

Abstract
The translocon is a protein-conducting channel conserved over all domains of life that serves to translocate proteins across or into membranes. Although this channel has been well studied for many years, the recent discovery of a high-resolution crystal structure opens up new avenues of exploration. Taking advantage of this, we performed molecular dynamics simulations of the translocon in a fully solvated lipid bilayer, examining the translocation abilities of monomeric SecYEβ by forcing two helices comprised of different amino acid sequences to cross the channel. The simulations revealed that the so-called plug of SecYEβ swings open during translocation, closing thereafter. Likewise, it was established that the so-called pore ring region of SecYEβ forms an elastic, yet tight, seal around the translocating oligopeptides. The closed state of the channel was found to block permeation of all ions and water molecules; in the open state, ions were blocked. Our results suggest that the SecYEβ monomer is capable of forming an active channel.
INTRODUCTION
As proteins are being synthesized by the ribosome, many of them require insertion into a membrane or transport across it. In the case of eukaryotes, this is the membrane of the endoplasmic reticulum where proteins are often initially stored; in prokaryotes, it is the cytoplasmic membrane. To accomplish the translocation, a membrane-protein complex, called SecYEG in bacteria, SecYEβ in archaea, and Sec61 in eukaryotes, is used. This complex, the so-called translocon, is able to recognize whether a protein belongs in the membrane or outside it, placing it in the appropriate location (1–4).
Structurally, the translocon is a heterotrimeric protein complex. The first two subunits, known respectively as the α- and γ-subunits, show significant sequence conservation across all domains of life. However, the third β-subunit has little similarity to bacteria and archaea/eukaryotes and, in fact, is not necessary for cell viability (1,5). The structure of the translocon taken from the archaeon Methanococcus jannaschii, SecYEβ (SecY corresponding to the α-subunit, SecE to γ, and Secβ to β), was recently solved by x-ray crystallography at a resolution of 3.5 Å (5). The structure is shown in Fig. 1.
FIGURE 1.

Simulated system of SecYEβ in a lipid bilayer/water environment. SecYEβ is shown in cartoon representation with SecY, SecE, and Secβ colored in gray, orange, and ochre, respectively. The plug, transmembrane domain 2a of SecY (residues Ile55 to Gly65) is presented in red, and TM2b (residues Gly76 to Ser91) and TM7 (residues Asn256 to Gly280) are shown in green (both also of SecY). The lipids are seen in yellow licorice representation with the phosphorus, nitrogen, and an oxygen of the headgroup highlighted as spheres colored in tan, blue, and red, respectively. The water box is drawn in transparent blue surface representation. (A) Side view of the simulated system. To display the protein more clearly, some lipids and water molecules have been removed, leaving a flat outward face. (B) Top view of the simulated system. The top solvation layer has been removed.
How the translocon actually places the nascent protein in the correct destination is still unclear. It is known that an N-terminal signal sequence is necessary for recognition by the channel (see, for example, Matlack et al. (1), Holland (3), Rapoport et al. (6), and Plath et al. (7)). This sequence is seen, through cross-linking experiments, to intercalate between helices 2b and 7 of the α-subunit (7,8), suggested by some to cause the channel to open, readying it for translocation of a water-soluble protein (9,10). The translocation requires the motion of a small helix, 2a, that, in the crystal structure, blocks the channel like a plug (see Fig. 1). This scenario provides a model for translocation of globular proteins, but how do proteins enter the membrane? One suggestion stipulates that the translocon has a lateral gate, located between helices 2b/3 and 7/8, through which polypeptides can move into the surrounding lipid phase; as recognized previously, the gate is the only location relatively free of lateral obstruction (5).
The translocon does not operate as the singular element required for protein localization but rather works with a large number of associated proteins. For instance, the driving force for the protein entering the translocon usually comes from a channel partner, the ribosome for cotranslational translocation or SecA for bacterial posttranslational translocation. Early experiments seemed to indicate that when the channel partner binds to the translocon, it makes close contact with the pore entrance, even possibly forming a seal (9,11). However, electron microscopy structures of a ribosome associated with the channel show a gap between them (12–17). If there is such a gap, how then are small molecules or ions prevented from crossing the pore? At the narrowest point of the mostly hydrophilic channel in SecY, there is a ring of hydrophobic residues that could act as a seal around the translocating polypeptide (5). If this is the case, flexibility as well as its ability to close on a translocating peptide are both important features. Studies have shown that disulfide-bonded loops (18) as well as large side-chain residues (19) are able to cross the translocon, indicating that some flexibility exists.
Although less characterized in archaea, known channel partners include SecDF homologs as well as signal peptidases (4). In eukaryotes and bacteria, a larger number of associated proteins has been discovered although the precise association with the channel is often unknown. Examples include in eukaryotes a signal peptidase, the oligosaccharyl transferase complex (OST), the translocating chain-associated membrane protein (TRAM), and the translocon-associated protein complex (TRAP) (6,16,20). The association if not function of these channel partners is beginning to come into focus due to improved imaging such as recent low resolution structures of TRAP seen with the translocon (16).
The role of oligomerization has also come into question due to the new monomer structure. Although known to exist as a dimer or tetramer in nature (12–15,21–24), it was originally believed that the association of monomers formed the functional channel (12,13,21,25) with a pore size of 40–60 Å (26). The crystal structure suggests, however, that a monomer can form the functional channel. Evidence for this is seen, for example, in experiments with detergent-solubilized SecA/SecYEG where a single copy of each is seen associated with a preprotein (24,27) as well as in an experiment demonstrating that the signal sequence can be cross-linked to residues in the center of a single SecY protein (28). However, a front-to-front arrangement of the dimer (with the lateral gates facing each other) has also been proposed that could allow for a larger pore to form (17). There is evidence, though, for a back-to-back model, such as a two-dimensional electron density map of a SecYEG dimer which, when docked with the three-dimensional structure, shows a back-to-back association with the transmembrane portions of SecE closest together (5). Cysteine cross-linking between SecEs also agrees with this arrangement (29). This would also keep the lateral gate exposed to the lipids as opposed to the other arrangement which would block it and, thus, require more complex motions for membrane insertion of proteins. The purpose of oligomerization in this arrangement has been suggested to be cooperative interactions between subunits that may alter the structure slightly, moving the plug farther out and widening the gap between helices 2b and 7, thereby “priming” it for translocation (30,31).
In this report, we present, to our knowledge, the first all-atom molecular dynamics simulations of SecYEβ in a fully solvated lipid bilayer. We explore the system in an equilibrated state without restraints. We also test the translocation abilities of the channel by pulling different polypeptides in helical form through the channel. With this, we see how the channel reacts to different amino acid sequences as well as how well the structure responds to this disturbance. We then simulate how the structure relaxes after the translocation. Finally, we examine a possible dimer arrangement.
METHODS
System assembly
The coordinates of the protein SecYEβ were obtained from the Protein Data Bank (PDB code 1RHZ) (5). The seven histidine residues (five in SecY, one each in SecE and Secβ) were singly protonated, making the residue neutral overall. No histidine residues are present inside the pore. Missing hydrogen atoms were added with the Psfgen plug-in of the Visual Molecular Dynamics (VMD) program (32). Missing residues (1, 434–436 of SecY; 1, 67–73 of SecE; 1–20, 53 of Secβ), all terminal, were not reconstructed.
The resulting structure was placed in a preequilibrated POPC bilayer membrane using the VMD plug-in Membrane by aligning hydrophilic residues with the lipid headgroups or membrane exterior and the hydrophobic surface with the interior of the bilayer. The location of the protein in the lipid bilayer agreed with that suggested in van den Berg et al. (5). Lipids overlapping the protein were removed. The lipid-protein combination was then solvated above and below the rectangular membrane patch using the VMD plug-in Solvate. Water molecules inside the pore were removed along with any water placed between the bilayer and the protein. Finally, the VMD plug-in Autoionize was used to add Na+ and Cl− ions at a concentration of 50 mM; the overall system was electrically neutral. At completion, the system consisted of 106,679 atoms including 251 lipids, 21,520 water molecules, 5 sodium ions, and 21 chloride ions. The preequilibration size was 111 × 109 × 106.5 Å3. The fully equilibrated system is shown in Fig. 1.
Molecular dynamics simulation
Simulations were performed using the parallel molecular dynamics program NAMD 2.5 along with the CHARMM27 force field for protein and lipids as well as the TIP3P model for water (33,34). Electrostatic interactions were evaluated based on a multiple-time-stepping algorithm where bonded interactions were computed every 1 fs, short-range nonbonded electrostatic and van der Waals interactions (12 Å cutoff with a smooth switching function beginning at 10 Å) every 2 fs, and long-range interactions every 4 fs. To compute long-range electrostatic interactions, the particle-mesh Ewald (PME) method was used with grid points no more than ∼1 Å apart. Simulations were performed on a variety of platforms including 128 1.6 GHz Itanium 2 processors with performance of ∼4.5 ns/day.
Constant temperature control, when used, involved Langevin dynamics coupled to all atoms except hydrogens with a 5 ps−1 damping coefficient (except when otherwise noted). When constant pressure was assumed, it was held at 1 atm by a Nose-Hoover Langevin piston with a decay period of 100 fs and a damping time of 50 fs.
All simulations employed periodic boundary conditions. The distance between protein images in adjacent periodic cells was never <17 Å and through the majority of simulations was ∼30 Å in all directions. When the system was held at constant pressure, a flexible cell was used, allowing the dimensions to vary independently.
System equilibration
The system was equilibrated in multiple steps over the course of 5 ns to most efficiently relax it. This step of our simulation will be collectively denoted here as simulation sim0. For the first part, “melting” of the lipid tails only was allowed and all other atoms were harmonically restrained to their original positions with a force constant of 2 kcal/(mol Å2). This phase consisted of 2000 steps of energy minimization and 0.5 ns of dynamics in the NVT ensemble. Throughout simulation sim0, the temperature was held at 300 K.
In a second step, the protein backbone was restrained to its crystallographic position with a force constant of 2 kcal/(mol Å2) whereas everything else was freed. This simulation, carried out in the NpT ensemble, was comprised of 1000 steps of energy minimization and 1 ns of dynamics. For the first 250 ps, water was pushed out of the pore and lipid tail region to allow the lipids to pack together and surround the protein. The remaining 750 ps had no extraneous forces on the water other than those used to control pressure and temperature.
The third and final step freed the backbone and allowed the entire system to equilibrate. This step lasted 3.5 ns in the NpT ensemble after another 1000 steps of minimization. The equilibrated system had a final size of 98.6 × 99.2 × 103.4 Å3.
For simulations sim1, sim1a, and sim2, the polypeptide of interest, a deca-alanine helix for simulations sim1 and sim1a and a 19 amino acid alanine/leucine helix for simulation sim2, was placed near the top of the channel, oriented along the z axis, attempting to place it in line with the plug while avoiding steric clashes with nearby SecYEβ. At this position, the polypeptide's backbone was restrained with a 2 kcal/(mol Å2) force constant for 1000 steps of minimization and 500 ps of dynamics. This allowed the water and protein to properly adapt to the introduction of the translocating helix. These short 0.5 ns equilibration steps were carried out in the NpT ensemble, whereas the further pulling simulations, sim1, sim1a, and sim2, were done in the NV ensemble, attempting to avoid any perturbations of the measured forces due to temperature or pressure control. The final simulations, sim3 and sim4, were again equilibration simulations and were carried out in the NpT ensemble.
Dimer assembly
A simulation (sim5) has also been performed to investigate the effect of oligomerization on protein translocation through SecYEβ. For this purpose, a dimer of SecYEβ was built in a back-to-back arrangement as seen in van den Berg et al. (5) and Bostina et al. (31) based on cryo-electron densities (22). In sim5, the two residues Ile50, one from each SecE, were placed close together (∼10 Å apart) in accordance with the efficient cross-linking seen in a previous experiment (29). The system was solvated and neutralized exactly as before for the monomer system except the ion concentration was increased to 100 mM. The system was equilibrated for 5 ns, in a manner similar to sim0 (see above).
Steered molecular dynamics
Steered molecular dynamics (SMD) is used in simulations sim1 and sim2 to pull a helix through the SecYEβ channel (35–40). In these cases, constant velocity SMD was employed. The method is implemented by means of an imaginary point traveling at constant velocity to which the center of mass of the atoms being pulled is coupled by a spring. The force is computed using
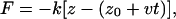 |
(1) |
which has been adapted from Isralewitz et al. (38) for pulling in the −z direction. This force is then distributed over all atoms involved, weighted by their masses.
Analysis
The root mean-square deviation (RMSD) was calculated using VMD. For each frame of the trajectory (taken every picosecond), the RMSD was computed based on the positions of the backbone atoms of SecYEβ as compared to their original crystallographic positions, after performing a best-fit alignment of each pair of structures.
The program HOLE was used to calculate local channel radii (41). HOLE maximizes the radii of spheres at various positions (in 0.5 Å increments) along the channel axis, using a Monte Carlo simulated annealing technique. For atom sizes, the AMBER van der Waals radii were used (42).
The center of mass of the backbone of deca-alanine and that of the last six residues of the 19 residue alanine/leucine helix and the force required to translocate them were tracked through simulations sim1 and sim2, respectively. In both cases, the data were adjusted, subtracting any center-of-mass motion of the lipid bilayer (motion of the bilayer implies motion of SecYEβ). The data were also shifted such that zero on the graph corresponds to the starting position of deca-alanine in sim1. Data points were taken every 100 fs and then averaged over every 5 ps and 10 ps for sim1 and sim2, respectively. Fluctuations in the force before averaging were on the order of 250 pN at most. All molecular images were prepared using VMD (32).
RESULTS
The results presented here are based on simulations summarized in Table 1. We describe first the equilibrated protein/lipid bilayer/water system. We then discuss the simulations translocating a deca-alanine helix as well as an alanine/leucine helix across SecYEβ. We follow with the simulated relaxation of SecYEβ after translocation where we demonstrate the protein's ability to return to its equilibrium structure. Finally, we conclude with a simulation of a SecYEβ dimer, examining the effects of dimerization on individual monomers.
TABLE 1.
List of the simulations performed. Simulations sim0 through sim5 are described in the text; simulations sim6 through sim11 are described in Supplementary Material
| Label | Length (ns) | Description |
|---|---|---|
| Sim0 | 5.0 | Equilibration of system |
| Sim1 | 1.4 | SMD simulation with deca-alanine |
| Sim1a | 7.4 | SMD simulation with deca-alanine |
| Sim2 | 1.9 | SMD simulation with alanine/leucine helix |
| Sim3 | 3.6 | Relaxation continuing from sim1 |
| Sim4 | 3.1 | Relaxation continuing from sim2 |
| Sim5 | 5.0 | Equilibration of back-to-back dimer |
Equilibration analysis
The equilibrated system was analyzed as resulting from simulation sim0 (see Table 1). We calculated the RMSD of the protein backbone as compared to the crystal structure over the entire 5 ns equilibration (see Fig. 2). After the initial restraints are released (see Methods), the RMSD climbs to a value of ∼2 Å before stabilizing. There were no large deformations of the protein observed during this simulation; the most noticeable changes involve the two loops in the cytoplasmic region (see Supplementary Fig. 5). The protein definitely appears stable after equilibration.
FIGURE 2.

Time dependence of the RMSD of the simulated system. The RMSD relative to the crystal structure (calculated for the protein backbone) is shown for simulation sim0 (see Table 1). Three stages of the simulation corresponding to different harmonic restraints (see Methods) are clearly discernable through jumps seen at t = 0 ns, t = 0.5 ns, and t = 1.5 ns.
We also examined the lipid packing both within the membrane itself and around the protein. By monitoring the penetration of water molecules into the lipid bilayer during the simulation, we determined how well the lipids stay together. Water molecules were not found to penetrate between lipids past the headgroups. Also no water molecules were seen at the hydrophobic protein-membrane interface.
The channel itself does permit the entrance of water molecules from the trans or cis sides. However, none traverse the channel entirely during equilibration (simulation sim0). In fact, water molecules are seen to come close to the pore ring (Ile75, Val79, Ile170, Ile174, Ile260, Leu406) (5), but no permeation events were observed. This was also confirmed by monitoring the positions of each water molecule over time. No water molecule is observed farther inward than the gating plug (helix 2a). Both pore and plug seem to be capable of blocking the channel from at least the flow of water. Although the same can be said for ions, the ion concentration was not sufficient to provide enough opportunities to examine ions near the pore. Channel crossing by ions and water molecules will be explored below for simulations sim1–sim4. Also, to better explore ion behavior, we repeated simulations sim1 and sim3 out to more than 9 ns at a higher ion concentration (increased from 50 mM to 150 mM). Further details are provided in the Supplementary Material.
Translocation of deca-alanine
After equilibration, the first simulation tested the response of SecYEβ to the introduction of deca-alanine, induced into crossing the channel. Deca-alanine, a stable helix in hydrophobic environments but not in an aqueous environment (43), was chosen for its size and uniformity (∼8.5 Å in diameter, 20 Å long, measured between atom centers). It was also chosen for its general hydrophobic character; the signal sequence of nascent proteins generally includes a small hydrophobic helix (7). The simulation, sim1, was performed at constant velocity using SMD (see Methods). The center of mass of the deca-alanine backbone was pulled at .05 Å/ps in the −z direction with a large spring constant (k = 5 kcal/(mol Å2), 〈Δz〉thermal = .35 Å) ensuring the helix followed the constraint closely. To counter the applied force, the lipid headgroups were restrained to their initial center-of-mass position in the z direction with a spring constant of 7 kcal/(mol Å2). Sim1 was run for 1.4 ns, allowing deca-alanine to traverse the entire channel; a longer simulation, sim1a (described below), was also run for 7.4 ns. Snapshots of simulation sim1 are shown in Fig. 3.
FIGURE 3.

Translocation of deca-alanine through SecYEβ. The figure shows the results of simulation sim1 (see Table 1). The representation and coloring of SecY, SecE, and Secβ is the same as in Fig. 1 with the exception that SecE and Secβ are rendered transparent. The plug (TM2a of SecY, residues Ile55 to Gly65) is shown in red. (A) Front view of SecYEβ and deca-alanine at t = 0. Deca-alanine is shown in blue cartoon representation together with its (transparent) surface and is positioned on the cytoplasmic side of SecYEβ before translocation. (B) Top view of the SecYEβ-deca-alanine system at t = 0.8 ns. The pore ring (residues Ile75, Val79, Ile170, Ile174, Ile260, and Leu406 of SecY), expanded from its equilibrium (t = 0) state, is shown in surface representation colored yellow with deca-alanine passing through it. Deca-alanine is partially unfolded at this point. (C) Final state of translocation (t = 1.4 ns). The plug has been pushed out into the solvent, and deca-alanine is seen unfolded next to the plug.
We first examined the behavior of the helix itself. One of the most notable features observed in sim1 is that deca-alanine did not move purely in the z direction, even though it was originally positioned directly above the plug (Fig. 3 A). Instead it followed a curved path through SecY. The center of mass of the helix deviated from a straight path by as much as 3.5 Å in the plane of the membrane at the center of the pore and up to 5.5 Å near the exit. We also observed the helix to partially unfold over the course of the simulation (Fig. 3, B and C). The leading turn of the helix unfolds first at ∼0.6 ns. Although the forces placed on deca-alanine certainly contribute to this, we can also not discount the fact that deca-alanine does not normally form stable helices in water (see Levy et al. (43) and references therein). Perhaps the most intriguing behavior of the translocating helix, though, is that it flips 180° near the beginning of the channel. Although this is not always observed (including in other simulations involving deca-alanine), here we see it happen due to interactions of the last two residues of deca-alanine with nearby residues of SecY, including specifically a highly conserved arginine, Arg104. Both hydrophobic as well as charge-dipole interaction between the two residues were present. This behavior could be due just to the initial starting position for deca-alanine, though Arg104 is positioned prominently at the mouth of the pore, suggesting a role as a binding site for channel partners or other proteins.
Blocking the translocation pathway is the “plug”, a small helix (helix 2a, approximately residues Ile55 to Gly65) of ∼10 residues connected to the larger body of SecY by two long coils (Fig. 3 A). Positioned approximately halfway through the channel, it has been proposed that the plug can be induced to move out of the pore into the periplasm, thereby leaving the channel in an open state (5,30). Due to the force of the translocating deca-alanine, the plug moved out of the pore allowing the deca-alanine to pass. This occurs without deformation of helix 2a or the rest of SecY; the coils allow the plug to pivot approximately about 2 glycine residues on opposite sides of the helix (Gly49 and Gly68). The final location of the helix can be seen in Fig. 3 C. Cysteine replacement experiments in Escherichia coli demonstrated that a residue in the plug can form a disulfide bridge in vivo with a residue in SecE (44). In sim1, we saw a general tendency of the plug to move toward SecE. At the point of closest approach, the plug and SecE were separated by ∼7.35 Å (between residues Ile55 in SecY and Lys66 in SecE). This is in contrast to the residues used in experiment; there, the bridge formed between residue 61 of SecY and residue 64 of SecE (both adjusted for their positions in M. jannaschii) (44). One can see in Fig. 3 C, though, that the plug is pushed quite far into the periplasm by the deca-alanine. In other simulations, the plug does not always move this far along the channel axis (see, for example, simulation sim2 below).
When the plug is no longer blocking the channel, something should still help prevent the flow of ions or other small solutes during translocation. Filling this role is the pore ring, located at the narrowest part of the channel. With residues located on multiple helices, we found the pore ring has the ability to expand and contract to control the passage of polypeptides. Deca-alanine was able to pass through the ring while maintaining a mostly helical character. The ring expanded from its original size of ∼3.5–5.5 Å to a size of 7–12 Å (the pore was not uniformly circular, so dimensions varied), large enough to accommodate even the full helix as shown in Fig. 3 B. To determine the quality of the seal around the deca-alanine, we counted the number of water molecules that crossed the pore ring during translocation. Following the helix closely were 10 water molecules crossing in the translocation direction. No ions came near the pore ring during the translocation. It has been suggested that flexible loops between helices 4 and 5 along with loops between helices 9 and 10 contribute to the ability of the pore ring to expand (5). We found, however, that the greatest motion occurred in helices 2b and 7. Given the asymmetric motion of the translocating polypeptide toward this side of the pore, this is not surprising, though differently sized or shaped polypeptides may disturb the pore in other ways. Although the pore ring appears broken in Fig. 3 B, we found that other residues not directly involved in the pore ring contributed to maintaining the translocation seal; specifically, Leu261, Ile257, Ile410, Val178, and Ile78 near the primary pore ring residues play such a role (see Supplementary Fig. 6). Four of these side groups, indeed, are well conserved across many species, at least in their hydrophobic character; the exception is Ile78, which is a tyrosine in some species. A similar suggestion that residues not originally defined as part of the pore ring play a role in maintaining the seal was also made recently based on the results of homology modeling (31).
We also measured the force placed on deca-alanine to enforce its translocation through the course of simulation sim1, shown in Fig. 4. The forces are large but in the expected range. For example, for the glycerol channel GlpF, forces on the order of 500 pN were encountered when translocation was enforced on a similar timescale, the forces acting however on a much smaller substrate in a constitutively open channel (unpublished work from the study in Jensen et al. (45)). However, because our simulation is far from in vivo conditions where translocation most likely occurs on the order of seconds (46), interpretation of specific quantities requires much caution. Since the plug lies immediately after the pore ring, it is difficult to say what contributions the plug and pore ring make to the total force profile. We attempted to examine their contributions separately in two new simulations. In the first one, described in the Supplementary Material (sim9), we pulled the plug out at the same (.05 Å/ps) speed without a translocating polypeptide. The maximum force required to remove the plug from the pore in this simulation was 1750 pN. Starting with this state, i.e., with the plug outside the pore, we performed another simulation (also described in the Supplementary Material) where deca-alanine was pulled through the channel and the force required measured. The maximum force in this simulation, at the pore ring, was nearly 1800 pN. Comparing this again to the quantities seen in Fig. 4, it is clear that the pore ring represents a large fraction of the peak force initially. Given the proximity of the plug to the pore ring, it is surprising that the maximum force is not somewhere closer to the sum of the two maxima, i.e., on the order of 3000 pN or more, instead of just 2200 pN; in fact, after deca-alanine clears the pore ring, the plug only represents a barrier of ∼1200 pN, although we cannot presume this is the largest barrier presented by the plug in sim1.
FIGURE 4.

Force profile along the SecYEβ translocation pathway. (Bottom) Force versus distance along the z axis for simulations sim1, sim1a, and sim2 shown in blue, black, and red, respectively. The shaded area represents the location of the hydrophobic core of the membrane. (Top) Side cut of SecY from a representative simulation shown in surface representation. The channel is positioned sideways with the cytoplasmic side to the left and the periplasmic side to the right. The scale of the figure corresponds to that of the channel distance shown and the dashed lines correlate specific positions between the top and bottom. Highlighted are the pore ring (yellow) as well as the plug (red). TM2b and TM7 are shown in green. The figure shows a snapshot of the channel during a translocation event. The pore is represented in blue and was calculated by the program HOLE (see Methods).
In analyzing these two events, namely opening the pore ring and displacing the plug, we first realized that opening the pore ring requires displacing four helices against the lateral pressure of the rest of the protein and membrane. Hydrophobic effects also play a role in opening the pore ring where the residues can become exposed to solvent. In contrast, the displacement of the plug is affected by solvent pressure but also by interactions with the central part of the pore; the disturbance caused by the translocating polypeptide could adversely affect these interactions that serve to hold the plug in place. In fact, it has been previously suggested that a bound signal sequence can cause the channel to open by destabilizing interactions of the plug with the larger body of SecY (5). We see in our results something similar to this suggestion, namely that the force required to cross the channel is less than the combined force required to open the two elements, the plug and pore ring, individually, suggesting that opening the pore ring destabilizes the plug.
In running a longer simulation, sim1a, we found results similar to those for sim1, although the speed was reduced by a factor of five. The path through the channel was the same; also, deca-alanine still unfolds partially when encountering the pore ring. The force profile, seen in Fig. 4 was similar, except it was reduced by ∼30%. Overall, the system was disturbed less (the RMSD of the protein is shown in Supplementary Fig. 8) with the exception of the plug. Increased interactions (specifically, a “dragging” force between deca-alanine and the side of the plug as opposed to the “pushing” force also seen) caused it to unfold partially, however not in a way that affects the channel's function.
Translocation of an alanine/leucine helix
In another SMD simulation, sim2, we translocated through SecYEβ a 19 amino acid alanine/leucine helix with the sequence ALAALALAALAALALAALA, from here on referred to as AL19. This sequence was chosen based on its known affinity for membrane insertion; although deca-alanine is hydrophobic, it actually is not likely to be inserted into the membrane's hydrophobic core (47). Sim2 was performed at the same constant velocity as sim1. The helix was positioned slightly off center in relation to the z axis running through the center of the plug. This was necessary to avoid steric clashes given the bulkier side chains of leucine as compared to alanine. The center of mass of the α-carbons of the last six residues was pulled at .05 Å/ps in the −z direction. The spring constants chosen for this simulation were 3 kcal/(mol Å2) for AL19 and 5 kcal/(mol Å2) restraining the lipid headgroups. Sim2 was run for 1.9 ns.
Like deca-alanine in sim1, AL19 unfolds during the translocation process, but it does so more quickly. Unfolding began at 0.6 ns and then continued until the entire helix was unfolded by 1.3 ns. The pore ring appears to be the primary cause for the unfolding. Although AL19 as constructed is rather stable as a helix in water (see the Supplementary Material), its larger size (as compared to deca-alanine) leads to more steric clashes with the surrounding protein. Also, because of the unfolding of AL19, the pore ring does not expand as much as in sim1. The pore reached a maximum diameter of between 6 and 10.5 Å. The greatest movement for pore ring residues involved Val79, located on helix 2b of SecY. Looking at water permeation of the pore ring, we see a similar number as before: 22 water molecules followed AL19 through the pore ring (no ion permeation occurred). Motion of the plug near the end of sim2 was not as pronounced as in sim1. Whereas in sim1 the plug moved 23 Å in the z direction away from the main body of SecY, in sim2, it moved only 7.5 Å in this direction. Due to its unfolding, AL19 does not make as broad a contact with the plug as deca-alanine, thereby mostly pushing the plug down and to the side instead of farther out into the solvent. The plug still remains a helix throughout the translocation process. Looking at contact with SecE, we actually see a closer approach of the plug as compared to sim1 with, at minimum, 4.6 Å separating residue Ile55 of the plug of SecY and residue Lys62 of SecE. Supplementary Fig. 7 shows the movement of the plug during this simulation.
We also examined the interactions between the translocating polypeptide and SecY. We had noticed a tendency of deca-alanine in sim1 to move along a curved path; such a path also materialized for AL19 as shown in Fig. 5. This path leads the translocating AL19 near helices 2b and 7 of SecY, the proposed signal sequence recognition center as well as the lateral gate (5). In Fig. 5 A, even before translocation the channel can be seen to form a pocket between helices 2b and 7. Therefore, we propose that steric effects are routing signal sequences to this area of the protein, the evident recognition site. We also see strong interactions between AL19 and helices 2b and 7 during translocation. Looking again at Fig. 4, why then is a lower maximum force required to pull AL19 through the channel as compared to deca-alanine? This is likely due to the early unfolding of AL19 as compared to deca-alanine; the pore ring opens more easily for the smaller (by lateral area) AL19. The difference in length also becomes clear in the last third of the simulation; the force required to pull AL19 through remains high compared to pulling deca-alanine through which, at the same center-of-mass position, has nearly exited the channel.
FIGURE 5.

Dynamics of pore formation in SecYEβ. Shown are results of simulation sim2 (see Table 1). Helices TM2b and TM7 are highlighted in green in surface representation; the SecY plug (see Fig. 1) is shown in red cartoon representation, whereas the rest of the protein is shown in transparent surface representation in the same colors as in Fig. 1. Blue spheres indicate the local pore radii as calculated by the program HOLE (see Methods). The center of the translocating helix is shown as a black sphere. (A) Channel state at t = 0. The blue spheres indicate the channel is closed at this stage, both by the pore ring and the plug. (B) Channel state at t = 1 ns. The pore is widened by the translocating helix; however the plug still partially blocks the channel. (C) Channel state at t = 1.9 ns. The plug is no longer blocking the channel. In all cases, the pore (blue) is directly adjacent to TM2b and TM7.
In experimental studies, translocating polypeptides as well as signal sequences have been successfully cross-linked to the lipid phase (7,48). Based on this, the suggested mechanism for membrane protein insertion strongly favors protein/lipid interactions (47,49–52). Experimental studies have shown that the probability for membrane insertion of certain amino acid sequences corresponds well to the previously determined Wimley-White free energy scale for amino acids in membranes, suggesting that direct interaction with the lipid phase is the determining factor for insertion (47,53). From our results, it seems clear that some protein/protein interaction occurs at least initially; whether this is still the case after signal sequence binding is indeterminate. We examined how close AL19 came to the hydrophobic lipid tails and found the closest approach was ∼7.6 Å, not sufficient to say the polypeptide truly samples the bilayer core. However given the large pulling velocities, AL19 had inadequate time to potentially diffuse toward lipid.
In support of the necessity of signal sequence interactions with helices 2b and 7 are studies of signal-sequence suppression mutants of SecY, also known as prl (protein localization) mutants (described in the review by Veenendaal et al. (2)). These prl mutants have specific residue changes, many of which are found to be located in helix 7, that restore the ability of the translocon to allow the passing of newly formed proteins with defective signal sequences (54–57). When we examined the translocation of AL19 (and to a lesser degree, deca-alanine as well), we found interactions with helix 7 included in particular hydrogen bonding with four channel-facing residues: Asn256, Ile260, Ala264, and Asn268, all corresponding to residues in E. coli that, when mutated, negate the need for a signal sequence (54). It should be noted, though, that prl mutants involving Ile260, a pore ring residue, may not be related to recognition but rather to channel opening (5).
Relaxation of states resulting from simulations sim1 and sim2
In simulations sim3 and sim4, we returned again to equilibration simulations. Picking up immediately where sim1 and sim2 left off (sim3 continuing sim1 and sim4 continuing sim2), we removed deca-alanine and AL19 from each simulated system (the enforced constant pressure led water to fill the empty space within picoseconds) and then continued simulations out to 5 ns. Sim3 was 3.6 ns long and sim4 was 3.1 ns long. The goal was to see how well SecYEβ returns to its original configuration, a key ability for preserving membrane integrity as well as for preparing the channel for another translocation cycle.
The plug was the portion of the protein most drastically affected by the previous simulations, evident by its location outside the pore as shown in Fig. 6 A. We found in both sim3 and sim4 that the plug moved back into the pore at least a large fraction of the way to its original position. In Fig. 6 C, taken from sim3, the plug is shown to be blocking the channel again although its orientation is slightly different. What is not obvious from Fig. 6 is that the plug has not retracted fully into the pore. In sim4, the opposite situation is seen; the plug retracts farther into the channel along the channel axis but less in the plane of the membrane. To quantify this movement, we measured the distance between the center of mass of the plug and the center of mass of the rest of SecY (to avoid measuring any net motion of the whole molecule). We found that the plug moved a maximum of 25 Å and 9.5 Å in sim1 and sim2, respectively, away from its equilibrium location. By the end of sim3 and sim4, the plug had moved back such that it was only 13 Å and 4 Å, respectively, away from its starting position, in each case a reversal of ∼50% of its total motion. However, the reversal was not equally distributed; most of it occurred during the first 1–2 ns after which period reversal was slow.
FIGURE 6.

Relaxation of SecYEβ after translocation shown in Fig. 4. The protein is viewed from the cytoplasmic side. Shown are the results of simulation sim3 (see Table 1). The representation of the protein and plug is the same as in Fig. 1. (A) SecYEβ, immediately after translocation of deca-alanine (sim1). The plug is still outside the channel. (B) SecYEβ, 1.4 ns after the translocation. The pore ring, shown in surface representation and colored yellow, is nearly closed. (C) SecYEβ 3.6 ns after helix translocation. The plug has retracted back into the channel, effectively blocking it again.
Our results based on sim1 and sim2 suggest the pore ring to be critical for maintaining the cytoplasm/periplasm barrier. Therefore, after the translocated polypeptide has left SecY, the pore ring should quickly close to prevent other molecules from passing across the membrane. This was found to be exactly the case in sim3. Fig. 6 B shows an almost closed pore ring after only 1.4 ns of relaxation in sim3. As another way to measure pore ring closure, we counted ion and water permeation events across the pore. As expected, we saw no ions cross the pore ring in either simulation. When we measured water conduction for sim3, we saw only three permeation events (all in the cytoplasmic direction). Because of its small size, deca-alanine during sim1 had actually already left the pore ring a full 0.4 ns before the end of the simulation, implying that the pore ring closes within about a nanosecond. In contrast to this, sim4 showed 15 water molecules crossing in the cytoplasmic direction and 12 toward the periplasm. Whereas AL19 in sim2 did not disturb the plug as much as deca-alanine did in sim1, AL19 did disturb the helices surrounding the pore ring through stronger interactions than deca-alanine did in sim1, possibly explaining why the pore was slower to close.
We calculated the RMSD (relative to the crystal structure) of the protein backbone for both pairs of simulations (sim1/3 and sim2/4) to monitor the overall change in the protein. The result is shown in Fig. 7, the first 1.4 ns and 1.9 ns representing sim1 and sim2, respectively, and the last 3.6 and 3.1 ns representing sim3 and sim4, respectively. The difference in maximal RMSD values is mainly due to the difference in plug motion in sim1 and sim2. Simulations sim1 and sim2 begin with an RMSD value of ∼2 Å, the RMSD at the end of sim0. Neither sim3 or sim4 relaxes completely back to the equilibrium RMSD value, but each decreases from its maximum RMSD by ∼30%.
FIGURE 7.

Deformation of SecYEβ during two translocation and relaxation cycles. Shown is the RMSD (in relation to the crystal structure) versus time for the backbone of SecYEβ in simulations sim1 and sim3 (shaded) and simulations sim2 and sim4 (solid). Sim1 ends and sim3 begins at 1.4 ns, whereas sim2 ends and sim4 begins at 1.9 ns.
Simulation of a SecYEβ dimer
A departure from the previous monomeric simulations, simulation sim5 is utilized to investigate a possible dimeric arrangement, the setup being described in Methods. The back-to-back assembly of the SecYEβ dimer was found to be stable, except for a dramatic change in each plug that became increasingly unstable. As shown in Fig. 8, there was both a change in plug position as well as in plug secondary structure. Specifically, the RMSD of the plug, defined here as residues 55–65, remained at ∼1 Å for the monomer structure (sim0) and assumed values of 2.25 Å and 3 Å for the two monomers of the dimer (sim5).
FIGURE 8.

Back-to-back dimerization of SecYEβ. (A) Dimer of SecYEβ viewed from the periplasm. Each monomer is shown in the same representation as in Fig. 1 with the exception of the color scheme of the left monomer (blue for SecY, yellow for SecE, and red for Secβ). The plugs in the dimer are without well-defined helical structure and are shown in green. (B) Destabilization of the plugs. The RMSD of the plugs (residues Ile55 to Gly65) is shown after fitting the entire SecY to the crystal structure at each point. Presented in black is the RMSD for the plug of the monomer during sim0 (see Table 1); shown in red and blue are the RMSD values for each plug from the dimer as evaluated during simulation sim5.
Simulation sim5 shows a cooperative pairing in the dimer where one monomer positively influences the other, preparing it for translocation through plug opening. This simulation assumed a back-to-back arrangement of the dimer's monomeric units; however, recent evidence can also be reconciled with a front-to-front arrangement in which the lateral gates, TMs 2b and 7, are aligned with each other (17). This arrangement could improve the functionality of the channel, allowing for a wider pore and preventing an influx of lipids when the lateral gate is open but would require large motions for placing membrane proteins into the bilayer.
DISCUSSION
Since the suggestion almost 30 years ago that a protein-conducting channel exists in intracellular membranes, a large body of data has been mounting in regard to what such a channel is and how it functions. As has been the case for many other proteins, the availability of a high resolution structure of SecYEβ has led to further insights, in particular permitting molecular dynamics simulations. Following this path, we have made some of the first explorations into the dynamical function of this channel, examining structural stability, translocation, gating, and the role of oligomerization.
Our simulations offer support for the idea that the SecY monomer can function as a translocation pore. During the simulated translocation of small polypeptides induced by pulling using SMD (see Methods), SecY showed flexibility in accommodating a translocating protein without greatly compromising its secondary and tertiary structure. We also saw SecY to relax back toward its equilibrium structure after a translocation event. The putative plug has been confirmed in its role through its observed ability to leave the channel as well as return. The pore ring is seen to provide a flexible, yet tight seal during translocation; at ion concentrations from 50 to 150 mM, no ion conduction across the channel was observed, neither before, during, nor after a translocation event. The simulations implicate new residues near the pore ring in maintaining the necessary seal; future experiments should confirm the role of these residues.
The purpose and nature of the observed oligomerization of SecYEβ remain unclear. Suggestions for the role of multimers include beneficial interactions between monomers as well as an increased number of binding sites for channel partners (5). The two conflicting models for the dimer, specifically those having a back-to-back and a front-to-front arrangement, are both supported by observation, yet another altogether different arrangement might be relevant as well. Our simulations offer evidence that a so-called back-to-back arrangement can serve a functional purpose by destabilizing the plug; such functionally relevant conformational changes were also suggested elsewhere (30,31). It should be noted that in both dimer models, in the case of cotranslational translocation of soluble proteins (the focus of this study), only one monomer serves as the active channel (17).
In future simulations, the role of various channel partners present during translocation should be considered. Although much can be learned from simulations of a single SecYEβ, one must be aware of factors that one cannot account for currently, including larger conformational changes on timescales yet inaccessible to molecular dynamics. Binding of channel partners such as the ribosome or SecA (in bacteria) that insert the nascent protein could fundamentally change the channel. In this respect, it has been suggested that the ribosome performs recognition of aqueous or membrane proteins and adjusts the translocon accordingly (58). Proteins like TRAP which have been seen associated with the channel (possibly more than one per channel) during translocation in eukaryotes could not yet be implicated in particular functions (16). Crystallographic structures of such proteins as well as high resolution structures of oligomers would be of great assistance in developing the larger picture of the protein translocation process.
Our current results have demonstrated the applicability of molecular dynamics simulations to SecYEβ, acting as an atomic resolution microscope into the channel's dynamics and paving the way for more detailed in silico investigations of the translocon in the future. The next simulations should examine the signal sequence/SecY interaction as well as the structural changes involved in lateral gating.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Supplementary Material
Acknowledgments
The authors thank Tom Rapoport for helpful discussions regarding this work as well as for his critical reading of the manuscript. The authors also thank Marcos Sotomayor for many helpful discussions and guidance.
This work was supported by the National Institutes of Health (NIH PHS-5-P41-RR05969 and NIH 1-R01-GM067887). The authors gratefully acknowledge computer time provided by the Pittsburgh Supercomputer Center and the National Center for Supercomputing Applications through the National Resources Allocation Committee (NRAC MCA93S028).
References
- 1.Matlack, K. E. S., W. Mothes, and T. A. Rapoport. 1998. Protein translocation: tunnel vision. Cell. 92:381–390. [DOI] [PubMed] [Google Scholar]
- 2.Veenendaal, A. K. J., C. van der Does, and A. J. M. Driessen. 2004. The protein-conducting channel SecYEG. Biochim. Biophys. Acta. 1694:81–95. [DOI] [PubMed] [Google Scholar]
- 3.Holland, I. B. 2004. Translocation of bacterial proteins—an overview. Biochim. Biophys. Acta. 1694:5–16. [DOI] [PubMed] [Google Scholar]
- 4.Eichler, J. 2000. Archaeal protein translocation: crossing membranes in the third domain of life. Eur. J. Biochem. 267:3402–3412. [DOI] [PubMed] [Google Scholar]
- 5.van den Berg, B., W. M. Clemons Jr., I. Collinson, Y. Modis, E. Hartmann, S. C. Harrison, and T. A. Rapoport. 2004. X-ray structure of a protein-conducting channel. Nature. 427:36–44. [DOI] [PubMed] [Google Scholar]
- 6.Rapoport, T. A., B. Jungnickel, and U. Kutay. 1996. Protein transport across the eukaryotic endoplasmic reticulum and bacterial inner membranes. Annu. Rev. Biochem. 65:271–303. [DOI] [PubMed] [Google Scholar]
- 7.Plath, K., W. Mothes, B. M. Wilkinson, C. J. Stirling, and T. A. Rapoport. 1998. Signal sequence recognition in posttranslational protein transport across the yeast ER membrane. Cell. 94:795–807. [DOI] [PubMed] [Google Scholar]
- 8.Wang, L., A. Miller, S. L. Rusch, and D. A. Kendall. 2004. Demonstration of a specific Escherichia coli SecY-signal peptide interaction. Biochemistry. 43:13185–13192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crowley, K. S., S. Liao, V. E. Worrell, G. D. Reinhart, and A. E. Johnson. 1994. Secretory proteins move through the endoplasmic reticulum membrane via an aqueous, gated pore. Cell. 78:461–471. [DOI] [PubMed] [Google Scholar]
- 10.Jungnickel, B., and T. A. Rapoport. 1995. A posttargeting signal sequence recognition event in the endoplasmic reticulum membrane. Cell. 82:261–270. [DOI] [PubMed] [Google Scholar]
- 11.Connolly, T., P. Collins, and R. Gilmore. 1989. Access of proteinase K to partially translocated nascent polypeptides in intact and detergent-solubilized membranes. J. Cell Biol. 108:299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beckmann, R., D. Bubeck, R. Grassucci, P. Penczek, A. Verschoor, G. Blobel, and J. Frank. 1997. Alignment of conduits for the nascent polypeptide chain in the ribosome-Sec61 complex. Science. 278:2123–2126. [DOI] [PubMed] [Google Scholar]
- 13.Ménétret, J. F., A. Neuhof, D. G. Morgan, K. Plath, M. Radermacher, T. A. Rapoport, and C. W. Akey. 2000. The structure of ribosome-channel complexes engaged in protein translocation. Mol. Cell. 6:1219–1232. [DOI] [PubMed] [Google Scholar]
- 14.Beckmann, R., C. M. T. Spahn, N. Eswar, J. Helmers, P. A. Penczek, A. Sali, J. Frank, and G. Blobel. 2001. Architecture of the protein-conducting channel associated with the translating 80S ribosome. Cell. 107:361–372. [DOI] [PubMed] [Google Scholar]
- 15.Morgan, D. G., J. F. Ménétret, A. Neuhof, T. A. Rapoport, and C. W. Akey. 2002. Structure of the mammalian ribosome-channel complex at 17 Å resolution. J. Mol. Biol. 324:871–886. [DOI] [PubMed] [Google Scholar]
- 16.Ménétret, J. F., R. S. Hedge, S. U. Heinrich, P. Chandramouli, S. J. Ludtke, T. A. Rapoport, and C. W. Akey. 2005. Architecture of the ribosome-channel complex derived from native membranes. J. Mol. Biol. 348:445–457. [DOI] [PubMed] [Google Scholar]
- 17.Mitra, K., C. Schaffitzel, T. Shaikh, F. Tama, S. Jenni, C. L. Brooks, N. Ban, and J. Frank. 2005. Structure of the E. coli protein-conducting channel bound to a translating ribosome. Nature. 438:318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tani, K., H. Tokuda, and S. Mizushima. 1990. Translocation of proOmpA possessing an intramolecular disulfide bridge into membrane vesicles of Escherichia coli. Effect of membrane energization. J. Biol. Chem. 265:17341–17347. [PubMed] [Google Scholar]
- 19.Kurzchalia, T. V., M. Wiedmann, H. Breter, W. Zimmermann, E. Bauschke, and T. A. Rapoport. 1988. tRNA-mediated labelling of proteins with biotin. A nonradioactive method for the detection of cell-free translation products. Eur. J. Biochem. 172:663–668. [DOI] [PubMed] [Google Scholar]
- 20.Clemons, W. M. Jr., J. F. Ménétret, C. W. Akey, and T. A. Rapoport. 2004. Structural insight into the protein translocation channel. Curr. Opin. Struct. Biol. 14:390–396. [DOI] [PubMed] [Google Scholar]
- 21.Manting, E. H., C. van der Does, H. Remigy, A. Engel, and A. J. M. Driessen. 2000. SecYEG assembles into a tetramer to form the active protein translocation channel. EMBO J. 19:852–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Breyton, C., W. Haase, T. A. Rapoport, W. Kühlbrandt, and I. Collinson. 2002. Three-dimensional structure of the bacterial protein-translocation complex SecYEG. Nature. 418:662–665. [DOI] [PubMed] [Google Scholar]
- 23.Mori, H., T. Tsukazaki, R. Masui, S. Kuramitsu, S. Yokoyama, A. E. Johnson, Y. Kimura, Y. Akiyama, and K. Ito. 2003. Fluorescence resonance energy transfer analysis of protein translocase. SecYE from Thermus thermophilus HB8 forms a constitutive oligomer in membranes. J. Biol. Chem. 278:14257–14264. [DOI] [PubMed] [Google Scholar]
- 24.Duong, F. 2003. Binding, activation and dissociation of the dimeric SecA ATPase at the dimeric SecYEG translocase. EMBO J. 22:4375–4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hanein, D., K. E. S. Matlack, B. Jungnickel, K. Plath, K.-U. Kalies, K. R. Miller, T. A. Rapoport, and C. W. Akey. 1996. Oligomeric rings of the Sec61p complex induced by ligands required for protein translocation. Cell. 87:721–732. [DOI] [PubMed] [Google Scholar]
- 26.Hamman, B. D., J. C. Chen, E. E. Johnson, and A. E. Johnson. 1997. The aqueous pore through the translocon has a diameter of 40–60 Å during cotranslational protein translocation at the ER membrane. Cell. 89:535–544. [DOI] [PubMed] [Google Scholar]
- 27.Yahr, T. L., and W. T. Wickner. 2000. Evaluating the oligomeric state of SecYEG in preprotein translocase. EMBO J. 19:4393–4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cannon, K. S., E. Or, W. M. Clemons Jr., Y. Shibata, and T. A. Rapoport. 2005. Disulfide bridge formation between SecY and a translocating polypeptide localizes the translocation pore to the center of SecY. J. Cell Biol. 169:219–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaufmann, A., E. H. Manting, A. K. J. Veenendaal, A. J. M. Driessen, and C. van der Does. 1999. Cysteine-directed cross-linking demonstrates that helix 3 of SecE is close to helix 2 of SecY and helix 3 of a neighboring SecE. Biochemistry. 38:9115–9125. [DOI] [PubMed] [Google Scholar]
- 30.Tam, P. C. K., A. P. Maillard, K. K. Y. Chan, and F. Duong. 2005. Investigating the SecY plug movement at the SecYEG translocation channel. EMBO J. 24:3380–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bostina, M., B. Mohsin, W. Kühlbrandt, and I. Collinson. 2005. Atomic model of the E. coli membrane-bound protein translocation complex SecYEG. J. Mol. Biol. 352:1035–1043. [DOI] [PubMed] [Google Scholar]
- 32.Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD—visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- 33.Phillips, J. C., R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale, and K. Schulten. 2005. Scalable molecular dynamics with NAMD. J. Comput. Chem. 26:1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.MacKerell, A. D. Jr., D. Bashford, M. Bellott, R. L. Dunbrack Jr., J. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, I. W. E. Reiher, B. Roux, M. Schlenkrich, J. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin, and M. Karplus. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- 35.Izrailev, S., S. Stepaniants, M. Balsera, Y. Oono, and K. Schulten. 1997. Molecular dynamics study of unbinding of the avidin-biotin complex. Biophys. J. 72:1568–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marszalek, P. E., H. Lu, H. Li, M. Carrion-Vazquez, A. F. Oberhauser, K. Schulten, and J. M. Fernandez. 1999. Mechanical unfolding intermediates in titin modules. Nature. 402:100–103. [DOI] [PubMed] [Google Scholar]
- 37.Isralewitz, B., M. Gao, and K. Schulten. 2001. Steered molecular dynamics and mechanical functions of proteins. Curr. Opin. Struct. Biol. 11:224–230. [DOI] [PubMed] [Google Scholar]
- 38.Isralewitz, B., J. Baudry, J. Gullingsrud, D. Kosztin, and K. Schulten. 2001. Steered molecular dynamics investigations of protein function. J. Mol. Graph. Model. 19:13–25. [DOI] [PubMed] [Google Scholar]
- 39.Wang, Y., K. Schulten, and E. Tajkhorshid. 2005. What makes an aquaporin a glycerol channel: a comparative study of AqpZ and GlpF. Structure. 13:1107–1118. [DOI] [PubMed] [Google Scholar]
- 40.Sotomayor, M., D. P. Corey, and K. Schulten. 2005. In search of the hair-cell gating spring: elastic properties of ankyrin and cadherin repeats. Structure. 13:669–682. [DOI] [PubMed] [Google Scholar]
- 41.Smart, O., J. Goodfellow, and B. Wallace. 1993. The pore dimensions of Gramicidin A. Biophys. J. 65:2455–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weiner, P. K., and P. A. Kollman. 1981. AMBER: assisted model building with energy refinement. A general program for modeling molecules and their interactions. J. Comput. Chem. 2:287–303. [Google Scholar]
- 43.Levy, Y., J. Jortner, and O. M. Becker. 2001. Solvent effects on the energy landscapes and folding kinetics of polyalanine. Proc. Natl. Acad. Sci. USA. 98:2188–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harris, C. R., and T. J. Silhavy. 1999. Mapping an interface of SecY (PrlA) and SecE (PrlG) by using synthetic phenotypes and in vivo cross-linking. J. Bacteriol. 181:3438–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jensen, M. Ø., S. Park, E. Tajkhorshid, and K. Schulten. 2002. Energetics of glycerol conduction through aquaglyceroporin GlpF. Proc. Natl. Acad. Sci. USA. 99:6731–6736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.White, S. H., and G. von Heijne. 2004. The machinery of membrane protein assembly. Curr. Opin. Struct. Biol. 14:397–404. [DOI] [PubMed] [Google Scholar]
- 47.Hessa, T., H. Kim, K. Bihlmaier, C. Lundin, J. Boekel, H. Andersson, I. Nilsson, S. H. White, and G. von Heijne. 2005. Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature. 433:377–381. [DOI] [PubMed] [Google Scholar]
- 48.McCormick, P. J., Y. Miao, Y. Shao, J. Lin, and A. E. Johnson. 2003. Cotranslational protein integration into the ER membrane is mediated by the binding of nascent chains to translocon proteins. Mol. Cell. 12:329–341. [DOI] [PubMed] [Google Scholar]
- 49.Martoglio, B., M. W. Hofmann, J. Brunner, and B. Dobberstein. 1995. The protein-conducting channel in the membrane of the endoplasmic reticulum is open laterally toward the lipid bilayer. Cell. 81:207–214. [DOI] [PubMed] [Google Scholar]
- 50.Mothes, W., S. U. Heinrich, R. Graf, I. Nilsson, G. von Heijne, J. Brunner, and T. A. Rapoport. 1997. Molecular mechanism of membrane protein integration into the endoplasmic reticulum. Cell. 89:523–533. [DOI] [PubMed] [Google Scholar]
- 51.Heinrich, S. U., W. Mothes, J. Brunner, and T. A. Rapoport. 2000. The Sec61p complex mediates the integration of a membrane protein by allowing lipid partitioning of the transmembrane domain. Cell. 102:233–244. [DOI] [PubMed] [Google Scholar]
- 52.Higy, M., S. Gander, and M. Spiess. 2005. Probing the environment of signal-anchor sequences during topogenesis in the endoplasmic reticulum. Biochemistry. 44:2039–2047. [DOI] [PubMed] [Google Scholar]
- 53.Wimley, W. C., and S. H. White. 1996. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 3:842–848. [DOI] [PubMed] [Google Scholar]
- 54.Osborne, R. S., and T. J. Silhavy. 1993. PrlA suppressor mutations cluster in regions corresponding to three distinct topological domains. EMBO J. 12:3391–3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Biecker, K. L., G. J. Phillips, and T. J. Silhavy. 1990. The Sec and Prl genes of Escherichia coli. J. Bioenerg. Biomembr. 22:291–310. [DOI] [PubMed] [Google Scholar]
- 56.Derman, A. I., J. W. Puziss, P. J. Bassford, and J. Beckwith. 1993. A signal sequence is not required for protein export in PrlA mutants of Escherichia coli. EMBO J. 12:879–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Flower, A. M., R. S. Osborne, and T. J. Silhavy. 1995. The allele-specific synthetic lethality of prlA-prlG double mutants predicts interactive domains of SecY and SecE. EMBO J. 14:884–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liao, S., J. Lin, H. Do, and A. E. Johnson. 1997. Both lumenal and cytosolic gating of the aqueous ER translocon pore are regulated from inside the ribosome during membrane protein integration. Cell. 90:31–41. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.