

Abstract
The lethal and oedema toxins produced by Bacillus anthracis, the aetiological agent of anthrax, are made by association of protective antigen with lethal and oedema factors and play a major role in the pathogenesis of anthrax. In the present paper, we describe the production of peptide-based specific inhibitors in branched form which inhibit the interaction of protective antigen with lethal and oedema factors and neutralize anthrax toxins in vitro and in vivo. Anti-protective antigen peptides were selected from a phage library by competitive panning with lethal factor. Selected 12-mer peptides were synthesized in tetra-branched form and were systematically modified to obtain peptides with higher affinity and inhibitory efficiency.
Keywords: anthrax in vivo inhibitor, anthrax toxin, multiple antigen peptide (MAP), oedema factor, peptide stability, phage peptide
Abbreviations: EF, oedema factor; Fmoc, fluoren-9-ylmethoxycarbonyl; HBS, Hepes-buffered saline; LF, lethal factor; MALDI–TOF, matrix-assisted laser-desorption ionization–time-of-flight; MAP, multiple antigen peptide; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide; PA, protective antigen
INTRODUCTION
Anthrax occurs after ingestion of spores of Bacillus anthracis, a toxigenic Gram-positive bacterium, which secretes two bipartite toxins known as oedema and lethal toxins. The dramatic increase in research for new anthrax therapeutics was prompted by the potential use of B. anthracis as a biological weapon. The main problem with anthrax therapy is the low efficiency of antibiotic treatment if it is not begun soon after exposure [1]. Once bacteria have secreted large amounts of anthrax toxins, treatment with antibiotics is largely ineffective. No specific therapy against anthrax toxins is currently available. New synthetic or recombinant molecules for combined use with antibiotics therefore have high priority. The possible use of genetically engineered antibiotic-resistant bacilli increases the need to develop effective anti-toxin treatment to counter the lethal effect of anthrax, failing antibiotic therapy.
Anthrax toxin consists of three proteins, PA (protective antigen), EF (oedema factor) and LF (lethal factor), which combine to form oedema and lethal toxins. PA is an 83 kDa protein which binds to PA cell-surface receptors [2] and is activated by proteases. Proteolytic removal of the N-terminal 20 kDa fragment allows the remaining receptor-bound portion (PA63) to oligomerize into a heptameric ring. The PA heptamer binds LF and EF and translocates them into the cytosol, where they produce major, often lethal, damage [3–8]. The different steps of the toxic process, mediated by protein interactions and enzymes are therefore the targets of therapeutic agents that are currently being studied worldwide [9].
With this aim, we selected anti-PA63 ligands from a large phage peptide library by competitive panning, using recombinant LF. This enabled single-step recovery of phages carrying peptides that interfere with PA–LF binding. We identified several sequences that inhibit PA–LF binding and also inhibit their toxicity towards cultured cells. Two peptide sequences were synthesized in linear and tetra-branched [MAP (multiple antigen peptide)] form [10], with four identical molecules linked to a lysine core. We demonstrated previously that MAP molecules are more effective as in vivo antidotes than linear peptides are [11] because of their higher stability against blood proteases and peptidases [12]. The two lead MAP sequences were systematically modified by alanine scanning, progressive shortening and randomization of residues found to be less critical for binding. Second-generation MAPs with higher affinity and inhibitory power were selected. One of the second-generation peptides was tested in an animal model and was found to completely inhibit anthrax toxin lethality. The same peptide also inhibited EF-mediated cAMP induction in different cell lines.
EXPERIMENTAL
Materials
All chemicals were obtained from Sigma unless specified otherwise.
Expression and purification of PA
Recombinant PA was produced with a C-terminal hexahistidine tag and was purified by Ni2+-chelate affinity chromatography. The expression vector pet20b+ containing the recombinant PA gene was transferred into Escherichia coli BL21(DE3) cells, and recombinant PA was recovered from bacterial inclusion bodies. The purity of PA was evaluated by SDS/12% PAGE.
Preparation of nicked PA (PA63)
Nicked PA was produced by treatment of purified recombinant PA with trypsin at a final trypsin/PA ratio of 1:1000 (w/w) for 45 min at room temperature (25 °C). The reaction was stopped by adding soya bean trypsin inhibitor at a final trypsin/inhibitor ratio of 1:20 (w/w). Oligomeric PA63 was prepared from activated PA by purification on a HiTrap Q FF column (Amersham Biosciences) in 20 mM Tris/HCl (pH 8.0) with a 0–1 M NaCl gradient. The protein was then dialysed overnight against PBS (pH 7.4) at 4 °C. The purity of PA63 was checked by SDS/12% PAGE.
Expression and purification of LF
A fusion protein of GST (glutathione S-transferase) and LF was expressed in E. coli strain BL21(DE3) transformed with the expression vector pGEX(2TK) (Amersham Biosciences) containing the LF gene. Purification of LF was performed by affinity chromatography on glutathione–Sepharose 4B (Amersham Biosciences) according to the manufacturer's instructions.
LF was eluted by adding 50 units of thrombin (Amersham Biosciences) in PBS for each millilitre of glutathione–Sepharose bed volume and incubating for 3 h at room temperature. The eluted solution was then dialysed against 20 mM Tris/HCl (pH 8.0) at 4 °C and was purified by anion-exchange chromatography using FPLC, which was performed to remove thrombin and to concentrate protein. Briefly, 2.0 mg of LF was loaded on a 1 ml Resource Q column (Amersham Biosciences) to obtain a 1.5 mg/ml protein solution by eluting with 0.5 M NaCl in 20 mM Tris/HCl (pH 8.0). The correct molecular-mass protein was then separated from the proteolytic fragments by FPLC, using a size-exclusion column (Superdex 200, 10/300 GL; Amersham Biosciences). The purity (>90%; results not shown) of LF was then tested by SDS/12% PAGE and Coomassie Blue staining.
Selection of anti-PA63 peptides
A random 12-mer peptide phage library Ph.D.12™ (New England Biolabs) was panned on the proteolytically activated form of anthrax protective antigen (PA63). Microtitre wells were coated overnight at 4 °C with 100 μl of purified 20 μg/ml PA63 in 0.1 M NaHCO3 (pH 8.6) and blocked with 0.1 M NaHCO3 (pH 8.6) and 0.5% (w/v) BSA. In the first round of biopanning, 10 μl of phage library [4×1010 pfu (plaque-forming units)] was added to the wells and allowed to bind for 1 h at room temperature with gentle rocking. After washing 10 times with TBS (50 mM Tris/HCl, pH 7.5, and 150 mM NaCl) containing 0.1% (v/v) Tween 20, bound phages were eluted by competition using 100 μl of 200 μg/ml purified LF with gentle rocking at room temperature for 1 h.
The amplified phages were titred and subsequently subjected to two further rounds of biopanning as described above, except that washing was performed using 0.5% (v/v) Tween 20.
Binding of selected clones to PA63 after the third round of panning was evaluated by phage ELISA in 96-well microtitre plates coated overnight at 4 °C with 100 μl of purified PA63 at 20 μg/ml.
Peptide synthesis
Monomeric peptides were synthesized as peptide amide by automated synthesizer (MultiSynTech) on Rink Amide MBHA resin (Nova Biochem) using Fmoc (fluoren-9-ylmethoxycarbonyl) chemistry and O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate/1,3-di-isopropylethylamine activation. MAPs were synthesized on Fmoc4-Lys2-Lys-βAla Wang resin. Side-chain-protecting groups were: t-butyl ester for glutamate; trityl for glutamine; t-butoxycarbonyl for lysine and tryptophan; 2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulphonyl for arginine; and t-butyl ether for serine. Peptides were then cleaved from the resin and deprotected by treatment with trifluoroacetic acid containing water and tri-isopropylsilane (95/2.5/2.5, by vol.). Crude peptides were purified by reversed-phase chromatography on a Vydac C18 column. Identity and purity of final products was confirmed by Ettan™ MALDI–TOF (matrix-assisted laser-desorption ionization–time-of-flight) MS (Amersham Biosciences).
Peptide processing in serum and plasma
A 150 μl volume of a 1 mg/ml solution of peptides was incubated at 37 °C with 40 μl of human serum or plasma. Samples withdrawn after 2 or 24 h were precipitated with 200 μl of methanol and were centrifuged at 10000 g for 1 min. The crude solution diluted in 0.1% (v/v) trifluoroacetic acid was analysed by HPLC using a C18 column. Controls for peptide retention time in the mixture were obtained by adding the same concentration of peptides to supernatants of plasma or serum treated with methanol and centrifuged as above, and running the mixture immediately. MS analysis of crude solutions and HPLC-eluted peaks (when detectable) was performed on an Ettan™ MALDI–TOF mass spectrometer. When HPLC peaks corresponding to uncleaved peptides were no longer detectable after incubation with plasma or serum, the HPLC eluent was collected at the appropriate retention time, allowing a window of ±2 min, and was analysed by MS to check for uncleaved peptides.
PA–LF binding (ELISA)
Nunc 96-well microtitre plates were coated with 5 μg/ml LF in PBS overnight at 4 °C. After PBS washing, plates were blocked with PBS and 3% (w/v) BSA for 1.5 h at 37 °C. Wells were then incubated with PA63 (5 μg/ml) and anti-PA63 peptides for 1 h at 37 °C. After washing with PBS containing 0.05% (v/v) Tween 20 and PBS, the plate was incubated with anti-His6–peroxidase antibody (Roche) diluted 1:500 in PBS containing 3% (w/v) BSA for 45 min at 37 °C.
Cytotoxicity assays
J774A.1 (ATCC) and RAW 264.7 cells (Istituto Zooprofilattico Sperimentale della Lombardia e dell'Emilia Romagna, Centre of Cell Culture, Brescia, Italy) were incubated in medium containing 0.5 μg/ml recombinant PA and 0.1 μg/ml recombinant LF with or without anti-PA63 MAPs. Cell viability was determined after 3.5 h of incubation using the MTT {3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide} dye assay. MTT was added to the cells to a final concentration of 0.5 mg/ml, and the cells were incubated for 1.5 h at 37 °C. The cells were then solubilized with 100 μl/well 10% (w/v) SDS and 45% (w/v) dimethylformamide (pH 4.5), and absorbance was measured at 595 nm with correction for absorption at 655 nm.
Intracellular cAMP was measured in lysates of Jurkat, HeLa and RAW cells treated with 2 μg/ml PA and 1 μg/ml EF in the presence of increasing concentrations of peptides. cAMP was quantified by EIA (enzyme-linked immunoassay) (Biotrak EIA, Amersham Biosciences).
Biacore
All Biacore experiments were performed on a BIACORE 1000/upgraded. All materials were purchased from Biacore International AB.
Biotinylated monomeric peptides, diluted to 100 μg/ml in HBS (Hepes-buffered saline; 10 mM Hepes, 150 mM NaCl, 3.4 mM EDTA and 0.005% polysorbate 20, pH 7.4), were immobilized on streptavidin-coated SA (streptavidin) sensor chip at a flow rate of 10 μl/min. For kinetic experiments, serial dilutions of PA63, diluted in HBS at concentrations ranging from 1 μM to 10 nM, were injected at a flow rate of 10 μl/min over immobilized peptides. Association and dissociation kinetic rate constants (kon and koff) and the equilibrium association constant Ka were calculated using BIAevaluation 3.0 software.
Inhibition of anthrax lethal toxin activity in vivo
Animal procedures were approved by the Ethical Committee of the Azienda Ospedaliera Universitaria Senese on 28 May 2004. Fisher 344 rats (average body mass 250 g) were anaesthetized by intraperitoneal injection of ketamine and acepromazine, and were inoculated in the tail vein with 40 μg of purified recombinant PA and 8 μg of purified recombinant LF diluted in PBS. Peptide powder was resuspended in PBS to a concentration of 2 mg/ml, diluted to 1 mg/ml (pH 7.5) and injected in the tail vein of rats 5 min after the lethal toxin injection. In some cases, a second injection of peptide inhibitor was given 1 h after the first injection. When anthrax symptoms were obvious, the rats were killed to avoid unnecessary distress. Surviving rats were monitored for 7 days.
RESULTS
Selection of peptide inhibitors
Selection of peptides that interfere with PA–LF binding was carried out by incubating the phage peptide library Ph.D.-12™ with purified recombinant PA63. Elution of specific anti-PA63 phage peptides was achieved by addition of recombinant LF which, by combining with PA63, selectively detaches peptides bound to PA63 on its LF-binding site, leaving all other phages that recognize different PA63 regions, attached to the protein (Figure 1a). Contrary to previous experiences [13–15], this approach turned out to be particularly efficient.
Figure 1. Production of peptides that inhibit PA–LF binding.

(a) Competitive selection of PA-binding phages. Phages from the library were allowed to bind to PA63-coated plastic wells. Selective elution of phage peptides recognizing the LF-binding site on PA was obtained by addition of purified LF. (b) From phage to MAP. Branched peptides in MAP form maintained the general features of phage-exposed peptides.
All sequenced clones shared the consensus motif YWWL (Tyr-Trp-Trp-Leu), although not always in the same sequence position. This short sequence is shared by previously described PA-binding peptides obtained from phage libraries by a different selection approach [14,15] and is not present in the sequence of LF, EF or PA20. Two phage peptides giving the highest inhibition of PA63–LF binding in a competition phage ELISA and carrying the sequences TLPYWWLTPSNP (p2) and NVMTYWWLDPPL (p3) respectively were chosen for chemical synthesis and further experiments.
Advantages of MAPs over linear peptides
The two selected 12-mer sequences were synthesized in monomeric and MAP forms. MAPs are dendrimeric molecules in which several peptide copies are synthesized on a lysine core [10]. We chose to synthesize tetra-branched MAP using a core of three lysine residues (Figure 1b).
Monomeric peptides did not significantly inhibit PA63–LF binding in ELISA, even when used at up to 200-fold molar excess with respect to PA63. Conversely, the MAPs gave significant inhibition even at a 2:1 molar ratio (Figure 2a).
Figure 2. Advantages of MAP over monomeric peptides.

(a) LF- (55 nM) coated ELISA plates were incubated with PA63 (80 nM) in the presence of peptides at three different concentrations. MAPs, but not their monomeric sequences, retained phage ability to inhibit PA63–LF binding in ELISA. Results are means±S.D. for at least three experiments. (b) MAPs were resistant to proteolysis in plasma and serum (which has stronger proteolytic activity). Panel 1, control HPLC profile of monomeric peptide p2 in the presence of plasma. Panel 2, monomeric peptide was degraded during 2 h of incubation in plasma at 37 °C. Only fragments derived from proteolytic cleavage were detected. Monomeric peptide was not detectable after 2 h of incubation in serum. Panel 3, control HPLC profile of MAP2 in the presence of serum. Panel 4, after 24 h of incubation in serum at 37 °C, the MAP was still detectable.
Stability of MAPs to proteolysis in human plasma and serum was tested by HPLC and MS as described in [12] and was compared with that of corresponding monomeric peptides. Serum has a stronger proteolytic activity owing to activation of the coagulation enzymes. As a general rule, peptides that were cleaved in plasma were also cleaved in serum in the same time. Those resistant in serum after 24 h were also resistant in plasma. Uncleaved MAPs could be detected after 24 h of incubation in plasma and even in serum, whereas monomeric peptides were cleaved after only 2 h of incubation in plasma. Monomeric peptides were not detectable after 24 h of incubation either in plasma or in serum (Figure 2b).
The use of peptides in MAP form offers several advantages. MAPs are stable to peptidases and proteases of biological fluids [12,16]. Moreover, synthesis in MAP form of sequences selected from a phage library, enables the activity of phage peptides to be retained. This may be due to the similarity between peptide structural arrangement in the MAP and in the phage-exposed form. In a MAP molecule, peptide sequences are linked to the lysine core by their C-terminus, as when expressed on the phage fusion protein (Figure 1b). Moreover, since MAPs contain more peptide copies, they enable multivalent binding, thus increasing binding efficiency, as in phage peptides.
We also synthesized the MAP form of the peptide having the sequence HTSTYWWLDGAP, which was reported to inhibit PA–LF binding and toxicity when covalently linked in multiple copies to a polyacrylamide backbone [15]. This MAP (MAPC1) was compared with MAP2 (TLPYWWLTPSNP) and MAP3 (NVMTYWWLDPPL) for efficiency in inhibiting PA63–LF binding in ELISA. The IC50 of MAP3 was approx. one order of magnitude lower than that of MAP2 and MAPC1 (Figure 3a). The MAPs were also tested for their ability to inhibit murine macrophage cell death, induced by incubation with PA83 and LF. The three MAPs produced a significant reduction in lethal-toxin-induced cell death. The resulting IC50 values were comparable with those obtained in the test for inhibition of PA63-LF binding (Figure 3b). MAP2 and MAP3 specifically bound PA63 and did not bind purified recombinant PA83 or LF in ELISA (results not shown).
Figure 3. Activity of phage library-derived MAPs.

(a) Inhibitory activity of MAPs on PA63–LF binding in ELISA. Purified oligomeric PA63 was incubated in LF-coated wells in the presence of MAPs. (b) Inhibitory activity of MAPs on lethal-toxin-induced cell mortality. J774A.1 cells were incubated with purified PA83 and LF in the presence of MAPs. Assays were performed in triplicate and IC50 values were calculated by non-linear regression analysis using GraphPad Prism 3.02 software. Results are means±S.D.
Pin-pointing critical residues of peptide inhibitors
A possible disadvantage of competitive elution of specific binders from a phage library is that concentration and affinity of the competitor may not be enough for recovery of high-affinity phages. Even when a large library is used, as in this case, it cannot therefore be assumed that selected peptides correspond to the best possible sequences. Thus we systematically modified the two sequences selected by competitive panning from the phage library, using alanine-scanning and progressive sequence shortening, followed by random re-elongation or substitution, with the aim of modifying less critical residues in a search to obtain peptides of higher affinity [11]. Unless otherwise indicated, all peptides were synthesized and tested in the tetra-branched MAP form.
Alanine scanning of MAP2 and MAP3 sequences showed that any residue in the YWWL consensus is critical and its replacement completely abolishes peptide inhibitory activity (Figures 4a and 4b). MAP3 has four additional residues whose substitution with alanine produced a sharp decrease in activity: Met3, Thr4, Pro10 and Pro11 (Figure 4b). The other residues (1, 2, 9 and 12) can be substituted without loss of activity. Alanine substitution in position 2 (NAMTYWWLDPPL) produced the best molecule in terms of inhibition (MAP3 V/A).
Figure 4. Pin-pointing critical residues of phage library-derived sequences.

Alanine scanning of MAP2 (a) and MAP3 (b) sequences. Twelve different tetra-branched MAPs, each carrying a single alanine substitution in the lead sequence, were tested for inhibition of PA63–LF binding by ELISA and compared with the activity of corresponding lead sequence in the same experiment. Results are means±S.D. for at least three experiments. (c) Summary of inhibitory activity of MAPs derived from progressive shortening of MAP2 (left-hand panel) and MAP3 (right-hand panel) sequences on PA63–LF binding in ELISA. Results are from at least three experiments for each peptide. Black squares, MAP inhibitory activity was more than 65% that of the lead MAP tested under the same conditions; grey squares, peptide activity was between 65 and 35% that of the lead MAP; white squares, peptide had less than 35% of the inhibitory activity of the lead peptide.
All of the residues in the MAP2 sequence can be replaced by alanine with moderate changes in peptide activity, with the notable exception of residues in the consensus sequence YWWL whose substitution produced complete loss of activity, and of Pro9 and Pro12 (Figure 4a).
Shortening of MAP2 and MAP3 essentially confirmed the results of alanine scanning. MAP3 can be shortened only by two residues at the N-terminus and one residue at the C-terminus with only a moderate decrease in activity. Further deletions produced a sharp decrease in peptide activity (Figure 4c). On the other hand, MAP2 can be shortened down to a six-residue core (YWWLTP) which is the minimum sequence inhibiting PA–LF binding in ELISA (Figure 4c).
Reconstructing peptide inhibitors
The six-residue minimum inhibiting sequence (YWWLTP) resulting from shortening of MAP2 was extended by systematically adding all natural amino acids (except cysteine, to avoid possible oxidation) at both termini. Extension at the N-terminus did not produce better peptides in terms of inhibition of PA63–LF binding in ELISA. On the contrary, extension at the C-terminus led to selection of the peptide YWWLTPP, which had a superior inhibitory capacity (Figure 5). Peptides with lysine and arginine at position 7 were completely inactive, like those having phenylalanine, tryptophan and lysine (results not shown). Further elongation of peptide YWWLTPP revealed the sequences YWWLTPPA, YWWLTPPQ and particularly YWWLTPPP with even greater inhibitory activity of PA63–LF binding. Addition of positively charged residues at position 8 again resulted in peptides with no inhibitory activity.
Figure 5. Inhibitory activity of lead and modified MAPs on PA63–LF binding in ELISA.

Results are means±S.D. for three independent experiments for each peptide.
The process of re-elongation of the minimum inhibitory sequence led to selection of octomer peptides having a PPX (Pro-Pro-Xaa) sequence at their C-terminus, as in 12-mer peptides with high activity (MAP3 and peptides derived from this lead sequence) and not in other lower-affinity peptides (MAP2 and MAPC1). From our results, we derived the consensus sequence YWWLXPPX for high-affinity binding to PA63. The critical role of the two proline residues is also confirmed by alanine scanning of MAP3, which showed that replacement of Pro10 and Pro11 led to a clear decrease in peptide activity and by alanine scanning of the sequence YWWLTP, which confirmed that the only residue that could be altered without a decrease in activity was threonine (results not shown).
In conclusion, systematic analysis of residues flanking the consensus sequence YWWL not only indicated peptides with higher affinity than the lead sequences selected from the library, but also provided a detailed picture of sequence–activity correlations of PA–LF peptide inhibitors.
All second-generation branched peptides were also compared with lead peptides MAP2 and MAP3 for ability to inhibit cell mortality caused by PA plus LF. Unlike what we found in experiments for inhibition of PA63–LF binding in ELISA, short peptides turned out to be less active than 12-mer peptides in inhibiting cell mortality (Figure 6a). Among 12-mer peptides, MAP3 V/A had an IC50 one order of magnitude lower than MAP3 (Figure 6b).
Figure 6. Inhibitory activity of lead and modified MAPs on lethal-toxin-induced cell death.

(a) J774A.1 cells were incubated with purified PA83 and LF in the presence of MAPs at concentrations of 8 μM (light grey bars) and 0.8 μM (dark grey bars). Results are means±S.D. for triplicate samples in three independent experiments. (b) Comparison of MAP3 and MAP3 V/A inhibitory activity. Assays were performed in triplicate, and IC50 values were calculated by non-linear regression analysis using GraphPad Prism 3.02 software. The broken line shows average cell viability obtained in the absence of peptides. Results are means±S.D. for at least three experiments.
Neutralization of anthrax oedema toxin
Recently, lethal toxin and oedema toxin were found to synergize in suppressing the activation and proliferation of dendritic and T-cells [17,18], which together are responsible for initiating adaptive immune responses. Thus specific and exclusive lethal toxin inhibitors may not be sufficient as antidotes for anthrax. We therefore tested the ability of MAP3 V/A and MAP YWWLTPPP to inhibit EF enzyme activity in different cell lines. MAP3 V/A completely inhibited EF-induced cAMP increase in different cell lines (Figure 7), whereas YWWLTPPP had no effect at the same concentrations.
Figure 7. Inhibitory activity of MAP3 V/A on oedema-toxin-induced cAMP increase.
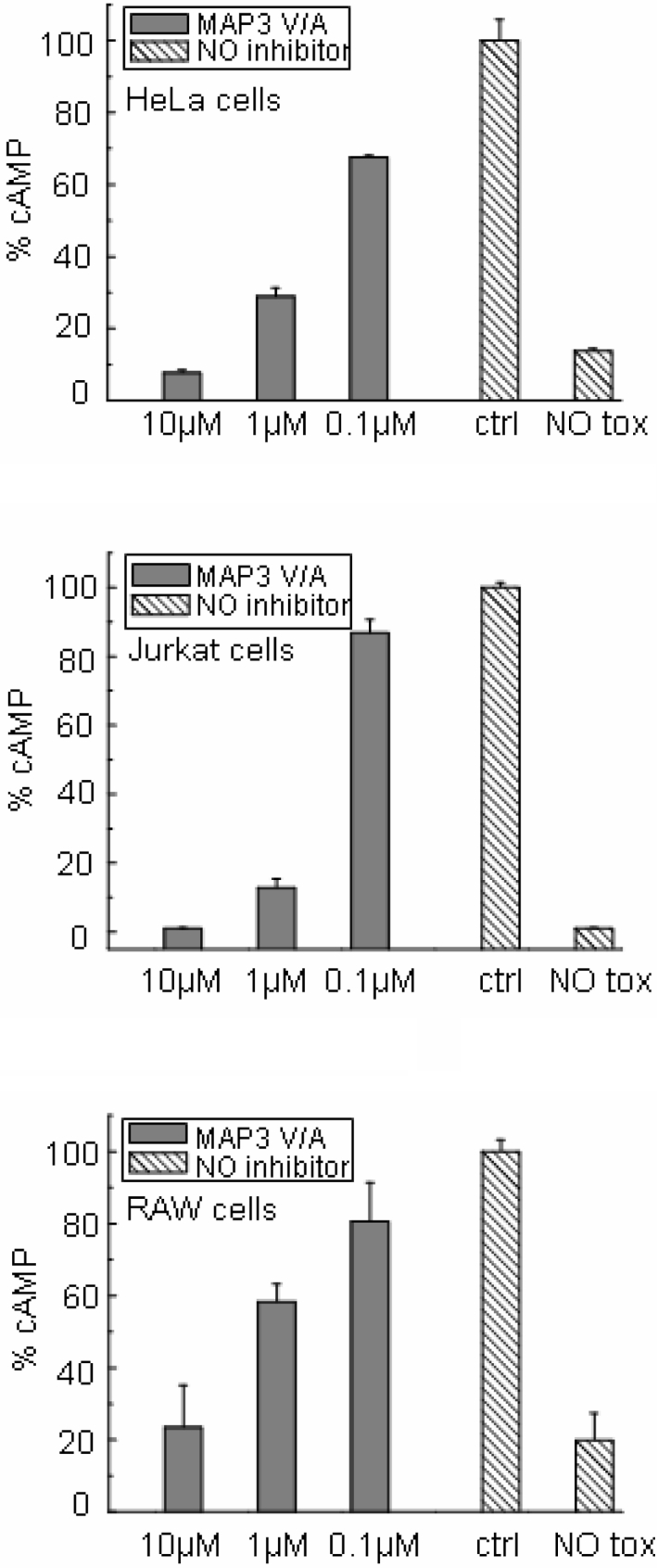
Quantification of cAMP in lysates of cells treated with 2 μg/ml PA and 1 μg/ml EF in the presence (grey bars) or absence (hatched bars) of peptide. NO inhibitor, cells with no peptide inhibitor. Basic levels of cAMP in the absence of toxin are indicated (NO tox). Results are means±S.D. for at least three experiments.
Binding kinetics of octomer and 12-mer peptides
The inhibitory activity of MAPs as measured by ELISA does not provide data on reaction rates that may be related to differences in peptide length or sequence. The binding kinetics of monomeric peptides p2, p3 and the octomer peptide YWWLTPPP with PA63 was therefore analysed by surface plasmon resonance (Figure 8, and Supplementary Figure 1 at http://www.BiochemJ.org/bj/395/bj3950157add.htm). Comparison of equilibrium constants (Ka) of peptide–PA63 binding essentially confirmed the IC50 ratios of corresponding MAPs, as calculated in PA63–LF binding experiments by ELISA. The Ka of the octomer peptide YWWLTPPP was 6.8×107 M−1, approx. 10-fold higher than that of the lead 12-mer p2 (7.2×106 M−1) and slightly better than that of the 12-mer peptide p3 (3.4×107 M−1). Analysis of reaction rates indicated that, although the increase in Ka of the 12-mer peptide p3 with respect to the 12-mer peptide p2 was due to an almost equally distributed improvement in kon and koff (kon were 5×103 Ms−1 and 1.4×104 Ms−1; koff values were 7×10−4 s−1 and 4×10−4 s−1 for p2 and p3 respectively), improvement of the octomer peptide Ka with respect to 12-mer peptides was mainly due to an increase in kon (8.8×104 Ms−1). The koff (1.3×10−3 s−1) of the octomer peptide was high than those of both 12-mer peptides.
Figure 8. Kinetics of PA63 binding.

Biotinylated monomeric peptides, diluted to 100 μg/ml in HBS, were immobilized on streptavidin-coated SA sensor chip at a flow rate of 10 μl/min. For kinetic experiments, serial dilutions of PA63 in HBS at concentrations ranging from 1 μM to 10 nM, were injected at a flow rate of 10 μl/min over immobilized peptides. Association and dissociation kinetic rate constants (kon and koff) and the equilibrium association constant Ka were calculated using BIAevaluation 3.0 software. The overlay of sensorgrams from BIAcore analysis of PA63 (300 nM) binding to biotinylated monomeric peptides bound to an SA sensor chip is shown.
This result suggests an explanation for the lower activity of the octomer compared with 12-mer peptides in neutralizing lethal toxin in cell lines. This difference in binding kinetics may render ELISA results different from those of cell assays, where concentrations of reagents are modified continuously by internalization of complexes. The higher koff of the octomer peptides could be responsible for their lower efficiency in the cell assay. Nonetheless, the increase in kon of the octomer peptide YWWLTPPP is promising for the design and selection of higher-affinity peptide inhibitors.
In vivo neutralization of anthrax lethal toxin
MAP2, MAP3, MAP3 V/A and MAP YWWLTPPP were tested for their ability to neutralize anthrax toxin in an animal model. Fisher 344 rats were injected with a mixture containing 40 μg of purified PA83 and 8 μg of purified LF. Under these conditions, rats died in approx. 2 h. At 5 min after injection of lethal toxin, rats were injected with peptide inhibitors. In some experiments, rats received a second boost of peptide 1 h after injection of the toxin. Injection of 1 mg of MAP3 V/A in a single shot or in two boosts of 500 μg each completely neutralized toxicity (Table 1). Identical amounts of the other peptides did not protect rats from the toxin.
Table 1. In vivo neutralization of anthrax toxin by MAP3 V/A.
| Number of rats | PA/LF (μg) | First boost (μg) | Second boost (μg) | Outcome |
|---|---|---|---|---|
| Ten | 40/8 | – | – | All dead after 116 (±13) min |
| Four | 40/8 | 250 | 250 | All dead after 140 (±1.4) min |
| Four | 40/8 | 500 | – | All dead after 140.5 (±2) min |
| Four | 40/8 | 500 | 500 | All alive |
| Seven | 40/8 | 1000 | – | All alive |
DISCUSSION
In this paper, we describe the selection and optimization of the activity in vitro and in vivo of new peptide-based inhibitors of anthrax lethal toxin. A competitive method was used for specific selection from a 12-mer peptide phage library of PA-binding phages that inhibited the interaction between PA and LF. Our system differs from previous methods for the isolation of anti-PA peptides [13–15] using libraries from the same commercial source. In these methods, specific phages directed to the LF-binding sites exposed by PA63 were recovered through a two-step elution after incubation of the phage library with PA63 oligomers. The first step was a subtractive procedure in which peptides binding to surfaces in whole PA83 or PA63 were removed by adding an excess of PA83 to the reaction tube, leaving peptides binding to specific regions of PA63 attached to the coated protein. In the second step, these specific anti-PA63 peptides were eluted by competition with an excess of the same PA63. This two-step procedure recovered peptides binding around or directly on to the LF- and EF-binding sites which are only present in oligomerized PA63. In contrast, our method uses a single elution step in which specific anti-PA63 peptides are detached by competition with the natural ligand LF. This only isolates peptides that interfere directly with binding of LF to PA63, which triggers the pathological mechanism of anthrax.
Peptide inhibitors isolated by LF competitive elution were then synthesized in a tetra-branched MAP form, which improved peptide biological activity by: (i) increasing peptide resistance to proteolytic degradation, and (ii) maintaining biological activity of phage peptides, thanks to their multimericity and chemical conformation.
The synthesis of sequences in MAP form is a substantial difference with respect to previous peptides [13–15]. Previously generated peptide inhibitors only neutralize anthrax toxin when coupled in multiple copies to polymeric supports such as acrylamide or poly(ethylene glycol). Compared with these polymeric molecules, MAPs have the advantage of a much lower molecular mass and being of definite chemical structure and stoichiometry. These features enable synthesis of molecules with highly reproducible activity and pharmacological properties, which cannot be guaranteed for polymeric molecules that have a variable number of peptide copies per molecule.
Since competitive selection from a combinatorial library does not guarantee the recovery of all of the high-affinity ligands, we systematically modified the length and sequence of peptides identified by phage selection, by synthesis and screening of mini-libraries of tetra-branched MAP molecules. This process enabled identification of residues that are critical for binding of PA and for inhibition of PA–LF binding, leading to selection of second-generation MAPs with higher efficiency. The second-generation 12-mer peptide MAP3 V/A has an IC50 at least one order of magnitude lower than any other similar lethal-toxin-inhibiting peptide identified so far and efficiently neutralizes anthrax toxin in vivo.
Apart from identification of at least one peptide inhibitor that is effective in vivo, which is a good candidate for a drug against anthrax toxins, our results are also of general utility for the design of novel peptide inhibitors of PA–LF binding.
Online data
Acknowledgments
We acknowledge grants from Istituto Superiore di Sanità, Rome, Italy (Italy–US Collaboration on Fighting Bioterrorism-Progetto Antrace, Tossine e terapia, convenzione 52/C2-4 04) and from the University of Siena (PAR 2003). Special thanks go to Cesare Montecucco for helpful discussion, Stefano Bindi for animal experiments and Silvia Scali for peptide synthesis.
References
- 1.Dixon T. C., Meselson M., Guillemin J., Hanna P. C. Anthrax. N. Engl. J. Med. 1999;341:815–826. doi: 10.1056/NEJM199909093411107. [DOI] [PubMed] [Google Scholar]
- 2.Bradley K. A., Mogridge J., Mourez M., Collier R. J., Young J. A. Identification of the cellular receptor for anthrax toxin. Nature (London) 2001;414:225–229. doi: 10.1038/n35101999. [DOI] [PubMed] [Google Scholar]
- 3.Chauhan V., Bhatnagar R. Identification of amino acid residues of anthrax protective antigen involved in binding with lethal factor. Infect. Immun. 2002;70:4477–4484. doi: 10.1128/IAI.70.8.4477-4484.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kurzchalia T. Anthrax toxin rafts into cells. J. Cell Biol. 2003;160:295–296. doi: 10.1083/jcb.200301032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leppla S. H. Anthrax toxin edema factor: a bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc. Natl. Acad. Sci. U.S.A. 1982;79:3162–3166. doi: 10.1073/pnas.79.10.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duesbery N. S., Webb C. P., Leppla S. H., Gordon V. M., Klimpel K. R., Copeland T. D., Ahn N. G., Oskarsson M. K., Fukasawa K., Paull K. D., Vande Woude G. F. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science. 1998;280:734–737. doi: 10.1126/science.280.5364.734. [DOI] [PubMed] [Google Scholar]
- 7.Vitale G., Pellizzari R., Recchi C., Napolitani G., Mock M., Montecucco C. Anthrax lethal factor cleaves the N-terminus of MAPKKs and induces tyrosine/threonine phosphorylation of MAPKs in cultured macrophages. Biochem. Biophys. Res. Commun. 1998;248:706–711. doi: 10.1006/bbrc.1998.9040. [DOI] [PubMed] [Google Scholar]
- 8.Vitale G., Bernardi L., Napolitani G., Mock M., Montecucco C. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem. J. 2000;352:739–745. [PMC free article] [PubMed] [Google Scholar]
- 9.Rainey G. J. A., Young J. A. T. Antitoxins: novel strategies to target agents of bioterrorism. Nat. Rev. Microbiol. 2004;2:721–726. doi: 10.1038/nrmicro977. [DOI] [PubMed] [Google Scholar]
- 10.Tam J. P. Synthetic peptide vaccine design: synthesis and properties of a high density multiple antigenic peptide system. Proc. Natl. Acad. Sci. U.S.A. 1988;85:5409–5413. doi: 10.1073/pnas.85.15.5409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lozzi L., Lelli B., Runci Y., Scali S., Bernini A., Falciani C., Pini A., Niccolai N., Neri P., Bracci L. Rational design and molecular diversity for the construction of anti-α-bungarotoxin antidotes with high affinity and in vivo efficiency. Chem. Biol. 2003;10:411–417. doi: 10.1016/s1074-5521(03)00094-2. [DOI] [PubMed] [Google Scholar]
- 12.Bracci L., Falciani C., Lelli B., Lozzi L., Runci Y., Pini A., De Montis M. G., Tagliamonte A., Neri P. Synthetic peptides in the form of dendrimers become resistant to protease activity. J. Biol. Chem. 2003;278:46590–46595. doi: 10.1074/jbc.M308615200. [DOI] [PubMed] [Google Scholar]
- 13.Mourez M., Collier R. J. Use of phage display and polyvalency to design inhibitors of protein–protein interactions. Methods Mol. Biol. 2004;261:213–228. doi: 10.1385/1-59259-762-9:213. [DOI] [PubMed] [Google Scholar]
- 14.Gujraty K., Sadacharan S., Frost M., Poon V., Kane R. S., Mogridge J. Functional characterization of peptide-based anthrax toxin inhibitors. Mol. Pharm. 2005;2:367–372. doi: 10.1021/mp050040f. [DOI] [PubMed] [Google Scholar]
- 15.Mourez M., Kane R. S., Mogridge J., Metallo S., Deschatelets P., Sellman B. R., Whitesides G. M., Collier R. J. Designing a polyvalent inhibitor of anthrax toxin. Nat. Biotechnol. 2001;19:958–961. doi: 10.1038/nbt1001-958. [DOI] [PubMed] [Google Scholar]
- 16.Falciani C., Lozzi L., Pini A., Bracci L. Bioactive peptides from libraries. Chem. Biol. 2005;12:417–426. doi: 10.1016/j.chembiol.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 17.Paccani S. R., Tonello F., Ghiottoni R., Natale M., Muraro L., D'Elios M. M., Tang W. J., Montecucco C., Baldari C. T. Anthrax toxins suppress T lymphocyte activation by disrupting antigen receptor signaling. J. Exp. Med. 2005;201:325–331. doi: 10.1084/jem.20041557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tournier J. N., Quesnel-Hellmann A., Mathieu J., Montecucco C., Tang W. J., Mock M., Vidal D. R., Goossens P. L. Anthrax edema toxin cooperates with lethal toxin to impair cytokine secretion during infection of dendritic cells. J. Immunol. 2005;174:4934–4941. doi: 10.4049/jimmunol.174.8.4934. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.