

Abstract
RNA interference (RNAi) is a RNA-mediated sequence-specific gene silencing mechanism. Recently, this mechanism has been used to down-regulate protein expression in mammalian cells by applying synthetic- or vector-generated small interfering RNAs (siRNAs). However, for the evaluation of this new knockdown technology, it is crucial to demonstrate biological consequences beyond protein level reduction. Here, we demonstrate that this new siRNA-based technology is suitable to analyse protein functions using the phosphatidylinositol (PI) 3-kinase signal transduction pathway as a model system. We demonstrate stable and transient siRNA-mediated knockdown of one of the PI 3-kinase catalytic subunits, p110β, which leads to inhibition of invasive cell growth in vitro as well as in a tumour model system. Importantly, this result is consistent with loss-of-function phenotypes induced by conventional RNase H-dependent antisense molecules or treatment with the PI 3-kinase inhibitor LY294002. RNAi knockdown of the downstream kinases Akt1 and Akt2 does not reduce cell growth on extracellular matrix. Our data show that synthetic siRNAs, as well as vector-based expression of siRNAs, are a powerful new tool to interfere with signal transduction processes for the elucidation of gene function in mammalian cells.
INTRODUCTION
RNA interference (RNAi) is a post-transcriptional gene-silencing mechanism initiated by double-stranded RNA (dsRNA) homologous in sequence to the targeted gene (1–3), [for review see (4)]. RNAi has been used extensively to determine gene function in a number of organisms, including plants (5), nematodes (6) and Drosophila (7,8). Until recently, RNAi in mammalian cells was not generally applicable, with the exception of embryonic cells (9,10). The discovery that transfection of short 21-nt RNA duplexes into mammalian cells interferes with gene expression and does not induce the unspecific anti-viral response that is usually obtained with long dsRNA opened new possibilities for application (2,11,12). Interestingly, these small interfering RNAs (siRNAs) resemble the processing products generated from longer dsRNAs. These siRNAs are generated by the so-called Dicer complex, a member of the RNase III family (13–16).
Depending on the particular gene function and assay systems used, a ‘gene-specific’ knockdown, induced by synthetic siRNA, might not be maintained for long enough to achieve a phenotypic change. In order to solve this problem we and others have developed vector-based expression systems to produce endogenous, functional siRNA molecules in vivo. RNA polymerase III (pol III) promoters seem to be well suited for this purpose, since they direct a high rate of transcription of small and stable RNA molecules such as U6 small nuclear RNA, tRNAs, H1 RNA or adenovirus-associated RNAs [for review see (17,18)]. In contrast to RNA polymerase II transcripts, these small RNA pol III-generated RNAs do not include mRNA-specific post-transcriptional modifications that can interfere with the function of the siRNA molecule. To demonstrate the feasibility of using siRNA expression systems to inhibit gene expression it is advantageous to analyse whether expected phenotypic changes are indeed induced after knockdown of endogenous genes in suitable model systems. Several recent reports have demonstrated the usefulness of RNA pol III promoters for the expression of siRNA molecules (19–25). In most cases, simply a reduction in protein expression was shown, however, a demonstration of the resulting biological consequences was missing. Very recently, two groups demonstrated siRNA-induced loss-of-function phenotypes using the tumour suppressor p53 as a target (19,20). One group analysed individual stable clones for their p53 expression and stated that >50% of stable siRNA plasmid transfectants showed a reduced p53 level (19). However, loss of p53 function is not detrimental to cell viability. Especially in vitro, cell culture systems often select for inactivation of p53 to enhance cell growth [for review see (26,27)]. Therefore, studies on the inhibition of p53 function in cells that are already immortal or even transformed might not reflect the general applicability of this technology.
The present report provides a more rigorous evaluation of this new technology by modulating multiple components of the phosphatidylinositol (PI) 3-kinase pathway. The PI 3-kinase pathway has been studied extensively in the past for its role in regulating cell growth, survival and transformation (28–30). A chronic activation of the PI 3-kinase pathway through loss of PTEN function appears to contribute to tumourigenesis indicating that this pathway represents an important checkpoint for controlled cell proliferation (31,32).
In order to demonstrate the efficacy and applicability of the siRNA knockdown technology we show the biological consequences after protein knockdown induced by established antisense molecules in comparison with siRNA-induced knockdown. We demonstrate that RNA pol III-dependent expression systems are highly efficient in generating functional hairpin siRNA molecules. Using this novel expression system we have identified the catalytic subunit of PI 3-kinase, p110β, which is essential for HeLa cell growth on extracellular matrix and for tumour growth in a mouse model. Thus, the expression system for siRNA molecules presented here is a first step in the development of more efficient tools for the generation of a sustained loss-of-function phenotypes in mammalian cells.
MATERIALS AND METHODS
Synthetic siRNAs and GeneBlocs
Synthetic siRNAs were purchased from Biospring (Frankfurt, Germany). The ribo-oligonucleotides were resuspended in RNase-free TE to a final concentration of 50 µM. In the case of bimolecular siRNA molecules, equal aliquots (100 µM) were combined to a final concentration of 50 µM. For the formation of intramolecular duplexes the siRNAs were incubated at 50°C for 2 min in annealing buffer (25 mM NaCl, 5 mM MgCl2) and were cooled down to room temperature. The synthetic siRNA molecules used in this study have the following sequences: 1A, (p110β 5′-aaauuccagugguucauucca-TT); 1B, (p110β 3′-TT-uuuaaggucaccaaguaaggu); 2A, (p110βMM 5′-auauuccugagcuugauacca-TT); 2B, (p110βMM 3′-TT-uauaaggacucgaacuauggu); 3A, p110β HIV-derived pA-loop (5′-aaauuccagugguucauucca-uaaagcuugcc-uggaaugaaccacuggaauuuuu-3′); 3B, p110β HIV-derived pA-loop (5′-uggaaugaaccacuggaauuu-uaaagcuugcc-aaauuccagugguucauuccauu-3′); 4A, (p110β (A)12-loop 5′-aaauuccagugguucauucca-aaaaaaaaaaa-uggaaugaaccacuggaauuuuu-3′); 4B, p110β (A)12-loop (5′-uggaaugaaccacuggaauuu-aaaaaaaaaaa-aaauuccagugguucauuccauu-3′); 5A, akt1 (A)12-loop (5′-acgaggggaguacaucaagac-aaaaaaaaaaa-gucuugauguacuccccucgu-3′); 6A, akt1 (A)4-loop (5′-acgaggggaguacaucaagac-aaaa-gucuugauguacuccccucgu-3′); 7A, akt1 (gaga)-loop (5′-acgaggggaguacaucaagac-gaga-gucuugauguacuccccucgu-3′); 8A, akt1 PEG-loop (5′-acgaggggaguacaucaagac-PEG-gucuugauguacuccccucgu-3′); 9A, akt2 (A)12-loop (5′-cuccuucauuggguacaagga-aaaaaaaaaaa-uccuuguacccaaugaaggag-3′); 10A, Akt1/2 (A)12-loop (5′-acgaggggaguacaucaagac-aaaaaaaaaaa-uccuuguacccaaugaaggag-3′); 10B, Akt1/2 (A)12-loop (5′-cuccuucauuggguacaagga-aaaaaaaaaaa-gucuugauguacuccccucgu-3′). The used p110β-specific GeneBloc (GB) represents the 3rd generation of antisense oligonucleotides and has the following schematic structure: cap-nnnnnnNNNNNNNNnnnnnn-cap. Cap represents inverted deoxy-abasic modifications, n stands for 2′-O-methyl ribonucleotides (A, G, U, C) and N represents phosphorothioate-linked deoxyribonucleotides (A, G, T, C) (33). These have the following sequences: p110β GB 5′-aaauucCAGTGGTTCauucca; p110β GBMM 5′-auauuc CTGAGCTTGauacca.
Cell culture and transfections
The particular HeLa cell line used in the experiments presented was a gift from M. Gossen (MDC Berlin, Germany) and was grown in minimum essential medium Eagle with 2 mM l-glutamine, Earle’s BSS, 1 mM sodium pyruvate, 0.1 mM non-essential amino acids, 10% fetal calf serum (FCS). Synthetic siRNA and antisense GB transfections were carried out in 96-well or 10 cm plates (at 30–50% confluency) by using cationic lipids such as Oligofectamine (Invitrogen, Carlsbad, CA) or NC388 (Atugen, Berlin) as reported previously (33). siRNA expression plasmids were transfected using Effectene™ (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Stable HeLa cells were established by selection in G418 (500 µg/ml) after transfection of cells in 5 µg of siRNA expression vectors. The next day the cells were trypsinised and seeded at a dilution of 1:20 into selective medium. The medium was renewed every 3 days; after 2 weeks the resistant colonies were trypsinised, combined to pools and cultured in selective medium.
Antibodies and immunoblotting
Cell lysates were prepared and aliquots of the cell extracts containing equal amounts of protein were analysed by immunoblotting as described previously. The murine monoclonal anti-p110α antibody has been described (34). Rabbit polyclonal anti-Akt and anti-phospho Akt (S473) antibodies were obtained from Cell Signalling Technology. The murine monoclonal anti-PTEN antibody was from Santa Cruz Biotechnology.
RNA preparation and northern blot
For the analysis of siRNAs after transfection total, cytoplasmic or nuclear RNA was prepared using RNAzol (WAK Chemie, Germany). For fractionated lysis, cells were washed with PBS and resuspended in lysis buffer (10 mM HEPES, 10 mM NaCl, 3 mM CaCl2, 0.5% NP-40) (35). Following centrifugation (800 g, 5 min, 4°C) the supernatant was collected as cytoplasmic fraction. Nuclear RNA was prepared from the remaining pellet. The RNA pellet was air dried and solved in DEPC-treated water. Concentration and purity was determined by OD260/280. RNAs were denatured by a DMSO/glyoxal (50% DMSO, 1 M glyoxal, 10 mM sodium phosphate buffer, pH 7.0) treatment for 1 h at 50°C. The glyoxal had been deionised previously by gently mixing with a bed resin (AG-510-X8, Bio-Rad, Germany) and aliquots were stored at –30°C. Denatured RNAs were separated in 3% Nusieve agarose (3:1) sodium phosphate-buffered gels. During electrophoresis the buffer was recirculated. The RNA was transferred onto nylon membranes (Nytran Supercharge, Schleicher & Schuell, Germany) and UV-crosslinked. After removal of glyoxal (washing in 20 mM Tris–HCl pH 7.4, 20 min, 65°C) pre-hybridisation was done in Church buffer (0.5 M sodium phosphate buffer pH 7.0, 7% SDS) at 37°C for 1 h. 32P 5′-End-labelled oligonucleotides (p110β 5′-ttttttttttttaaattccagtggttcattcca-3′, p110β MM 5′-ttttttttttttatattcctgagcttgatacca-3′, U6 probe 5′-gctaatcttctctgtatcgttccaatttt-3′, tRNAVal probe 5′-gaacgtgataaccactacactacggaaac-3′) were used for hybridisation overnight at 37°C in 5× SSC (0.15 M NaCl and 0.015 M sodium citrate), 1% SDS. Blots were washed several times under stringent conditions with 5× SSC, 1.0% SDS and with 2× SSC, 1.0% SDS at 37°C and exposed to phosphorimager screens (Molecular Dynamics).
Construction of siRNA expression plasmids
The pol III promoter cassettes U6+2 and U6+27 were PCR-generated using synthetical oligonucleotides and cloned into an EcoRI/XhoI restriction site of a pUC-derived vector. The specific siRNA insert was cloned using a non-palindromic restriction enzyme (BsmBI with 5′ overhang TTTT, 3′ overhang GGCA). Inserts were generated by annealing two synthetic oligonucleotides with 5′-CCGT and 3′-AAAA overhangs (Table 1).
Table 1. U6+2-derived gene-specific siRNAs.

Gene-specific 20–21 nt sequences from the target transcript separated by a 12A spacer sequence. In the case of PTEN loop (2) the spacer sequence is uaugucugccgc. Mismatches are shown in italics. The putative transcription start (gu) is shown in bold. In the case of PTEN, the two nucleotides gu at the start site are sequence specific.
Quantitation of mRNA by Taqman analysis
The RNA of cells transfected in 96 wells was isolated and purified using the Invisorb RNA HTS 96 kit (InVitek GmbH, Berlin). Inhibition of targeted mRNA expression was detected by real-time RT–PCR (Taqman) analysis using 300 nM 5′ forward primer, 300 nM 3′ reverse primer and 100 nM of the Taqman probe Fam-Tamra labelled. The gene-specific primer sequences can be obtained on request. The reaction was carried out in 50 μl and assayed on the ABI PRISM 7700 Sequence Detector (Applied Biosystems) according to the manufacturer’s instructions under the following conditions: 48°C for 30 min, 95°C for 10 min, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C.
In vitro growth on matrigel matrix
HeLa cells transfected with GB, synthetic siRNAs or siRNA expressing U6+2 plasmids were trypsinised 48 h post-transfection. The cells were washed in medium and seeded into duplicate 24 wells (100 000 cells per well) pre-coated with 250 µl matrigel basement membrane matrix (Becton Dickinson). After incubation for 24–72 h photographs were taken at 5× magnification with an Axiocam camera attached to an Axiovert S100 microscope (Zeiss).
Subcutaneous tumour model
Five- to six-week-old female immune-deficient nude mice (Shoe:NMRI-nu/nu) or SCID mice (C.B-17/Icr/BlnA-scid/scid) were injected subcutaneously with 5 × 106 HeLa cells [cell number was determined by CASY®1 cell counter (Schärfe System, Reutlingen) and manually] in a volume of 0.1 ml PBS. The mice were monitored daily and tumour volume was measured with a calliper, using the formula volume = length × width2 /2.
RESULTS
Activities of synthetic siRNA molecules with different stem–loop structures
To test the capability of synthetic and expressed siRNA molecules for modulating biological processes we have chosen the PI 3-kinase pathway as a model system (Fig. 1). Growth factor receptor-induced activation of PI 3-kinase (consisting of a regulatory p85 and a catalytic p110 subunit) results in activation and phosphorylation of its downstream effector Akt, thereby supporting cellular responses such as proliferation, survival or migration [for review see (30,36, 37)]. Most cells express two of the four known p110 isoforms, p110α and p110β.
Figure 1.

Modulation of the PI 3-kinase pathway by RNAi. A schematic representation of growth factor-induced activation of the PI 3-kinase pathway is shown. Growth factor stimulation of cells leads to activation of their cognate receptors at the cell membrane, which in turn associate with and activate intracellular signalling molecules such as PI 3-kinase. PTEN interferes with PI 3-kinase-mediated downstream responses and ensures that activation of the pathway occurs in a transient manner. Three known potential downstream effectors including Akt, PDK and JNK are indicated. RNA-mediated gene silencing was attempted for five components of the PI 3-kinase pathway, the PI 3-kinase catalytic subunits p110α, p110β, the downstream kinases Akt1 and Akt2 and the phosphatase PTEN.
At first we wanted to establish the structural requirements for monomolecular siRNA molecules with self-complementary structures, since these molecules could be expressed from a single expression cassette. First we tested whether synthetic siRNA molecules with self-complementary structures can inhibit gene expression as efficiently as standard double-stranded siRNA molecules. For these experiments we used a p110β mRNA-specific sequence that has been successfully used in a conventional antisense molecule (33). p110β-specific synthetic siRNAs with different structures were transfected into HeLa cells and the reduction in p110β mRNA was measured by real-time PCR (TaqMan). A dose-dependent titration showed no significant difference in efficiency of mRNA knockdown achieved by the standard bimolecular double-strand 21mer and the corresponding monomolecular molecules as analysed by real-time PCR (Taqman) (Fig. 2A). Two different loop structures, a (A)12 loop and an HIV-derived pA-loop (38), were tested in parallel with similar results. A comparison of the relative position of the antisense sequence and the loop structure revealed an improved knockdown efficiency with the antisense sequence being located 3′ to the loop (Fig. 2A; compare construct 3A and 3B with 4A and 4B).
Figure 2.


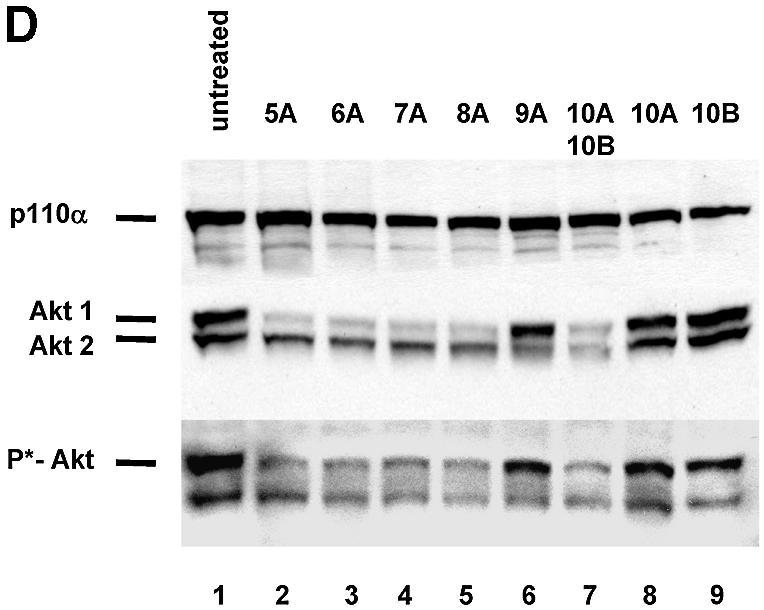
Synthetic siRNAs with different loops are functional in reducing p110β, Akt1 and Akt 2 expression. (A) Inhibition of p110β mRNA expression in siRNA-transfected HeLa cells. Samples were analysed by real-time RT–PCR (Taqman) for the level of p110β mRNA expression 24 h after transfection of the indicated siRNAs. p110β mRNA levels are shown relative to the mRNA levels of p110α, which serve as an internal reference. The transfected bimolecular siRNAs (21mer with 3′ TT overhangs, molecule 1AB) or the monomolecular siRNAs with loop structures are schematically shown. Note that the position of the loops [HIV-derived pA-loop; (A)12-loop] relative to the antisense sequence is reversed in 3A and 4A relative to 3B and 4B. The 2AB siRNA molecule contains six mismatches in the 21mer duplex and serves as a negative control together with the untreated sample. Each bar represents triplicate transfections (± standard deviation). (B) Inhibition of Akt1 mRNA expression in siRNA-transfected HeLa cells. Samples were analysed in parallel for the level of Akt1 and Akt2 mRNA expression 24 h after transfection of the indicated siRNAs. The different loops [A-loops, GAGA-loop and a PEG-linker] and their putative secondaries are shown schematically. The siRNA molecule 9A is specific for Akt2 and serves as a negative control. Note that 10A and 10B do not contain self-complementary sequences and are transfected in combination. (C) Inhibition of Akt2 mRNA expression in HeLa cells transfected with the indicated siRNA molecules. Akt2 mRNA levels are shown relative to the mRNA levels of p110β. The Akt1-specific molecule 7A serves here as a negative control. (D) Inhibition of Akt1 and Akt2 protein expression analysed by immunoblot. The cells were harvested 48 h after transfection of the indicated hairpin siRNAs (20 nM). Cell extracts were separated by SDS–PAGE and analysed by immunoblotting using anti-p110 antibody, anti-Akt 1/2. Similar results were obtained with an antibody specific for the phosphorylated form of Akt1. The positions of p110α, another catalytic subunit of PI 3-kinase, which was used as a loading control, and of Akt1, Akt2 and phosphorylated Akt (P*-Akt) are indicated on the left.
As a next step we tested the influence of different loop structures on inhibition of mRNA and protein expression. For these experiments we chose to inhibit the PI 3-kinase effectors Akt1 and Akt2 (also known as PKB or Rac-protein kinase) as targets. Significantly, the reduction in Akt1 mRNA as well as Akt1 protein levels was completely independent of the loop structure tested (Fig. 2B and D; compare molecules 5A, 6A, 7A and 8A). Even a molecule containing a rather unphysiological structure, such as a polyethyleneglycol (PEG) linker as a loop, efficiently reduced Akt1 expression indicating that size and nucleotide sequence of the loop is not crucial (Fig. 2B and D, molecule 8A). A synthetic siRNA molecule specific for Akt2 (9A) was used to control for specificity, and had no effect on Akt1 levels (Fig. 2B). This molecule, however, efficiently silenced Akt2 expression (Fig. 2C and D). Complementary RNA molecules with loop structures have the possibility to anneal as double strands in monomolecular or bimolecular structures under physiological hybridisation conditions (Fig. 2B, loop or bubble structure). To address the question of whether the siRNA molecules exert their function via adapting an intramolecular loop or an intermolecular ‘bubble’ (schematically shown in Fig. 2B) we transfected two molecules not capable of folding back on themselves. These constructs contained Akt1- and Akt2-specific sequences within the same molecule (Fig. 2B, constructs 10A and 10B) and were designed to be restricted to form a bimolecular duplex (‘bubble’). Surprisingly, this molecule efficiently mediated Akt1 and Akt2 mRNA knockdown as well as protein knockdown when transfected after annealing of both strands (Fig. 2B and D). Whether loop and bubble structures are indeed substrates for RNA-processing enzymes, e.g. Dicer, is not clear at this point. A recent study by Paddison et al. (20) suggests that hairpin-containing siRNAs are more dependent on Dicer activity than double-stranded siRNAs. However, our data demonstrating RNAi activity using a PEG linker molecule indicate that the linker sequence is likely to be irrelevant.
Knockdown of the catalytic subunit p110β, Akt1, Akt2 by synthetic hairpin siRNA molecules has different consequences in cell-based assays
Having demonstrated that synthetic monomolecular siRNA molecules are as potent as bimolecular siRNA duplexes, we wanted to evaluate the use of these molecules by comparing the effect of established GB antisense molecules with that of synthetic siRNA molecules on PI 3-kinase signalling and invasive HeLa cell growth on matrigel (33). The successful inhibition of PI 3-kinase function by synthetic antisense or siRNA molecules was monitored through reduction of phospho-Akt levels (37), p110α served as a loading control (Fig. 3A). A reduction of Akt1 and Akt2 levels was also detected after transfection of siRNA molecules specific for these proteins. Interestingly, the reduction in phospho-Akt levels was comparable in cells treated with siRNA specific for Akt1 or p110β suggesting that Akt1 is the predominantly phosphorylated form in these cells. Inhibition of the PI 3-kinase pathway, either by reducing p110β expression or by the small molecule inhibitor LY29004, causes a substantial reduction of HeLa cell growth on matrigel (Fig. 3B). Cells with invasive growth potential exhibit enhanced growth on basement membranes such as a matrigel matrix (39,40). We therefore assayed cell growth on this type of semi-solid surface to measure changes in response to siRNA-induced modulation of the PI 3-kinase pathway. p110β-specific hairpin siRNAs (molecule 4B, Fig. 2A) and p110β-specific GB (33) molecules were equally efficient, indicating a consistent biological consequence. The catalytic p110β subunit seems to be the more relevant isoform in this particular HeLa cell line since knockdown of p110α had no inhibitory effect on cell growth on matrigel (data not shown, see also Fig. 5). Inhibition of Akt1 or Akt2 expression individually did not interfere with cell growth. Also the combined inhibition of both molecules had no effect (data not shown, see also Fig. 5) suggesting that neither Akt1 nor Akt2 is necessary for proliferation on extracellular matrix. Taken together these experiments demonstrate that synthetic monomolecular siRNA molecules with loop structures are efficient tools for the evaluation of gene function in cell-based assays.
Figure 3.

p110β-specific siRNA and p110β antisense (GB) molecules inhibit HeLa cell growth on matrigel. (A) Inhibition of Akt1, Akt2 and p110β protein expression by use of siRNA and antisense (GB) molecules as analysed by immunoblot. Due to the lack of a p110β-specific antibody the p110β knockdown was demonstrated indirectly by the reduction of the phospho-Akt signal. The amount of p110α was used as a loading control. (B) HeLa cell growth on extracellular matrix after siRNA and GB transfection. Web-like cell structures are indicative of normal cell growth, spotted cell clustering signifies poor cell growth. HeLa cells were incubated with 30 nM of siRNA specific for Akt1, Akt2 and p110β. p110β-specific GB with the respective inactive mismatch controls (MM) were transfected using the same concentration (30 nM) as controls. Cells were trypsinised 48 h later and then seeded in duplicate samples on matrigel in 24 wells (100 000 cells per well). Untransfected control HeLa cells were seeded on matrigel in the presence of 10 µM PI 3-kinase inhibitor LY294002 or with the vehicle DMSO. After 24 h on matrigel the cells were photographed at 5× magnification. Sizing bars of 100 µm are shown in the left upper corner of the first picture. The experiment was repeated in several independent transfections; representative pictures are shown.
Figure 5.


Knockdown of p110β by siRNA expression reduces HeLa growth on matrigel. (A) HeLa cell growth on matrigel after transfection with Akt1-, Akt2-, Akt1&2-, PTEN-, p110β- and p110α-specific U6+2 siRNA expression plasmids. Cells were trypsinised 48 h post-transfection and seeded on matrigel in duplicate samples in 24 wells (100 000 cells per well). After 48 h on matrigel the cells were photographed at 5× magnification. The transfected U6+2 siRNA expression plasmids and their targets are indicated (for sequence of the expressed siRNAs see Table 1; MM, mismatch). Note that plasmid Akt1&2 contains a sequence which is specific for both Akt1 and Akt2. Untreated cells and cells transfected with a GFP expression plasmid were used as controls. A sizing bar of 200 µm is shown in the left upper corner of the first picture. The experiment was reproduced in several independent transfections; representative pictures are shown. (B) Inhibition of protein expression analysed by immunoblot. An aliquot of the trypsinised cells seeded on matrigel were plated on 10 cm dishes (800 000 cells). Cells were lysed after 24 h and cell extracts analysed by immunoblotting using the indicated antibodies. The positions of the specific signals for p110α, PTEN, Akt1, Akt2 and phospho-Akt are indicated on the left.
Development of vector systems for the expression of PI 3-kinase pathway-specific siRNA molecules
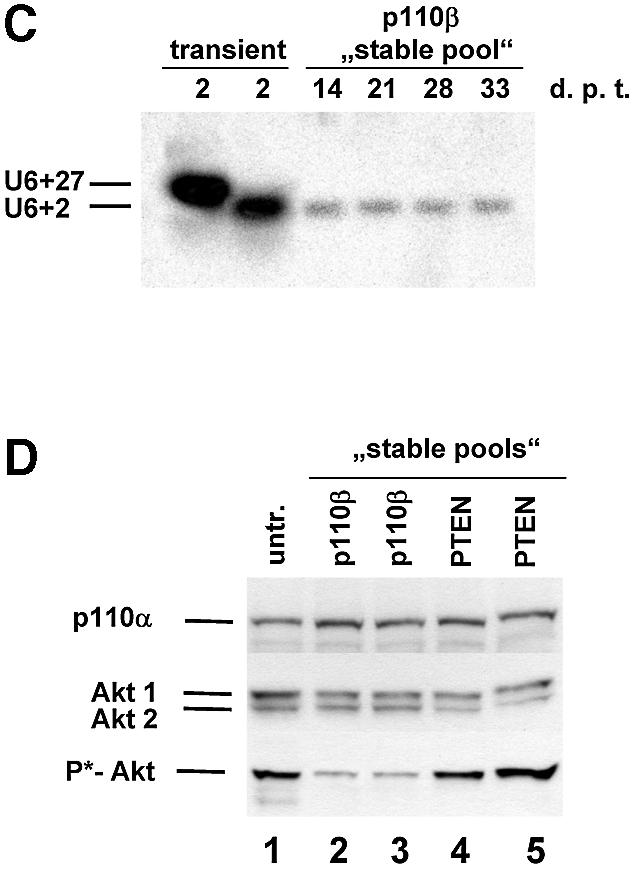
We next attempted to express stem–loop structure siRNAs using a RNA pol III promoter system. The U6 promoter has been widely used to express small RNAs such as antisense, ribozymes or aptamers for therapeutic purposes (17,18,41). Most recently it has been reported that the endogenous U6 or H1 promoter is efficient in directing the expression of hairpin siRNAs (19–25,42). Here we designed two truncated synthetic promoter constructs, U6+2 and U6+27, which differ in the length of the 5′ leader for expression of siRNA molecules (Fig. 4A). These short synthetic promoter constructs (120 bp) are restricted to the absolutely required RNA pol III cis-regulatory elements, including the SPH-, OCT- elements and TATA-box (43). This approach will facilitate the optimisation of expression levels and the development of inducible systems in the future. The so-generated pol III-dependent transcripts all exhibit a defined transcription start point (G+1T), do not contain a poly-adenosine tail that might obstruct RNAi activity and contain a termination signal consisting of just four to five thymidines (Fig. 4A). The respective expression cassettes were designed to position the 21mer sense strand 5′ to the loop (12 adenosine nucleotides) followed by the 21mer antisense strand. This appeared to be the most effective structural composition (see also Fig. 2, construct 4B). Consequently, transcripts have the potential to fold back on themselves, but might also form a bimolecular structure (‘bubble’, see Fig 2B). Transfection of these U6+2 and U6+27 expression plasmids encoding a p110β-specific siRNA sequence caused a dramatic reduction in p110β mRNA as analysed by real-time PCR (Fig. 4B). A corresponding p110β mismatch construct (Table 1) was used as a negative control. The induced reduction of p110β mRNA levels was slightly improved using the U6+2 construct. The successful expression of the siRNA molecules was verified by northern blot analysis using synthetic p110β siRNA molecules with loops as standards for expression (Fig. 4C). For this purpose it is crucial to disrupt the secondary structures of the RNA by stringent denaturing conditions (see Material and Methods) to allow for efficient probe hybridisation on hairpin RNA. The similar size of the synthetic siRNA molecules (54 nt) and the expressed siRNA molecules suggested that the stem–loop structure is not processed by cleavage in the cell. In our northern blot analysis we were not able to detect any processed siRNA molecules (data not shown). This result is in conflict with a recent report on hairpin siRNAs, which states that hairpin molecules are rapidly processed in cells to siRNAs (21–22 nt) (19). A possible explanation for our success in detecting unprocessed hairpin molecules could be a more efficient RNA denaturing step used in our protocol that precludes the ‘zipper-like’ refolding of the hairpin and therefore allows the detection of the respective hairpin structures by a labelled probe. However, at this point we cannot exclude that small amounts of processed siRNA molecules are present and mediating the silencing activity. In addition, we have analysed a series of randomised linker sequences so far without observing differences in potency suggesting that in general the specificity of the loop sequence may not represent a critical parameter (data not shown). This is confirmed by the previous finding that a molecule with a synthetic PEG linker can be active in gene silencing (Fig. 2B and D), indicating that the loop sequence itself is not crucial for generating active siRNAs. To determine the localisation of the expressed siRNA molecules we performed a fractionated lysis (Fig. 4D). To control for our fractionation procedure we monitored probes specific for a tRNA (cytoplasmic) and for endogenous U6 RNA (nuclear). The U6+2 as well as the U6+27 promoter-directed siRNA molecules (Fig. 4A) were found to be localised in the cytoplasm indicating that, in this context, the +27 leader sequence is not functional as a nuclear retention signal (44). Whether the cytoplasmatic localisation is an absolute requirement for RNA silencing activity is not entirely clear. However, at least one enzyme associated with RNAi, the Dicer complex, has been reported to be localised in the cytoplasm of mammalian cells (45). Quantitative northern blot analysis demonstrated that expression levels driven by transient transfection of the U6+2 promoter plasmid (10 µg) were comparable to the intracellular amount of siRNA molecules present after transfection of synthetic siRNA molecules (Fig. 4E, lower panel). The p110β mRNA knockdown achieved after plasmid or synthetic siRNA transfection was dose-dependent in both cases and correlated with the amount of intracellular siRNA molecules (Fig. 4E, upper panel). In the next step we have analysed how long the knockdown in protein expression can be maintained after transient transfection of siRNA encoding plasmids. Using PTEN as a target we were able to verify protein reduction in cells up to 8 days post transfection of U6+2 siRNA expression plasmids (data not shown). The knockdown mediated by single transfection of synthetic, unmodified siRNA molecules inhibited protein expression for only up to 2–4 days. Taken together, the data indicate that hairpin siRNA molecules can be functionally expressed using a RNA pol III-dependent promoter.
Figure 4.



U6+2 and U6+27 RNA pol III-promoter expression cassettes express functional hairpin siRNA molecules. (A) Schematic structure of the synthetic U6+2 and U6+27 siRNA expression constructs. The regulatory elements SPH, OCT, TATA box and the putative transcription start (G+1T) of the synthetic U6 promoter constructs are shown. In this study we used 12 nt-long loop sequences [(A)12 or (N)12] flanked by 20–21 nt-long complementary gene-specific sequences (Table 1). Note that the U6+27 construct contains a 27 nt-long transcribed leader sequence. (B) Inhibition of p110β mRNA expression in HeLa cells transfected with the indicated siRNA expression plasmids U6+2 and U6+27. Samples were analysed in parallel for the level of p110β mRNA expression 24 h after transfection by real-time RT–PCR (Taqman). p110β mRNA levels are shown relative to the mRNA levels of p110α, which served as an internal reference. The sequence of the p110β and p110β MM (mismatch) siRNAs are shown in Table 1. Each bar represents triplicate transfections (± standard deviation). (C) Northern blot analysis demonstrating the expression of hairpin siRNA directed by the U6+2 and U6+27 promoter constructs. Lanes 1–4 represent the signal of different amounts of synthetic p110β siRNA (4B see Fig. 2) spiked into 10 µg total RNA from untreated cells; lanes 5 and 6, and 7 and 8, represent 10 µg total RNA 72 h post-transfection with the indicated constructs. (D) Cytoplasmic localisation of expressed siRNA. Northern analysis of fractionated RNA (T, total; C, cytosolic; N, nuclear) using endogenous tRNAVal, endogenous U6 RNA and p110β siRNA-specific probes. (E) Inhibition of p110β mRNA expression in HeLa cells transiently transfected with the indicated siRNA expression plasmids U6+2 or with different concentrations of synthetic siRNA molecules (molecule 4B see Fig. 2). Samples were analysed in parallel for the level of p110β mRNA expression 72 h after transfection. Cells transfected with a GFP expression plasmid served as a negative control. RNA was prepared from HeLa cells 72 h after transfection und subjected to real-time RT–PCR (Taqman) analysis or northern blot analysis. p110β mRNA levels are shown relative to the mRNA levels of PTEN, which served as internal reference. Each bar represents triplicate transfections (± standard deviation). Shown in the bottom panel is a northern blot analysis comparing the relative amounts of synthetic siRNA or expressed siRNA molecules in cells. Total RNA (10 µg) was loaded in each lane. Lanes 1–4 represent the signal obtained after transfection of the U6+2 expression plasmids; in lanes 6–9, RNA was loaded from cells transfected with different amounts of synthetic siRNA, lane 5 contains RNA from cells after transfection of a GFP expression plasmid.
Application of vector-generated siRNA molecules in cell based assays and mouse models
After demonstrating that it is possible to use expressed siRNA molecules for gene function analyses we started to investigate the role of individual members of the PI 3-kinase pathway in cellular growth using this recombinant knockdown technology. We first wanted to test whether expressed siRNA-induced reduction of the expression of the two PI 3-kinase catalytic subunits, p110β or p110α, was sufficient to decrease HeLa cell growth on matrigel. Analogous to the results obtained with synthetic siRNA or antisense molecules, expressed siRNAs specific for p110β inhibited HeLa cell growth (Fig. 5A, middle panel). We used three different p110β-specific siRNAs (Table 1) with similar inhibitory effects. The inhibition obtained was as efficient as that observed with the small molecule PI 3-kinase inhibitor, LY294002 (compare Figs 3B and 5A). Neither a p110α, PTEN, PTEN mismatch nor p110β mismatch construct interfered with cell growth indicating that this is specific for p110β siRNA constructs. These results suggest that p110β is the predominant catalytic isoform essential for growth on matrigel in this HeLa cell line, whereas p110α can act as the predominant form in other cells (33) (data not shown). Inhibition of Akt1, Akt2 or a combination of both did not decrease the ability of HeLa cells to grow on matrigel despite causing an efficient protein knockdown (Fig. 5A, bottom panel, and B). For this experiment we used, next to Akt1- or Akt2-specific siRNA expression plasmids, an expression vector simultaneously inhibiting expression of both Akt1 and Akt2 with equal efficiency (Akt1&2, Table 1). To assess the transfection efficiency and the persistence of expression we used a GFP expression plasmid (Fig. 5A, bottom right panel). As expected the GFP plasmid showed no inhibition in this assay. An aliquot of the cells seeded on matrigel was used for immunoblot analysis to demonstrate a reduction in PTEN, p110α, Akt1 and Akt2 expression (Fig. 5B, lanes 2, 4, 7 and 8). The Akt protein knockdowns were comparable to the results obtained earlier using the synthetic siRNA molecules (Fig. 5B, see also Fig. 2C). As mentioned before, inhibition of p110β expression could not be tested directly since no suitable antibody was available. However, a dramatic inhibition of Akt phosphorylation was observed in cells transfected with p110β-specific siRNA expression plasmids (Fig. 5B, lane 5). It is important to note that the reduction of phospho-Akt was similar when comparing cells with knockdown of p110β, Akt1 or Akt1&2 (Fig. 5B, compare lane 5 with lanes 7, 10 and 12). We conclude from these data that both downstream kinases, Akt1 and Akt2, do not seem to be essential for proliferation in our assay, which suggests that PI 3-kinase regulates this response via Akt-independent additional functions. Due to the experimental set up, which included replating in fresh growth factor-containing medium, we did not detect a significant stimulation in Akt-phosphorylation after PTEN knockdown. Taken together the data indicate that knockdown of the catalytic subunit of PI 3-kinase, p110β induced by vector-derived siRNA can decrease proliferation and inhibits the potential to form network growth structures on matrigel. The inhibition of network structure formation observed in this assay might also reflect a decrease in motility of cells with reduced p110β activity. The involvement of PI 3-kinase signalling in cell motility, chemotaxis and actin-cytoskeleton organisation has been demonstrated in a variety of cell types (46–49). To test whether a transient transfection of siRNA plasmids is sufficient to change the growth of HeLa cells in vivo, we analysed tumour growth of transiently transfected cells in immune-deficient mice (Fig. 6A). Identical cell numbers were implanted subcutaneously into nude mice. The untreated control groups grew faster in animals indicating that the transfection procedure and the presence of the siRNA expression plasmids themselves slowed down proliferation to a certain extent. However, HeLa cells transfected with a p110β-specific siRNA plasmid grew even more slowly compared with the PTEN siRNA control in nude mice. Similar results were obtained using SCID mice as recipients (data not shown). The data were reproduced with plasmids encoding different siRNA sequences specific for both targets, p110β or PTEN, (data not shown; see Table 1). Due to the transient nature of the experiment, the difference in cell growth ceased about 8 days after implantation. Figure 6B shows the analysis of an aliquot of the cells that had been transplanted into mice and were seeded in parallel on matrigel. The growth inhibitory effect on extracellular matrix confirmed the successful knockdown of p110β. There was no difference in the rate of proliferation observed between different pools of transfectants grown on plastic before the in vivo implantation (data not shown). Therefore, the difference in proliferation of PTEN and p110β siRNA-treated cells appears to be solely dependent on the extracellular environment. A similarly stringent requirement for interaction with basement membrane to distinguish between growth of normal and malignant epithelial cells has been described previously (39). The data indicate that loss-of-function studies, where extended incubation times are required for a phenotypic analysis, are more practicable using transient expression of siRNA plasmids.
Figure 6.

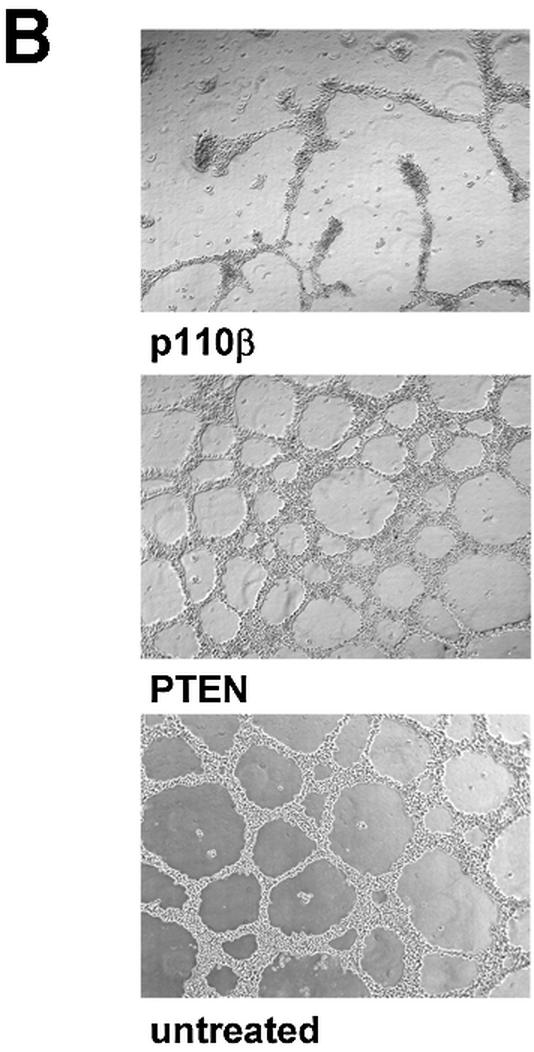

Tumour growth studies of HeLa cells transfected with siRNA expression plasmids. (A) 5 × 106 HeLa cells transiently transfected with the indicated plasmids were injected subcutaneously into nude mice. Tumour volumes were determined at regular time intervals. Curves represent mean values obtained from eight nude mice. (B) Aliquots of the transplanted cells were analysed on matrigel and photographed 24 h later. (C) Northern blot analysis of stably and transiently transfected cells to determine the siRNA expression level. RNA was prepared from pools of transfected cells at different days post-transfection (d.p.t.). (D) Status of Akt phosphorylation in HeLa cell pools stably expressing the indicated siRNA molecules. Extracts from untransfected (untr.) HeLa cells (lane 1 ), 4 weeks (lanes 2 and 4) or 8 weeks (lanes 3 and 5) post-transfection were analysed. The positions of p110α, which was used as a loading control, and of Akt1, Akt2 and phosphorylated Akt (P*-Akt) are indicated on the left. (E) Growth curves of cells stably transfected with PTEN- and p110β-specific siRNA expression plasmids in nude mice. HeLa cells (5 × 106), stably transfected with the indicated plasmids, were injected subcutaneously into nude mice. HeLa cells stably transfected with a GFP expression plasmid were used as control. Curves represent mean values obtained from eight nude mice.
The ultimate benefit of endogenously expressed siRNA molecules will be in establishing stable cell lines to permit the study of loss-of-function phenotypes. In order to test this we established pools of cells stably expressing PTEN or p110β-specific siRNA molecules. Comparison of the siRNA expression levels of transiently transfected versus stable pools revealed a 50–100 times lower expression level in stable transfectants (Fig. 6C). We found that individual stable clones and pools showed similar siRNA expression levels suggesting that most cells in the pool are positive for siRNA expression (data not shown). However, despite the low expression level of siRNA a reduction in Akt phosphorylation was detected in stable pools of cells expressing p110β siRNA when compared with cells expressing PTEN-specific siRNA (Fig. 6D, compare lanes 2 and 3 with 4 and 5). We wanted to analyse whether the relatively modest stable expression level of p110β siRNA was sufficient for causing a phenotypic difference in the mouse model. GFP expression, directed from the same vector backbone, was used as a control for G418 selection. Remarkably, it was indeed possible to detect a reduced degree of tumour growth after implantation of cells stably transfected with p110β siRNA compared with cells transfected with PTEN siRNA or GFP expression plasmids (Fig. 6E). In contrast to the experiment with transiently transfected cells the difference in growth was not very prominent but was persistent up to the end of the experiment. Whether selection of individual stable clones with higher siRNA expression levels might result in more striking phenotypes remains to be elucidated but most likely will depend on the particular target and cell line employed.
DISCUSSION
Several studies using synthetic siRNA molecules and siRNA expression systems have demonstrated the successful reduction in protein expression (12,45,50). However, since siRNA-mediated knockdown of gene expression can never be as complete as a ‘knockout’, it is important to determine whether the resulting protein knockdown is indeed sufficient to cause changes in phenotype. We have successfully inhibited PI 3-kinase activity in HeLa cells by applying synthetic siRNA molecules or siRNA expression systems specific for the catalytic subunit p110β. Importantly, we obtain the identical knockdown phenotype in our cell-based assay after treatment with a chemical inhibitor or transfection of conventional antisense molecules. Furthermore the inhibition of downstream effectors of PI 3-kinase, such as Akt, correlated with the inhibition of cell growth on extracellular matrix independent of the used knockdown method (Figs 3 and 5). Inhibiting p110α had no effect on signal-transduction or on matrigel growth, although the respective siRNA molecules, as well as antisense molecules, efficiently inhibited p110α mRNA expression (Fig. 5 and data not shown). In the cells used for our study the β isoform is likely to be the predominantly expressed form of the PI 3-kinase catalytic subunit. In cells where p110α is the predominant isoform or where both isoforms are equally important, knockdown of p110α expression efficiently interferes with PI 3-kinase signalling (33). This indicates that unlike small molecule PI 3-kinase inhibitors, this sequence-specific approach can selectively down-regulate specific isoforms of target molecules and determine their individual contribution to biological processes. Based on our siRNA-induced loss-of-function analysis of downstream effectors of p110 we conclude that Akt1 and Akt2 may not be required for HeLa growth on extracellular matrix (Figs 3 and 5). Even the simultaneous inhibition of both molecules had no effect under conditions where p110β knockdown reproducibly blocked matrigel growth. We cannot entirely rule out that the knockdown achieved was insufficient. However, the protein levels for both Akt1 and Akt2 were reduced to >90% which caused a more substantial inhibition in Akt activation than that observed after p110β knockdown. Additional inhibition of Akt3 did not change the result either (data not shown). The contribution of Akt3 is likely to be negligible in this cell type due to the low expression level of this isoform relative to Akt1 and Akt2. The result was also independent of whether Akt expression was inhibited by using siRNA or GB molecules. The failure of Akt to play a pivotal role in regulating the invasive growth potential was corroborated in various human cancer cell lines (data not shown). This result is rather surprising, since Akt is commonly perceived as ‘the’ mediator of PI 3-kinase signalling. E.g. an essential role of Akt-1 in tumourigenesis has been described for embryonic PTEN–/– (ES) cells (51). However, previous reports have indicated that Akt may be responsible for mediating some, but not all PI 3-kinase-induced responses (see also Fig. 1). For example, activated forms of Akt were found to be less potent in inducing proliferative and invasive cellular responses as the corresponding activated forms of p110 (52,53).
Endogenous expression of siRNA molecules can be achieved by two approaches. In the first approach two individual promoter constructs are used to express both strands separately (23,24). We and others have employed a single promoter construct for the expression of monomolecular, hairpin-containing siRNA molecules (19–22,25). Both techniques employ RNA pol III-dependent promoters to express functional siRNA molecules [for review see (42)]. It has been proposed that hairpin siRNA molecules have to be processed into bimolecular siRNAs to become functional (19,42). However, our northern blot analysis revealed that exclusively unprocessed hairpin molecules are present intracellularly, thus questioning the necessity for an obligatory processing step (Fig. 4C). In agreement with this hairpin molecules with synthetic loops (e.g. PEG) that are refractory to processing are equally efficient in gene silencing (Fig. 2). Therefore, the loop structure itself cannot be a crucial substrate for processing enzymes including nucleases involved in siRNA function. However, we observed differences in RNAi activity depending on the relative position of the loop with respect to the antisense strand (Fig. 2A), which indicated that directionality of the reaction is a mechanistic requirement. Furthermore, we have shown that efficient gene silencing of two genes can occur simultaneously with siRNAs that cannot form intramolecular loop structures but have the potential to form a bimolecular ‘bubble’ structure (Fig. 2). This result implies that alternative bimolecular structures are also formed and are capable of inducing RNAi, in addition to the monomolecular hairpin molecules, which were previously suggested to be the functional structure.
It has been hypothesised that expressed siRNA molecules and the mRNA target need to be co-localised in the cytoplasm for efficient gene silencing. We found high expression of U6+2- and U6+27-derived siRNA molecules restricted to the cytoplasm (Fig. 4D), which is in contrast to earlier reports showing that the U6 snRNA promoter leads primarily to nucleoplasmic expression (17,25). Our data, however, suggest that the U6 siRNA transcripts do not include nuclear retention signals and are therefore suitable for the expression of functional hairpin siRNAs.
Transient as well as stable expression of siRNA molecules allowed us to study the phenotypic consequences of a sustained p110β knockdown in cell-based assays and in animal models. The growth inhibitory effect in nude mice was marginal in the case of stable expression of p110β-specific siRNAs despite a continuously observed reduction in Akt phosphorylation (Fig. 6). A possible explanation is the relatively low expression level of siRNA in stable cell pools compared with transiently transfected cells, which might be insufficient to reduce the PI 3-kinase activity beyond a critical threshold. Therefore, it will be necessary to develop more efficient siRNA expression cassettes to ensure high siRNA expression levels after stable integration in the genome. Nevertheless, the selection of individual cell clones with high levels of expressed siRNA will be particularly difficult in cases where the target genes confer essential cellular functions such as proliferation and survival. In these instances it will be an advantage to apply inducible systems for the regulated expression of siRNAs. Interestingly, a tetracycline-responsive derivative of the human U6 promoter has already been described for a vector-directed expression of antisense RNA (54).
Taken together we demonstrate that synthetic as well as vector-derived siRNA molecules are promising new potent tools to analyse and modulate signal transduction cascades, and thus represent true alternatives to conventional loss-of-function approaches for validation of gene function.
Acknowledgments
ACKNOWLEDGEMENTS
We thank the members of the Atugen team for many helpful comments during the experiments. We are grateful to Manfred Gossen, Bert Pronk and Katharina Ahrens for critical reading of the manuscript. This study was supported by a grant from the Bundesministerium für Forschung und Technologie (No. 0311830/9).
REFERENCES
- 1.Fire A. (1999) RNA-triggered gene silencing. Trends Genet., 15, 358–363. [DOI] [PubMed] [Google Scholar]
- 2.Elbashir S.M., Harborth,J., Lendeckel,W., Yalcin,A., Weber,K. and Tuschl,T. (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature, 411, 494–498. [DOI] [PubMed] [Google Scholar]
- 3.Waterhouse P.M., Wang,M.B. and Lough,T. (2001) Gene silencing as an adaptive defence against viruses. Nature, 411, 834–842. [DOI] [PubMed] [Google Scholar]
- 4.Sharp P.A. (2001) RNA interference—2001. Genes Dev., 15, 485–490. [DOI] [PubMed] [Google Scholar]
- 5.Baulcombe D.C. (1999) Fast forward genetics based on virus-induced gene silencing. Curr. Opin. Plant Biol., 2, 109–113. [DOI] [PubMed] [Google Scholar]
- 6.Montgomery M.K., Xu,S. and Fire,A. (1998) RNA as a target of double-stranded RNA-mediated genetic interference in Caenorhabditis elegans. Proc. Natl Acad. Sci. USA, 95, 15502–15507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kennerdell J.R. and Carthew,R.W. (1998) Use of dsRNA-mediated genetic interference to demonstrate that frizzled and frizzled 2 act in the wingless pathway. Cell, 95, 1017–1026. [DOI] [PubMed] [Google Scholar]
- 8.Kennerdell J.R. and Carthew,R.W. (2000) Heritable gene silencing in Drosophila using double-stranded RNA. Nat. Biotechnol., 18, 896–898. [DOI] [PubMed] [Google Scholar]
- 9.Wianny F. and Zernicka-Goetz,M. (2000) Specific interference with gene function by double-stranded RNA in early mouse development. Nature Cell Biol., 2, 70–75. [DOI] [PubMed] [Google Scholar]
- 10.Paddison P.J., Caudy,A.A. and Hannon,G.J. (2002) Stable suppression of gene expression by RNAi in mammalian cells. Proc. Natl Acad. Sci. USA, 99, 1443–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tuschl T. (2001) RNA interference and small interfering RNAs. Chembiochem., 2, 239–245. [DOI] [PubMed] [Google Scholar]
- 12.Harborth J., Elbashir,S.M., Bechert,K., Tuschl,T. and Weber,K. (2001) Identification of essential genes in cultured mammalian cells using small interfering RNAs. J. Cell Sci., 114, 4557–4565. [DOI] [PubMed] [Google Scholar]
- 13.Bernstein E., Caudy,A.A., Hammond,S.M. and Hannon,G.J. (2001) Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature, 409, 363–366. [DOI] [PubMed] [Google Scholar]
- 14.Grishok A., Pasquinelli,A.E., Conte,D., Li,N., Parrish,S., Ha,I., Baillie,D.L., Fire,A., Ruvkun,G. and Mello,C.C. (2001) Genes and mechanisms related to RNA interference regulate expression of the small temporal RNAs that control C. elegans developmental timing. Cell, 106, 23–34. [DOI] [PubMed] [Google Scholar]
- 15.Ketting R.F., Fischer,S.E., Bernstein,E., Sijen,T., Hannon,G.J. and Plasterk,R.H. (2001) Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes Dev., 15, 2654–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knight S.W. and Bass,B.L. (2001) A role for the RNase III enzyme DCR-1 in RNA interference and germ line development in Caenorhabditis elegans. Science, 293, 2269–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Good P.D., Krikos,A.J., Li,S.X., Bertrand,E., Lee,N.S., Giver,L., Ellington,A., Zaia,J.A., Rossi,J.J. and Engelke,D.R. (1997) Expression of small, therapeutic RNAs in human cell nuclei. Gene Ther., 4, 45–54. [DOI] [PubMed] [Google Scholar]
- 18.Medina M.F. and Joshi,S. (1999) RNA-polymerase III-driven expression cassettes in human gene therapy. Curr. Opin. Mol. Ther., 1, 580–594. [PubMed] [Google Scholar]
- 19.Brummelkamp T.R., Bernards,R. and Agami,R. (2002) A system for stable expression of short interfering RNAs in mammalian cells. Science, 296, 550–553. [DOI] [PubMed] [Google Scholar]
- 20.Paddison P.J., Caudy,A.A., Bernstein,E., Hannon,G.J. and Conklin,D.S. (2002) Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev., 16, 948–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sui G., Soohoo,C., Affar el,B., Gay,F., Shi,Y. and Forrester,W.C. (2002) A DNA vector-based RNAi technology to suppress gene expression in mammalian cells. Proc. Natl Acad. Sci. USA, 99, 5515–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu J.Y., DeRuiter,S.L. and Turner,D.L. (2002) RNA interference by expression of short-interfering RNAs and hairpin RNAs in mammalian cells. Proc. Natl Acad. Sci. USA, 99, 6047–6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee N.S., Dohjima,T., Bauer,G., Li,H., Li,M.J., Ehsani,A., Salvaterra,P. and Rossi,J. (2002) Expression of small interfering RNAs targeted against HIV-1 rev transcripts in human cells. Nat. Biotechnol., 20, 500–505. [DOI] [PubMed] [Google Scholar]
- 24.Miyagishi M. and Taira,K. (2002) U6 promoter driven siRNAs with four uridine 3′ overhangs efficiently suppress targeted gene expression in mammalian cells. Nat. Biotechnol., 20, 497–500. [DOI] [PubMed] [Google Scholar]
- 25.Paul C.P., Good,P.D., Winer,I. and Engelke,D.R. (2002) Effective expression of small interfering RNA in human cells. Nat. Biotechnol., 20, 505–508. [DOI] [PubMed] [Google Scholar]
- 26.Prives C. (1998) Signaling to p53: breaking the MDM2-p53 circuit. Cell, 95, 5–8. [DOI] [PubMed] [Google Scholar]
- 27.Sherr C.J. (1998) Tumor surveillance via the ARF-p53 pathway. Genes Dev., 12, 2984–2991. [DOI] [PubMed] [Google Scholar]
- 28.Stein R.C. and Waterfield,M.D. (2000) PI3-kinase inhibition: a target for drug development? Mol. Med. Today, 6, 347–357. [DOI] [PubMed] [Google Scholar]
- 29.Vazquez F. and Sellers,W.R. (2000) The PTEN tumor suppressor protein: an antagonist of phosphoinositide 3-kinase signaling. Biochim. Biophys. Acta, 1470, M21–M35. [DOI] [PubMed] [Google Scholar]
- 30.Roymans D. and Slegers,H. (2001) Phosphatidylinositol 3-kinases in tumor progression. Eur. J. Biochem., 268, 487–498. [DOI] [PubMed] [Google Scholar]
- 31.Cantley L.C. and Neel,B.G. (1999) New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc. Natl Acad. Sci. USA, 96, 4240–4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ali I.U. (2000) Gatekeeper for endometrium: the PTEN tumor suppressor gene. J. Natl Cancer Inst., 92, 861–863. [DOI] [PubMed] [Google Scholar]
- 33.Sternberger M., Schmiedeknecht,A., Kretschmer,A., Gebhardt,F., Leenders,F., Czauderna,F., von Carlowitz,I., Engle,M., Giese,K., Beigelman,L. and Klippel,A. (2002) GeneBlocs are powerful tools to study and delineate signal transduction processes which regulate cell growth and transformation. Antisense Nucleic Acid Drug Dev., 12, 131–143. [DOI] [PubMed] [Google Scholar]
- 34.Klippel A., Escobedo,J.A., Hirano,M. and Williams,L.T. (1994) The interaction of small domains between the subunits of phosphatidylinositol 3-kinase determines enzyme activity. Mol. Cell Biol., 14, 2675–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang Y. and Carmichael,G.C. (1996) Role of polyadenylation in nucleocytoplasmic transport of mRNA. Mol. Cell Biol., 16, 1534–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wymann M.P. and Pirola,L. (1998) Structure and function of phosphoinositide 3-kinases. Biochim. Biophys. Acta, 1436, 127–150. [DOI] [PubMed] [Google Scholar]
- 37.Coffer P.J., Jin,J. and Woodgett,J.R. (1998) Protein kinase B (c-Akt): a multifunctional mediator of phosphatidylinositol 3-kinase activation. Biochem. J., 335, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berkhout B., Vastenhouw,N.L., Klasens,B.I. and Huthoff,H. (2001) Structural features in the HIV-1 repeat region facilitate strand transfer during reverse transcription. RNA, 7, 1097–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Petersen O.W., Ronnov-Jessen,L., Howlett,A.R. and Bissell,M.J. (1992) Interaction with basement membrane serves to rapidly distinguish growth and differentiation pattern of normal and malignant human breast epithelial cells. Proc. Natl Acad. Sci. USA, 89, 9064–9068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lochter A., Srebrow,A., Sympson,C.J., Terracio,N., Werb,Z. and Bissell,M.J. (1997) Misregulation of stromelysin-1 expression in mouse mammary tumor cells accompanies acquisition of stromelysin-1-dependent invasive properties. J. Biol. Chem., 272, 5007–5015. [DOI] [PubMed] [Google Scholar]
- 41.Das G., Henning,D., Wright,D. and Reddy,R. (1988) Upstream regulatory elements are necessary and sufficient for transcription of a U6 RNA gene by RNA polymerase III. EMBO J., 7, 503–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tuschl T. (2002) Expanding small RNA interference. Nat. Biotechnol., 20, 446–448. [DOI] [PubMed] [Google Scholar]
- 43.Kunkel G.R. and Hixson,J.D. (1998) The distal elements, OCT and SPH, stimulate the formation of preinitiation complexes on a human U6 snRNA gene promoter in vitro. Nucleic Acids Res., 26, 1536–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grimm C., Lund,E. and Dahlberg,J.E. (1997) In vivo selection of RNAs that localize in the nucleus. EMBO J., 16, 793–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Billy E., Brondani,V., Zhang,H., Muller,U. and Filipowicz,W. (2001) Specific interference with gene expression induced by long, double-stranded RNA in mouse embryonal teratocarcinoma cell lines. Proc. Natl Acad. Sci. USA, 98, 14428–14433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kundra V., Escobedo,J.A., Kazlauskas,A., Kim,H.K., Rhee,S.G., Williams,L.T. and Zetter,B.R. (1994) Regulation of chemotaxis by the platelet-derived growth factor receptor-beta. Nature, 367, 474–476. [DOI] [PubMed] [Google Scholar]
- 47.Kotani K., Yonezawa,K., Hara,K., Ueda,H., Kitamura,Y., Sakaue,H. Ando,A., Chavanieu,A., Calas,B., Grigorescu,F. et al. (1994) Involvement of phosphoinositide 3-kinase in insulin- or IGF-1-induced membrane ruffling. EMBO J., 13, 2313–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wennstrom S., Hawkins,P., Cooke,F., Hara,K., Yonezawa,K., Kasuga,M., Jackson,T., Claesson-Welsh,L. and Stephens,L. (1994) Activation of phosphoinositide 3-kinase is required for PDGF-stimulated membrane ruffling. Curr. Biol., 4, 385–393. [DOI] [PubMed] [Google Scholar]
- 49.Martin S.S., Haruta,T., Morris,A.J., Klippel,A., Williams,L.T. and Olefsky,J.M. (1996) Activated phosphatidylinositol 3-kinase is sufficient to mediate actin rearrangement and GLUT4 translocation in 3T3-L1 adipocytes. J. Biol. Chem., 271, 17605–17608. [DOI] [PubMed] [Google Scholar]
- 50.Elbashir S.M., Lendeckel,W. and Tuschl,T. (2001) RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev., 15, 188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stiles B., Gilman,V., Khanzenzon,N., Lesche,R., Li,A., Qiao,R., Liu,X. and Wu,H. (2002) Essential role of AKT-1/protein kinase B alpha in PTEN-controlled tumorigenesis. Mol. Cell. Biol., 22, 3842–3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shaw L.M., Rabinovitz,I., Wang,H.H., Toker,A. and Mercurio,A.M. (1997) Activation of phosphoinositide 3-OH kinase by the alpha6beta4 integrin promotes carcinoma invasion. Cell, 91, 949–960. [DOI] [PubMed] [Google Scholar]
- 53.Klippel A., Escobedo,M.A., Wachowicz,M.S., Apell,G., Brown,T.W., Giedlin,M.A., Kavanaugh,W.M. and Williams,L.T. (1998) Activation of phosphatidylinositol 3-kinase is sufficient for cell cycle entry and promotes cellular changes characteristic of oncogenic transformation. Mol. Cell. Biol., 18, 5699–5711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ohkawa J. and Taira,K. (2000) Control of the functional activity of an antisense RNA by a tetracycline-responsive derivative of the human U6 snRNA promoter. Hum. Gene Ther., 11, 577–585. [DOI] [PubMed] [Google Scholar]