

Abstract
The gastric pathogen Helicobacter pylori translocates the CagA protein into epithelial cells by a type IV secretion process. Translocated CagA is tyrosine phosphorylated (CagAP-Tyr) on specific EPIYA sequence repeats by Src family tyrosine kinases. Phos phorylation of CagA induces the dephosphorylation of as yet unidentified cellular proteins, rearrangements of the host cell actin cytoskeleton and cell scattering. We show here that CagAP-Tyr inhibits the catalytic activity of c-Src in vivo and in vitro. c-Src inactivation leads to tyrosine dephosphorylation of the actin binding protein cortactin. Concomitantly, cortactin is specifically redistributed to actin-rich cellular protrusions. c-Src inactivation and cortactin dephosphorylation are required for rearrangements of the actin cytoskeleton. Moreover, CagAP-Tyr-mediated c-Src inhibition downregulates further CagA phosphorylation through a negative feedback loop. This is the first report of a bacterial virulence factor that inhibits signalling of a eukaryotic tyrosine kinase and on a role of c-Src inactivation in host cell cytoskeletal rearrangements.
Keywords: actin cytoskeleton/molecular pathogenesis/type IV secretion/tyrosine phosphorylation
Introduction
Type III and type IV secretion systems mediate the direct injection of virulence factors into host cells and play a key role in the pathogenesis of numerous bacteria that infect plants or animals (Finlay and Falkow, 1997; Lee and Schneewind, 2001). Type III secretion systems translocate protein effectors through a syringe-like device and have been detected in pathogens such as Yersinia and Salmonella species, Shigella flexneri, enteropathogenic Escherichia coli (EPEC) and Pseudomonas aeruginosa (Hueck, 1998; Kubori et al., 1998; Galan and Collmer, 1999; Cornelis and van Gijsegem, 2000). Type IV secretion systems are functionally related but evolutionary distinct from type III machineries and mediate the transfer of DNA and/or proteins into the host cell cytoplasm (Burns, 1999; Christie and Vogel, 2000). The prototypic member of the latter transporter family is that of Agrobacterium tumefaciens, consisting of assembled VirB and VirD proteins that drive the translocation of the oncogenic transfer (T)-DNA–protein complex into plant cells. Until now, only a few secreted protein effectors of type IV transporters have been described, such as pertussis toxin of Bordetella pertussis (Burns, 1999), DotA and RalF from Legionella pneumophila (Nagai and Roy, 2001; Nagai et al., 2002) and CagA from the gastric pathogen Helicobacter pylori (Segal et al., 1999; Asahi et al., 2000; Backert et al., 2000; Odenbreit et al., 2000; Stein et al., 2000).
In H.pylori, type I strains harbour the cag (cytotoxin-associated genes) pathogenicity island (cagPAI), whereas type II strains lack the entire cagPAI (Censini et al., 1996; Akopyants et al., 1998). Among several H.pylori virulence determinants, like VacA or NapA (Montecucco and Rappuoli, 2001), the cagPAI has raised special interest because cagPAI positive-isolates significantly increase the risk of infected patients developing severe gastritis, peptic ulcer disease or even gastric cancer (Peek and Blaser, 2002). Accordingly, WHO has classified H.pylori as a class I carcinogen (IARC, 1994). The cagPAI encodes homologs of a VirB/D complex known from other type IV secretion systems, the 120–150 kDa immuno-dominant antigen CagA and several other proteins with unknown functions (Censini et al., 1996). It is now well established that H.pylori actively injects CagA into target cells in a cagPAI-dependent manner (Montecucco and Rappuoli, 2001). Furthermore, this H.pylori type IV secretion system stimulates the production of pro-inflammatory cytokines and chemokines by infected host cells in a CagA/VirD4-independent manner, possibly by translocating another as yet unknown factor or by direct activation of a cell surface receptor (Crabtree et al., 1995; Censini et al., 1996; Selbach et al., 2002a). Systematic mutagenesis has revealed that many genes throughout the whole cagPAI are essential for both CagA translocation and the induction of pro-inflammatory responses (Fischer et al., 2001; Selbach et al., 2002a).
Upon translocation into the host cell cytosol, CagA undergoes tyrosine phosphorylation (Segal et al., 1999; Asahi et al., 2000; Backert et al., 2000; Odenbreit et al., 2000; Stein et al., 2000). Phosphorylation of CagA occurs within the C-terminus of the protein and is mediated by members of the Src family of tyrosine kinases (Selbach et al., 2002b; Stein et al., 2002). The major phosphorylation motif is a cluster of Glu-Pro-Ile-Tyr-Ala (EPIYA) sequence repeats that share homology to c-Src consensus phosphorylation sites. The number of these EPIYA motifs varies from 1–5 repeat units depending on the individual CagA protein species (Covacci et al., 1993; Selbach et al., 2002b; Stein et al., 2002). For example, Y972 was identified as the most important phosphorylation site in CagA from the TIGR H.pylori strain (Backert et al., 2001). CagA phosphorylation was found to be a prerequisite for the induction of actin cytoskeletal rearrangements in AGS gastric epithelial cells (Backert et al., 2001; Stein et al., 2002). The characteristic morphology of infected cells has been referred to as the ‘hummingbird phenotype’ (Segal et al., 1999). This phenotype resembles hepatocyte growth factor (HGF)-induced scattering of Madin–Darby Canine Kidney (MDCK) cells. HGF binds to the HGF receptor c-Met and activates a signalling cascade which ultimately leads to the dissociation of epithelial cells (Weidner et al., 1990; Stella and Comoglio, 1999). However, the mechanism by which H.pylori induces scattering of AGS cells is not understood. Recently, the protein tyrosine phosphatase (PTPase) Shp-2 was shown to bind specifically to transiently expressed CagAP-Tyr via its src homology 2 (SH2) domain followed by the activation of the Shp-2 PTPase activity (Higashi et al., 2002). Independent reports have demonstrated that CagAP-Tyr initiates the dephosphorylation of several as yet unidentified host cell proteins (Backert et al., 2000; Püls et al., 2002). How ever, whether the latter events are linked to the activation of Shp-2 and the induction of cytoskeletal rearrangements or if actin binding proteins like the Arp2/3 (actin related protein) complex and N-WASP might play a role in this scenario remains to be clarified (Stein et al., 2002).
Here we identify cortactin, an actin binding protein and c-Src substrate, to be dephosphorylated in a CagAP-Tyr-dependent manner. Significantly, the subcellular location of cortactin changes upon H.pylori infection, implicating an important role of this protein for the CagA-mediated rearrangement of the actin cytoskeleton. Moreover, we show that phosphorylation of CagA leads to inhibition of c-Src resulting in cortactin dephosphorylation. Since activated c-Src prevents both cortactin dephosphorylation and cytoskeletal rearrangements, these events are critically involved in CagAP-Tyr-induced signalling to the host cell cytoskeleton.
Results
CagAP-Tyr induces cytoskeletal rearrangements and host protein dephosphorylation
AGS gastric epithelial cells acquire an elongated shape with needle-like protrusions upon infection with wild-type H.pylori. We followed these changes in morphology by live cell imaging (Figure 1A). After bacterial attachment the cells lose their cell–cell contacts and start migrating. This cell scattering effect was fully established after 240 min infection with wild-type bacteria. Induction of these cytoskeletal rearrangements were not observed in cells infected with isogenic P1ΔcagA mutant (Figure 1B). Complementation of our cagA mutant with wild-type cagA (P1ΔcagA/cagA) restored the cellular phenotype (Backert et al., 2001). However, H.pylori P1ΔcagA expressing cagA mutated at the known phosphorylation site (P1ΔcagA/cagAY972F) did not induce cytoskeletal rearrangements (Figure 1B, lower panel), indicating that both CagA translocation and phosphorylation at Y972 are essential.

Fig. 1. Helicobacter pylori-induced cytoskeletal rearrangements depend on CagA tyrosine phosphorylation and are associated with dephosphorylation of p80. (A) Live cell imaging of an AGS cell cluster infected by wild-type H.pylori reveals cell elongation and scattering. (B) An isogenic cagA gene mutant does not stimulate this response. Complementation with wild-type cagA (P1ΔcagA/cagA) but not cagA lacking the tyrosine phosphorylation site (P1ΔcagA/cagAY972F) restores the phenotype. (C) HGF (20 pM) induced scattering of MDCK but not AGS cells. Treatment of cells with cycloheximide (10 µg/ml) prevents HGF-induced MDCK cell scattering but not the H.pylori-induced phenotype in AGS cells. (D) The time course of CagA tyrosine phosphorylation (filled arrowhead) parallels dephosphorylation of p80 (open arrowhead). In order to distinguish CagAP-Tyr from a 120 kDa host cell protein (asterisk) CagA was also immunoprecipitated (lower panels). (E) Effect of different H.pylori mutants on the tyrosine phosphorylation pattern of AGS cells. Dephosphorylation of p80 strictly correlates with CagA phosphorylation. α-tubulin blots served as loading controls. Scale bars: 20 µm.
The morphology of infected AGS cells is reminiscent of cell scattering induced by HGF receptor (c-Met) signalling. In MDCK cells, the morphogenic properties of HGF depend on gene transcription and translation (Rosen et al., 1990). Thus, we sought to explore whether CagAP-Tyr stimulates an HGF-like response in AGS cells. As expected, cell scattering was induced in HGF-treated MDCK control cells, however, exposure of AGS cells to 20–100 pmol HGF did not induce cell scattering (Figure 1C, middle panel). Cell scattering was blocked by addition of the protein translation inhibitor cycloheximide in HGF-stimulated MDCK cells but not in AGS cells infected with H.pylori (Figure 1C, lower panel). Thus, scattering of infected AGS cells does not require protein biosynthesis. Therefore, the cellular mechanisms of H.pylori-induced scattering of AGS cells and HGF-induced scattering of MDCK cells are considerably different.
Our findings suggested that the CagAP-Tyr-induced cellular phenotype involves an early host response that is independent of de novo protein synthesis. In order to pinpoint early host signalling events upon accumulation of intracellular CagAP-Tyr, we first investigated tyrosine phosphorylation in AGS cells infected with wild-type H.pylori in a time course (Figure 1D). CagA phosphorylation was determined both in whole cell lysates (upper panel) and in anti-CagA immunoprecipitates (lower panel). We found that accumulating amounts of CagAP-Tyr were temporally correlated with the dephosphorylation of an ∼80 kDa host cell protein p80 (upper panel, open arrowhead) upon infection with wild-type H.pylori. Like the cytoskeletal rearrangements, dephosphorylation of p80 required CagA phosphorylation on Y972 because the P1ΔcagA/cagAY972F mutant had no effect (Figure 1E). These results raised the possibility that CagA phosphorylation, dephosphorylation of p80 and rearrangements of the actin cytoskeleton might be functionally related to each other.
Cortactin is the dephosphorylated host protein p80
We hypothesized that p80 dephosphorylation might be crucial for H.pylori-induced actin cytoskeletal rearrangements to occur and sought to identify this protein. Cortactin is an 80 kDa actin binding protein that has recently emerged as a central regulator of the actin cytoskeleton since it has been shown to stimulate the actin nucleation activity of the Arp2/3 complex (Uruno et al., 2001; Weaver et al., 2001). To test whether cortactin is the dephosphorylated host cell protein, AGS cells were infected with wild-type H.pylori and cagPAI mutant strains for 6 h followed by immunoprecipitation of cortactin and western blotting using an anti-phosphotyrosine antibody. Indeed, wild-type H.pylori but not the P1ΔcagA and P1ΔvirB11 mutants induced the dephosphorylation of cortactin (Figure 2A). As a control, reprobing of the blot with an anti-cortactin antibody revealed similar amounts of cortactin present in the immunoprecipitates (Figure 2B). Respective phosphotyrosine patterns of whole cell lysates are given for comparison (Figure 2C). Thus, CagAP-Tyr induces the dephosphorylation of the actin binding protein cortactin.
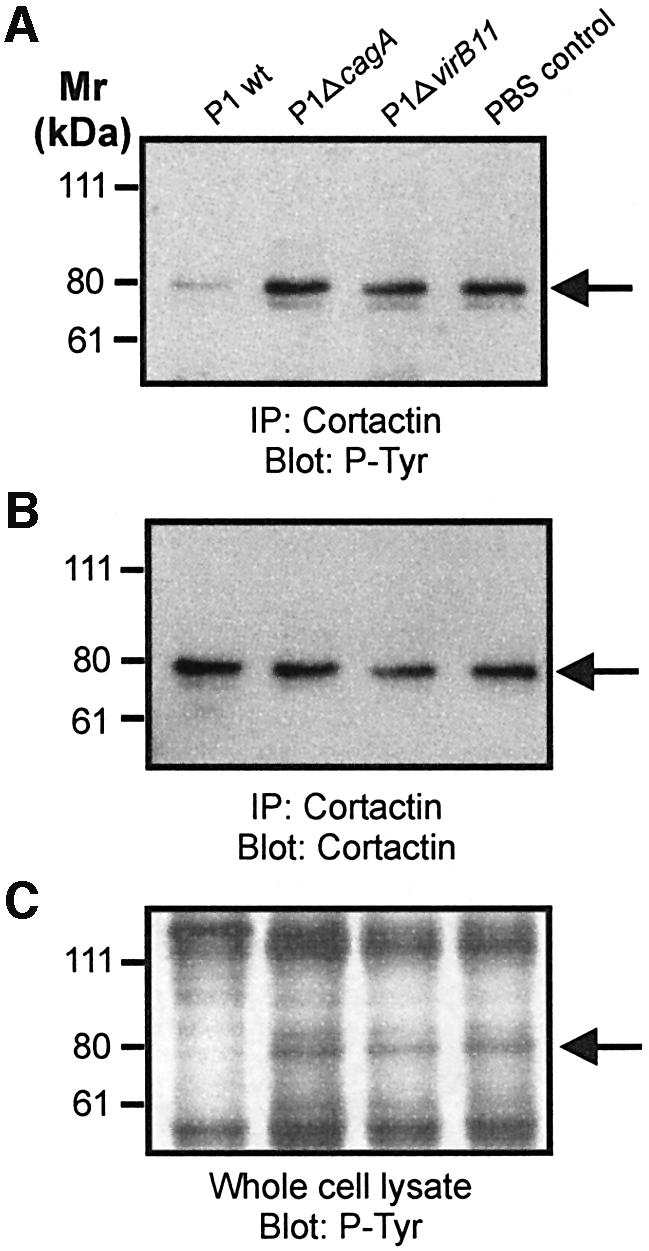
Fig. 2. Cortactin is specifically dephosphorylated upon infection with wild-type H.pylori. Cortactin was immunoprecipitated from infected AGS cells with a monoclonal anti-cortactin antibody. Probing with a phosphotyrosine-specific antibody reveals dephosphorylation of cortactin (A). The anti-cortactin blot shows that equal amounts of cortactin were precipitated (B). Tyrosine phosphorylation pattern of whole cell lysates are shown as control (C). Arrows indicate the position of cortactin on the gels.
Cortactin is recruited to the tips of needle-like protrusions in infected AGS cells
In order to investigate the cellular localization of cortactin in AGS cells upon infection with H.pylori, confocal laser scanning microscopy was applied. In AGS cells infected with wild-type bacteria, cortactin predominantly co-localized with filamentous actin (F-actin) at the tip and at the base of the characteristic cellular protrusions (Figure 3A, C and G, arrowheads). In contrast, in non-infected AGS cells (data not shown) or cells infected with the cagA mutant, cortactin was predominantly detected in a spot-like pattern which did not extensively co-localize with F-actin (Figure 3B, D and H, arrows). Thus, cortactin is significantly redistributed in AGS cells and co-localizes with F-actin upon wild-type infection.

Fig. 3. Cytoskeletal rearrangements are associated with cortactin re-localization. AGS cells infected with wild-type H.pylori (A, C, E and G) or the isogenic cagA mutant (B, D, F and H) were stained with anti-cortactin antibody (A and B), phalloidin (C and D) and anti-H.pylori antiserum (E and F). Confocal laser scanning microscopy reveals cortactin co-localization with F-actin at the tip and the base of the protrusions (G, arrowheads). In cells infected with the cagA mutant, cortactin has a spot-like distribution throughout the cytoplasm (H, arrows). Scale bar: 10 µm.
Dephosphorylation of cortactin proceeds in a Shp-2-independent manner
Overexpressed CagAP-Tyr in AGS cells has been reported to co-immunoprecipitate with Shp-2 (Higashi et al., 2002). This suggested that CagAP-Tyr-induced activation of Shp-2 could mediate the dephosphorylation of cortactin. In order to test whether Shp-2 is involved in the dephosphorylation of cortactin, we specifically inhibited Shp-2 by overexpression of a dominant-negative Shp-2 construct (shp-2ΔPTPase). This construct encodes a truncated Shp-2 protein deleted in the catalytic PTPase domain and has been shown to inhibit the activity of endogenous Shp-2 in a dominant-negative manner (Tang et al., 1995). We have used this PTPase deletion construct, rather than serine substitution of the catalytic cysteine, because Cys to Ser mutants can sequester substrates (Bliska et al., 1992). Moreover, transiently transfected Shp-2ΔPTPase (open arrowhead) can be readily distinguished from endogenous Shp-2 (filled arrowhead) due to its smaller size (Figure 4A, upper panel). Importantly, we found that expression of Shp-2ΔPTPase had no significant effect on the amount of CagAP-Tyr in infected samples (Figure 4A, arrow) nor on tyrosine phosphorylation of cortactin (Figure 4B, arrow). As a control for the dominant-negative function of this construct we analysed the basal levels of active p44/p42 mitogen-activated protein kinases (MAPK) with an activation-specific antibody (Cai et al., 2002). In agreement with its dominant-negative function, we found that Shp-2ΔPTPase expression significantly reduced MAPK activation in AGS cells (Figure 4C). Collectively, these results indicated that CagAP-Tyr-induced cortactin dephosphorylation is independent of Shp-2 catalytic activity.

Fig. 4. Cortactin dephosphorylation is independent of the PTPase Shp-2. Cells were transfected with a shp-2 construct deleted in the phosphatase domain (shp-2ΔPTPase) that acts in a dominant-negative manner. (A) Shp-2ΔPTPase is expressed in AGS cells (upper panel, open arrowhead) and migrates below endogenous Shp-2 (filled arrowhead). Shp-2ΔPTPase does not significantly affect CagA tyrosine phosphorylation (lower panels, arrow). (B) Helicobacter pylori induces cortactin dephosphorylation (arrow) irrespective of Shp-2ΔPTPase expression. (C) As a control for dominant-negative function Shp-2ΔPTPase-induced repression of basal MAPK activity was assayed with an activation specific antibody. α-tubulin blots served as loading controls. (D) Cells were infected with H.pylori, transfected with a CagA expression construct or transfected and infected simultaneously. Only CagAP-Tyr translocated by live bacteria (open arrowhead), but not transfected CagAP-Tyr (closed arrowhead), induced cortactin dephosphorylation (arrow). Conversely, only transfected but not translocated CagAP-Tyr co-immunoprecipitated with Shp-2 (right panels).
In order to analyse whether translocated CagAP-Tyr interacts with Shp-2 as has been reported for transfected CagA by Higashi et al. (2002), we compared whole cell lysates from infected and transfected cells. We were able to distinguish the two CagA species due to their different sizes (Figure 4D, left panel). While both translocated (open arrowhead) and transfected CagA (closed arrowhead) were tyrosine phosphorylated, only translocated CagA induced cortactin dephosphorylation (arrow). However, despite the higher amount of translocated CagAP-Tyr, only transfected CagAP-Tyr co-immunoprecipitated with Shp-2 (right panels). These results strongly suggest that translocated CagA from the H.pylori strain P1 and the TIGR strain 26695 (not shown) induces cortactin dephosphorylation in a Shp-2-independent manner.
AGS cells infected with H.pylori revealed strongly reduced c-Src activity
Protein tyrosine phosphorylation is regulated by both protein tyrosine kinases and PTPases. We did not detect PTPase activity in immunoprecipitated CagA nor in CagAP-Tyr (data not shown). Thus, we considered the possibility that H.pylori might induce the dephosphorylation of cortactin by inactivation of its cellular kinase. As cortactin has been reported to be a c-Src substrate (Wu et al., 1991), we wanted to determine the catalytic activity of c-Src during infection. For this purpose, c-Src was immunoprecipitated from whole cell lysates with a c-Src-specific antibody. The catalytic activity of precipitated c-Src was determined by an in vitro autophosphorylation assay in the presence of [γ-32P]ATP (Figure 5A). We found that infection with wild-type H.pylori, but not P1ΔcagA and P1ΔvirB11 mutants inhibited the catalytic activity of c-Src (Figure 5A, upper panel, arrow). As a control, reprobing of the blot with a c-Src-specific antibody verified that equal amounts of c-Src were present (Figure 5A, lower panel, arrow). Thus, H.pylori inhibits the catalytic activity of c-Src in a CagA-dependent manner.

Fig. 5. CagAP-Tyr-specific inactivation of c-Src. (A) The catalytic activity of immunoprecipitated c-Src was determined by an in vitro kinase assay with [γ-32P]ATP. Helicobacter pylori wild-type infection strongly reduces c-Src autophosphorylation (upper panel, arrow). Blotting with a c-Src-specific antibody shows that similar amounts of c-Src were precipitated (lower panel, arrow). The arrowhead marks the immunoglobulin heavy chain. (B) Western blotting with phosphospecific anti-c-Src antibodies revealed that H.pylori induces c-Src inactivation by dephosphorylation of Y418 (upper panel) and phosphorylation of Y527 (lower panel). (C) Effect of CagA phosphorylation (filled arrowhead) on c-Src inactivation. Only bacteria complemented with wild-type cagA (P1ΔcagΑ/cagA), but not cagA mutated in the phosphorylation site (P1ΔcagΑ/cagAY972F) induced Y418 dephosphorylation in c-Src (open arrowhead).
Phosphorylation of CagA at Y972 results in the inactivation of c-Src
c-Src contains two important tyrosine phosphorylation sites with opposing effects on protein conformation and catalytic activity (Hunter, 1987). The activity of c-Src is repressed through intramolecular interactions between its SH2 and SH3 domains with a C-terminal tyrosine residue (Y527) and the SH2-kinase linker sequence, respectively (Hubbard, 1999). Autophosphorylation occurs at Y418 and leads to the activation of the kinase, whereas phosphorylation of Y527 (numbering according to chicken c-Src) by Csk inhibits Src kinase activity (Nada et al., 1991). In order to investigate the molecular mechanism of CagA-induced Src inactivation, we determined the phosphorylation status of Y418 and Y527 with phosphospecific antibodies. We found that wild-type H.pylori induced both dephosphorylation of Y418 and phosphorylation of Y527 (Figure 5B, arrowheads). Thus, H.pylori-mediated c-Src inactivation involves both dephosphorylation of the Src autophosphorylation site and phosphorylation of the inhibitory C-terminal residue Y527. In order to determine whether CagA phosphorylation at Y972 is essential for the inhibition of c-Src, we infected AGS cells with cagA mutants that have been complemented either with wild-type cagA (P1ΔcagA/cagA) or with cagA lacking the phosphorylation site (P1ΔcagA/cagAY972F). Again, infection with P1ΔcagA/cagA induced Src inactivation (Figure 5C, open arrowhead). In contrast, the CagA phosphorylation-deficient mutant P1ΔcagA/cagAY972F did not inactivate Src. Therefore, we concluded that the inactivation of c-Src is specifically induced by CagA phosphorylated on Y972.
CagA phosphorylation is regulated through c-Src inactivation via a negative feedback loop
Since c-Src is the cellular kinase of CagA (Selbach et al., 2002b; Stein et al., 2002) and is subsequently inactivated by CagAP-Tyr, we might have detected a negative feedback loop which could explain how phosphorylation of injected CagA is regulated. In order to test this hypothesis, we analysed the temporal correlation between CagA phosphorylation, Src inactivation, and dephosphorylation of cortactin (Figure 6A). The time course shows gradually increasing amounts of injected CagAP-Tyr between 1–3 h of AGS cell infection with wild-type H.pylori (Figure 6A, filled arrowhead). Again, cortactin was dephosphorylated over time (open arrowhead). Reprobing of the same membrane with a phosphospecific antibody recognizing Src phosphorylated on Y418 (active Src) revealed that the amount of active Src decreased drastically between 1–3 h after infection with wild-type H.pylori (Figure 6A, arrow). This suggests that there are two processes in infected AGS cells which occur in parallel: (i) CagA is phosphorylated by active Src at early stages of infection; and (ii) c-Src is gradually inactivated by the accumulation of CagAP-Tyr within the host cell.

Fig. 6. CagAP-Tyr-induced c-Src inactivation inhibits succeeding CagA phosphorylation. (A) A time course reveals that accumulation of CagAP-Tyr (filled arrowhead) in the host cell correlates with the degree of cortactin dephosphorylation (open arrowhead) and c-Src inactivation (arrow). The asterisk denotes a 120 kDa host cell protein. (B) CagAP-Tyr inhibits succeeding CagA phosphorylation. Cells infected with the P1 strains were lysed, combined with an excess of lysate derived from the H.pylori strain P282 and in vitro phosphorylation reactions were performed. Tyrosine phosphorylation of CagA from the strain P282 (filled arrow) and P1 (open arrow) was analysed. P1 wild-type infection strongly reduced phosphorylation of CagA from strain P282 (second lane). The AGS cell control marked with an asterisk was not incubated with P282 lysate (last lane). Similar amounts of CagA and host cell lysates were present in anti-CagA and anti-α-tubulin blots (lower panels).
To test whether a negative feedback loop regulates CagA phosphorylation, we investigated the effect of CagAP-Tyr-induced Src inhibition on the Src-mediated phosphorylation of a second CagA protein species from another H.pylori strain. For this purpose, AGS cells were infected with wild-type H.pylori strain P1, P1ΔcagA, and P1ΔvirB11 mutants at an m.o.i. of 100. After 6 h, the infected cells were harvested, lysed and incubated with an excess of H.pylori lysate prepared from strain P282 in an in vitro phosphorylation reaction. These two H.pylori strains have been chosen because their CagA is phosphorylated to a similar extent during infection (data not shown) and the individual CagA protein species can be easily distinguished by their different sizes; 130 kDa (strain P1, open arrow) and 150 kDa (P282, filled arrow), respectively (Figure 6B). The results show that lysates from non-infected AGS cells or cells infected with P1ΔcagA or P1ΔvirB11 mutants strongly phosphorylated CagA from strain P282 in vitro. In contrast, a markedly reduced phosphorylation of CagA from strain P282 was observed when lysate of AGS cells infected with wild-type P1 was used. Anti-CagA and anti-tubulin blots verified that identical amounts of P282 and host cell lysates were present. Thus, CagAP-Tyr inhibits further CagA phosphorylation by modulating c-Src activity through a negative feedback loop.
Purified CagA inhibits c-Src in vitro
In order to determine whether CagA can inactivate c-Src in the absence of other cellular factors we performed in vitro experiments. For this purpose, we purified CagA and incubated it with recombinant c-Src. The catalytic activity of c-Src was determined by autophosphorylation. When CagA was incubated with c-Src in the presence of [γ-32P]ATP, c-Src activity was reduced as compared with incubation of c-Src without CagA (Figure 7A, open arrowhead). Densitometric analysis revealed that this reduction was ≤3-fold. Next, we wanted to know whether this inhibitory function depends on CagA phosphorylation. Therefore we tyrosine phosphorylated CagA in vitro by pre-incubation with c-Src and non-radioactive ATP. After 25 min, [γ-32P]ATP was added and c-Src activity was determined by autophosphorylation. We found that previous in vitro phosphorylation of CagA significantly enhanced its inhibitory effect on c-Src activity (Figure 7B, open arrowhead). As a control, c-Src was pre-incubated with non-radioactive ATP alone for 25 min before the CagA/[γ-32P]ATP mix was added. Coomassie Blue staining of the exposed gels shows that identical amounts of CagA were loaded. Collectively, these results demonstrate that CagAP-Tyr can directly inhibit c-Src, thereby providing a direct molecular link between both proteins.

Fig. 7. Purified CagA inhibits recombinant c-Src activity in vitro. (A) Purified CagA significantly reduced autophosphorylation of recombinant c-Src (open arrowhead). (B) Purified CagA was phosphorylated (CagAP-Tyr) with nonradioactive ATP before addition of [γ-32P]ATP (first lane). CagAP-Tyr had a marked inhibitory effect on c-Src activity. The lower panels show Coomassie Blue stains of the exposed gels to ensure equal amounts of CagA were loaded.
c-Src inactivation is essential for cortactin dephosphorylation
Because CagA translocation and phosphorylation resulted in both c-Src inactivation and dephosphorylation of the Src substrate cortactin, it seems plausible to assume that CagA induces cortactin dephosphorylation by inactivating c-Src. To prove this hypothesis, we transiently expressed an activated Src construct (srcY527F) in AGS cells, infected these cells with H.pylori and analysed cortactin tyrosine phosphorylation and the Src kinase activation status (Figure 8). Our data show that while H.pylori induced the dephosphorylation of cortactin in mock-transfected cells, expression of SrcY527F prevented cortactin dephosphorylation (Figure 8, upper panel, open arrowhead). Helicobacter pylori inhibited endogenous Src but was unable to inactivate the transiently expressed c-SrcY527F (Figure 8, second panel, arrow). Therefore, H.pylori induces the dephosphorylation of cortactin by inhibiting the tyrosine kinase c-Src. Interestingly, transfection of a wild-type c-Src construct did not lead to a significant increase in cortactin phosphorylation. Moreover, in AGS cells expressing the srcY527F construct, CagA became hyperphosphorylated (Figure 8, upper panel, filled arrowhead), indicating that the feedback inhibition which under normal conditions regulates CagA phosphorylation was disrupted. Blots were stripped and reprobed with anti-c-Src, anti-cortactin, anti-CagA and anti-tubulin antibodies as loading controls, thereby excluding artefacts (Figure 8, lower panels).

Fig. 8. CagAP-Tyr induces cortactin dephosphorylation through inactivation of c-Src. AGS cells were transiently transfected with c-src or activated src (src Y527F) constructs. The cells were either infected with the strain P1 or left uninfected. Activated src (i) induced the hyperphosphorylation of CagA (filled arrowhead) and Src (arrow); (ii) prevented cortactin dephosphorylation (open arrowhead); and (iii) prevented Src inactivation (second panel, arrow). c-Src, cortactin, CagA and α-tubulin blots were performed as controls.
c-Src inactivation is essential for cytoskeletal rearrangements
In order to determine the role of Src inactivation and cortactin dephosphorylation for the rearrangement of the actin cytoskeleton, AGS cells were transfected with c-src constructs (wild-type and srcY527F), infected with H.pylori and analysed by immunofluorescence microscopy (Figure 9). For the staining procedure we chose a low concentration of the c-Src-specific antibody that did not notably stain those cells expressing only endogenous c-Src levels. In this way we were able to distinguish between non-transfected cells and cells expressing the respective construct. To ensure that all AGS cells were equally infected, wild-type H.pylori were centrifuged onto the cells. We found that infected AGS cells overexpressing c-Src typically had the same elongated morphology as non-transfected control cells (Figure 9A, B and C). In contrast, infected AGS cells expressing the active Src construct had a rounded phenotype (arrow) that was clearly distinct from the elongated shape (arrowheads) of neighbouring non-transfected cells (Figure 9D, E and F). As a control, transfection of AGS cells with the src constructs without infection did not induce the formation of cellular protrusions (data not shown). Thus, Src inhibition by H.pylori is essential for rearrangement of the actin cytoskeleton.

Fig. 9. c-Src inactivation is essential for cytoskeletal rearrangements. AGS cells were transiently transfected with wild-type c-Src or constitutively active SrcY527F. Subsequently, the cells were infected with H.pylori, stained for F-actin (B and E) and Src expression (C and F) followed by epifluorescence microscopy. While cells expressing c-Src showed characteristic actin-rich protrusions (A, B and C), constitutively active Src prevented these H.pylori- induced cytoskeletal rearrangements (D, E and F, arrow). Arrowheads indicate neighbouring non-transfected cells as internal control. Scale bar: 10 µm.
Discussion
Direct injection of bacterial virulence factors into host cells has been described for two secretion machineries, termed type III and type IV (Finlay and Falkow, 1997; Galan and Collmer, 1999; Christie and Vogel, 2000). After translocation, these effector molecules target various components of eukaryotic signal transduction pathways in order to mediate bacterial attachment or entry, to transform the host cell or to block bacterial uptake by phagocytosis. Pathogenic H.pylori strains translocate CagA into gastric epithelial cells, thereby inducing cytoskeletal rearrangements that are thought to be involved in gastric disease (Montecucco and Rappuoli, 2001; Peek and Blaser, 2002). CagA has been shown to be phosphorylated on specific tyrosine residues by Src family kinases (Selbach et al., 2002b; Stein et al., 2002), to interact with the PTPase Shp-2 (Higashi et al., 2002), and to induce dephosphorylation of as yet unidentified host proteins (Backert et al., 2000; Püls et al., 2002). Here we report that CagA targets eukaryotic tyrosine kinase signalling to induce cytoskeletal rearrangements. To our knowledge, this is the first report of a bacterial virulence factor that specifically inhibits tyrosine kinase signalling.
The model depicted in Figure 10 summarizes the experimental data of this study. Once H.pylori has attached to gastric epithelial cells, CagA is translocated into the host cell cytosol by the type IV transporter encoded in the cagPAI (Segal et al., 1999; Asahi et al., 2000; Backert et al., 2000; Odenbreit et al., 2000; Stein et al., 2000). In this study, we show that phosphorylation of CagA at Y972 results in a marked inactivation of Src activity by a mechanism that involves both c-Src dephosphorylation at Y418 and phosphorylation at Y527. Phosphorylation of Y527 creates a binding site for the Src SH2 domain which stabilizes intramolecular interactions within the Src molecule that lead to a closed and catalytically inactive conformation (Hunter, 1987; Hubbard, 1999). Importantly, Src inactivation results in the dephosphorylation and redistribution of the actin binding protein cortactin. As the cytoskeletal rearrangements of AGS cells are independent of de novo host cell protein synthesis, c-Src inactivation, but not nuclear signalling, controls AGS cell scattering by modulating the organization of the actin cytoskeleton.

Fig. 10. Model for CagA-induced signalling leading to the cytoskeletal rearrangements of gastric epithelial cells. Helicobacter pylori translocates CagA by a type IV secretion dependent process. CagA is tyrosine-phosphorylated by Src which also phosphorylates cortactin. CagAP-Tyr inactivates c-Src by a mechanism involving phosphorylation of Y527 and dephosphorylation of Y418. Src inactivation prevents succeeding CagA phosphorylation and leads to cortactin dephosphorylation. Dephosphorylated cortactin has enhanced actin cross-linking and/or nucleation activity and may induce the characteristic rearrangements of the actin cytoskeleton involved in cell scattering, designated as the hummingbird phenotype.
Interestingly, Src plays two separate roles in CagA-induced host cell signalling. First, c-Src is the kinase of CagA and activates the CagA molecule by phosphorylation of the EPIYA sequence motif. Secondly, phosphorylated CagA induces defined phosphorylation and dephosphorylation events within the c-Src molecule which lead to the inactivation of c-Src. Both processes are distinct from each other because non-phosphorylated CagA is the substrate for c-Src while only CagAP-Tyr induces c-Src inactivation. Both processes together constitute a classical negative feedback loop, which provides an explanation for how the level of CagAP-Tyr accumulation within the host cell is regulated. Since CagA phosphorylation appears to be a prerequisite for its pathogenic effects, this feedback loop presumably plays an important role for the control of CagA cytotoxicity.
The molecular mechanism underlying CagAP-Tyr-dependent inhibition of c-Src remains unclear. CagA has no significant sequence homology to any known protein. A direct involvement of the C-terminal Src kinase Csk appears unlikely, as dominant-negative Csk had no apparent effect on CagAP-Tyr function (our unpublished data). However, CagAP-Tyr could possibly interact with SH2 domain-containing proteins. Furthermore, CagA contains a PxxP motif which constitutes a potential binding site for SH3 domains (Backert et al., 2001). Src family tyrosine kinases contain both a SH2 and a SH3 domain (Hunter, 1987). However, stable association of CagAP-Tyr with the Src SH2 and/or SH3 domains seems unlikely because this would stabilize the open conformation and therefore activate Src (Xu et al., 1999). On the other hand, our observation that purified CagA can inhibit recombinant c-Src in vitro suggests that there is a direct interaction between both proteins. We suggest that the interaction between both proteins is transient and therefore difficult to investigate with conventional methods like co-immunoprecipitation. We propose that CagAP-Tyr associates with c-Src and shifts the equilibrium from the open conformation of the kinase to the closed and inactive form. In the living cell, this inactive conformation is subsequently stabilized through phosphorylation of the negative regulation site in c-Src (Y527) by the C-terminal Src kinase Csk (Nada et al., 1991) or through intermolecular autophosphorylation of the same site (Osusky et al., 1995). It would be interesting to reveal the crystal structure of CagAP-Tyr in order to assess the structural basis of the interaction with c-Src.
It has been reported previously that overexpressed CagAP-Tyr interacts with the PTPase Shp-2 (Higashi et al., 2002). Here we demonstrate that translocated CagAP-Tyr induces Src inactivation and cortactin dephosphorylation in a Shp-2-independent manner. The reason for the discrepancy between these observations is not clear. Possibly CagA derived from the strain NCTC11637 used by Higashi et al. (2002) has different properties compared with CagA from the strain P1 used in this study. Alternatively, translation of transfected CagA in the eukaryotic cytoplasm could effect post-translational modification, conformation or subcellular localization of the protein. It is therefore unclear whether transfected CagA and injected CagA interact with the same host cell proteins. In any case, we observed that translocated CagA from the strains P1 and 26695 (TIGR strain) can induce cytoskeletal rearrangements although it does not co-precipitate with Shp-2. This indicates that Shp-2-independent mechanisms can also induce phenotypical changes. Interestingly, another group has recently reported a Shp-2-independent interaction of CagA from a different H.pylori strain with the adaptor protein Grb-2 (Mimuro et al., 2002). In contrast to c-Src inactivation, however, Grb-2 binding is independent of CagA tyrosine phosphorylation. Thus, CagA may have multiple functions and interfere with different eukaryotic signalling pathways.
The phenotype of H.pylori-infected AGS cells resembles cell scattering observed for HGF-treated MDCK cells (Weidner et al., 1990; Stella and Comoglio, 1999). Consequently, it has been suggested that CagA might interfere with or mimic HGF-induced intracellular signalling (Segal et al., 1999; Backert et al., 2001). In this context, it is interesting to note that cortactin co-immunoprecipitates with the HGF receptor c-Met and associates with c-Met in vitro (Crostella et al., 2001). Perhaps even more relevant, Src has been shown to be required for HGF-induced cell motility (Cutrupi et al., 2000). However, we demonstrate here that cycloheximide, an inhibitor of eukaryotic protein translation and HGF-induced cell scattering of MDCK cells, did not abolish the H.pylori-induced cytoskeletal rearrangements within AGS cells. This suggests that AGS cell scattering involves an early host response that is independent of nuclear signalling events and de novo protein biosynthesis.
Apart from regulating the level of CagA phosphorylation, H.pylori-induced Src inactivation leads to a specific dephosphorylation of the Src substrate cortactin. Several other bacterial pathogens have been reported to modify cortactin phosphorylation and/or localization. For example, invasion of epithelial cells by Shigella flexneri induces the tyrosine phosphorylation of cortactin by a c-Src-mediated signalling pathway (Dehio et al., 1995). Similarly, invasion of endothelial cells by Neisseria meningitidis stimulates cortactin phosphorylation in an ErbB2/Src-dependent manner (Hoffmann et al., 2001). On the other hand, enteropathogenic and enterohemorrhagic Escherichia coli (EPEC and EHEC, respectively) recruit cortactin to the bacterial adhesion site without apparent changes of its tyrosine phosphorylation (Cantarelli et al., 2000). Consequently, phosphorylation of cortactin seems to play a role in bacterial invasion (Neisseria and Shigella) while cortactin recruitment without tyrosine phosphorylation is a common feature during attachment of extracellular bacteria (EPEC and EHEC). To our knowledge, H.pylori is the first known pathogen that specifically induces the dephosphorylation of cortactin. Similarly to Shigella and Neisseria, the activity of Src in infected cells determines the level of cortactin phosphorylation. In contrast to the latter pathogens, however, cortactin does not appear to be involved in H.pylori invasion or attachment. Although H.pylori can specifically enter host cells, this bacterium is a major extracellular pathogen with only few intracellular bacteria observed (Su et al., 1999; Kwok et al., 2002). We suggest that H.pylori-induced dephosphorylation and re-localization of cortactin may orchestrate more global rearrangements of the actin cytoskeleton within the entire host cell associated with AGS cell scattering.
Cortactin binds F-actin and localizes to the sites of dynamic actin assembly (Weed et al., 2000), but the mechanism by which cortactin tyrosine phosphorylation and dephosphorylation events modulate the architecture of the actin cytoskeleton is unknown. Dephosphorylation of cortactin has been reported to increase its actin crosslinking activity in vitro (Huang et al., 1997). Therefore, dephosphorylated cortactin could cross-link actin filaments into bundles found within the characteristic cellular extensions. In addition, cortactin has been shown to activate actin polymerization by the Arp2/3 complex (Uruno et al., 2001; Weaver et al., 2001). It will be interesting to determine whether tyrosine phosphorylation/dephosphorylation of cortactin plays a role in this process. However, CagAP-Tyr-mediated inactivation of c-Src may also affect cytoskeletal rearrangements through cortactin-independent mechanism(s). For example, Src activity is required for the turnover of focal adhesions during cell migration (Fincham and Frame, 1998). Delayed focal adhesion disassembly could lead to the formation of F-actin-containing protrusions that remain attached to the extracellular matrix at the trailing edge of moving cells. Therefore, the combined effects of CagAP-Tyr-mediated c-Src inactivation and H.pylori-induced motogenic responses (Churin et al., 2001) can explain the formation of the cellular phenotype.
In summary, the characteristic changes in morphology of infected AGS cells resemble the process of oncogenic transformation. Since cagA-positive H.pylori are associated with the onset of gastric cancer (Montecucco and Rappuoli, 2001; Peek and Blaser, 2002), it is tempting to speculate that CagAP-Tyr contributes to oncogenic transformation of infected cells by interfering with c-Src signalling to cortactin. Interestingly, the gene encoding cortactin is amplified in some human cancers and cortactin is suspected to play a major role in tumour invasion (Schuuring et al., 1993; Patel et al., 1998). It will be interesting to find out how CagAP-Tyr-mediated disruption of Src signalling to cortactin could contribute to the onset of gastric cancer in vivo.
Materials and methods
Helicobacter pylori strains and cell culture
The generation of isogenic H.pylori P1 mutants and cagA complementation constructs has been described previously (Backert et al., 2001). The H.pylori strain P282 was isolated from a duodenal ulcer patient. Helicobacter pylori were cultivated on horse serum agar plates under microaerophilic conditions by standard procedures. AGS and MDCK cells were cultivated in RPMI 1640 medium (Invitrogen) supplemented with 25 mM HEPES buffer and 10% fetal bovine serum (Gibco) for 2 days to reach monolayers of ∼70% confluence. For microscopic studies, cells were seeded on acid-washed glass coverslips.
Synchronized infection assays
Helicobacter pylori were suspended in PBS and added to AGS cells at an m.o.i. of 100. Bacteria were centrifuged onto the cells for 5 min at 800 g at room temperature. After incubation at 37°C in a 5% CO2/95% air incubator for 5–12 h, cells were washed once with PBS and harvested in ice-cold PBS containing 1 mM Na3VO4 (Sigma-Aldrich) and pelleted together with attached bacteria at 800 g for 5 min at 4°C.
Transfection of AGS cells
Cells were transiently transfected in 12- or 6-well tissue culture dishes using Lipofectamine 2000 according to the manufacturer’s instructions (Invitrogen). Transfection rates were >70% as determined by co-transfection of a GFP construct. Dominant-negative Shp-2 with a deletion in the catalytic PTPase domain (Shp-2ΔPTPase) was expressed from the vector pEBB (Oh et al., 1999). The construct was a gift of B.Neel. Murine c-Src and constitutively active Src in the vector pNeoMSV (Broome and Hunter, 1996) was provided by T.Hunter. Haemagglutinin-tagged CagA from the strain NCTC11637 in the vector pSP65SRα was given to us by M.Hatakeyama (Higashi et al., 2002). Transfected cells were incubated at 37°C and 5% CO2 for 24–48 h before infection.
Immunoprecipitation
1 × 107 AGS cells were lysed in Ripa buffer [25 mM HEPES, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100, 125 mM NaCl, 1 mM EDTA, 1 mM Na3VO4, Complete protease inhibitors (Roche)] by 20 passages through a 20-gauche syringe. Insoluble material was removed by centrifugation at 12 000 g for 10 min. Lysates were pre-cleared with protein G–Sepharose (Pharmacia) for 30 min at 4°C. Two micrograms of monoclonal anti-cortactin or anti-Src (Upstate) antibodies were added to the supernatants and incubated overnight. Immune complexes were precipitated by the addition of protein G–Sepharose for 1 h and washed four times in Ripa buffer. Precipitates were either used for in vitro kinase assays or analysed by SDS–PAGE.
In vitro phosphorylation assays
In order to determine the c-Src activity in infected AGS cells, c-Src immunoprecipitates were washed once in kinase buffer [25 mM HEPES pH 7.0, 150 mM NaCl, 10 mM MgCl2, 1% NP-40, 5 mM DTT, 1 mM Na3VO4, Complete protease inhibitors] and resuspended in 40 µl kinase buffer with 10 µCi [γ-32P]ATP (Pharmacia). Reactions were incubated at 30°C for 30 min and stopped by the addition of reducing sample buffer and boiling. c-Src autophosphorylation was detected by SDS–PAGE and autoradiography. In order to determine the effect of H.pylori infection of AGS cells on further phosphorylation of CagA, 1 × 107 infected AGS cells (m.o.i. of 100) were lysed in 1 ml of ice-cold kinase buffer by 20 passages through a 20-gauche syringe. In parallel, 4 × 109 H.pylori cells from the strain P282 were resuspended in 2 ml of kinase buffer and lysed in the same manner. Forty microlitres of AGS cell lysates were combined with 60 µl of H.pylori lysate and 40 µM ATP was added. Reactions were incubated for 5 min at 30°C and analysed by SDS–PAGE and immunoblotting.
CagA purification
For analysis of direct interaction with c-Src in vitro, CagA was extracted from a 2 l liquid broth culture (OD = 0.8) of the H.pylori TIGR strain 26695 with 8 M Urea/20 mM Tris–HCl pH 8.0 and purified under denaturing conditions by column chromatography on an anion exchange resin (Source Q15, Pharmacia) followed by gel filtration on a Superose12 HR10/30 column in the presence of 8 M Urea, 150 mM NaCl, 20 mM Tris–HCl pH 8.0. Renaturation was performed by dialysis against kinase buffer (25 mM HEPES pH 7.0, 150 mM NaCl, 10 mM MgCl2, 1% NP-40, 5 mM DTT). Reactions with recombinant human c-Src (Upstate) and [γ-32P]ATP were performed as described above.
Immunoblotting
SDS–PAGE, protein transfer to PVDF membranes and probing with primary and secondary antibodies was performed by standard procedures. We used monoclonal antibodies against phosphotyrosine (Santa Cruz), c-Src (Upstate), cortactin (Upstate), Shp-2 (Santa Cruz), α-tubulin (Sigma) and phospho-p44/42 MAPK (Cell Signaling). Rabbit polyclonal phosphospecific antisera against phosphorylated tyrosines 418 or 527 of Src were obtained from Biosource. CagA was detected with a polyclonal rabbit anti-CagA antibody (Schützendeller Biochemica, Tübingen, Germany). Horseradish peroxidase-conjugated anti-mouse or anti-rabbit secondary antibodies (Amersham) were used and detected with the Renaissance western blot kit system for ECL immunostaining (ICN).
Microscopy
Immunostaining was performed using standard procedures. Polyclonal anti-H.pylori antiserum was obtained from Biomeda. Filamentous actin was detected with Alexa 488-conjugated phalloidin (MoBiTec). All secondary antibodies were purchased from Jackson Immunoresearch. Samples were analysed by confocal laser scanning microscopy using a Leica TCS SP microscope equipped with an argon/krypton mixed gas laser source (Leica) or by epifluorescence microscopy with a Leica DM/R microscope.
Acknowledgments
Acknowledgements
We thank T.Hunter (Salk Institute, La Jolla, CA), B.Neel (Harvard Medical School, Boston, MA) and M.Hatakeyama (Hokkaido University, Sapporo, Japan) for expression constructs. This work was supported by grants from the Fonds der Chemischen Industrie to T.F.M. and from the BMBF to C.R.H.
Note added in proof
After completing the review process of this manuscript, Tsutsumi and co-workers [Tsutsumi,R., Higashi,H., Higuchi,M., Okada,M. and Hatakeyama,M. (2002) Attenuation of Helicobacter pylori CagA-SHP-2 signaling by interaction between CagA and C-terminal Src kinase. J. Biol. Chem. (epub ahead of print)] reported that CagA is capable of interacting with C-terminal Src kinase (Csk) when both proteins were overexpressed from constructs. They further showed that transfection of CagA stimulated Csk, which in turn inactivated Src kinase. Although we observed a direct inhibition of Src by CagAP-Tyr in vitro, activation of Csk could nicely explain our finding that Src is phosphorylated on Y527. However, activation of Csk during H.pylori infection needs to be demonstrated in future experiments.
References
- Akopyants N.S. et al. (1998) Analyses of the cag pathogenicity island of Helicobacter pylori. Mol. Microbiol., 28, 37–53. [DOI] [PubMed] [Google Scholar]
- Asahi M. et al. (2000) The Helicobacter pylori CagA protein can be tyrosine phosphorylated in gastric epithelial cells. J. Exp. Med., 191, 593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backert S., Ziska,E., Brinkmann,V., Zimny-Arndt,U., Fauconnier,A., Jungblut,P.R., Naumann,M. and Meyer,T.F. (2000) Translocation of the Helicobacter pylori CagA protein in gastric epithelial cells by a type IV secretion apparatus. Cell Microbiol., 2, 155–164. [DOI] [PubMed] [Google Scholar]
- Backert S., Moese,S., Selbach,M., Brinkmann,V. and Meyer,T.F. (2001) Phosphorylation of tyrosine 972 of the Helicobacter pylori CagA protein is essential for induction of a scattering phenotype in gastric epithelial cells. Mol. Microbiol., 42, 631–644. [DOI] [PubMed] [Google Scholar]
- Bliska J.B., Clemens,J.C., Dixon,J.E. and Falkow,S. (1992) The Yersinia tyrosine phosphatase: specificity of a bacterial virulence determinant for phosphoproteins in the J774A.1 macrophage. J. Exp. Med., 176, 1625–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broome M.A. and Hunter,T. (1996) Requirement for c-Src catalytic activity and the SH3 domain in platelet-derived growth factor BB and epidermal growth factor mitogenic signaling. J. Biol. Chem., 271, 16798–16806. [DOI] [PubMed] [Google Scholar]
- Burns D.L. (1999) Biochemistry of type IV secretion. Curr. Opin. Microbiol., 2, 25–29. [DOI] [PubMed] [Google Scholar]
- Cai T., Nishida,K., Hirano,T. and Khavari,P.A. (2002) Gab-1 and Shp-2 promote Ras/MAPK regulation of epidermal growth and differentiation. J. Cell Biol., 159, 103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantarelli V.V., Takahashi,A., Akeda,Y., Nagayama,K. and Honda,T. (2000) Interaction of enteropathogenic or enterohemorrhagic Escherichia coli with HeLa cells results in translocation of cortactin to the bacterial adherence site. Infect. Immun., 68, 382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Censini S., Lange,C., Xiang,Z., Crabtree,J.E., Ghiara,P., Borodovsky,M., Rappuoli,R. and Covacci,A. (1996) cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc. Natl Acad. Sci. USA, 93, 14648–14653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie P.J. and Vogel,J.P. (2000) Bacterial type IV secretion: conjugation systems adapted to deliver effector molecules to host cells. Trends Microbiol., 8, 354–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churin Y., Kardalinou,E., Meyer,T.F. and Naumann,M. (2001) Pathogenicity island-dependent activation of Rho GTPases Rac1 and Cdc42 in Helicobacter pylori infection. Mol. Microbiol., 40, 815–823. [DOI] [PubMed] [Google Scholar]
- Cornelis G.R. and Van Gijsegem,F. (2000) Assembly and function of type III secretory systems. Annu. Rev. Microbiol., 54, 735–774. [DOI] [PubMed] [Google Scholar]
- Covacci A. et al. (1993) Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proc. Natl Acad. Sci. USA, 90, 5791–5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree J.E., Xiang,Z., Lindley,I.J., Tompkins,D.S., Rappuoli,R. and Covacci,A. (1995) Induction of interleukin-8 secretion from gastric epithelial cells by a cagA negative isogenic mutant of Helicobacter pylori. J. Clin. Pathol., 48, 967–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crostella L., Lidder,S., Williams,R. and Skouteris,G.G. (2001) Hepatocyte growth factor/scatter factor-induces phosphorylation of cortactin in A431 cells in a Src kinase-independent manner. Oncogene, 20, 3735–3745. [DOI] [PubMed] [Google Scholar]
- Cutrupi S. et al. (2000) Src-mediated activation of α-diacylglycerol kinase is required for hepatocyte growth factor-induced cell motility. EMBO J., 19, 4614–4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehio C., Prevost,M.C. and Sansonetti,P.J. (1995) Invasion of epithelial cells by Shigella flexneri induces tyrosine phosphorylation of cortactin by a pp60c-src-mediated signalling pathway. EMBO J., 14, 2471–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fincham V.J. and Frame,M.C. (1998) The catalytic activity of Src is dispensible for translocation to focal adhesions but controls the turnover of these structures during cell motility. EMBO J., 17, 81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay B.B. and Falkow,S. (1997) Common themes in microbial pathogenicity revisited. Microbiol. Mol. Biol. Rev., 61, 136–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer W., Püls,J., Buhrdorf,R., Gebert,B., Odenbreit,S. and Haas,R. (2001) Systematic mutagenesis of the Helicobacter pylori cag pathogenicity island: essential genes for CagA translocation in host cells and induction of interleukin-8. Mol. Microbiol., 42, 1337–1348. [DOI] [PubMed] [Google Scholar]
- Galan J.E. and Collmer,A. (1999) Type III secretion machines: bacterial devices for protein delivery into host cells. Science, 284, 1322–1328. [DOI] [PubMed] [Google Scholar]
- Higashi H., Tsutsumi,R., Muto,S., Sugiyama,T., Azuma,T., Asaka,M. and Hatakeyama,M. (2002) SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science, 295, 683–686. [DOI] [PubMed] [Google Scholar]
- Hoffmann I., Eugene,E., Nassif,X., Couraud,P.O. and Bourdoulous,S. (2001) Activation of ErbB2 receptor tyrosine kinase supports invasion of endothelial cells by Neisseria meningitidis. J. Cell Biol., 155, 133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C., Ni,Y., Wang,T., Gao,Y., Haudenschild,C.C. and Zhan,X. (1997) Down-regulation of the filamentous actin cross-linking activity of cortactin by Src-mediated tyrosine phosphorylation. J. Biol. Chem., 272, 13911–13915. [DOI] [PubMed] [Google Scholar]
- Hubbard S.R. (1999) Src autoinhibition: let us count the ways. Nat. Struct. Biol., 6, 711–714. [DOI] [PubMed] [Google Scholar]
- Hueck C.J. (1998) Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol. Mol. Biol. Rev., 62, 379–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter T. (1987) A tail of two src’s: mutatis mutandis. Cell, 49, 1–4. [DOI] [PubMed] [Google Scholar]
- IARC (1994) Schistomes, Liver Flukes and Helicobacter pylori. IARC Monographs on the Evaluation of Carcinogenesis Risks to Humans. 61. IARC (International Agency for Research on Cancer), Lyon, France.
- Kubori T., Matsushima,Y., Nakamura,D., Uralil,J., Lara-Tejero,M., Sukhan,A., Galan,J.E. and Aizawa,S.I. (1998) Supramolecular structure of the Salmonella typhimurium type III protein secretion system. Science, 280, 602–605. [DOI] [PubMed] [Google Scholar]
- Kwok T., Backert,S., Schwarz,H., Berger,J. and Meyer,T.F. (2002) Specific entry of Helicobacter pylori into cultured gastric epithelial cells via a zipper-like mechanism. Infect. Immun., 70, 2108–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee V.T. and Schneewind,O. (2001) Protein secretion and the pathogenesis of bacterial infections. Genes Dev., 15, 1725–1752. [DOI] [PubMed] [Google Scholar]
- Mimuro H., Suzuki,T., Tanaka,J., Asahi,M., Haas,R. and Sasakawa,C. (2002) Grb2 is a key mediator of Helicobacter pylori CagA protein activities. Mol. Cell, 10, 745–755. [DOI] [PubMed] [Google Scholar]
- Montecucco C. and Rappuoli,R. (2001) Living dangerously: how Helicobacter pylori survives in the human stomach. Nat. Rev. Mol. Cell. Biol., 2, 457–466. [DOI] [PubMed] [Google Scholar]
- Nada S., Okada,M., MacAuley,A., Cooper,J.A. and Nakagawa,H. (1991) Cloning of a complementary DNA for a protein-tyrosine kinase that specifically phosphorylates a negative regulatory site of pp60c-src. Nature, 351, 69–72. [DOI] [PubMed] [Google Scholar]
- Nagai H. and Roy,C.R. (2001) The DotA protein from Legionella pneumophila is secreted by a novel process that requires the Dot/Icm transporter. EMBO J., 20, 5962–5970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai H., Kagan,J.C., Zhu,X., Kahn,R.A. and Roy,C.R. (2002) A bacterial guanine nucleotide exchange factor activates ARF on Legionella phagosomes. Science, 295, 679–682. [DOI] [PubMed] [Google Scholar]
- Odenbreit S., Püls,J., Sedlmaier,B., Gerland,E., Fischer,W. and Haas,R. (2000) Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science, 287, 1497–1500. [DOI] [PubMed] [Google Scholar]
- Oh E.S. et al. (1999) Regulation of early events in integrin signaling by protein tyrosine phosphatase Shp-2. Mol. Cell. Biol., 19, 3205–3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osusky M., Taylor,S.J. and Shalloway,D. (1995) Autophosphorylation of purified c-Src at its primary negative regulation site. J. Biol. Chem., 270, 25729–25732. [DOI] [PubMed] [Google Scholar]
- Patel A.S., Schechter,G.L., Wasilenko,W.J. and Somers,K.D. (1998) Overexpression of Ems1/cortactin in NIH 3T3 fibroblasts causes increased cell motility and invasion in vitro. Oncogene, 16, 3227–3232. [DOI] [PubMed] [Google Scholar]
- Peek R.M. Jr and Blaser,M.J. (2002) Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat. Rev. Cancer, 2, 28–37. [DOI] [PubMed] [Google Scholar]
- Püls J., Fischer,W. and Haas,R. (2002) Activation of Helicobacter pylori CagA by tyrosine phosphorylation is essential for dephosphorylation of host cell proteins in gastric epithelial cells. Mol. Microbiol., 43, 961–969. [DOI] [PubMed] [Google Scholar]
- Rosen E.M., Meromsky,L., Goldberg,I., Bhargava,M. and Setter,E. (1990) Studies on the mechanism of scatter factor. Effects of agents that modulate intracellular signal transduction, macromolecule synthesis and cytoskeleton assembly. J. Cell Sci., 96, 639–649. [DOI] [PubMed] [Google Scholar]
- Schuuring E., Verhoeven,E., Litvinov,S. and Michalides,R.J. (1993) The product of the ems1 gene, amplified and overexpressed in human carcinomas, is homologous to a v-src substrate and is located in cell-substratum contact sites. Mol. Cell. Biol., 13, 2891–2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E.D., Cha,J., Lo,J., Falkow,S. and Tompkins,L.S. (1999) Altered states: involvement of phosphorylated CagA in the induction of host cellular growth changes by Helicobacter pylori. Proc. Natl Acad. Sci. USA, 96, 14559–14564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selbach M., Moese,S., Meyer,T.F. and Backert,S. (2002a) Functional analysis of the Helicobacter pylori cag pathogenicity island reveals both VirD4-CagA-dependent and VirD4-CagA-independent mechanisms. Infect. Immun., 70, 665–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selbach M., Moese,S., Hauck,C.R., Meyer,T.F. and Backert,S. (2002b) Src is the kinase of the Helicobacter pylori CagA protein in vitro and in vivo. J. Biol. Chem., 277, 6775–6778. [DOI] [PubMed] [Google Scholar]
- Stein M., Rappuoli,R. and Covacci,A. (2000) Tyrosine phosphorylation of the Helicobacter pylori CagA antigen after cag-driven host cell translocation. Proc. Natl Acad. Sci. USA, 97, 1263–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein M., Bagnoli,F., Halenbeck,R., Rappuoli,R., Fantl,W.J. and Covacci,A. (2002) c-Src/Lyn kinases activate Helicobacter pylori CagA through tyrosine phosphorylation of the EPIYA motifs. Mol. Microbiol., 43, 971–980. [DOI] [PubMed] [Google Scholar]
- Stella M.C. and Comoglio,P.M. (1999) HGF: a multifunctional growth factor controlling cell scattering. Int. J. Biochem. Cell Biol., 31, 1357–1362. [DOI] [PubMed] [Google Scholar]
- Su B., Johansson,S., Fallman,M., Patarroyo,M., Granstrom,M. and Normark,S. (1999) Signal transduction-mediated adherence and entry of Helicobacter pylori into cultured cells. Gastroenterology, 117, 595–604. [DOI] [PubMed] [Google Scholar]
- Tang T.L., Freeman,R.M.Jr, O’Reilly,A.M., Neel,B.G. and Sokol,S.Y. (1995) The SH2-containing protein-tyrosine phosphatase SH-PTP2 is required upstream of MAP kinase for early Xenopus development. Cell, 80, 473–483. [DOI] [PubMed] [Google Scholar]
- Uruno T., Liu,J., Zhang,P., Fan,Yx., Egile,C., Li,R., Mueller,S.C. and Zhan,X. (2001) Activation of Arp2/3 complex-mediated actin polymerization by cortactin. Nat. Cell Biol., 3, 259–266. [DOI] [PubMed] [Google Scholar]
- Weaver A.M., Karginov,A.V., Kinley,A.W., Weed,S.A., Li,Y., Parsons,J.T. and Cooper,J.A. (2001) Cortactin promotes and stabilizes Arp2/3-induced actin filament network formation. Curr. Biol., 11, 370–374. [DOI] [PubMed] [Google Scholar]
- Weed S.A., Karginov,A.V., Schafer,D.A., Weaver,A.M., Kinley,A.W., Cooper,J.A. and Parsons,J.T. (2000) Cortactin localization to sites of actin assembly in lamellipodia requires interactions with F-actin and the Arp2/3 complex. J. Cell Biol., 151, 29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidner K.M., Behrens,J., Vandekerckhove,J. and Birchmeier,W. (1990) Scatter factor: molecular characteristics and effect on the invasiveness of epithelial cells. J. Cell Biol., 111, 2097–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H., Reynolds,A.B., Kanner,S.B., Vines,R.R. and Parsons,J.T. (1991) Identification and characterization of a novel cytoskeleton-associated pp60src substrate. Mol. Cell. Biol., 11, 5113–5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W., Doshi,A., Lei,M., Eck,M.J. and Harrison,S.C. (1999) Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol. Cell, 3, 629–638. [DOI] [PubMed] [Google Scholar]