


Abstract
Cdc13 is an essential protein from Saccharomyces cerevisiae that caps telomeres by protecting the C-rich telomeric DNA strand from degradation and facilitates telomeric DNA replication by telomerase. In vitro, Cdc13 binds TG-rich single-stranded telomeric DNA with high affinity and specificity. A previously identified domain of Cdc13 encompassing amino acids 451–694 (the 451–694 DBD) retains the single-stranded DNA-binding properties of the full-length protein; however, this domain contains a large unfolded region identified in heteronuclear NMR experiments. Trypsin digestion and MALDI mass spectrometry were used to identify the minimal DNA-binding domain (the 497–694 DBD) necessary and sufficient for full DNA-binding activity. This domain was completely folded, and the N-terminal unfolded region removed was shown to be dispensable for function. Using affinity photocrosslinking to site-specifically modified telomeric single-stranded DNA, the 497–694 DBD was shown to contact the entire 11mer required for high-affinity binding. Intriguingly, both domains bound single-stranded telomeric DNA with much greater affinity than the full-length protein. The full-length protein exhibited the same rate of dissociation as both domains, however, indicating that the full-length protein contains a region that inhibits association with single-stranded telomeric DNA.
INTRODUCTION
Telomeres are nucleoprotein complexes that form the ends of eukaryotic chromosomes. They are composed of repetitive tracts of DNA and a suite of proteins that specifically recognize both the double-stranded region and the G-rich single-stranded 3′ overhang of telomeric DNA (1). Telomeres perform various functions in the cell, including capping the end of the chromosome, protecting it from degradation and end-to-end fusion, and serving as a substrate for telomerase, the specialized reverse transcriptase that replicates telomeres (2).
Several strategies have been identified for telomere capping by telomere end-binding proteins (TEBPs) (3). For example, in the hypotrichous ciliate Oxytricha nova, the heterodimeric TEBP specifically recognizes and buries the single-stranded overhang (4,5). An end-binding protein with limited sequence similarity to O.nova TEBP has been identified both in the fission yeast Schizosaccharomyces pombe and in humans (6). Deletion of this protein (Pot1) in fission yeast results in loss of telomeric DNA, chromosomal mis-segregation, and reduced growth that could be bypassed by circularization of the chromosome. In mammalian cells, a TRF2-mediated duplex lariat structure at the terminus of the chromosome called a t-loop has been proposed to sequester the end of the chromosome (7).
Cdc13 is an essential telomere-capping protein from the budding yeast Saccharomyces cerevisiae that protects the C-rich telomeric strand from degradation (8–11). In vivo, the temperature sensitive mutant allele cdc13-1 causes resection of the C-rich strand at the non-permissive temperature, along with cell-cycle arrest and lethality (12). In independent genetic studies, CDC13 has also been shown to be a positive and negative regulator of telomere length (13). Mutation of residue 252 of Cdc13 causes a failure in telomere replication, even though the catalytic function of telomerase is not impaired in such mutant strains (14–16). These mutant alleles of CDC13 can be reciprocally suppressed by certain mutations of the EST1 subunit of telomerase (16), suggesting that the positive regulatory role of Cdc13 in vivo is recruitment of the enzyme to telomeric chromatin. Consistent with these activities, Cdc13 is believed to be localized to the 3′ single-stranded overhang at the telomere as it binds single-stranded yeast telomeric DNA with both high affinity and specificity (14,17). Cdc13 does not bind double-stranded telomeric DNA or single- or double-stranded DNA of random sequence, and it does not require a free 3′ end for binding. Cdc13 has the same affinity for binding a free single-stranded 3′ end, however, such that it could bind and localize to the very end of the 3′ overhang in vivo. In fact, the DNA-binding function of Cdc13 in isolation has been shown to be active in vivo. Tethering of the telomerase components Est1p or Est3p to the telomere by fusion with the Cdc13 DNA-binding domain (DBD) restores immortality to senescing mutants of Cdc13 and results in longer telomeres, respectively (18,19). In addition, fusion of the DBD to the end-protection factor Stn1p restores cell viability in the absence of full-length, functional Cdc13, although these cells still undergo senescence since they are unable to recruit telomerase components (16). These experiments indicate that Cdc13 functions to localize key proteins to the telomere that are involved in telomere end protection and replication.
A DBD of Cdc13 (451–694 DBD) was previously identified within the 924-residue full-length protein by deletion mapping and limited proteolysis, facilitating biochemical studies of Cdc13 bound to single-stranded telomeric DNA (20,21). The DBD has no similarity to any sequence in the database. Notably, no detectable sequence similarity can be detected between the 451–694 DBD and the other telomeric DNA end-binding proteins S.pombe and human Pot1 (6) or the heterodimeric O.nova TEBP (22–24). Even in the complete absence of sequence similarity, the recent solution structure of the Cdc13 DBD characterized here revealed that this domain adopts the same fold as both TEBP and the predicted fold of Pot1 (25). This result suggests that these telomere-binding proteins are evolutionarily related and that structure–function studies of Cdc13 are directly relevant to telomere maintenance in other organisms.
Although the 451–694 DBD retains DNA-binding properties comparable to that of the full-length protein, as assessed by both biochemical (20,21) and genetic (16,18) studies, it does not represent the minimal, independently folded structural domain. To better understand structure–function relationships governing single-stranded DNA binding at telomeres, we have further characterized the biochemical and structural properties of the minimal DBD.
MATERIALS AND METHODS
Production of recombinant DBDs
DNA encoding the Cdc13 single-stranded telomeric DBDs (amino acids 451–694 and 497–694) was PCR-amplified from a genomic clone (generously provided by the Lundblad laboratory, Baylor College of Medicine) and subcloned into the T7 expression vector pET21a (Novagen) between the NdeI and XhoI restriction sites. Electrotransformed (1.8 V, 400 Ω, 25 µF) BL21(DE3) Escherichia coli were grown in LB medium with 50 µg/l ampicillin at 37°C to an OD600 of 0.6 and induced with 1 mM IPTG at 22°C for 4–5 h. Cells were harvested by centrifugation, resuspended in buffer A (50 mM potassium phosphate, pH 7.0, 50 mM NaCl, 0.5 mM Na2EDTA, 0.02% NaN3 and 2 mM DTT), and lysed by two passes through a French press (Aminco). Cell extract was cleared of nucleic acids by precipitation with 0.1% polyethylenimine at 4°C for 30 min with stirring and then centrifugation. The addition of a PEI precipitation step in the purification protocol, which was included in the purification of every protein studied here, was necessary in order to remove endogenous E.coli nucleic acids that were non-specifically bound to the recombinant protein. The amount of non-specifically bound nucleic acid could be readily followed by monitoring the A260/A280 ratio in the UV/Vis absorption spectrum. Failure to remove the bound nucleic acids resulted in spurious binding features. The cleared supernatant was purified by ion-exchange chromatography over a 5 ml HiTrap SP–Sepharose column (Pharmacia) by gradient elution with buffer B (buffer A with 1 M NaCl). The protein eluted at ∼60% B and was over 95% pure as estimated by Coomassie-stained SDS–PAGE. The yield of protein was typically 15–20 mg per liter of cells.
Production of recombinant His-tagged 497–694 DBD
DNA encoding the 497–694 DBD was PCR-amplified and subcloned into the expression vector pET21a (Novagen), between the NdeI and XhoI restriction sites, in-frame with the C-terminal His-tag. BL21(DE3) E.coli were transformed, grown, and induced as described above. Cells were pelleted by centrifugation, resuspended in lysis buffer (50 mM sodium phosphate, pH 8.0, 300 mM NaCl, 10% glycerol, 0.5% Tween 20, 10 mM imidazole, 5 mM β-mercaptoethanol, 1 mM PMSF, 5 µM pepstatin A, 10 µM leupeptin, 100 µM antipain and 200 µM chymostatin) and lysed by French press. Cellular debris was removed by centrifugation, and the supernatant was cleared of DNA by precipitation with 0.1% polyethylenimine at 4°C for 30–45 min with stirring, followed by centrifugation. The supernatant was purified by affinity chromatography using a 5 ml HiTrap Chelating HP column (Pharmacia) charged with nickel chloride. Equilibration/wash buffer was the same as lysis buffer but without protease inhibitors. Bound protein was eluted with a linear gradient of imidazole from 10 to 500 mM. Protein was estimated to be 95% pure as measured by Coomassie-stained SDS–PAGE.
Production of recombinant full-length Cdc13
Frozen SF9 cells infected with baculovirus expressing full-length His6-Cdc13 (amino acids 1–924) were provided by the Lundblad Laboratory. The protein was purified using nickel-NTA chromatography as described previously (14).
Limited trypsin cleavage of the 451–694 DBD
The 451–694 DBD (30 µM in 200 µl), alone or with 1 molar equivalent telomeric 11mer (dGTGTGGGTGTG), was incubated with 0.4% w/w trypsin (Sigma) at room temperature in 50 mM potassium phosphate, pH 7.0, 350 mM NaCl, 0.25 mM Na2EDTA, 0.02% NaN3 and 2 mM DTT. At various time points, 8 µl aliquots of the reaction were withdrawn, diluted with 12 µl of water and 5 µl of SDS loading dye, and run on 15% SDS–PAGE visualized with Coomassie brilliant blue stain.
MALDI mass spectrometry of the limited trypsin cleavage products
For MALDI mass spectrometric analysis, the 451–694 DBD (30 µM in 200 µl) was incubated with 0.4% w/w trypsin in 10 mM Tris–HCl, 2 mM DTT at room temperature. Aliquots were withdrawn as described above for SDS–PAGE analysis. At the 10 min time point, 2 µl of the reaction mixture was mixed with 2 µl of α-cyano-4-hydroxycinnamic acid (CHCA) matrix. The samples were analyzed in positive ion mode on a Voyager-DE STR mass spectrometer (Perseptive Biosystems). Internal sample calibration was achieved with a mixture of insulin, thioredoxin and myoglobin standards.
Preparation of NMR samples
Uniformly 15N isotopically labeled DBD (451–694 and 497–694 DBD without a His-tag) was produced by expression in minimal media containing 6.7 g/l Na2HPO4, 3 g/l KH2PO4, 1.5 g/l NaCl, 2 g/l glucose, 10 ml/l basal medium Eagle vitamin solution (Gibco-BRL), 162.2 µg/l FeCl3, 2.86 mg/l H3BO4, 15 mg/l CaCl2·2H2O, 40 µg/l CoCl2·6H2O, 200 µg/l CuSO4·5H2O, 208 mg/l MgCl2·6H2O, 2 µg/l MoO3, 208 µg/l ZnCl2 and 1.5 g/l (15NH4)2SO4. Growth and purification was as described above except for a 7 h induction time with IPTG, yielding typically 10–15 mg protein per liter of medium. The 451–694 DBD was concentrated to 400 µM in 50 mM potassium phosphate, pH 7.0, 50 mM NaCl, 0.02% NaN3, 2 mM DTT-d10 and 10% D2O. This domain was insoluble at concentrations above 400 µM. The 497–694 DBD was concentrated to 700 µM in 50 mM imidazole-d4, pH 7.0, 150 mM NaCl, 100 mM Na2SO4, 0.02% NaN3, 2 mM DTT-d10 and 10% D2O.
NMR methods
NMR data were collected at 20 or 25°C on a Varian Unity Inova 600 MHz spectrometer. 1H-15N HSQC spectra were obtained with 2048 points and 128 t1 increments using a gradient sensitivity-enhanced pulse sequence (26). Spectra were processed with the NMRPipe/NMRDraw programs using a cosine apodization function and one round of zero filling (27).
Modified photoactive DNA oligonucleotides
The 5-iodo-2′deoxyuridine-containing single-stranded DNA oligonucleotides (Operon; Table 1) were resuspended in 10 mM triethylammonium acetate buffer, pH 6.0, and purified by acetonitrile gradient on a semi-preparative C4 reversed-phase column at 4 ml/min (Vydac). Solutions of the purified oligonucleotides were prepared in deionized water and stored at –20°C. The purity of the DNA was determined to be >99% by MALDI mass spectrometry obtained in negative ion mode using hydroxypicolinic acid (HPA) as the crystallization matrix.
Table 1. Yields of crosslinked complexes with photoactive DNAs.
| Name | Sequence | % Yield |
|---|---|---|
| WT | dGTGTGGGTGTG | 0 |
| DNA1 | dGIUGTGGGTGTG | 19 |
| DNA2 | dGTGIUGGGTGTG | 22 |
| DNA3 | dGTGTGGGIUGTG | 40 |
| DNA4 | dGTGTGGGTGIUG | 19 |
IU, 5-iodouracil substituted for the base thymine.
Protein–DNA photocrosslinking
Crosslinking reactions (500 µl of 100 µM protein and DNA) were performed in 50 mM potassium phosphate buffer, pH 7.0, 50 mM NaCl and 1 mM DTT. The reactions were transferred to a 1 ml polymethylmethacrylate cuvette with a 1 cm path-length and irradiated at 325 nm, stirring, for 3 h with an Omnichrome Series 74 He–Cd laser operating at 25–27 mW. The reactions were analyzed by SDS–PAGE.
Equilibrium binding assays by gel shift and filter binding
The 11mer dGTGTGGGTGTG was 5′-end labeled using T4 DNA kinase according to the Gibco-BRL protocol, with 5 µM DNA and 150 mCi/ml [γ-32P]ATP. A 25 µl labeling reaction was incubated at 37°C for 30 min. Unincorporated 32P was removed using microspin G25 columns (Pharmacia). All assays were conducted in 5 mM HEPES, pH 7.8, 75 mM KCl, 2.5 mM MgCl2, 0.1 mM Na2EDTA, 1 mM DTT and 0.1 mg/ml BSA. Equilibrium binding reactions were performed with 32P-11mer at concentrations 10-fold below the dissociation constant and serial dilutions of protein. The reactions were incubated on ice for 30–60 min to equilibrate. For gel-shift assays, 5 µl of each reaction with a small amount of bromophenol blue tracking dye were loaded on a 20 × 20 cm × 1.5 mm, 5% acrylamide, non-denaturing gel. Gels were equilibrated at a constant 200 V for 30–45 min before the samples were loaded. Gels were dried and visualized by a PhosphorImager (Molecular Dynamics). For filter binding, 80 µl of each binding reaction was filtered through a 96-well MultiScreen MAHA N4550 filter plate using a MultiScreen Resist Vacuum Manifold (Millipore). The wells were pre-washed with 80 µl of binding buffer without BSA, washed with 2 × 200 µl after the samples had been filtered, and the filter allowed to dry before being exposed to a PhosphorImager screen. For both assays, spots were quantified (Imagequant) and plots were normalized and fit with a standard two-state binding model: y = (ymax) / [1 + (Kd / x)] where x is the concentration of protein, y is the fraction of DNA bound and ymax is total protein bound, normalized. Equilibrium dissociation constants (Kds) are reported as an average value plus or minus the standard deviation of at least three measurements determined on different days or with different protein preparations.
Off rates measured by native PAGE
Protein (5.55 nM) was incubated for 60 min on ice with 555 pM 32P-labeled DNA in 200 µl of binding buffer containing bromophenol blue dye. Aliquots of 18 µl were removed over the time course; 2 µl of 1 µM unlabeled DNA was added to each aliquot (final concentrations of 5 nM protein, 500 pM 32P-labeled DNA, and 100 nM unlabeled DNA). Time points were analyzed by a 5% native acrylamide gel run at 200 V. Plots of protein-bound 32P-labeled DNA versus time were fit to single exponential decay curves: y = C1[exp(–koff ∗ x)] + C2 where x is time in hours, y is the fraction of DNA bound, C1 is the span (Ymax – C2), and C2 is the asymptote or plateau of non-specific binding.
Binding titrations to determine the fraction of active protein
32P-labeled DNA (1 nM) and unlabeled DNA (100 nM) were mixed and heated to 90°C for 10 min and cooled quickly on ice. This mixture was incubated on ice for 1.5 h with varying amounts of protein (full-length Cdc13, the 451–694 DBD or the 497–694 DBD) ranging from 0 to 3 molar equivalents. The samples were analyzed by a 5% native acrylamide gel.
RESULTS
Limited proteolysis reveals the minimal Cdc13 DBD
Deletion mapping and limited proteolysis were used previously to map a DBD (451–694 DBD) within the 924 amino acid full-length Cdc13 protein (20,21). This domain has a strong tendency to precipitate at high concentrations which is somewhat alleviated at reduced temperatures (15–20°C). The 1H-15N HSQC spectrum of the 451–694 DBD (Fig. 1A) reveals that while a folded species is present with well dispersed chemical shifts in both proton and nitrogen dimensions, the spectrum is dominated by cross-peaks with chemical shifts clustering between 8 and 9 p.p.m. in the proton dimension. The lack of chemical shift dispersion and the presence of sharp linewidths in these peaks indicate the presence of an unfolded species. The features of this spectrum could be due to an unfolded region of polypeptide in an otherwise folded domain or the presence of both folded and unfolded proteins at equilibrium.
Figure 1.

Comparison of Cdc13 DBD 1H-15N HSQC spectra. (A) 1H-15N HSQC spectrum of 400 µM 451–694 DBD at 600 MHz, 20°C. The sample was prepared in 50 mM potassium phosphate, pH 7.0, 50 mM NaCl, 0.02% NaN3, 2 mM DTT-d10 and 10% D2O. (B) 1H-15N HSQC spectrum of 700 µM 497–694 DBD at 600 MHz, 25°C. The sample was prepared in 50 mM imidazole-d4, pH 7.0, 150 mM NaCl, 100 mM Na2SO4, 0.02% NaN3, 2 mM DTT-d10 and 10% D2O.
To test the possibility of an unfolded region in an otherwise folded domain, the 451–694 DBD was subjected to limited trypsin digestion at room temperature. Figure 2 illustrates the time course of the reaction analyzed by SDS–PAGE. Upon exposure to trypsin, the 28 kDa 451–694 DBD almost immediately formed a smaller, 22 kDa stable fragment which was remarkably resistant to further cleavage. This reaction pattern did not change in the presence of 1 molar equivalent of the single-stranded DNA ligand dGTGTG GGTGTG, indicating that the unfolded region did not become structured upon binding DNA (data not shown).
Figure 2.
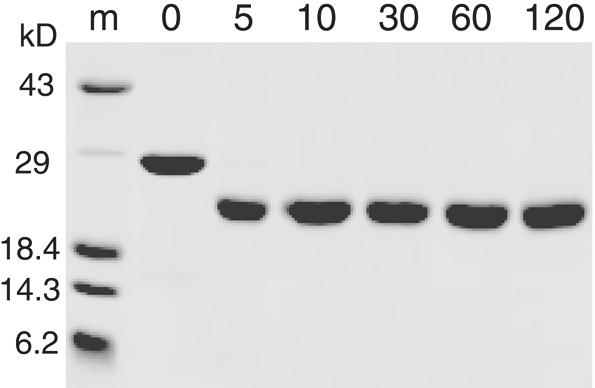
SDS–PAGE time course of limited trypsin digestion of the 451–694 DBD. Reactions were performed as described in Materials and Methods. Lane m, protein markers; other lanes in minutes of time of the reaction.
MALDI mass spectrometry was used to specifically determine the boundaries of the smaller, stable domain. An identical trypsin digest product was obtained in low salt conditions (10 mM Tris–HCl), which facilitated direct analysis of the reaction. The major product by MALDI (Fig. 3) is shown to be a MH(+1) of 21 985 ± 72 Da. Examination of the map of predicted trypsin cut sites reveals that this fragment corresponds to one of two predicted fragments: amino acids 504–692 [MH(+1) 21 971] or amino acids 496–685 [MH(+1) 21 979]. This result indicates that approximately 50 amino acids at the N-terminus of the 451–694 DBD are particularly susceptible to trypsin cleavage, presumably because this region is unfolded.
Figure 3.

MALDI-TOF mass spectrum of trypsin digest products. (a) Minor digestion product; (b–e) protein internal calibration standards as follows: b, insulin +1; c, myoglobin +2; d, thioredoxin +1; e, myoglobin +1. (f) major digestion product; (g) trypsin.
The 497–694 DBD forms a stable structural domain
Several shorter candidates of the domain were subcloned for recombinant expression in E.coli. A construct of the domain comprising amino acids 497–694 expressed in high yield as a soluble protein. This domain exhibited several favorable features relative to the 451–694 DBD. It was considerably more soluble than the longer domain and remained soluble at higher temperatures (25–30°C). A comparison of the 1H-15N HSQC spectrum of the parent domain (451–694 DBD) and of the minimal domain (497–694 DBD) is shown in Figure 1. The NMR spectrum was dramatically improved by the removal of the unfolded region. The dispersed resonances of the folded species are almost identical between the two domains. However, the random coil resonances (Fig. 1A) have completely disappeared (Fig. 1B), consistent with removal of a large region of unfolded polypeptide that does not affect the structure of the folded domain.
The 497–694 DBD contacts the entire minimal 11-nt DNA
The minimal DNA required for high-affinity binding by full-length Cdc13 is an 11mer, dGTGTGGGTGTG. This sequence of DNA is complementary to the yeast telomerase RNA template and is representative of yeast telomeric sequence (20,28). Four variants of this minimal DNA were used for photocrosslinking, with the chromophore 5-iodouracil substituted for each of the four thymine bases of the molecule (Table 1). The iodine atom of 5-iodouracil is approximately the same size as the methyl group of thymine; therefore, this substitution is not likely to perturb binding of the protein–DNA complex. Indeed, the Kd of each of the substituted DNAs for binding to the 497–694 DBD was identical to the unsubstituted DNA (data not shown). The long wavelength of 5-iodouracil’s absorption and photocrosslinking chemistry (325 nm) disfavors non-specific excitation of the DNA and protein chromophores (29). As expected, no covalent products were formed and protein degradation was not observed upon irradiation of the 497–694 DBD complexed with unsubstituted DNA (Fig. 4A). A study of the time course of photocrosslinking revealed that each substituted DNA formed a specific covalent adduct consistent with 1:1 stoichiometry of the protein and DNA (Fig. 4B–E). The yields of crosslinked species determined by densitometry ranged from 20 to 40% (Table 1), which are typical for systems using this chromophore (29–31). Identification of the exact site of crosslinking on the protein was precluded by the inability to generate complete proteolytic digests of the protein–DNA adduct. However, we have recently obtained more detailed information on the binding interface of the protein by NMR structural analysis using experiments that measure intermolecular contacts (25). Several aromatic amino acids are located along the interface that would be expected to form crosslinks to 5-iodouracil. These data indicate that the 497–694 DBD contains all the contacts needed for binding the minimal DNA 11mer.
Figure 4.

SDS–PAGE of photo-crosslinked 497–694 DBD/DNA at 100 µM each. Shown are time courses for irradiation at 325 nm, with lanes labeled in minutes and the first lane containing size markers. The conditions are stated in Materials and Methods. (A) WT DNA of sequence dGTGTG GGTGTG; (B) DNA1; (C) DNA2; (D) DNA3; (E) DNA4.
The 451–694 DBD and the 497–694 DBD bind telomeric DNA more tightly than the full-length protein
To test whether the smaller 497–694 DBD was both necessary and sufficient for function, gel-shift and filter-binding assays were used to determine the equilibrium binding dissociation constant (Kd) of this domain with the single-stranded telomeric DNA substrate dGTGTGGGTGTG. Figure 5 compares the binding curves determined by filter-binding assays of full-length Cdc13, the 451–694 DBD and the 497–694 DBD. Dissociation constants (Kds) measured by gel-shift assay yielded the same results (data not shown). The measured Kd of 310 ± 50 pM for full-length Cdc13 confirms the result of previous studies (14,20). However, we observed that the Kd for 451–694 DBD was 5 ± 1 pM, which is substantially lower than previously reported (370 pM) (20) and ranging from 72 to 240 nM for similar substrates (32). The Kd for the 497– 694 DBD (both with and without the His-tag) was 3 ± 1 pM, or ∼100-fold tighter than for full-length Cdc13 and similar to that of the 451–694 DBD. Binding titrations well above the Kd indicated that protein preparations were over 75% active under these conditions with a binding stoichiometry of 1:1 (data not shown). Therefore, the differences that we observed in binding were not due to a difference in the fraction of active protein.
Figure 5.

Fraction of DNA bound as a function of protein concentration used to determine the equilibrium dissociation binding constant. Diamonds, full-length Cdc13; squares, 451–694 DBD; and circles, 497–694 DBD bound to dGTGTGGGTGTG. Each curve is an average of at least three separate experiments conducted by filter binding. Plots were fit with a standard two-state binding model. Equilibrium dissociation constants (Kds) are 310 ± 50, 5 ± 1 and 3 ± 1 pM, respectively. Full-length protein is His-tagged, 451–694 DBD is not His-tagged, and 497–694 DBD was measured with and without a His-tag (no difference in binding was observed).
The dissociation rates of the three proteins are similar
Dissociation rates were measured by competition assay for the three proteins and are presented in Figure 6. These dissociation rates are quite similar, with rate constants of 2.8 × 10–4 min–1 for full-length Cdc13, 3.3 × 10–4 min–1 for 451–694 DBD, and 4.3 × 10–4 min–1 for 497–694 DBD. This gives a half-life for the complex of ∼41 h for full-length Cdc13, 27 h for the 497–694 DBD, and 35 h for the 451– 694 DBD. Based on the similarity of the dissociation rates, the large difference in Kd between full-length protein and the two domains must be due to an association rate effect. Association rates were too fast to measure accurately by gel-shift assay directly. From the measured Kds and dissociation rates, the calculated association rates are as follows: 9.0 × 105 M–1 min–1 for full-length Cdc13, 6.6 × 107 M–1 min–1 for the 451–694 DBD, and 1.4 × 108 M–1 min–1 for the 497– 694 DBD.
Figure 6.

Fraction of labeled DNA bound as a function of time as measured by native PAGE, used to determine the dissociation rate constant. Diamonds, full-length Cdc13; squares, 451–694 DBD; and circles, 497– 694 DBD. The experiments were conducted as stated in Materials and Methods. Full-length protein is C-terminally His-tagged, while both domains are not His-tagged.
DISCUSSION
Single-stranded nucleic acids are involved in a wide array of fundamental biological functions, including telomere regulation, DNA replication, repair and recombination, transcriptional mechanisms, translation, and RNA splicing (33–35). Recognition of single-stranded nucleic acids has been implicated in many pathological processes in humans, ranging from cancer and aging to various infectious diseases. Therefore, the sequence-specific and non-specific recognition of single-stranded nucleic acids by proteins is crucial for maintaining, manipulating and utilizing the genetic material contained in cells. Relatively little is known concerning the requirements, either structural or functional, for sequence recognition in a single-stranded context. Cdc13 is an essential protein in S.cerevisiae that regulates telomere capping and telomeric replication (36). The single-stranded telomeric DBD is central to these functions and can substitute for full-length protein when fused to appropriate binding partners (16,18). The high affinity and sequence specificity of single-stranded DNA recognition exhibited by Cdc13 (14,20) make this an ideal system to further our understanding of sequence-specific single-stranded DNA binding.
Previous identification of an independent DBD of Cdc13 (451–694 DBD) has facilitated biochemical studies of its single-stranded DNA-binding activity (20,32). Although the DBD defined by amino acids 451–694 is competent for single-stranded telomeric DNA binding in vitro and can function in vivo, the NMR data presented here clearly show that it is not a completely folded domain. The 1H-15N HSQC fingerprint spectrum of this domain (Fig. 1A) contains several intense, poorly dispersed resonances indicative of an unstructured state superpositioned on the well dispersed resonances indicative of a folded protein. Protein domains involved in a diverse array of cellular processes have been found to be intrinsically unstructured and only fold upon binding to their protein or nucleic acid partners (37). Thus, we considered the possibility that the unfolded region is directly involved in contacting DNA. However, in this case the unfolded region does not fold upon binding and does not affect single-stranded telomeric DNA binding by Cdc13 directly. The 451–694 DBD also exhibits poor solubility and is therefore unsuitable for in vitro characterization. Presumably, a construct of this domain expressed in yeast may have unexpected, variable effects as well.
In this work, the Cdc13 DBD was refined by limited trypsin digestion and MALDI mass spectrometry (Figs 2 and 3), producing a domain (497–694 DBD) that is both structurally and functionally independent. This represents the true, minimal DBD. In contrast to the 451–694 DBD, the 1H-15N HSQC spectrum of the 497–694 DBD reveals the presence of a completely folded species (Fig. 1B). Careful examination of the spectrum obtained on the 451–694 DBD reveals that the well dispersed resonances of the 497–694 DBD are present within the spectrum of the 451–694 DBD, clearly demonstrating that the folded region is also present in the longer domain. Further biochemical characterization using affinity photocrosslinking revealed that the smaller, folded domain contacts the entire 11mer of single-stranded DNA required for high affinity and specificity binding (20). Affinity photocrosslinking has previously been used successfully as a probe of contacts between protein and single-stranded DNA (38). In particular, crosslinks found between chromophore-containing DNA and the O.nova TEBP corresponded to sites of protein–DNA contacts in the high resolution structure (22,29,39). Similarly high-yielding protein–DNA crosslinks were obtained in this system with use of the 5-iodouracil chromophore. We have shown that sites throughout the DNA 11mer crosslink and therefore interact with the 198 amino acid minimal binding domain defined here. This result has been subsequently confirmed by mapping the protein–DNA interface using NMR spectroscopy, which revealed that several amino acids capable of crosslinking do contact DNA (25).
Interestingly, both DBDs bind DNA with tighter affinity (Kd ≈ 3–5 pM) than the full-length protein (Kd = 310 pM) (Fig. 5). Thus, the smaller, 497–694 DBD contains the essential region for contacting single-stranded telomeric DNA. The difference in affinity between the isolated domains and full-length protein could simply be an artifact of extracting the domain from the full-length protein. However, enhanced binding of functional domains relative to full-length protein has been observed in the other TEBPs, O.nova TEBP α subunit (40) and S.pombe Pot1p (6), perhaps indicating that this is a general feature of this family of TEBPs. In the case of Pot1 protein, deletion of a large C-terminal segment increased binding affinity by ∼10-fold, while in O.nova TEBP α subunit several different truncations at the C-terminus increased binding. Additionally, several splice variants of human Pot1 protein (hPot1) have been identified and appear to interact differentially with single-stranded human telomeric DNA (P.Baumann, E.Podell and T.R.Cech, personal communication). The full-length protein may contain a region inhibitory to binding which could play a regulatory role in vivo by attenuating the extremely tight binding of the DBD.
Our characterization of the 451–694 DBD has generated results that are in contrast to previous studies of the same domain (20,32). In our study we have used highly purified, soluble preparations of the DBDs, taking care to ensure that over 75% of the protein was active. We have observed that it is critical to include a small amount of BSA (or Nonidet P-40 detergent) in the binding reactions to prevent loss of the protein to surfaces and aggregation or precipitation of the protein. When this protocol was not followed we obtained low and irreproducible binding, presumably due to non-specific loss of protein and/or protein inactivation (data not shown). Previous studies intended to determine the binding affinity of the 451–694 DBD were not performed in the presence of BSA or detergent (20,32; V.Lundblad, personal communication). This may explain the discrepancy between previous results and the binding affinities reported here.
The rates of dissociation of the DNA ligand for full-length Cdc13, 451–694 DBD and 497–694 DBD are uniformly slow, yet similar. The calculated rate of association of the 451– 694 DBD and the 497–694 DBD with DNA is two to three orders of magnitude faster than for full-length protein. Thus, the differences in binding affinity are primarily due to differences in the rates of association. We have not determined in vitro if the attenuation of binding in the full-length protein is due to regions N- or C-terminal to the DBD. Further study of this phenomenon is underway to determine what effect this attenuation has on yeast telomeres in vivo.
Cdc13 performs critical functions with partner protein complexes both in capping the telomere, protecting it from degradation and fusion, and in regulating telomeric replication and length. The single-stranded telomeric DBD of Cdc13 is central to its function and exhibits unusually high affinity and specificity for its DNA target. We have delineated the true minimal folded domain that is both necessary and sufficient for high-affinity binding to single-stranded telomeric DNA. The present work provides insights into this important mode of DNA recognition.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to gratefully acknowledge and thank members of the Wuttke laboratory, especially Douglas Theobald for performing titration binding experiments, Thorsten Schäfer for contributions to the photocrosslinking studies, and Corey Mandel for subcloning the 497–694 DBD His-tagged construct. We thank Rachel Mitton-Fry, Leslie Glustrom, Art Pardi and Fiona Jucker for careful reading of the manuscript. We thank Vicki Lundblad and Timothy Hughes for access to data before publication, for plasmids encoding both full-length Cdc13 and the 451–694 DBD, and for SF9 cell pellets containing full-length Cdc13. We thank Tad Koch for help with photocrosslinking and use of his He-Cd laser. We are thankful for all sources of funding for this research, which has been provided by the NIH (GM59414), the American Cancer Society, a University of Colorado Junior Faculty Development Award, and a pre-doctoral fellowship from the U.S. Army Breast Cancer Research Program (E.M.A.) (DAMD17-99-1-9150). The NMR instrumentation was purchased with partial support from NIH RR11969 and NSF 9602941. We also thank the W. M. Keck Foundation for support of the Molecular Structure Program on the Boulder campus.
REFERENCES
- 1.Shore D. (2001) Telomeric chromatin: replicating and wrapping up chromosome ends. Curr. Opin. Genet. Dev., 11, 189–198. [DOI] [PubMed] [Google Scholar]
- 2.Blackburn E.H. (2001) Switching and signaling at the telomere. Cell, 106, 661–673. [DOI] [PubMed] [Google Scholar]
- 3.de Lange T. (2001) Telomere capping—one strand fits all. Science, 292, 1075–1076. [DOI] [PubMed] [Google Scholar]
- 4.Gottschling D.E. and Zakian,V.A. (1986) Telomere proteins: specific recognition and protection of the natural termini of Oxytricha macronuclear DNA. Cell, 47, 195–205. [DOI] [PubMed] [Google Scholar]
- 5.Price C.M. and Cech,T.R. (1987) Telomeric DNA–protein interactions of Oxytricha macronuclear DNA. Genes Dev., 1, 783–793. [DOI] [PubMed] [Google Scholar]
- 6.Baumann P. and Cech,T.R. (2001) Pot1, the putative telomere end-binding protein in fission yeast and humans. Science, 292, 1171–1175. [DOI] [PubMed] [Google Scholar]
- 7.Griffith J.D., Comeau,L., Rosenfield,S., Stansel,R.M., Bianchi,A., Moss,H. and de Lange,T. (1999) Mammalian telomeres end in a large duplex loop. Cell, 97, 503–514. [DOI] [PubMed] [Google Scholar]
- 8.Weinert T.A. and Hartwell,L.H. (1993) Cell cycle arrest of cdc mutants and specificity of the RAD9 checkpoint. Genetics, 134, 63–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garvik B., Carson,M. and Hartwell,L. (1995) Single-stranded DNA arising at telomeres in cdc13 mutants may constitute a specific signal for the RAD9 checkpoint. Mol. Cell. Biol., 15, 6128–6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diede S.J. and Gottschling,D.E. (1999) Telomerase-mediated telomere addition in vivo requires DNA primase and DNA polymerases alpha and delta. Cell, 99, 723–733. [DOI] [PubMed] [Google Scholar]
- 11.Diede S.J. and Gottschling,D.E. (2001) Exonuclease activity is required for sequence addition and Cdc13p loading at a de novo telomere. Curr. Biol., 11, 1336–1340. [DOI] [PubMed] [Google Scholar]
- 12.Booth C., Griffith,E., Brady,G. and Lydall,D. (2001) Quantitative amplification of single-stranded DNA (QAOS) demonstrates that cdc13-1 mutants generate ssDNA in a telomere to centromere direction. Nucleic Acids Res., 29, 4414–4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chandra A., Hughes,T.R., Nugent,C.I. and Lundblad,V. (2001) Cdc13 both positively and negatively regulates telomere replication. Genes Dev., 15, 404–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nugent C.I., Hughes,T.R., Lue,N.F. and Lundblad,V. (1996) Cdc13p: a single-strand telomeric DNA-binding protein with a dual role in yeast telomere maintenance. Science, 274, 249–252. [DOI] [PubMed] [Google Scholar]
- 15.Lingner J., Cech,T.R., Hughes,T.R. and Lundblad,V. (1997) Three Ever Shorter Telomere (EST) genes are dispensible for in vitro yeast telomerase activity. Proc. Natl Acad. Sci. USA, 94, 11190–11195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pennock E., Buckley,K. and Lundblad,V. (2001) Cdc13 delivers separate complexes to the telomere for end protection and replication. Cell, 104, 387–396. [DOI] [PubMed] [Google Scholar]
- 17.Lin J.-J. and Zakian,V.A. (1996) The Saccharomyces CDC13 protein is a single-strand TG1–3 telomeric DNA-binding protein in vitro that affects telomere behavior in vivo. Proc. Natl Acad. Sci. USA, 93, 13760–13765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evans S.K. and Lundblad,V. (1999) Est1 and Cdc13 as comediators of telomerase access. Science, 286, 117–120. [DOI] [PubMed] [Google Scholar]
- 19.Hughes T.R., Evans,S.K., Weilbaecher,R.G. and Lundblad,V. (2000) The Est3 protein is a subunit of yeast telomerase. Curr. Biol., 10, 809–812. [DOI] [PubMed] [Google Scholar]
- 20.Hughes T.R., Weilbaecher,R.G., Walterscheid,M. and Lundblad,V. (2000) Identification of the single-strand telomeric DNA binding domain of the Saccharomyces cerevisiae Cdc13 protein. Proc. Natl Acad. Sci. USA, 97, 6457–6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang M.-J., Lin,Y.-C., Pang,T.-L., Lee,J.-M., Chou,C.-C. and Lin,J.-J. (2000) Telomere-binding and Stn1p-interacting activities are required for the essential function of Saccharomyces cerevisiae Cdc13p. Nucleic Acids Res., 28, 4733–4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horvath M.P., Schweiker,V.L., Bevilacqua,J.M., Ruggles,J.A. and Schultz,S.C. (1998) Crystal structure of the Oxytricha nova telomere end binding protein complexed with single strand DNA. Cell, 95, 963–974. [DOI] [PubMed] [Google Scholar]
- 23.Horvath M.P. and Schultz,S.C. (2001) DNA G-quartets in a 1.86 Å resolution structure of an Oxytricha nova telomeric protein–DNA complex. J. Mol. Biol., 310, 367–377. [DOI] [PubMed] [Google Scholar]
- 24.Classen S., Ruggles,J.A. and Schultz,S.C. (2001) Crystal structure of the N-terminal domain of Oxytricha nova telomere end-binding protein alpha subunit both uncomplexed and complexed with telomeric ssDNA. J. Mol. Biol., 314, 1113–1125. [DOI] [PubMed] [Google Scholar]
- 25.Mitton-Fry R.M., Anderson,E.M., Hughes,T.R., Lundblad,V. and Wuttke,D.S. (2002) Conserved structure for single-stranded telomeric DNA recognition. Science, 296, 145–147. [DOI] [PubMed] [Google Scholar]
- 26.Kay L.E., Keifer,P. and Saarinen,T. (1992) Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J. Am. Chem. Soc., 114, 10663–10665. [Google Scholar]
- 27.Delaglio F., Grzesiek,S., Vuister,G.W., Zhu,G., Pfeifer,J. and Bax,A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR, 6, 277–293. [DOI] [PubMed] [Google Scholar]
- 28.Singer M.S. and Gottschling,D.E. (1994) TLC1: template RNA component of Saccharomyces cerevisiae telomerase. Science, 266, 404–409. [DOI] [PubMed] [Google Scholar]
- 29.Willis M.C., Hicke,B.J., Uhlenbeck,O.C., Cech,T.R. and Koch,T.H. (1993) Photocrosslinking of 5-iodouracil-substituted RNA and DNA to proteins. Science, 262, 1255–1257. [DOI] [PubMed] [Google Scholar]
- 30.Steen H., Petersen,J., Mann,M. and Jensen,O.N. (2001) Mass spectrometric analysis of a UV-cross-linked protein–DNA complex: tryptophans 54 and 88 of E. coli SSB cross-link to DNA. Protein Sci., 10, 1989–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wong D.L. and Reich,N.O. (2000) Identification of tyrosine 204 as the photo-cross-linking site in the DNA–EcoRI DNA methyltransferase complex by electrospray ionization mass spectrometry. Biochemistry, 39, 15410–15417. [DOI] [PubMed] [Google Scholar]
- 32.Lin Y.-C., Hsu,C.-L., Shih,J.-W. and Lin,J.-J. (2001) Specific binding of single-stranded telomeric DNA by Cdc13p of Saccharomyces cerevisiae. J. Biol. Chem., 276, 24588–24593. [DOI] [PubMed] [Google Scholar]
- 33.Swamynathan S.K., Nambiar,A. and Guntaka,R.V. (1998) Role of single-stranded DNA regions and Y-box proteins in transcriptional regulation of viral and cellular genes. FASEB J., 12, 515–522. [DOI] [PubMed] [Google Scholar]
- 34.Broomfield S., Hryciw,T. and Xiao,W. (2001) DNA postreplication repair and mutagenesis in Saccharomyces cerevisiae. Mutat. Res., 486, 167–184. [DOI] [PubMed] [Google Scholar]
- 35.Handa N., Nureki,O., Kurimoto,K., Kim,I., Sakamoto,H., Shimura,Y., Muto,Y. and Yokoyama,S. (1999) Structural basis for the recognition of the tra mRNA precursor by the Sex-lethal protein. Nature, 398, 579–585. [DOI] [PubMed] [Google Scholar]
- 36.Lustig A.J. (2001) Cdc13 subcomplexes regulate multiple telomere functions. Nature Struct. Biol., 8, 297–299. [DOI] [PubMed] [Google Scholar]
- 37.Wright P.E. and Dyson,H.J. (1999) Intrinsically unstructured proteins: re-assessing the protein structure–function paradigm. J. Mol. Biol., 293, 321–331. [DOI] [PubMed] [Google Scholar]
- 38.Meisenheimer K.M. and Koch,T.H. (1997) Photocross-linking of nucleic acids to associated proteins. Crit. Rev. Biochem. Mol. Biol., 32, 101–140. [DOI] [PubMed] [Google Scholar]
- 39.Hicke B.J., Willis,M.C., Koch,T.H. and Cech,T.R. (1994) Telomeric protein–DNA contacts identified by photo-cross-linking using 5-bromodeoxyuridine. Biochemistry, 33, 3364–3373. [DOI] [PubMed] [Google Scholar]
- 40.Fang G., Gray,J.T. and Cech,T.R. (1993) Oxytricha telomere-binding protein: separable DNA-binding and dimerization domains of the α-subunit. Genes Dev., 7, 870–882. [DOI] [PubMed] [Google Scholar]