

Abstract
Free radical-induced cellular stress contributes to cancer during chronic inflammation. Here, we investigated mechanisms of p53 activation by the free radical, NO. NO from donor drugs induced both ataxia-telangiectasia mutated (ATM)- and ataxia-telangiectasia mutated and Rad3-related-dependent p53 posttranslational modifications, leading to an increase in p53 transcriptional targets and a G2/M cell cycle checkpoint. Such modifications were also identified in cells cocultured with NO-releasing macrophages. In noncancerous colon tissues from patients with ulcerative colitis (a cancer-prone chronic inflammatory disease), inducible NO synthase protein levels were positively correlated with p53 serine 15 phosphorylation levels. Immunostaining of HDM-2 and p21WAF1 was consistent with transcriptionally active p53. Our study highlights a pivotal role of NO in the induction of cellular stress and the activation of a p53 response pathway during chronic inflammation.
Keywords: posttranslational modification‖phosphorylation
An increased cancer risk occurs in tissues of the body undergoing chronic inflammation (1). Although it appears that the onset of carcinogenesis associated with inflammation is mediated by free radical species, identification of specific free radicals and their targets remains vague. NO is a candidate free radical, and the p53 tumor suppressor is a candidate molecular target. Our recent finding of increased p53 mutation load in inflamed colon tissue from patients with ulcerative colitis (UC, a cancer-prone inflammatory bowel disease) (2), Wilson's disease, and hemochromatosis (3), along with elevated inducible NO synthase (iNOS) levels, is consistent with p53 as a molecular target of free radicals.
p53 mutations contribute to clonal cellular expansion and genomic instability because of diminished regulation of cell cycle checkpoints, DNA repair, and apoptosis (4–6). However, before somatic mutations, p53 mediates these anticarcinogenic cellular functions through a DNA damage-response pathway involving phosphorylation and acetylation posttranslational modifications (4–6). We report here several mechanisms through which NO induces p53 activation in vitro (cells exposed to NO-generating drugs and NO-releasing macrophages) and in UC.
Methods
Comet Assay.
For all treatments, an alkali comet assay was performed according to instructions provided by the manufacturer (Comet Assay, Trevigen, Gaithersburg, MD). Cells treated with hydrogen peroxide (200 μM, 20 min) were used as positive controls. Fifty comets per treatment were quantified after being captured with the IPLab Spectrum Scientific Image Processing program (Signal Analytics, Fairfax, VA) and transferred to the NIH image (Version 1.62, Wayne Rasband, National Institutes of Health) analysis program.
Cell Culture and Treatment.
The MCF-7 human cancer cell line (containing wild-type p53) was provided by T. L. Woodward and S. Z. Haslam (Michigan State University, East Lansing, MI). These cells, as well as the murine macrophage cell line, ANA-1 (used in coculture experiments), and mouse embryonic fibroblasts (MEFs) were cultured in DMEM (GIBCO/BRL). HCT 116, HCT 116 p53−/−, and HCT 116 p21−/− colon carcinoma cells (provided by B. Vogelstein and K. Kinzler, Johns Hopkins Medical Institutions) were maintained in McCoy's 5A medium. Epstein–Barr virus-transformed C3ABR (normal) and L3 (ataxia-telangiectasia) lymphoblastoid cells (provided by K. K. Khanna, Queensland Institute of Medical Research) were grown in RPMI 1640 medium. Media were supplemented with 10% FBS (Biofluids, Rockville, MD), 4 mM glutamine (Biofluids), penicillin (10 units/ml), and streptomycin (10 μg/ml, Biofluids). For MCF-7 cells, 1 nM estradiol (Sigma) also was added.
Coculture Conditions.
MCF-7 cells were seeded at 2.5 × 106 cells per 150-mm culture dish 24 h before exposure to macrophages. Before adding to MCF-7 cells, log-phase ANA-1 murine macrophages were activated with IFN-γ (100 units/ml; R & D Systems) and lipopolysaccharide (10 ng/ml, Escherichia coli, 0111:B4; Sigma) in a two-step paradigm, as described (7). Cells (2.5 × 107) were then added to the actively growing MCF-7 cells, and the coculture was incubated for 8 h before harvest.
Immunoprecipitation, Western Blot Analysis, and Antibodies.
Cells or homogenized tissue samples were lysed in a buffer containing 50 mM Tris⋅HCl, pH 7.5, 5 mM EDTA, 150 mM NaCl, 1% triton X-100, 50 mM sodium fluoride, 10 mM sodium pyrophosphate, 25 mM β-glycerophosphate, 1 mM sodium ortho vanadate, 1 mM sodium molybdate, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 5 μg/ml pepstatin A, 0.5 mM PMSF, and 5 μM Trichostatin A plus a recommended dose (three tablets per 50 ml of buffer) of Complete protease inhibitor mixture tablets (Roche Diagnostics). p53 protein was isolated by using double immunoprecipitation with mouse monoclonal anti-p53 antibodies (human cells or tissue: agarose conjugated-Pab 1801 and -DO-1; Santa Cruz Biotechnology). Briefly, after protein quantification, an aliquot of complete lysate was saved. For the remaining lysate, conjugated antibodies were added to 2 mg of whole cell extract (5 μg of each conjugated antibody) or 20 mg of tissue extract (20 μg of each conjugated antibody) and incubated while rotating for 2 h at 4°C. After centrifugation (2,300 × g, 1 min), the agarose–Ab complex was again added to the supernatant and incubated for 1 h at 4°C. The pellet (agarose–antibody–antigen complex) was washed five times with cold lysis buffer, then a buffer containing 63 mM Tris⋅HCl, 10% glycerol, 2% SDS, and 0.0025% bromophenol blue was added to the sample. When separating whole cell lysates, the same sample buffer containing β-mercaptoethanol was added. Three hundred micrograms of the immunoprecipitated cellular protein, 2 mg of the immunoprecipitated tissue extract, or 30 μg of cell lysate were loaded per well, separated by SDS/PAGE (Invitrogen), and electrotransferred onto poly(vinylidene difluoride) membranes (Immobilon-P, Millipore). The following primary antibodies were used for protein analysis by Western blotting procedures: monoclonal anti-human p53 (clone DO-1, Oncogene Research), polyclonal anti-human p53 phosphoserine 15 (Cell Signaling, Beverly, MA) and acetylated lysine 382 (Oncogene Research). Polyclonal anti-human p53 phosphoserines 20, 33, 46, 315, 392 were generated in E.A.'s lab. Other antibodies used were monoclonal anti-human actin (clone C4, Roche Diagnostics), monoclonal anti-human p21 (WAF1, Oncogene Research), monoclonal anti-human MDM-2 (HDM-2, Oncogene Research), polyclonal anti-human iNOS (Cayman Chemical, Ann Arbor, MI) or monoclonal anti-human iNOS (Transduction Laboratories, Lexington, KY). Positive and negative controls used are indicated in the figures.
Nitrate and Nitrite Assay.
Nitrite and nitrate are the stabile end products of NO metabolism and were measured in culture media with a fluorometric assay kit (Cayman Chemical).
FACS and Mitotic Index Assays.
FACS analysis and the mitotic index assays have been described (8, 9).
Immunohistochemistry.
Serial sections were incubated in monoclonal anti-human iNOS (Transduction Laboratories), monoclonal anti-human p21WAF1 (Oncogene Research), monoclonal anti-human MDM-2 (HDM-2, Oncogene Research), or polyclonal anti-human p53 phosphoserine 15 (Cell Signaling). Signals were amplified with a biotinylated anti-mouse or anti-rabbit IgG (DAKO), followed by horseradish peroxidase-conjugated avidin–biotin complex (Vector Laboratories); the chromogen was diaminobenzidine tetrahydrochloride (Pierce).
Results
NO from Drug Donors and Macrophages Initiates a DNA Damage-Induced p53 Response Pathway.
First, to identify whether NO initiates a DNA damage-induced p53 response pathway, MCF-7 cells (containing wild-type p53) were exposed to NO donors [S-nitrosoglutathione (GSNO; half-life, 48 min) or spermine NONOate (SPER/NO; half-life, 37 min)]. NO donors [0.5 mM, chosen because this dose induced maximal levels of p53 posttranslational modifications (Fig. 5c, which is published as supporting information on the PNAS web site, www.pnas.org) and is consistent with endogenous NO concentrations found in chronic active inflammation (10, 11)], induced DNA damage, dose-dependent transient p53 phosphorylation (serines 15, 20, 33, 46, 315, and 392), and acetylation (lysine 382), leading to p53 accumulation (Fig. 1 a and b and Fig. 5 a, b, and d).
Figure 1.

(a) DNA damage is induced in MCF-7 cells after exposure to 0.5 mM SPER/NO. Cells were exposed for 4 h as indicated, then processed for the alkaline comet assay. At least 50 comets were quantified per treatment. (b) Time-course increase in p53 posttranslational modifications and p53 levels after exposure to 0.5 mM SPER/NO. Equal amounts of protein were immunoprecipitated, and Western blot assays were performed. HCT 116 cells genetically engineered to be p53−/− were used as a negative control. UV treatment (25 J/m2, 24 h) was used as a positive control. Purified p53 from baculovirus infected cells was loaded onto the gel to identify the specific p53 band (not shown). (c) p53 accumulated and was posttranslationally modified in MCF-7 cells cocultured with NO-releasing ANA-1 macrophages. MCF-7 cells were either unexposed or exposed to unactivated or activated ANA-1 macrophages as described in Methods. Appropriate controls used are indicated. After 8 h of coincubation, cells were lysed, and Western blot assays were performed. Lane 1, MCF-7 cells only; lane 2, MCF-7 cells + unstimulated ANA-1 cells; lane 3, MCF-7 cells + stimulated ANA-1 cells; lane 4, unstimulated ANA-1 cells only; lane 5, stimulated ANA-1 cells only; lane 6, MCF-7 cells + cytokine stimulation; lane 7, MCF-7 cells + cytokine stimulation + l-NMMA (250 μM); lane 8, MCF-7 cells + stimulated ANA-1 cells + l-NMMA (250 μM); lane 9, HCT 116 p53−/− cells; lane 10, MCF-7 cells + UV. Relative nitrate plus nitrite levels are also shown.
To model conditions of active inflammation, a coculture system with p53-null murine macrophages and wild-type p53 MCF-7 cells (10 macrophages:1 MCF-7 cell) was used. Cytokines stimulated iNOS and the macrophages to release NO, resulting in p53 accumulation and posttranslational modifications in neighboring MCF-7 cells (Fig. 1c, lane 3 and Fig. 5f). The p53 residues modified most markedly after iNOS induction in neighboring macrophages were those modified most by the NO donors. These modifications were prevented by inhibition of NO synthesis with l-N-monomethyl arginine (Fig. 1c, lane 8). The observation that the coculture system was less potent in inducing p53 modifications appears to reflect a dose/rate effect of NO (Fig. 5 e and f).
NO-Induced Phosphorylation of p53 at Serine 15 Is Ataxia-Telangiectasia Mutated (ATM) and ATM- and Rad3-Related (ATR)-Dependent.
The serine 15 phosphorylation on p53 (P-Ser-15), a principal residue modified in vitro (Fig. 1 b and c; Fig. 5d), mediates p53 accumulation and activation, and is modified by specific kinases in vivo (4–6). We, therefore, focused on this residue to identify possible mechanisms involved in NO-associated p53 posttranslational modification. We examined the ATM, ATR, p38, and DNA-PK kinases, because they were previously shown to be either involved in P-Ser-15 after cellular stress or activated by NO (4–6, 12). We found a role of ATM in NO-induced p53 phosphorylation and accumulation in both isogenic human cell lines and MEFs from gene knockout mice. ATM−/− MEFs exposed to SPER/NO (0.5 mM) had a reduced P-Ser-18 (murine equivalent of the P-Ser-15 in human cells) signal compared with ATM+/+ MEFs (Fig. 2 a and b). Five-Gy γ-irradiation was a positive DNA-damaging agent control, which also resulted in reduced P-Ser-18 in ATM−/− MEFs; the negative control was 25 J/m2 UV, which resulted in no observable change compared with ATM+/+ MEFs, consistent with results from previous studies (13). An early but reduced response also occurred in NO-exposed human ataxia-telangiectasia lymphoblastoid cell lines (Fig. 2 c and d). This is in contrast to a previous study that did not find a difference in p53 response to 1 mM GSNO in ATM heterozygous vs. homozygous human diploid fibroblasts (14).
Figure 2.

(a) ATM partially mediates NO-induced P-Ser-18 in MEFs. ATM+/+ or ATM−/− MEFs were exposed to 0.5 mM SPER/NO, 5 Gy γ-irradiation, or 25 J/m2 UV for indicated time points (hr). Cells were lysed, and Western blot assays were performed. (b) Bar graphs representing quantitative densitometry of Western blot bands shown in a. (c) ATM and ATR mediate NO-induced P-Ser-15 in human cells. Human lymphoblastoid cells from a healthy individual (C3ABR) or an individual with ataxia telangiectasia (AT) were exposed to 0.5 mM SPER/NO ± caffeine (1 mg/ml) for indicated time points (hr). The calpain inhibitor, ALLN (20 μM), which inhibits the proteasome, was used as a negative control for posttranslational modifications; MCF-7 cells exposed to 25 J/m2 UV were used as a positive antibody control. Cells were lysed, and Western blot assays were performed. (d) Bar graphs representing quantitative densitometry of Western blot bands shown in c.
Overexpression of its homologue ATR can complement the radioresistant DNA synthesis defect of cells lacking ATM (15); thus, we tested whether ATR was responsible for this early and diminished P-Ser-15 signal. Therefore, the experiment was repeated with the addition of caffeine, which inhibits ATM and ATR kinases, but not DNA-PK kinase (16). P-Ser-15 was further diminished with caffeine pretreatment (Fig. 2 c and d). We did not find significant involvement of either DNA-PK or p38 kinase in NO-induced p53 P-Ser-15 posttranslational modification (Fig. 6, which is published as supporting information on the PNAS web site). Taken together, both ATM and ATR are responsible for p53 P-Ser-15 after cellular exposure to NO.
NO-Induced p53 Phosphorylation Activates p53 Targets and Engages a G2/M Checkpoint.
We and others have reported there is a relationship between NO and p53 in cellular physiology. For example, NO can modulate p53-mediated cellular events involved in carcinogenesis and tumor progression, and p53 transrepresses the transcription of iNOS in a negative feedback loop (17–19). Consistent with these findings, GSNO or SPER/NO induced cell cycle changes (G1 checkpoint, an early decrease in the percentage of cells in active S-phase, and a G2/M checkpoint) and modulated the expression of p53 transcriptionally regulated proteins in MCF-7 cells (Fig. 7 a and b, which is published as supporting information on the PNAS web site). To determine whether these functional changes were due to p53 activation and were not cell type specific, we exposed HCT 116, HCT 116 p53−/−, and HCT 116 p21−/− isogenic colon cell lines to 1 mM SPER/NO. This resulted in similar P-Ser-15 levels to those found with 0.5 mM SPER/NO in MCF-7 cells (Fig. 7c). The NO-induced reduction in active S-phase cells and G1 cell cycle checkpoint was p53- and p21-independent (Fig. 7d). The inhibition of ribonucleotide reductase is a candidate molecule for these observations, because this has previously been shown to be inhibited by NO, resulting in a rapid decrease in the incorporation of thymidine into DNA during S-phase of the cell cycle (8, 20). Interestingly, the G2/M checkpoint was p53- and p21-dependent (Fig. 3b). This checkpoint was confirmed by measuring the reduction in mitotic index (Fig. 3b) and has been shown previously with other drugs (21, 22). The p53-dependent increase in the proapoptotic Bax protein (Fig. 3a) is consistent with the requirement of p53 in NO-induced apoptosis. The decrease in HDM-2 at 2 h (Fig. 3a) is also consistent with a recent report demonstrating a decrease of the MDM-2 protein by 2 h with 1 mM GSNO in MEFs (14).
Figure 3.

(a) Specific proteins are induced in a p53-dependent manner after exposure of colon cancer cells to 1 mM SPER/NO. HCT 116 and HCT 116 p53−/− cells were exposed to the 0.5 mM SPER/NO for indicated time points (hr), then lysed. Western blot assays were performed. Numbers above the bands indicate densitometry values as a ratio relative to control values. (b) There is a p53- and p21-dependent G2/M arrest in colon cancer cells exposed to NO. HCT 116 cells (●), HCT 116 p53−/− (○), or HCT 116 p21−/− (▴) cells were exposed to 1 mM SPER/NO for indicated time periods (hr). Cells were harvested at the same time, then FACS analysis was performed. Data are presented as a percentage relative to untreated cells. Gating was done according to Fig. 7e.
p53 Is Phosphorylated, Accumulates, and Is Active in UC, a Chronic Inflammatory Disease.
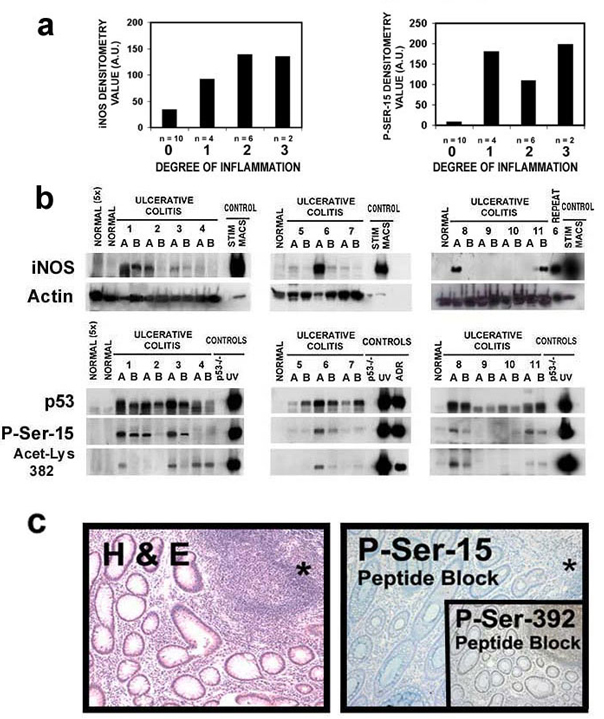
Having established that NO is a key inflammatory species involved in p53 posttranslational modification and activation in cell culture, we examined colon tissues from patients with the colon cancer-prone (23) chronic inflammatory disease, UC (Fig. 4; Fig. 8, which is published as supporting information on the PNAS web site). Because the degree of inflammation is dynamic and varies within the UC colon, two samples (A and B) were taken from each surgical specimen. Nine of eleven UC cases had detectable levels of iNOS protein. In contrast, iNOS levels were undetectable in normal colon tissues from non-UC donors. This was apparent even when five times the amount of protein from normal tissue was loaded onto the gel (Fig. 4a). Nonparametric regression analysis revealed a significant increase (P < 0.05) in iNOS levels with increasing degree of inflammation (Fig. 8a). p53 protein levels and posttranslational modifications also were undetectable in normal colon tissues, even when 5-fold higher protein was loaded. Results were the same in tissues derived from five separate non-UC colon tissues (data not shown). In contrast, 9 of 11 UC patients had detectable P-Ser-15 levels, and all 11 UC patients had detectable p53. There also was a significant increase in P-Ser-15 levels (P < 0.05) with increasing degree of inflammation (Fig. 8a). The immediate rise in P-Ser-15 levels with minimal inflammation (inflammation index of 1, Fig. 8a) is consistent with the hypothesis that P-Ser-15 is a sensitive biomarker of inflammation. There also were detectable levels of acetylated lysine 382 (Acet-Lys-382) (Fig. 8b). Highly significant positive correlations between iNOS and P-Ser-15 (rs = 0.75, P < 0.001) and between P-Ser-15 and p53 levels (rs = 0.83, P < 0.001) were observed (Fig. 4b), consistent with the hypothesis that P-Ser-15 is a modification involved in NO-induced p53 accumulation in the UC colon.
Figure 4.

(a) iNOS, P-Ser-15, and p53 protein levels are elevated in colon tissues from UC patients. Two samples (A and B) were taken from surgical specimens derived from each of 11 UC patients. Results from four patients (eight samples) are shown here. Results from the other seven patients are shown in Fig. 8b. Representative colon postmortem tissues from five donors without disease (control samples) also were included (two samples are shown). (b) iNOS and P-Ser-15 levels, and P-Ser-15 and p53, are correlated in UC colon tissues. After densitometry, the Spearman rank correlation coefficient was calculated to examine the relationship among iNOS levels, p53 levels, and levels of p53 posttranslational modifications. (c) iNOS, P-Ser-15, and p53 downstream protein levels are elevated in UC patients. Serial tangential sections of crypts of Leiberkuhn were exposed to indicated antibodies, as described in Methods. (Left) Normal colon. (Top to Bottom) Anti-iNOS, antiphosphoserine-15, anti- p21WAF1, anti-HDM-2. (Right) UC colon. (Top to Bottom) Anti-iNOS (*, a lymphocytic aggregate, consistent with an inflammatory infiltrate; E, epithelial; and S, stromal cells of the UC tissue), antiphosphoserine-15, anti-p21WAF1, anti-HDM-2. Antibody controls are shown in Fig. 8b. All are ×100; Inset is ×400.
Fig. 4c shows representative serial tangential sections of crypts of Lieberkuhn from the colon of a healthy patient (“Normal”) and from a UC patient (“Ulcerative Colitis”). Except for a few cells, P-Ser-15 immunostaining was undetectable in the normal colon. In UC, staining occurred in both epithelial (E) and stromal (S) cells. Staining was specific because there was no signal with an IgG antibody control and the signal was eliminated by preincubating the anti-P-Ser-15 antibody with its epitope-containing blocking peptide (Fig. 8c). In UC, but not in normal tissue, both epithelial and stromal cells were positively stained after incubation with the anti-iNOS antibody. Although not detectable in normal colon tissue, p53 transcriptional targets such as p21WAF1 and HDM-2 were positive in UC colon, consistent with p53 activation.
Discussion
Exposure to exogenous γ-irradiation, UV, and some chemotherapeutic agents activates a DNA damage-response pathway, resulting in phosphorylation, acetylation, and activation of p53 (4–6). We have demonstrated that this critical DNA damage-response pathway is induced by NO in cell culture (through ATM and ATR) and in UC, a colon cancer-prone chronic inflammatory disease.
Advances in the last decade have identified many of the genetic and epigenetic events and their functional significance in the hyperplasia–adenoma–carcinoma pathogenesis of sporadic human colon cancer (24–26). Less is known, however, about the molecular mechanisms involved in the more common transition from chronic ulceration to dysplasia to carcinoma found in UC. UC is characterized by sustained nitrosative/oxidative stress and DNA damage generated during chronic inflammation (2, 27, 28). Our cell culture data, in combination with the positive correlation between inflammatory index, iNOS, and P-Ser-15 levels, and in turn, P-Ser-15 and p53 levels, suggest that P-Ser-15 is a critical event in NO-induced p53 activation. Previous in vitro studies have shown that P-Ser-15 modulates p53 function (4–6). P-Ser-15 p53 also has been shown to have a reduced binding affinity for HDM-2 (4–6). Because HDM-2 targets p53 for ubiquitination and shuttles it to the cytoplasm for proteasome degradation, this reduced binding results in elevated p53 accumulation and transcriptional activity (29–31). Similarly, P-Ser-15 has been shown to inhibit the nuclear export of p53, leading to p53 nuclear accumulation (32). Whether this and other modifications influence p53 functions in the UC colon remains to be determined. Our results indicating an increase in p21WAF1 (Fig. 4c), a G2/M cell cycle checkpoint gene (Fig. 3b) (21) downstream of p53, are consistent with this hypothesis. The identification of residues modified on p53 by NO exposure and in UC tissue, then, enhances our understanding of the mechanisms by which tissues, undergoing chronic inflammation, protect themselves against genomic insult from free radicals such as NO.
Although our study established NO in this DNA damage-mediated p53 adaptive response, other free radicals may also lead to p53 activation in chronic inflammation. However, the findings that ANA-1 macrophages used in our coculture model do not release superoxide nor H2O2 under our stimulating conditions (7), as well as the relatively long half-life of NO (33), establish the significance of NO in this process. Ongoing experiments to address this issue in vivo involve examining the p53 pathway in response to inflammation in animal models.
The mechanism of activation of ATM and ATR by NO is unknown. NO has previously been shown to directly activate kinases or inactivate phosphatases that target p53 (12). Our study indicates that DNA damage activation of ATM and ATR kinases is responsible, at least in part, for NO-mediated P-Ser-15 modification on p53. Because ATM−/− mice respond with attenuated p21WAF1 induction after whole body irradiation (34), and we (Fig. 4c) and others (35) have shown p21WAF1 staining in the colonic epithelium of UC tissue, the ATM/ATR kinase pathway is worthy of further study in this and other chronic inflammatory diseases.
On the basis of this and other studies (1, 2), we propose a model of colon carcinogenesis in UC patients that highlights a paradoxical role of NO in this process. During chronic inflammation, NO and other free radicals are involved in repeated genomic insult. A key role of NO is supported by experiments showing trinitrobenzene-treated (which induces chronic colitis) iNOS knockout mice have reduced long-term inflammatory damage to the colon compared with wild-type mice (36). However, NO can also protect from cellular damage by (i) abrogating cytotoxicity by reactive oxygen species (37); and (ii) as shown here, by inducing an adaptive activation of p53 through ATM- and ATR-mediated P-Ser-15 and cell cycle checkpoints, as supported by the observed p21WAF1 positive staining in UC tissue (Fig. 4c). This transient cell cycle arrest would allow DNA repair of free radical-induced DNA damage. However, NO can inhibit certain DNA repair enzymes (38) that would partially oppose this protective effect of p53 activation of cell cycle checkpoints. Induction of apoptosis is also a cellular defense to DNA damage. Although others have found that apoptosis is slightly elevated in lesional areas (39), we did not detect significant levels of terminal deoxynucleotidyltransferase-mediated dUTP end-labeling staining in epithelial cells of colonic crypts from any UC cases (data not shown). In addition, UC samples used in this study did not show detectable p53 serine 46 phosphorylation, a modification involved in p53-mediated apoptosis (40). Because macrophage migration inhibitory factor (MIF) can inhibit p53-mediated apoptosis (41, 42), possibly through PGE-2 production by COX-2 (41, 43), and levels of MIF are increased in UC when compared with normal colon samples (44, 45), free radical-damaged cells could escape apoptosis and clonally expand. In addition, NO can further attenuate apoptosis by directly inhibiting caspases via nitrosylation (46, 47). Albeit rarely in the large population of colon cells at risk, p53 mutations occur within inflammatory lesions of the UC colon (2). Continued genomic damage from NO and other free radicals, generated from activated macrophages and the stressed colonic epithelium, could drive clonal selection and expansion of these p53 mutant cells that are resistant to free radical-induced growth arrest and apoptosis. Continued genomic instability may then lead to morphologically detectable dysplastic and neoplastic cells that progress over a prolonged period to a detectable tumor. Thus, we propose a model of UC colon carcinogenesis that may apply more broadly to other cancer-prone chronic inflammatory diseases.
Supplementary Material
Acknowledgments
We thank Sharlyn Mazur for insightful discussions, Dorothea Dudek for editorial assistance, and Karen MacPherson for assistance in manuscript preparation. We also thank Anthony Wynshaw-Boris (National Human Genome Institute, Bethesda) for providing the mice from which the ATM−/− MEFs were derived.
Abbreviations
- UC
ulcerative colitis
- iNOS
inducible NO synthase
- ATM
ataxia-telangiectasia mutated
- ATR
ATM- and Rad3-related
- MEF
mouse embryonic fibroblast
- P-Ser-15
serine 15 phosphorylation on p53
- SPER/NO
spermine NONOate
- GSNO
S-nitrosoglutathione
References
- 1.Ames B N, Gold L S, Willett W C. Proc Natl Acad Sci USA. 1995;92:5258–5265. doi: 10.1073/pnas.92.12.5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hussain S P, Amstad P, Raja K, Ambs S, Nagashima M, Bennett W P, Shields P G, Ham A J, Swenberg J A, Marrogi A J, et al. Cancer Res. 2000;60:3333–3337. [PubMed] [Google Scholar]
- 3.Hussain S P, Raja K, Amstad P A, Sawyer M, Trudel L J, Wogan G N, Hofseth L J, Shields P G, Billiar T R, Trautwein C, et al. Proc Natl Acad Sci USA. 2000;97:12770–12775. doi: 10.1073/pnas.220416097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vogelstein B, Lane D, Levine A J. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 5.Wahl G M, Carr A M. Nat Cell Biol. 2001;3:E277–E286. doi: 10.1038/ncb1201-e277. [DOI] [PubMed] [Google Scholar]
- 6.Appella E, Anderson C W. Pathol Biol (Paris) 2000;48:227–245. [PubMed] [Google Scholar]
- 7.Espey M G, Miranda K M, Pluta R M, Wink D A. J Biol Chem. 2000;275:11341–11347. doi: 10.1074/jbc.275.15.11341. [DOI] [PubMed] [Google Scholar]
- 8.Linke S P, Clarkin K C, Di Leonardo A, Tsou A, Wahl G M. Genes Dev. 1996;10:934–947. doi: 10.1101/gad.10.8.934. [DOI] [PubMed] [Google Scholar]
- 9.Wang X W, Zhan Q, Coursen J D, Khan M A, Kontny H U, Yu L, Hollander M C, O'Connor P M, Fornace A J, Jr, Harris C C. Proc Natl Acad Sci USA. 1999;96:3706–3711. doi: 10.1073/pnas.96.7.3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boughton-Smith N K, Evans S M, Hawkey C J, Cole A T, Balsitis M, Whittle B J, Moncada S. Lancet. 1993;342:338–340. doi: 10.1016/0140-6736(93)91476-3. [DOI] [PubMed] [Google Scholar]
- 11.Chazotte-Aubert L, Hainaut P, Ohshima H. Biochem Biophys Res Commun. 2000;267:609–613. doi: 10.1006/bbrc.1999.2003. [DOI] [PubMed] [Google Scholar]
- 12.Xu W, Liu L, Smith G C, Charles L. Nat Cell Biol. 2000;2:339–345. doi: 10.1038/35014028. [DOI] [PubMed] [Google Scholar]
- 13.Siliciano J D, Canman C E, Taya Y, Sakaguchi K, Appella E, Kastan M B. Genes Dev. 1997;11:3471–3481. doi: 10.1101/gad.11.24.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang X, Michael D, de Murcia G, Oren M. J Biol Chem. 2002;277:15697–15702. doi: 10.1074/jbc.M112068200. [DOI] [PubMed] [Google Scholar]
- 15.Cliby W A, Roberts C J, Cimprich K A, Stringer C M, Lamb J R, Schreiber S L, Friend S H. EMBO J. 1998;17:159–169. doi: 10.1093/emboj/17.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sarkaria J N, Busby E C, Tibbetts R S, Roos P, Taya Y, Karnitz L M, Abraham R T. Cancer Res. 1999;59:4375–4382. [PubMed] [Google Scholar]
- 17.Moncada S, Erzurum S C. Nat Rev Mol Cell Biol. 2002;3:214–220. doi: 10.1038/nrm762. [DOI] [PubMed] [Google Scholar]
- 18.Nakaya N, Lowe S W, Taya Y, Chenchik A, Enikolopov G. Oncogene. 2000;19:6369–6375. doi: 10.1038/sj.onc.1204100. [DOI] [PubMed] [Google Scholar]
- 19.Ambs S, Ogunfusika M O, Merriam W G, Bennett W P, Billiar T R, Harris C C. Proc Natl Acad Sci USA. 1998;95:8823–8828. doi: 10.1073/pnas.95.15.8823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hamad A M, Knox A J. FEBS Lett. 2001;506:91–96. doi: 10.1016/s0014-5793(01)02883-6. [DOI] [PubMed] [Google Scholar]
- 21.Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown J P, Sedivy J M, Kinzler K W, Vogelstein B. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 22.Sandri M I, Isaacs R J, Ongkeko W M, Harris A L, Hickson I D, Broggini M, Vikhanskaya F. Nucleic Acids Res. 1996;24:4464–4470. doi: 10.1093/nar/24.22.4464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lashner B A, Silverstein M D, Hanauer S B. Dig Dis Sci. 1989;34:1536–1541. doi: 10.1007/BF01537106. [DOI] [PubMed] [Google Scholar]
- 24.Fearon E R, Vogelstein B. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 25.Kinzler K W, Vogelstein B. Cell. 1996;87:159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 26.Shih I M, Wang T L, Traverso G, Romans K, Hamilton S R, Ben Sasson S, Kinzler K W, Vogelstein B. Proc Natl Acad Sci USA. 2001;98:2640–2645. doi: 10.1073/pnas.051629398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loeb K R, Loeb L A. Am J Pathol. 1999;154:1621–1626. doi: 10.1016/S0002-9440(10)65415-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rachmilewitz D, Stamler J S, Bachwich D, Karmeli F, Ackerman Z, Podolsky D K. Gut. 1995;36:718–723. doi: 10.1136/gut.36.5.718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haupt Y, Maya R, Kazaz A, Oren M. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 30.Kubbutat M H, Jones S N, Vousden K H. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- 31.Tao W, Levine A J. Proc Natl Acad Sci USA. 1999;96:3077–3080. doi: 10.1073/pnas.96.6.3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Xiong Y. Science. 2001;292:1910–1915. doi: 10.1126/science.1058637. [DOI] [PubMed] [Google Scholar]
- 33.Lancaster J R., Jr Nitric Oxide. 1997;1:18–30. doi: 10.1006/niox.1996.0112. [DOI] [PubMed] [Google Scholar]
- 34.Wang S, Guo M, Ouyang H, Li X, Cordon-Cardo C, Kurimasa A, Chen D J, Fuks Z, Ling C C, Li G C. Proc Natl Acad Sci USA. 2000;97:1584–1588. doi: 10.1073/pnas.97.4.1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arai N, Mitomi H, Ohtani Y, Igarashi M, Kakita A, Okayasu I. Mod Pathol. 1999;12:604–611. [PubMed] [Google Scholar]
- 36.Zingarelli B, Szabo C, Salzman A L. Gut. 1999;45:199–209. doi: 10.1136/gut.45.2.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wink D A, Hanbauer I, Krishna M C, DeGraff W, Gamson J, Mitchell J B. Proc Natl Acad Sci USA. 1993;90:9813–9817. doi: 10.1073/pnas.90.21.9813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jaiswal M, LaRusso N F, Nishioka N, Nakabeppu Y, Gores G J. Cancer Res. 2001;61:6388–6393. [PubMed] [Google Scholar]
- 39.Iwamoto M, Koji T, Makiyama K, Kobayashi N, Nakane P K. J Pathol. 1996;180:152–159. doi: 10.1002/(SICI)1096-9896(199610)180:2<152::AID-PATH649>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 40.Oda K, Arakawa H, Tanaka T, Matsuda K, Tanikawa C, Mori T, Nishimori H, Tamai K, Tokino T, Nakamura Y, et al. Cell. 2000;102:849–862. doi: 10.1016/s0092-8674(00)00073-8. [DOI] [PubMed] [Google Scholar]
- 41.Mitchell R A, Liao H, Chesney J, Fingerle-Rowson G, Baugh J, David J, Bucala R. Proc Natl Acad Sci USA. 2002;99:345–350. doi: 10.1073/pnas.012511599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hudson J D, Shoaibi M A, Maestro R, Carnero A, Hannon G J, Beach D H. J Exp Med. 1999;190:1375–1382. doi: 10.1084/jem.190.10.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roberts P J, Morgan K, Miller R, Hunter J O, Middleton S J. Gut. 2001;48:468–472. doi: 10.1136/gut.48.4.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murakami H, Akbar S M, Matsui H, Onji M. Eur J Clin Invest. 2001;31:337–343. doi: 10.1046/j.1365-2362.2001.00796.x. [DOI] [PubMed] [Google Scholar]
- 45.Shkolnik T, Livni E, Reshef R, Lachter J, Eidelman S. Am J Gastroenterol. 1987;82:1275–1278. [PubMed] [Google Scholar]
- 46.Mannick J B, Hausladen A, Liu L, Hess D T, Zeng M, Miao Q X, Kane L S, Gow A J, Stamler J S. Science. 1999;284:651–654. doi: 10.1126/science.284.5414.651. [DOI] [PubMed] [Google Scholar]
- 47.Kim Y M, Talanian R V, Billiar T R. J Biol Chem. 1997;272:31138–31148. doi: 10.1074/jbc.272.49.31138. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}