2 cells (Figure 2), and probably in leaf epidermal cells of A.thaliana, N.tabacum and N.benthamiana (Figure 1; data not shown). The membrane structures visualized by the GFP-tagged AtTOM proteins were also labeled with FM4-64 (Figure 3), supporting the hypothesis that they are tonoplasts. Because the fractionation patterns of AtTOM1–GFP and GFP–AtTOM2A in membrane flotation analysis showed a close resemblance to those of non-tagged NtTOM1 and NtTOM2A, respectively (Figure 4), it is unlikely that tagging with GFP altered their intracellular localization. In iodixanol gradients, the TOM proteins and their GFP-tagged derivatives were all abundantly present in the tonoplast-rich low-buoyant-density membrane fractions (Fraction T). This confirms that TOM1 and TOM2A are mainly localized to the tonoplasts. The simultaneous appearance of NtTOM1, NtTOM2A and the tonoplast marker V-PPase in fractions 4 and 5 in ‘Infect 2’ (Figure 5C) is in good agreement with the assumption that the TOM proteins are tonoplast proteins. However, since At- and NtTOM1 were also found in Fraction M, where membranes derived from various organelles were fractionated, it seems that TOM1 possesses another localization in addition to the tonoplasts. The TOM1-bearing organelle(s) in Fraction M is yet to be determined. Since most tonoplast proteins are synthesized on the ER membranes and transported to the tonoplasts via the Golgi stacks and other compartments (Jiang and Rogers, 1998; 1999), TOM1 found in Fraction M may represent the protein in such intermediate compartments.
Subcellular localization of TMV-L replication proteins and replication complex
The accumulation of the negative-strand RNA of TMV-L reaches the maximal level at ∼6 h.p.i. in infected BY-2 protoplasts (Ishikawa et al., 1991). Therefore the number of replication complexes of TMV-L sequestering the negative-strand RNA is expected to reach the maximal level by 6 h.p.i., and the excess 130K/180K proteins produced after 6 h.p.i. may not participate in the formation of the active replication complex. Based on this assumption, we investigated subcellular distribution of the 130K/180K proteins and the viral RdRp activity at 6 h.p.i.
The 130K/180K proteins and the RdRp activity were all abundantly detected in Fraction T. As discussed above, Fraction T contained large amounts of NtTOM1 and NtTOM2A as well as the tonoplast marker proteins. Furthermore, in fractions 4 and 5 of several TMV-L-inoculated protoplast samples (Figure 5C, ‘Infect 2’), the signals for NtTOM1, NtTOM2A, the tonoplast marker V-PPase, the 130K/180K proteins and the viral RdRp activity were stronger than in the corresponding fractions of ‘Infect 1’ sample (Figure 5B). These results strongly suggest that the replication complex of TMV-L is associated with the tonoplasts or tonoplast-derived membranes where NtTOM1 and NtTOM2A coexist. Remodeling or proliferation of intracellular membranes of host cells has been reported for many positive-strand RNA viruses (e.g. Schlegel et al., 1996; Schaad et al., 1997; Reichel and Beachy, 1998; Carette et al., 2000). Therefore the signals detected in fractions 4 and 5 of ‘Infect 2’ may result from a reorganization of the tonoplasts occurring upon infection with TMV-L. Since the alteration of the fractionation patterns of the tonoplast-associated proteins was not observed in all TMV-L-inoculated protoplast samples, the reorganization may take place during a very short period of time or the altered patterns may reflect slight differences of experimental conditions.
The 130K/180K proteins and viral RdRp activity were also detected in Fraction M containing the membranes derived from multiple organelles (Figure 5B and C). This observation suggests that the active tobamovirus replication complex is also associated with non-tonoplast membranes. It is noteworthy that Fraction M contained NtTOM1 but not NtTOM2A at significant levels (see the final section of Discussion).
While part of the 130K/180K proteins was associated with the membranes, they were also abundantly detected in the loading layer of the iodixanol gradients (Fraction L) containing non-membrane-bound proteins. This observation is in contrast with the fact that most of the 1a replication proteins of BMV and AMV are strongly associated with membranes (den Boon et al., 2001; van der Heijden et al., 2001). This may represent a difference in the number of molecules that tether the viral replication proteins to the relevant membranes, or it may be due to a difference of affinity of the viral replication proteins to the surface of the membranes. Since Fraction L did not show a detectable level of viral RdRp activity (Figure 5B and C), tobamoviral RNA synthesis probably takes place only in association with membranes.
Previous studies with immunofluorescent microscopy suggested that the 130K/180K proteins of a tobamovirus are localized on the ER membranes (Heinlein et al., 1998; Más and Beachy, 1999). It was demonstrated by immunoelectron microscopy that the 130K/180K proteins accumulate in cytoplasmic inclusions (Hills et al., 1987; Saito et al., 1987). The inclusion bodies contained tubular membrane structures probably derived from the ER (Hills et al., 1987). Therefore TMV-L replication complexes detected in Fraction M may be associated with the ER membranes. However, these precedent studies failed to find the tonoplast-associated 130K/180K proteins. This might have been because the 130K/180K proteins scattered on the surface of the tonoplasts were difficult to detect by immunofluorescent or immunoelectron microscopy. In addition, since most of the previous studies were performed using cells at later stages of tobamovirus infection, it is possible that the excess 130K/180K proteins produced in the infected cells obstructed the detection of the tonoplast-bound replication proteins.
Possible roles of TOM1 and TOM2A in tobamovirus replication complex formation
This study identified the tonoplast-associated tobamovirus replication complex fractionated into Fraction T and the replication complex associated with non-tonoplast uncharacterized membranes distributed into Fraction M. The importance of AtTOM1 in tobamovirus multiplication in vivo (Ishikawa et al., 1993; Yamanaka et al., 2002), and the interaction of At- and NtTOM1 with the 130K/180K proteins of tobamoviruses (Yamanaka et al., 2000; 2002; unpublished data) are consistent with the observation that both Fraction T and Fraction M also contained a large amount of TOM1. Taken together, these results suggest that it is very highly likely that TOM1 is an essential component of the tobamovirus replication complex.
Although the involvement of AtTOM2A in the multiplication of tobamoviruses has been demonstrated genetically (Ohshima et al., 1998; Tsujimoto et al., 2003), the fractionation pattern of NtTOM2A was not perfectly parallel to that of TMV-L RdRp activity; NtTOM2A was abundantly present in Fraction T but not in Fraction M, while both fractions showed high levels of TMV-L RdRp activity. A plausible explanation for the presence of the RdRp activity in Fraction M where NtTOM2A is almost absent, is that the formation of the tobamovirus replication complex is facilitated by, but does not absolutely require, TOM2A. When the intracellular concentration of the 130K/180K proteins is very low during the early stages of infection, tobamoviruses may require both TOM1 and TOM2A to build the replication complex efficiently. Hence the tonoplasts bearing both TOM1 and TOM2A would be preferentially selected as a site of the tobamovirus replication. On the other hand, when the intracellular concentration of the 130K/180K proteins becomes high in later stages of infection, the replication complex would also be formed without TOM2A on the TOM1-bearing membranes that are fractionated into Fraction M. This possibility will be tested by investigating a temporal change of the fractionation patterns of the tobamovirus 130K/180K proteins and the RdRp activity and/or by analyzing their fractionation patterns using tom2a mutant cells.
Materials and methods
Plasmid construction
Plasmid psmRS-GFP harboring modified GFP sequence (Davis and Vierstra, 1996) was utilized to generate plasmids to express GFP-tagged AtTOM1 and AtTOM2A derivatives. A BamHI restriction enzyme recognition site existing upstream of the start codon of the GFP open reading frame (ORF) was used to construct pAtTOM1-GFP and pAtTOM2A-GFP. Because of the presence of a BamHI site in the coding sequence of the AtTOM1 gene, the polymerase chain reaction (PCR) was used to introduce a BglII site upstream of the start codon and to replace the stop codon with a linker sequence encoding four glycine codons followed by a BglII site. The resulting PCR products were digested with BglII and inserted into BamHI-digested psmRS-GFP to yield pAtTOM1-GFP. The sequence containing the AtTOM2A ORF was amplified by PCR using primers designed to create the BamHI site upstream of the start codon and to replace the stop codon with a sequence containing the BamHI site. The PCR product was digested with BamHI and cloned into psmRS-GFP, yielding pAtTOM2A-GFP.
To facilitate the construction of pGFP-AtTOM1 and pGFP-AtTOM2A, plasmid psmRS-GFP-NoStop was generated as follows. The 3′-terminal region of the GFP ORF was amplified by PCR using primers sRGFP314F (5′-CTCTTTCAAGGACGACGGGAAC-3′) and sRGFPG4ScR (5′-ATGAGCTCTCCACCTCCACCTTTGTATAGTTCATCCATC-3′) to remove the termination codon of the GFP sequence and to introduce four glycine codons immediately downstream of the GFP coding sequence and upstream of the SacI site. The PCR product was cloned back into psmRS-GFP using the ClaI and SacI sites, yielding psmRS-GFP-NoStop. AtTOM1 and AtTOM2A sequences were amplified by PCR using primers containing the sequence of SacI site immediately upstream of the start codons and downstream of the stop codons of AtTOM1 and AtTOM2A ORFs. The SacI-digested PCR products were cloned into psmRS-GFP-NoStop to generate pGFP-AtTOM1 and pGFP-AtTOM2A.
Culture conditions of BY-2 cells
Tobacco BY-2 (N.tabacum cv. Bright Yellow 2) cells were grown in a BY-2 growth medium (modified Linsmaier and Skoog medium; Nagata et al., 1992) at 26°C in the dark with constant shaking. Cells were maintained by subculturing into fresh medium at a dilution of ∼1:65 every 7 days. Cells at 3 days after subculturing were used for experiments.
Agrobacterium-mediated transformation of BY-2 cells
BY-2 cells stably expressing a Golgi marker, GmMan1–GFP, were kindly provided by Andreas Nebenführ (Nebenführ et al., 1999). To generate the transgenic BY-2 cell lines expressing AtTOM1–GFP and GFP– AtTOM2A, the expression cassettes of pAtTOM1-GFP and pGFP-AtTOM2A were cloned into pBI121 (Clontech) using the HindIII and EcoRI sites. The resulting constructs were transformed into Agrobacterium tumefaciens (strain EHA105; Hood et al., 1993) by electroporation. The transformed bacteria were co-cultivated with BY-2 cells for 2 days, and the transgenic BY-2 cells were selected on BY-2 agar medium containing 200 mg/l kanamycin and 500 mg/l carbenicillin. The transgenic BY-2 cells expressing GFP-tagged VM23 and PAQ1 were generated by Agrobacterium-mediated transformation with plasmids pMAT-GV and pMAT-GP, respectively (T.Tsuchiya, S.Suga and M.Maeshima, unpublished). The BamHI–SacI fragment harboring the GFP gene fused with the ER-targeting and the ER-retention signal sequences was obtained from plasmid pBImGFP5ER (Tamai and Meshi, 2001). This gene cassette was cloned between the BamHI and SacI sites of the pBI121 vector, and the resulting construct was used to generate the transgenic BY-2 expressing mGFP5ER.
Plant materials, microprojectile bombardment and confocal laser scanning microscopy
Microprojectile bombardment was performed using a Biolistic PDS-1000/He Particle Delivery System (Bio-Rad), essentially according to the manufacturer’s instructions. Fully expanded leaves of A.thaliana (L.) Heynh. ecotype Columbia (Col-0), N.benthamiana and N.tabacum (cv Xanthi nn), ∼4 weeks old, were harvested, placed on 0.4% agar plates and bombarded with a rupture-disk pressure of 900 p.s.i. at a target distance of ∼10 cm. In bombardment of BY-2 cells, the method described by Flynn et al. (1998) was used with some modifications. Briefly, BY-2 cells (3 days after subculturing) were spread on filter papers soaked with the fresh BY-2 growth medium in Petri dishes. After incubation for 1 h, the cells were bombarded with a rupture-disk pressure of 450 p.s.i. at a target distance of ∼10 cm. For each bombardment, 0.6 mg of gold particles (1 µm in diameter) coated with 1 µg of plasmid DNA were used. Bombarded leaves and BY-2 cells were incubated at 23°C in the dark. At 18 to 24 h after bombardment, they were analyzed for the GFP fluorescence with the 488 nm argon laser of a confocal laser scanning microscope LSM410 (Carl Zeiss), and the acquired images were processed using Photoshop 5.0 (Adobe).
FM4-64 staining of transgenic BY-2 cells
Transgenic BY-2 cells (3 days after subculturing) were incubated with 16 µM FM4-64 (Molecular Probes; 2 mM stock solution in dimethylsulfoxide) in the BY-2 growth medium for 5 min, washed twice with the fresh medium and incubated for ∼20 min or 10 h. The fluorescence of FM4-64 and GFP in the cells was analyzed with a 543 nm helium-neon laser and a 488 nm argon laser, respectively, using a confocal laser scanning microscope LSM410 (Carl Zeiss). The acquired images were processed using Photoshop 5.0 (Adobe).
Protoplast inoculation
Protoplasts were prepared from BY-2 cells and inoculated with TMV-L RNA as described previously (Watanabe et al., 1987).
Iodixanol density gradient analysis
BY-2 cells or protoplasts were collected and washed twice with 0.4 M mannitol. The cells were suspended in Homogenization Buffer (0.25 M sucrose, 1 mM EDTA, 10 mM HEPES-NaOH pH 7.4 and 10 mM DTT) containing protease inhibitor (one tablet of Complete™, Mini, EDTA-free, Roche Diagnostics, per 10 ml). BY-2 cells and protoplasts were homogenized using a Potter–Elvehjem (glass) homogenizer (15 strokes at ∼750 r.p.m.) and a tight-fitting Dounce homogenizer (50 strokes), respectively. The lysates were centrifuged at 400 g for 10 min in a microcentrifuge to pellet and remove the nuclei and cell debris. The lysates were immediately used for the following fractionation analysis or stored at –80°C.
Iodixanol (OptiPrep™, Nycomed, Roskilde, Denmark) density gradient analysis was performed essentially according to the manufacturer’s instructions. A working solution of 50% iodixanol (5:1 v/v mixture of OptiPrep™ and 0.25 M sucrose, 6 mM EDTA and 60 mM HEPES–NaOH pH 7.4) was diluted with the Homogenization Buffer to prepare a 30% iodixanol solution. Iodixanol continuous gradients were formed with 4.3 ml each of 0% (the Homogenization Buffer containing the protease inhibitor) and 30% iodixanol solutions in open-top centrifuge tubes (Seton Scientific; Part No. 7031) using Gradient Mate™ (BioComp Instruments Inc.) at 80°C and 20 r.p.m. for 2 min. Then, 1.2 ml of the cell lysates was mixed with 2.8 ml of the 50% iodixanol working solution to adjust the iodixanol concentration to 35% (v/v) and loaded into the bottom of the gradients, followed by ultracentrifugation in a Beckman SW40Ti rotor (114 000 g for 16 h at 4°C). The gradients were fractionated into 18 fractions of 570 µl each from the top using a piston gradient fractionator (BioComp Instruments Inc.). Fractions were stored at –80°C or analyzed immediately by standard SDS–PAGE and immunoblot analysis. Quantification of protein band intensities was performed using ImageJ (NIH; http://rsb.info.nih.gov/ij/).
Antibodies
The TOM1-specific rabbit antiserum was produced by injecting rabbits with a synthetic polypeptide corresponding to amino acids 203–220 of AtTOM1 (CRFPIESKGRRKKLHEVGS; the N-terminal extra cystein residue was added to facilitate conjugation) conjugated with keyhole limpet hemocyanin. Complementary DNA (cDNA) fragments of AtTOM2A mRNA corresponding to the C-terminal 97 amino acids and TMV-L RNA corresponding to amino acids 647–1116 of the 130K protein were cloned into pET16b and pET32c(+) (Novagen), respectively. The fusion polypeptides containing regions encoded by the cDNA fragments were expressed in Escherichia coli strains BL21 pLysS and Origami (DE3) pLysS (Novagen), respectively, by induction with isopropyl-β-d-thiogalactopyranoside. The expressed polypeptides carrying His6 tags were purified using Ni-NTA Superflow resin (Qiagen) and were used to immunize rabbits for the production of antisera against AtTOM2A and the TMV-L replication proteins. Rabbit polyclonal anti-GFP antibody was purchased from Chemicon International Inc. Rabbit polyclonal antibody against vacuolar H+-PPase (Takasu et al., 1997; Maeshima, 2000) was kindly provided by Masayoshi Maeshima. Rabbit polyclonal antibody against plant Sec61p, which was synthesized against a synthetic peptide corresponding to the N-terminal region of the Sec61p homologs of several plant species and recognizes an ∼40 kDa ER protein of various plant species (K.Matsuoka, unpublished result) was kindly provided by Ken Matsuoka (RIKEN, Yokohama, Japan). An anti-rabbit secondary antibody conjugated with horseradish peroxidase was purchased from Amersham Pharmacia Biotech.
RdRp assay
Forty microliters each of iodixanol gradient fractions were directly used for the assay. RdRp reaction mixture (50 µl) contained 1 mM ATP, 1 mM GTP, 1 mM UTP, 0.025 mM CTP, 20 µCi [α-32P]CTP, 10 mM DTT, 5 µg actinomycin D, 10 mM Mg(OAc)2 and 40 U RNasin (Promega). The reaction was performed at 28°C for 1 h. The RNA products were purified by phenol extraction and ethanol precipitation. The RNA samples were treated with S1 nuclease at 37°C for 20 min, purified again as above and then analyzed by 8 M urea–2.4% PAGE. The 32P signals were detected and quantified using a Bio Imaging Analyzer (BAS 1000, Fuji Photo Film).
Acknowledgments
Acknowledgements
We thank Masayoshi Maeshima and Shinobu Suga for plasmids pMAT-GV and pMAT-GP and the anti-vacuolar H+-PPase antibody, Ken Matsuoka for the anti-Sec61 antibody, Andreas Nebenführ for GmMan1–GFP-expressing BY-2 cells, Yasuo Niwa for psmRS-GFP, Akihiko Nakano and Ikuko Nishimura for helpful discussions, Ingrid Lambein for critical reading of the manuscript and Kumi Fujiwara for general assistance. We used the facilities of the Biopolymer Analysis Laboratory in the Faculty of Agriculture, Hokkaido University, and the Research Center for Molecular Genetics at Hokkaido University. This work was supported in part by grants from the Japan Society for the Promotion of Science (JSPS) to M.I. Y.H. is a JSPS fellow.
References
- Buck K.W. (1996) Comparison of the replication of positive-stranded RNA viruses of plants and animals. Adv. Virus Res., 47, 159–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carette J.E., Stuiver,M., van Lent,J., Wellink,J. and van Kammen,A.B. (2000) Cowpea mosaic virus infection induces a massive proliferation of endoplasmic reticulum but not Golgi membranes and is dependent on de novo membrane synthesis. J. Virol., 74, 6556–6563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis S.J. and Vierstra,R.D. (1996) Soluble derivatives of green fluorescent protein (GFP) for use in Arabidopsis thaliana. Weeds World, 3, 43–48. [Google Scholar]
- den Boon J.A., Chen,J.B. and Ahlquist,P. (2001) Identification of sequences in brome mosaic virus replicase protein 1a that mediate association with endoplasmic reticulum membranes. J. Virol., 75, 12370–12381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger D., Teterina,N., Ehrenfeld,E. and Bienz,K. (2000) Formation of the poliovirus replication complex requires coupled viral translation, vesicle production and viral RNA synthesis. J. Virol., 74, 6570–6580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn C.R., Mullen,R.T. and Trelease,R.N. (1998) Mutational analyses of a type 2 peroxisomal targeting signal that is capable of directing oligomeric protein import into tobacco BY-2 glyoxysomes. Plant J., 16, 709–720. [DOI] [PubMed] [Google Scholar]
- Froshauer S., Kartenbeck,J. and Helenius,A. (1988) Alphavirus RNA replicase is located on the cytoplasmic surface of endosomes and lysosomes. J. Cell Biol., 107, 2075–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haseloff J. and Siemering,K.R. (1998) The uses of green fluorescent protein in plants. In Chalfie,M. and Kain,S. (eds), Green Fluorescent Protein: Properties, Applications and Protocols. Wiley–Liss, New York, NY, pp. 191–220.
- Heinlein M., Padgett,H.S., Gens,J.S., Pickard,B.G., Casper,S.J., Epel,B.L. and Beachy,R.N. (1998) Changing patterns of localization of the tobacco mosaic virus movement protein and replicase to the endoplasmic reticulum and microtubules during infection. Plant Cell, 10, 1107–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hills G.J., Plaskitt,K.A., Young,N.D., Dunigan,D.D., Watts,J.W., Wilson,T.M. and Zaitlin,M. (1987) Immunogold localization of the intracellular sites of structural and nonstructural tobacco mosaic virus proteins. Virology, 161, 488–496. [DOI] [PubMed] [Google Scholar]
- Hood E.E., Gelvin,S.B., Melchers,L.S. and Hoekema,A. (1993) New Agrobacterium helper plasmids for gene-transfer to plants. Transgenic Res., 2, 208–218. [Google Scholar]
- Ishikawa M., Meshi,T., Motoyoshi,F., Takamatsu,N. and Okada,Y. (1986) In vitro mutagenesis of the putative replicase genes of tobacco mosaic virus. Nucleic Acids Res., 14, 8291–8305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa M., Meshi,T., Ohno,T. and Okada,Y. (1991) Specific cessation of minus-strand RNA accumulation at an early stage of tobacco mosaic virus infection. J. Virol., 65, 861–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa M., Naito,S. and Ohno,T. (1993) Effects of the tom1 mutation of Arabidopsis thaliana on the multiplication of tobacco mosaic virus RNA in protoplasts. J. Virol., 67, 5328–5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L.W. and Rogers,J.C. (1998) Integral membrane protein sorting to vacuoles in plant cells: evidence for two pathways. J. Cell Biol., 143, 1183–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L.W. and Rogers,J.C. (1999) Sorting of membrane proteins to vacuoles in plant cells. Plant Sci., 146, 55–67. [Google Scholar]
- Kim J., Huang,W.P., Stromhaug,P.E. and Klionsky,D.J. (2002) Convergence of multiple autophagy and cytoplasm to vacuole targeting components to a perivacuolar membrane compartment prior to de novo vesicle formation. J. Biol. Chem., 277, 763–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujala P., Ikäheimonen,A., Ehsani,N., Vihinen,H., Auvinen,P. and Kääriäinen,L. (2001) Biogenesis of the Semliki Forest virus RNA replication complex. J. Virol., 75, 3873–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutsuna N. and Hasezawa,S. (2002) Dynamic organization of vacuolar and microtubule structures during cell cycle progression in synchronized tobacco BY-2 cells. Plant Cell Physiol., 43, 965–973. [DOI] [PubMed] [Google Scholar]
- Maeshima M. (1992) Characterization of the major integral protein of vacuolar membrane. Plant Physiol., 98, 1248–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeshima M. (2000) Vacuolar H+-pyrophosphatase. Biochim. Biophys. Acta, 1465, 37–51. [DOI] [PubMed] [Google Scholar]
- Más P. and Beachy,R.N. (1999) Replication of tobacco mosaic virus on endoplasmic reticulum and role of the cytoskeleton and virus movement protein in intracellular distribution of viral RNA. J. Cell Biol., 147, 945–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meshi T., Watanabe,Y., Saito,T., Sugimoto,A., Maeda,T. and Okada,Y. (1987) Function of the 30 kd protein of tobacco mosaic virus: involvement in cell-to-cell movement and dispensability for replication. EMBO J., 6, 2557–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller D.J., Schwartz,M.D. and Ahlquist,P. (2001) Flock house virus RNA replicates on outer mitochondrial membranes in Drosophila cells. J. Virol., 75, 11664–11676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata T., Nemoto,Y. and Hasezawa,S. (1992) Tobacco BY-2 cell-line as the HeLa-cell in the cell biology of higher plants. Int. Rev. Cytol., 132, 1–30. [Google Scholar]
- Nebenführ A., Gallagher,L.A., Dunahay,T.G., Frohlick,J.A., Mazurkiewicz,A.M., Meehl,J.B. and Staehelin,L.A. (1999) Stop-and-go movements of plant Golgi stacks are mediated by the acto-myosin system. Plant Physiol., 121, 1127–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshima K., Taniyama,T., Yamanaka,T., Ishikawa,M. and Naito,S. (1998) Isolation of a mutant of Arabidopsis thaliana carrying two simultaneous mutations affecting tobacco mosaic virus multiplication within a single cell. Virology, 243, 472–481. [DOI] [PubMed] [Google Scholar]
- Osman T.A.M. and Buck,K.W. (1996) Complete replication in vitro of tobacco mosaic virus RNA by a template-dependent, membrane-bound RNA polymerase. J. Virol., 70, 6227–6234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prod’homme D., Le Panse,S., Drugeon,G. and Jupin,I. (2001) Detection and subcellular localization of the turnip yellow mosaic virus 66K replication protein in infected cells. Virology, 281, 88–101. [DOI] [PubMed] [Google Scholar]
- Reichel C. and Beachy,R.N. (1998) Tobacco mosaic virus infection induces severe morphological changes of the endoplasmic reticulum. Proc. Natl Acad. Sci. USA, 95, 11169–11174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restrepo-Hartwig M.A. and Ahlquist,P. (1996) Brome mosaic virus helicase- and polymerase-like proteins colocalize on the endoplasmic reticulum at sites of viral RNA synthesis. J. Virol., 70, 8908–8916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restrepo-Hartwig M.A. and Ahlquist,P. (1999) Brome mosaic virus RNA replication proteins 1a and 2a colocalize and 1a independently localizes on the yeast endoplasmic reticulum. J. Virol., 73, 10303–10309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust R.C., Landmann,L., Gosert,R., Tang,B.L., Hong,W.J., Hauri,H.P., Egger,D. and Bienz,K. (2001) Cellular COPII proteins are involved in production of the vesicles that form the poliovirus replication complex. J. Virol., 75, 9808–9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T., Hosokawa,D., Meshi,T. and Okada,Y. (1987) Immunocytochemical localization of the 130K and 180K proteins (putative replicase components) of tobacco mosaic virus. Virology, 160, 477–481. [DOI] [PubMed] [Google Scholar]
- Schaad M.C., Jensen,P.E. and Carrington,J.C. (1997) Formation of plant RNA virus replication complexes on membranes: role of an endoplasmic reticulum-targeted viral protein. EMBO J., 16, 4049–4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlegel A., Giddings,T.H., Ladinsky,M.S. and Kirkegaard,K. (1996) Cellular origin and ultrastructure of membranes induced during poliovirus infection. J. Virol., 70, 6576–6588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz M., Chen,J.B., Janda,M., Sullivan,M., den Boon,J. and Ahlquist,P. (2002) A positive-strand RNA virus replication complex parallels form and function of retrovirus capsids. Mol. Cell, 9, 505–514. [DOI] [PubMed] [Google Scholar]
- Suga S., Imagawa,S. and Maeshima,M. (2001) Specificity of the accumulation of mRNAs and proteins of the plasma membrane and tonoplast aquaporins in radish organs. Planta, 212, 294–304. [DOI] [PubMed] [Google Scholar]
- Takamatsu N., Ishikawa,M., Meshi,T. and Okada,Y. (1987) Expression of bacterial chloramphenicol acetyltransferase gene in tobacco plants mediated by TMV-RNA. EMBO J., 6, 307–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasu A., Nakanishi,Y., Yamauchi,T. and Maeshima,M. (1997) Analysis of the substrate binding site and carboxyl terminal region of vacuolar H+-pyrophosphatase of mung bean with peptide antibodies. J. Biochem., 122, 883–889. [DOI] [PubMed] [Google Scholar]
- Tamai A. and Meshi,T. (2001) Tobamoviral movement protein transiently expressed in a single epidermal cell functions beyond multiple plasmodesmata and spreads multicellularly in an infection-coupled manner. Mol. Plant Microbe Interact., 14, 126–134. [DOI] [PubMed] [Google Scholar]
- Tritel M. and Resh,M.D. (2000) Kinetic analysis of human immunodeficiency virus type 1 assembly reveals the presence of sequential intermediates. J. Virol., 74, 5845–5855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto Y., Numaga,T., Ohshima,K., Yano,M., Ohsawa,R., Goto,D.B., Naito,S. and Ishikawa,M. (2003) Arabidopsis TOBAMOVIRUS MULTIPLICATION (TOM) 2 locus encodes a transmembrane protein that interacts with TOM1. EMBO J., 22, 335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda T., Yamaguchi,M., Uchimiya,H. and Nakano,A. (2001) Ara6, a plant-unique novel type Rab GTPase, functions in the endocytic pathway of Arabidopsis thaliana. EMBO J., 20, 4730–4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Heijden M.W., Carette,J.E., Reinhoud,P.J., Haegi,A. and Bol,J.F. (2001) Alfalfa mosaic virus replicase proteins P1 and P2 interact and colocalize at the vacuolar membrane. J. Virol., 75, 1879–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vida T.A. and Emr,S.D. (1995) A new vital stain for visualizing vacuolar membrane dynamics and endocytosis in yeast. J. Cell Biol., 128, 779–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y., Meshi,T. and Okada,Y. (1987) Infection of tobacco protoplasts with in vitro transcribed tobacco mosaic virus RNA using an improved electroporation method. FEBS Lett., 219, 65–69. [Google Scholar]
- Yamanaka T., Ohta,T., Takahashi,M., Meshi,T., Schmidt,R., Dean,C., Naito,S. and Ishikawa,M. (2000) TOM1, an Arabidopsis gene required for efficient multiplication of a tobamovirus, encodes a putative transmembrane protein. Proc. Natl Acad. Sci. USA, 97, 10107–10112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka T. et al. (2002) Complete inhibition of tobamovirus multiplication by simultaneous mutations in two homologous host genes. J. Virol., 76, 2491–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]