

Abstract
The E2F family is composed of at least eight E2F and two DP subunits, which in cells exist as E2F/DP heterodimers that bind to and regulate E2F target genes. While DP-1 is an essential and widespread component of E2F, much less is known about the DP-3 subunit, which exists as a number of distinct protein isoforms that differ in several respects including the presence of a nuclear localisation signal (NLS). We show here that the NLS region of DP-3 harbours a binding site for 14-3-3ɛ, and that binding of 14-3-3ɛ alters the cell cycle and apoptotic properties of E2F. DP-3 responds to DNA damage, and the interaction between DP-3 and 14-3-3ɛ is under DNA damage-responsive control. Further, 14-3-3ɛ is present in the promoter region of certain E2F target genes, and reducing 14-3-3ɛ levels induces apoptosis. These results identify a new level of control on E2F activity and, at a more general level, suggest that 14-3-3 proteins integrate E2F activity with the DNA damage response.
Keywords: cancer, cell cycle, E2F, transcription, 14-3-3
Introduction
The E2F family of transcription factors is inextricably linked with cell cycle progression and apoptosis, through co-ordinating the expression of a large body of genes involved in regulating the G1 to S phase transition, as well as others involved with apoptosis (Stevens and La Thangue, 2003). During the cell cycle, E2F activity is controlled through the temporal association of ‘pocket' proteins, the prototype member being the retinoblastoma protein (pRb), which physically interacts with E2F. In turn pRb is regulated at the level of phosphorylation through the activity of cyclin-dependent kinases. Overall, E2F is intimately connected with the cell cycle, and its activity contributes to the timely expression of genes required for early cell cycle progression.
The E2F family is composed of at least eight E2F and two DP subunits, which physically associate as E2F/DP heterodimers that bind to and regulate E2F target genes (Trimarchi and Lees, 2002). E2F-1, the founding member of the family, displays the properties of both an oncogene and tumour suppressor (La Thangue, 2003; Stevens and La Thangue, 2003). For example, while E2F-1 can advance quiescent cells into S phase (Johnson et al, 1993; Qin et al, 1994), the level of apoptosis in Rb−/− mice is suppressed by E2F-1 deficiency (Tsai et al, 1998; Yamasaki et al, 1998), and E2F-1−/− mice display defects in apoptosis together with an increased incidence of tumours (Field et al, 1996; Yamasaki et al, 1996).
Much less is known about the DP family of proteins. The first identified member, DP-1, is a widespread component of the E2F heterodimer (Girling et al, 1993; Bandara et al, 1994). It is an essential gene since mice that lack DP-1 activity are embryonically lethal, in part due to defects in the development of extra-embryonic tissue (Kohn et al, 2003). The other member of the DP family, murine DP-3 (which has a protein isoform that is equivalent to human DP-2), is also quite widely expressed (Ormondroyd et al, 1995; Zhang and Chellappan, 1995; Rogers et al, 1996).
A notable feature of DP-3 is its complex regulation, which distinguishes it from other members of the DP and E2F family (Ormondroyd et al, 1995). DP-3 RNA is subject to extensive alternative splicing that results in at least four distinct protein isoforms, referred to as α, β, γ and δ (Figure 2A). A region, referred to as the extra (E) region, present in the α and δ isoforms (Figure 2A), contributes to a bi-partite nuclear localisation signal (NLS) in conjunction with a juxta-positioned stretch of basic residues. Thus, DP-3 isoforms that contain the E region are localised to nuclei, whereas isoforms devoid of the E region are present mostly in the cytoplasm (de la Luna et al, 1996; Allen et al, 1997). The N-terminal extension, present in the α isoform, binds DIP, which is a BTB/POZ domain protein that influences the activity of the α isoform (de la Luna et al, 1999).
Figure 2.
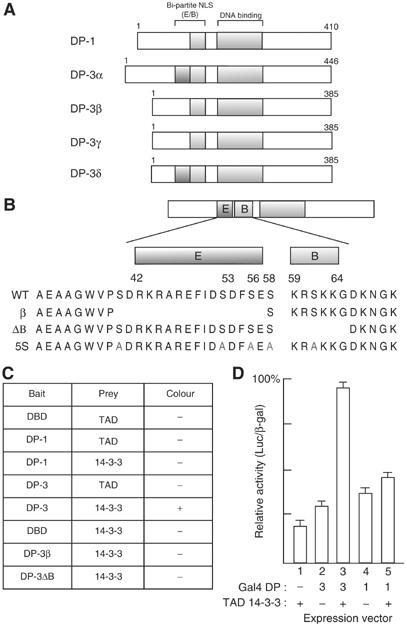
Identification of a 14-3-3ɛ binding site in DP-3. (A) Schematic representation of the DP family of proteins. The position of the bi-partite NLS, composed of the E and B region is indicated, together with the conserved DNA-binding domain. The four known isoforms of DP-3, α, β, γ and δ (Ormondroyd et al, 1995), are indicated. DP-1 is shown for comparison. The human DP-2 protein (Zhang and Chellappan, 1995) is equivalent to the murine DP-3α isoform. (B) Protein sequence of the E and B region, together with the sequence absent in the natural protein isoform DP-3β and the mutant derivative DP-3ΔB (de la Luna et al, 1996), and the serine (S) residues mutated to alanines (A; indicated in red) in DP-3 5S. (C) Summary of results from the yeast two-hybrid assay with the indicated baits and prey using 14-3-3ɛ fused to the VP16 trans-activation domain (TAD). (D) Mammalian two-hybrid assay in U20S cells with DP-1 or DP-3δ fused to the Gal4 DNA-binding domain and 14-3-3ɛ fused to the TAD of VP16. All plates were co-transfected with pCMV-β-gal (2 μg) as an internal control. Values are given as the ratio of luciferase to β-gal activities.
The 14-3-3 proteins represent an evolutionarily conserved family that are involved in a wide range of biological processes (Fu et al, 2000; van Hemert et al, 2001). They bind to and alter the activity of target proteins through, for example, sequestration, re-localisation and conformational alterations (Fu et al, 2000; Tzivion et al, 2001). There are seven 14-3-3 isoforms that exist as dimers in cells, which recognise target molecules predominantly through phosphorylated serine residues (Muslin et al, 1996; Rittinger et al, 1999).
It is known that 14-3-3 proteins play an important role in cell cycle control. For example, phosphorylation of Cdc25 phosphatase, which activates Cdc2 at mitosis by de-phosphorylating tyrosine 15, creates a 14-3-3 binding site that subsequently leads to the cytoplasmic sequestration of Cdc25 (Peng et al, 1997; Blasina et al, 1999; Kumagai and Dunphy, 1999). This may involve an alteration in the activity of the Cdc25 NLS by affecting its interaction with importin α/β (Kumagai and Dunphy, 1999; Zeng and Piwnica-Worms, 1999). Further, the interaction between 14-3-3 and p53 enhances DNA-binding activity and the ensuing p53 response (Hermeking et al, 1997;Waterman et al, 1998; Chehab et al, 2000), and the proapoptotic activity of Bad is regulated by binding to 14-3-3, which prevents formation of a heterodimer with the Bcl2 oncoprotein (Xing et al, 2000).
In order to gain further insight into the role of DP-3 and the regulation of E2F activity, we have explored the control of DP-3 during the DNA damage response. Our results indicate that DP-3 is induced in response to DNA damage, and suggest that 14-3-3ɛ plays an important role in this process. Specifically, 14-3-3ɛ binds to the NLS region of DP-3, and this interaction alters the cell cycle and apoptotic properties of E2F. Moreover, the interaction between 14-3-3ɛ and DP-3 is under DNA damage control. 14-3-3ɛ is present in the promoter region of E2F target genes, and further altering the level of 14-3-3ɛ induces apoptosis. These results define a new and intricate level of control on the DP-3 subunit and, at a more general level, suggest that 14-3-3 proteins integrate E2F activity with the DNA damage response.
Results
DP-3 is DNA damage responsive
Previous studies suggested that E2F activity plays a role in the DNA damage response (Blattner et al, 1999; Hofferer et al, 1999; O'Connor and Lu, 2000). Specifically, E2F-1 is a DNA damage-responsive protein (Stevens et al, 2003). We reasoned that DP subunits may in a similar manner respond to DNA damage and therefore investigated the effect of DNA damage on DP-1 and DP-3. In cells treated with bleomycin, which acts as a radiomimetic drug (Demonacos et al, 2004), a clear increase in DP-3 levels was apparent, whereas DP-1 was not affected (Figure 1). DP-3 underwent a similar response to UV light and etoposide treatment (data not shown). As expected, the levels of E2F-1 increased under DNA damage. Therefore, DP-3 is a DNA damage-responsive DP subunit.
Figure 1.
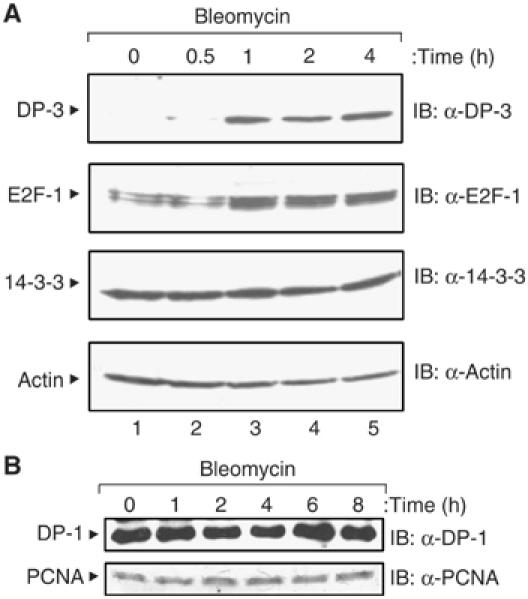
DNA damage induction of DP-3. SiHa cells were treated with bleomycin (5 μg/ml) for the indicated time (in hours), harvested and immunoblotted with anti-DP-3 (A), anti-DP-1 (A), anti-E2F-1 (A), anti-14-3-3ɛ (A), anti-PCNA (B) or anti-actin (A) as indicated.
DP-3 binds to 14-3-3ɛ
To investigate whether the response of DP-3 to DNA damage involved protein–protein interactions, we employed a yeast two-hybrid screen to identify proteins that interact with DP-3 (Figure 2A and B). We expressed a Lex A fusion protein fused to the N-terminal 110 residues of DP-3δ and screened for interacting proteins in a 10.5-day mouse embryo cDNA library (Figure 2). The sequence of one positive clone revealed it to be the epsilon (ɛ) isoform of the 14-3-3 family (Roseboom et al, 1994).
The specificity of the interaction was assessed in yeast where the interaction between 14-3-3ɛ and DP-3 was found to be dependent upon the presence of both the E and B region, since the natural DP-3β isoform (which lacks the E region) and the DP-3ΔB mutant derivative (lacking the stretch of basic residues) failed to interact (Figure 2B and C). It is consistent with this idea that DP-1, which lacks sequence that exhibits similarity to the E region (Ormondroyd et al, 1995), also failed to interact with 14-3-3ɛ (Figure 2C). The interaction was tested in mammalian U2OS cells using the two-hybrid assay with VP16-tagged 14-3-3ɛ and Gal4-DNA-binding domain-tagged DP-3δ or DP-1 where significant stimulation of activity was apparent when VP16-14-3-3ɛ was co-expressed with Gal4-DP-3δ but not with Gal4-DP-1 (Figure 2D).
To further characterise the interaction, we pursued in vitro binding assays and thereafter assessed the interaction in mammalian cells. Purified GST-DP-3δ (Figure 3A) was incubated in an extract prepared from transfected COS7 cells expressing myc-tagged 14-3-3ɛ. Relative to GST alone, a specific interaction occurred between GST-DP-3 and 14-3-3ɛ and, as expected, DP-3 bound to the E2F subunit (Figure 3B). The interaction was also studied using GST-14-3-3ɛ and extracts prepared from COS7 cells expressing DP-3 and E2F proteins, where a similar level of specificity was seen (data not shown).
Figure 3.
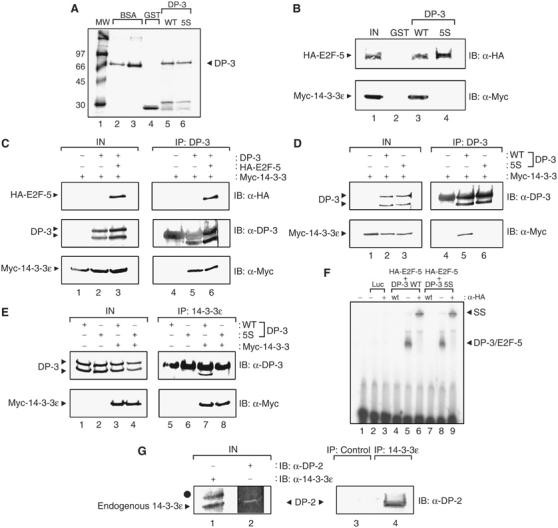
14-3-3ɛ binds to DP-3. (A) Coomassie stain showing bacterially expressed GST (track 4), GST-DP-3δ wt (track 5), GST-DP-3 5S (track 6) and 2 and 5 μg BSA (track 2 and 3, respectively) as protein standards. Molecular weight markers are shown in track 1. (B) COS7 cells were transiently transfected with expression vectors encoding myc-14-3-3ɛ (50 μg) and HA-E2F-5 (50 μg). The cells were harvested and used in a binding assay with either 5 μg of GST (track 2), GST-DP-3δ wt (track 3) or GST-DP-3 5S (track 4). The input levels of E2F-5 and 14-3-3ɛ are shown in track 1. (C) COS7 cells were transiently transfected with expression vectors encoding HA-E2F-5 (20 μg), DP-3δ wt (20 μg), myc-14-3-3ɛ (40 μg) and β-gal (5 μg) as an internal control. Cells were harvested and subjected to immunoprecipitation with anti-DP-3 antibody and immunoblotted with either anti-HA, anti-DP-3 or anti-myc as indicated. The immunoglobulin heavy chain (*) obscures the upper DP-3 polypeptide in the IB anti-DP-3 treatment. (D, E) COS7 cells were transiently transfected with the following amounts of the indicated expression vectors encoding DP-3δ wt (20 μg), DP-3 5S mutant (20 μg), myc-14-3-3ɛ (40 μg) and β-gal (5 μg) as an internal control. Cells were harvested and subjected to immunoprecipitation with either anti-DP-3 antibody (D) or anti-14-3-3ɛ antibody (E) and immunoblotted with either anti-DP-3 or anti-myc as indicated. The immunoglobulin heavy chain (*) obscures the upper DP-3δ polypeptide in the IB anti-DP-3 treatment. (F) In vitro-translated luciferase (tracks 2 and 3), HA-E2F-5 and DP-3δ wt (tracks 4–6) or HA-E2F-5 and DP-3 5S (tracks 7–9) were assessed for DNA binding on the E2F site taken from the E2A promoter as described. Track 1 shows the probe alone; wt indicates the presence of nonradiolabelled wild-type E2F site (100-fold excess) and − and + indicate the absence or presence of anti-HA antibody. The DP-3/E2F-5 complex and the supershift (SS) that occurs upon the addition of anti-HA are indicated. (G) Human ML-1 cells were harvested and subjected to immunoprecipitation with either anti-14-3-3ɛ or a control antibody, and then immunoblotted with either anti-DP-2 or anti-14-3-3ɛ as indicated. Endogenous 14-3-3ɛ and DP-2 are shown in tracks 1 and 2. The symbol • indicates a nonspecific band in the input extracts recognised by the anti-14-3-3ɛ antibody. Note that human DP-2 is equivalent to murine DP-3.
We next studied the interaction between DP-3δ and 14-3-3ɛ in cells. By immunoprecipitation followed by immunoblotting, DP-3δ and 14-3-3ɛ bound specifically to each other, and the interaction was not altered by the presence of the E2F subunit (Figure 3C). A similar level of specificity was apparent when antibodies against either DP-3δ or 14-3-3ɛ were used in the primary immunoprecipitation step (Figure 3C–E). Most importantly, we established that 14-3-3ɛ and DP-3 interact under physiological conditions by immunoprecipitating 14-3-3ɛ from ML-1 cells, where human DP-2 (the human equivalent of murine DP-3) was specifically detected in the immunocomplex (Figure 3G). Thus, DP-3 and 14-3-3ɛ interact under physiological conditions.
The DP-3 5S mutant derivative cannot bind to 14-3-3ɛ
To explore the role of the 14-3-3ɛ in regulating DP-3 activity, we sought to identify a mutant derivative of DP-3 that failed to bind 14-3-3ɛ. The results derived from the two-hybrid assays indicated that the E and B region are necessary for the interaction between DP-3 and 14-3-3ɛ, suggesting that residues within these domains, or in close proximity, are likely to be involved in binding 14-3-3ɛ. We undertook a mutagenesis strategy in which we altered residues in DP-3 that were likely to influence the interaction between the two proteins. Our attention focussed on serine (S) residues in the E and B region, given the role of S residues in the 14-3-3 recognition of target proteins (Muslin et al, 1996; Rittinger et al, 1999).
A panel of mutant derivatives where individual or groups of S residues were altered to alanine (A) was prepared and assessed for in vitro binding to 14-3-3ɛ. The DP-3 5S mutant, where five serine residues in DP-3δ had been changed to alanine (Figure 2B), failed to interact with 14-3-3ɛ, in contrast to the other mutant derivatives 2S, 3S and 4S, which retained binding activity (Figure 3B, and data not shown). Similarly, in cells there was very reduced binding to 14-3-3ɛ (Figure 3D and E), suggesting that DP-3 5S is a mutant derivative devoid of 14-3-3ɛ binding activity.
To determine the specificity of the effect seen in DP-3 5S for 14-3-3ɛ binding, we investigated several established properties of DP proteins. DP-3 5S behaved like wild-type DP-3 in all assays tested. Thus, DP-3 5S bound to an E2F partner (Figure 3B), retained DNA-binding activity (Figure 3F), and accumulated in nuclei (Figure 6) like its wild-type counterpart. The inability of DP-3 5S to bind to 14-3-3ɛ did not therefore reflect a general and nonspecific loss of activity. Given the specific loss-of-function in DP-3 5S for 14-3-3ɛ binding activity, we compared wild-type DP-3δ to the properties of DP-3 5S in order to gain a clearer understanding of the role that 14-3-3ɛ might play in regulating E2F activity.
Figure 6.
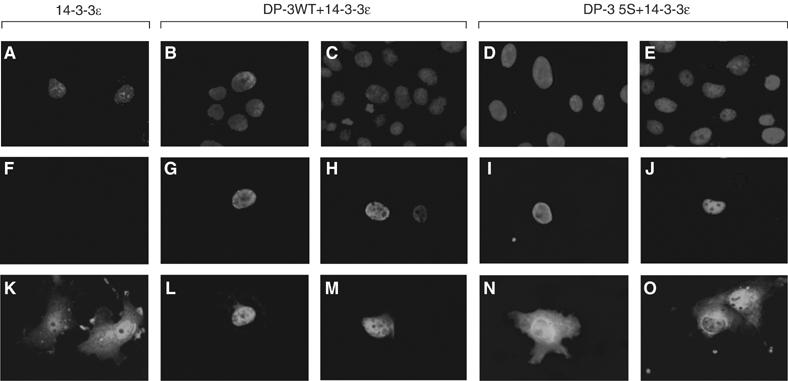
Intracellular location of 14-3-3ɛ and DP-3δ. COS7 cells were transfected as indicated with the following expression vectors encoding HA-E2F-5 (2 μg), DP-3 wt (2 μg), DP-3 5S (2 μg) and 14-3-3ɛ (4 μg) as indicated. Around 65 h post-transfection, the cells were fixed and subjected to immunofluorescence. Cellular DNA was stained with DAPI (A--E), while detection of the exogenously expressed 14-3-3ɛ (K--O) and DP-3δ (F--J) was carried out using a monoclonal anti-myc antibody and the polyclonal anti-DP-3 antibody, 7.2, respectively. The assay was carried out blind and about 200 cells were assessed. Data shown are typical of three independent experiments.
Functional consequences of 14-3-3ɛ on E2F activity
As E2F regulates transcription, we surmised that 14-3-3ɛ may impart altered transcriptional activity on E2F. To investigate this possibility we co-expressed 14-3-3ɛ with E2F, where the DP partner was provided by DP-3δ, and the E2F subunit by E2F-5, and measured the activity of the E2F-responsive cyclin E promoter (Botz et al, 1996). There was a titratable increase in the activity of the cyclin E promoter upon the expression of 14-3-3ɛ (Figure 4A), suggesting that 14-3-3ɛ augments E2F activity. In contrast, a similar activity profile was not apparent when wild-type DP-3δ was replaced by DP-3 5S (Figure 4A), arguing that the ability of 14-3-3ɛ to influence E2F activity is dependent upon its interaction with DP-3.
Figure 4.
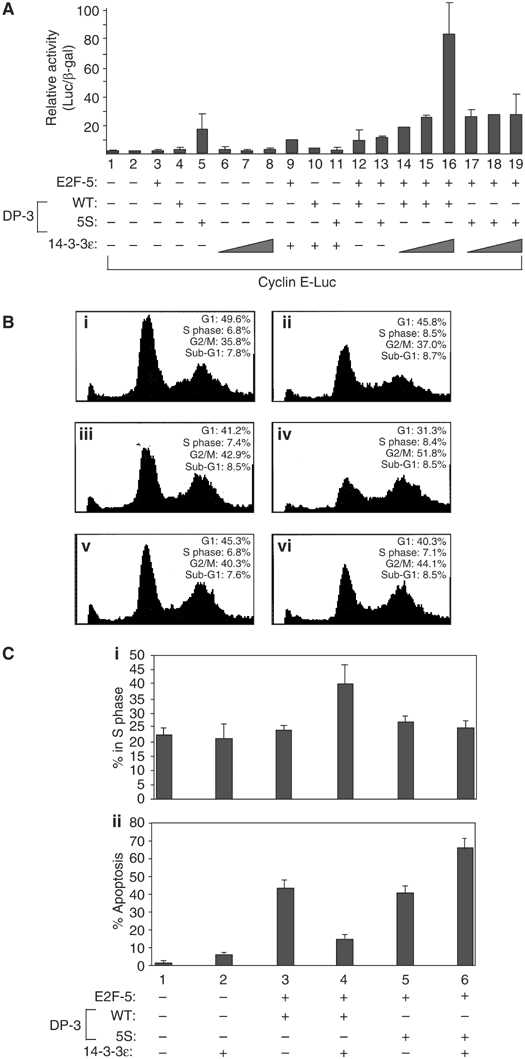
Growth-regulating and apoptotic control of E2F activity by 14-3-3ɛ. (A) COS7 cells were transfected with cyclin E-luc (0.5 μg), and expression vectors encoding HA-E2F-5 (1 μg), DP-3δ wt (1 μg), DP-3 5S (1 μg) and 14-3-3ɛ (0.5, 1 or 2 μg) as indicated and pCMV-β-gal (1 μg) as an internal control; where no 14-3-3ɛ titration is indicated, 1 μg of the 14-3-3ɛ vector was transfected. Cells were harvested and treated as described. The data shown is representative of three independent experiments and the values depict the relative level of luciferase to β-gal expression. (B) COS7 cells were transfected with pcDNA3 (20 μg, i), CD20 (5 μg, i–vi), HA-E2F-5 (5 μg, iii–vi), DP-3δ wt (5 μg, iii–iv), DP-3 5S (5 μg, v and vi) and 14-3-3ɛ (10 μg, ii, iv and vi) as indicated. Cells were harvested and subjected to FACS analysis as described. The data shown are representative of three independent experiments; the percentage of transfected cells in sub-G1, G1, S phase and G2/M percentages are indicated. (C) (i) COS7 cells were transfected with the following plasmids: pcDNA3 (2 μg), HA-E2F-5 (2 μg), DP-3δ wt (2 μg), DP-3 5S (2 μg), 14-3-3ɛ (4 μg) and RFP (1 μg) as indicated. Cells were harvested and measured for Brdu incorporation as described. The assay was carried out blind and 200 cells were counted, and the values shown represent the average of three separate experiments. (C, ii) COS7 cells were treated as described in (i) and subjected to the TUNEL assay.
As E2F has an established role in regulating the cell cycle and apoptosis (Stevens and La Thangue, 2003), we asked if the interaction with 14-3-3ɛ and consequent effect on transcription had any impact on these properties. The expression of the E2F heterodimer, E2F-5/DP-3δ, reduced the size of the G1 population and increased the number of G2/M phase cells, which, in the presence of 14-3-3ɛ, was significantly enhanced (Figure 4B(iii) and (iv)). Moreover, while the consequence of E2F-5/DP-3 5S expression was similar, the subsequent effect of co-expressing 14-3-3ɛ was much reduced (Figure 4B(v) and (vi)), suggesting that 14-3-3ɛ augments the cell cycle effects that result from E2F activity.
To substantiate this idea, we measured the proportion of cells undergoing DNA synthesis by incorporation of 5-bromo-2′-deoxyuridine (Brdu). While 14-3-3ɛ was capable of augmenting DNA synthesis in the presence of the E2F heterodimer, enhanced DNA synthesis was not apparent with DP-3 5S (Figure 4C(i)). Based upon the cell cycle analysis and Brdu incorporation results (Figure 4B and C(i)), we conclude that 14-3-3ɛ facilitates cell cycle progression, and that the physical interaction between DP-3 and 14-3-3ɛ is likely to be required for this effect.
We assessed the effect of 14-3-3ɛ on E2F-dependent apoptosis (Qin et al, 1994). While apoptosis was induced by the expression of E2F-5/DP-3δ, it was less significant in the presence of 14-3-3ɛ (Figure 4C(ii)). DP-3 5S possessed similar apoptotic activity to wild-type DP-3δ, although the level of apoptosis was not reduced upon the expression of 14-3-3ɛ; in fact, there was a reproducible increase in apoptotic activity (Figure 4C(ii)), perhaps reflecting a dominant-negative effect of DP-3 5S on apoptosis. Overall, these results show that 14-3-3ɛ can influence E2F apoptotic activity.
We considered that 14-3-3ɛ may impart biochemical changes on E2F, such as an influence on protein stability. Consequently, cells expressing DP-3δ, DP-3 5S, E2F-5 and 14-3-3ɛ proteins were treated with cycloheximide and thereafter the half-life measured. Both DP-3δ and E2F-5 possessed similar stabilities, each subunit exhibiting a half-life of about 2 h (Figure 5A and C). A marked reduction in the stability of both subunits occurred in the presence of 14-3-3ɛ, approaching a half-life of about 60 min (Figure 5A and C). This effect was dependent upon the interaction with 14-3-3ɛ since the stability of DP-3 5S was significantly greater than wild-type DP-3 in the presence of 14-3-3ɛ, with a half-life of greater than 2.5 h (Figure 5B and C). Further, the co-expression of 14-3-3ɛ had minimal effect on the half-life of DP-3 5S (Figure 5B and C), supporting the idea that 14-3-3ɛ influences the stability of DP-3.
Figure 5.
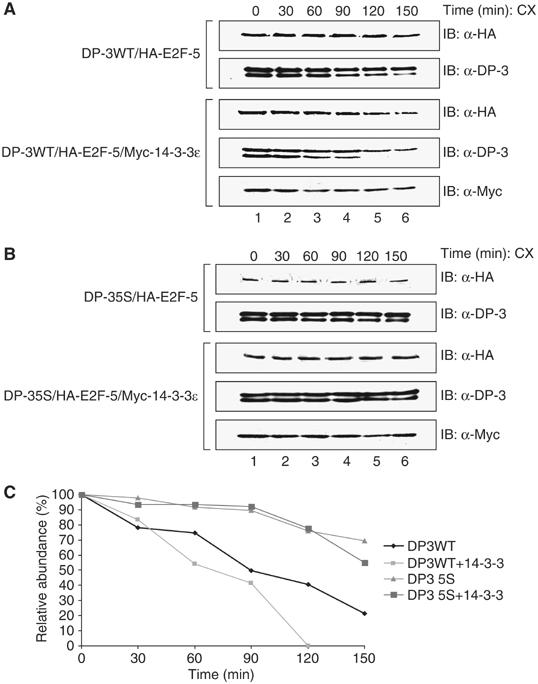
14-3-3ɛ affects the stability of the DP-3δ/E2F-5 heterodimer. (A, B) COS7 cells were transfected with expression vectors (5 μg) for HA-E2F-5, DP-3δ wt, DP-3 5S and (10 μg) myc-14-3-3ɛ as indicated. The cells were treated with cycloheximide (CX) and subsequently harvested at the indicated time points. DP-3δ was detected using the polyclonal antibody 7.2, E2F-5 with anti-HA and 14-3-3ɛ with anti-myc. (C) Quantitation of the stability of DP-3δ wt and DP-3 5S in the presence or absence of 14-3-3ɛ.
As 14-3-3 proteins regulate the activity of target proteins in part by altering the intracellular location (Fu et al, 2000; Muslin and Xing, 2000), we reasoned that intracellular location of one of DP-3δ or 14-3-3ɛ may be affected upon their interaction. Consistent with previous studies, DP-3δ was predominantly nuclear (Figure 6G and H) reflecting the presence of a bi-partite NLS (de la Luna et al, 1996; Allen et al, 1997). In contrast, 14-3-3ɛ was distributed throughout the cell (Figure 5K). In cells expressing both 14-3-3ɛ and DP-3δ, there was a clear shift in the localisation of 14-3-3ɛ, which became mostly nuclear (Figure 6L and M).
To substantiate that this effect was due to the interaction between 14-3-3ɛ and DP-3δ, we studied the effect of DP-3 5S on 14-3-3ɛ. Although DP-3 5S was nuclear (Figure 6I and J), it failed to affect the localisation of 14-3-3ɛ (Figure 6N) and, while the cytoplasmic distribution remained similar to 14-3-3ɛ alone, in a small number of cells an accumulation of 14-3-3ɛ in the perinuclear region was evident (Figure 6O). Thus, the interaction between DP-3δ and 14-3-3ɛ results in an altered nuclear distribution of 14-3-3ɛ.
The 14-3-3ɛ/DP-3 interaction is DNA damage responsive, and 14-3-3ɛ localises to the promoters of E2F target genes
As DP-3 is DNA damage responsive, and because 14-3-3ɛ can regulate DP-3 activity, we assessed whether the physical interaction between 14-3-3ɛ and DP-3δ is regulated by DNA damage. In cells treated with the DNA-damaging agent etoposide, there was a marked reduction in the level of 14-3-3ɛ relative to untreated cells that bound to DP-3δ (Figure 7). Combined with the earlier results on the increased stability of DP-3 5S, the reduced interaction with 14-3-3ɛ could account for the increased levels of DP-3 seen under DNA damage conditions (Figure 1).
Figure 7.
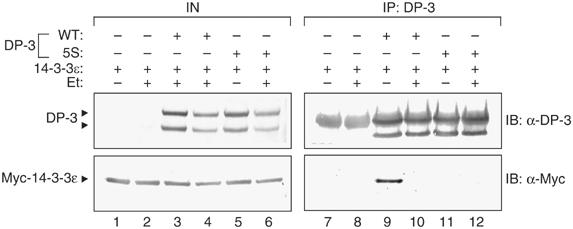
The interaction between 14-3-3ɛ and DP-3δ is regulated by DNA damage. COS7 cells were transfected with expression vectors encoding DP-3δ wt (20 μg), DP-3 5S (20 μg), myc-14-3-3ɛ (40 μg) and β-gal (5 μg) as an internal control. At 48 h post-transfection, cells were treated with 10 μM etoposide (Et) and 12 h later harvested and subjected to immunoprecipitation with anti-DP-3 antibody and immunoblotted with either anti-DP-3 or anti-myc as indicated. The immunoglobulin heavy chain (*) obscures the upper DP-3δ polypeptide in the IB anti-DP-3 treatment.
To investigate whether 14-3-3ɛ could locate to the promoter region of E2F target genes, we performed chromatin immunoprecipitation (ChIP) on chromatin purified from SiHa cells, which express high levels of DP-3 (data not shown). As expected from its role as a subunit of the E2F heterodimer, DP-3 was readily detected on the E2F binding site of different E2F target genes, including APAF-1, p73 and E2F-1 (Figure 8A and B). Interestingly, 14-3-3ɛ was also detected on the E2F binding site in chromatin prepared from unperturbed cells (Figure 8A and B), a result consistent with the presence of an endogenous DP-3/14-3-3ɛ complex (Figure 3G).
Figure 8.
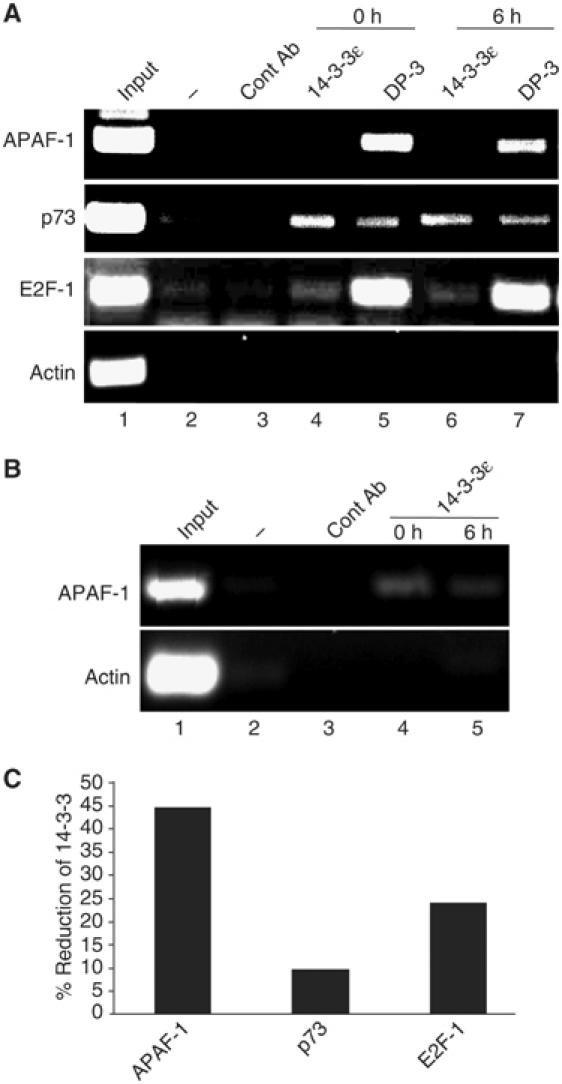
14-3-3ɛ and DP-3 locate to the promoters of E2F target genes. (A) Chromatin immunoprecipilation (ChIP) was performed as described using chromatin prepared from SiHa cells pre- and postbleomycin treatment (5 μg/ml for 6 h) and primers encompassing the E2F sites in APAF-1, p73 and E2F-1 together with the indicated antibodies (see Materials and methods). β-Actin served as a negative control. (B) ChIP was performed as described using chromatin prepared from SiHa cells using the APAF-1 E2F binding site and 14-3-3ɛ antibody. Twice the amount of chromatin was used in (B) to that in (A). (C) Quantitation of 14-3-3ɛ binding by ChIP to the E2F sites in the APAF-1, p73 and E2F-1 promoters. Quantitation of the binding of 14-3-3ɛ to APAF-1 was taken from the results shown in (B).
When ChIP was performed on chromatin purified from DNA-damaged cells (bleomycin treatment for 6 h), while the levels of DP-3 remained similar to untreated cells, there was a significant reduction in the amount of 14-3-3ɛ associated with each gene, which was particularly evident for APAF-1 (Figure 8C). These results indicate that endogenous 14-3-3ɛ co-exists with DP-3 on the promoters of E2F target genes, and that the level of 14-3-3ɛ is regulated during the DNA damage response.
To explore the role of endogenous 14-3-3ɛ in the control of DP-3, we undertook an siRNA approach to reduce the levels of 14-3-3ɛ, and thereafter studied the effect on DP-3 and E2F target genes. We identified an siRNA oligonucleotide, which in cells caused in a specific reduction in 14-3-3ɛ immunostaining (Figure 9A). The reduced levels were specific because a number of unrelated proteins, such as zyxin (Griffith et al, 2005), were not affected even though zyxin could be knocked down with a zyxin-specific siRNA (Figure 9A).
Figure 9.
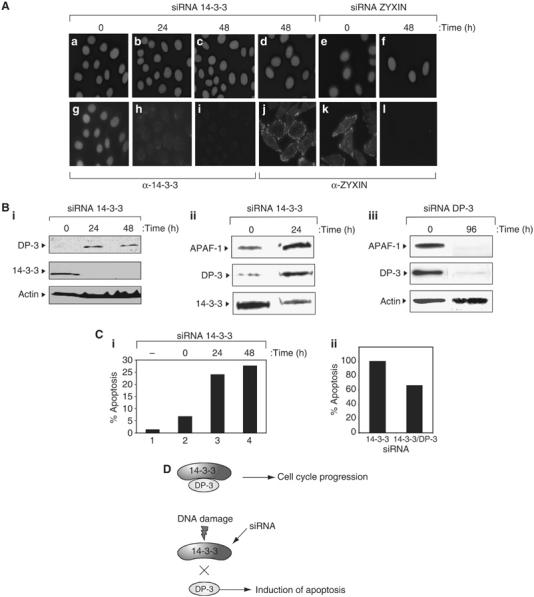
14-3-3ɛ regulates DP-3 in intact cells. (A) SiHa cells were treated with 14-3-3ɛ siRNA or zyxin siRNA (as a positive control) as described for the indicated times and immunostained with anti-14-3-3ɛ or anti-zyxin (bottom), or DAPI (top). Note the reduced intensity of 14-3-3ɛ at 24 and 48 h after treatment with 14-3-3ɛ siRNA. (B) SiHa cells were treated with 14-3-3ɛ (i and ii) or DP-3 (iii) siRNA and immunoblotted with anti-DP-3, anti-14-3-3ɛ, anti-APAF-1 or anti-actin as indicated. Note the increase in DP-3 coincides with a decrease in the level of 14-3-3ɛ (i), and that reduced DP-3 levels causes a concomitant reduction in APAF-1 levels (iii). (C) SiHa cells were treated as described above with 14-3-3 siRNA and assayed by TUNEL at 24 and 48 h post-treatment (i). The level of TUNEL-positive cells, namely apoptotic, is indicated. In (ii), SiHa cells were treated with either 14-3-3 siRNA alone or together with DP-3 siRNA. The graph shows the relative increase in apoptosis (sub-G1 cells) with 14-3-3 siRNA compared to co-treatment with DP-3 siRNA. The level of apoptosis with 14-3-3 siRNA was arbitrarily set to 100%. (D) The model for control of DP-3 was by 14-3-3ɛ. The interaction of 14-3-3ɛ with DP-3 maintains low levels of DP-3 and thereby facilitates cell cycle progression. Reducing the interaction of 14-3-3ɛ with DP-3 either by DNA damage or siRNA increases apoptosis, which correlates with increased levels of DP.
The effect of the 14-3-3ɛ siRNA was recapitulated at the protein level where reduced levels of 14-3-3ɛ were apparent upon treatment with the siRNA (Figure 9B). However, the knockdown of 14-3-3ɛ coincided with an induction of DP-3 and target genes such as APAF-1 (Figure 9B). Under conditions in which DP-3 levels were reduced by introducing a DP-3 siRNA, there was a concomitant reduction in the expression of proteins encoded by E2F target genes, such as APAF-1 (Figure 9B). These observations are consistent with the earlier results on the increased stability seen with DP-3 5S and suggest that 14-3-3ɛ regulates DP-3 in cells, and that the APAF-1 gene is one of the E2F target genes regulated through this mechanism.
As DP-3 5S exhibits increased apoptotic activity (Figure 4C), we reasoned that the knockdown of 14-3-3ɛ and the consequent induction of DP-3 (Figure 9B) may correlate with increased levels of apoptosis. We tested this idea by measuring apoptosis in cells treated with siRNA against 14-3-3ɛ. Under these conditions, there was a clear induction of apoptosis in siRNA treated cells (Figure 9C), indicating that the induction of DP-3 caused by the knockdown of 14-3-3ɛ correlates with higher levels of apoptosis. Moreover, when cells were co-treated with siRNA 14-3-3ɛ and siRNA DP-3, there was a significant reduction in apoptotic activity (Figure 9C). These results suggest that the apoptosis induced by siRNA 14-3-3ɛ requires DP-3 activity.
Discussion
14-3-3 proteins regulate DP-3
The 14-3-3 family represents a group of proteins that function in diverse cellular pathways and affect processes including cell cycle progression and apoptosis (Fu et al, 2000; Muslin and Xing, 2000; Tzivion et al, 2001; van Hemert et al, 2001). The results described here define a new role for 14-3-3 proteins in targeting the DP family member, DP-3, which enables 14-3-3ɛ to influence and alter the functional properties of the E2F pathway. Given the established role for E2F in regulating cell cycle events, these results define a pathway through which 14-3-3 activity is integrated with cell cycle control.
Our evidence suggests that the interaction between 14-3-3ɛ and DP-3 is of functional importance during the DNA damage response. Thus, DP-3 accumulates under DNA damage conditions, the interaction between 14-3-3ɛ and DP-3 is regulated under DNA damage conditions and reducing the levels of 14-3-3ɛ causes increased levels of DP-3. Taken together, the results suggest that the interaction between 14-3-3ɛ and DP-3 is regulated during the DNA damage response. Moreover, the analysis of DP-3 5S, which cannot bind to 14-3-3ɛ and compared to wild-type DP-3 exhibits compromised transcriptional and cell cycle activity in the presence 14-3-3ɛ, suggests that 14-3-3ɛ augments E2F activity under normal conditions. During DNA damage, when DP-3 increases upon 14-3-3ɛ release, DP-3 may be able to influence apoptosis (Figure 4C). At a physiological level, the increased apoptosis is entirely compatible with the effects of the DNA damage response, which often causes the activation of genes and proteins involved in cell death.
DP-3 subunit specificity of 14-3-3ɛ
The DP-3 subunit occurs as at least four distinct protein isoforms (Stevens and La Thangue, 2003). Alternative splicing of DP-3 RNA determines the presence of the E region, which participates as one-half of a bi-partite NLS, the other part being located within the closely positioned basic (B) domain (de la Luna et al, 1996; Allen et al, 1997). Our results suggest that the bi-partite NLS region is a recognition site for 14-3-3ɛ, and further that this interaction controls DP-3 activity. In this respect, it is interesting to note that signals controlling the intracellular location of other target proteins are regulated by 14-3-3 proteins (Muslin and Xing, 2000). For example, the phosphorylation of Cdc25 phosphatase by checkpoint kinase during the DNA damage response leads to its association with 14-3-3 and subsequent cytoplasmic sequestration of (Peng et al, 1997; Blasina et al, 1999; Kumagai and Dunphy, 1999). The cytoplasmic location is believed to prevent Cdc25 reaching a critical target, Cdc2, and thus delays cell cycle progression by retaining Cdc2 in an inactive state. As binding of 14-3-3 occurs close to the Cdc25 NLS, altered intracellular location of Cdc25 may in part result from interference with NLS activity (Kumagai and Dunphy, 1999; Zeng and Piwnica-Worms, 1999). In a similar manner, histone deacetylase (HDAC) 4 and 5, which require a nuclear location to exert their effects on chromatin, upon phosphorylation bind to 14-3-3 and thereafter acquire a cytoplasmic location (Grozinger and Schreiber, 2000). As with Cdc25, 14-3-3 binding may impede the function of the HDAC NLS (Grozinger and Schreiber, 2000).
The interaction between DP-3 and 14-3-3ɛ shows some similarities to the aforementioned examples. Specifically, 14-3-3ɛ recognition overlaps the bi-partite NLS in DP-3, and the interaction regulates DP-3 activity. Moreover, and despite the fact that the recognition site within DP-3 contains five S residues, none of these residues appear to fit either the RSXpSXP or the RXY/FXpSP sequence, which represent two previously identified consensus recognition sequences for 14-3-3 proteins within target proteins (Yaffe et al, 1997). There are, however, examples of 14-3-3 targets that do not fit either of these consensus sequences. The proto-oncogene product Cbl contains the variation RX1–2SX2–3S (Liu et al, 1997), while keratin 18 contains a novel 14-3-3 recognition site (Ku et al, 1998). The forkhead transcription factor FKHRL1 also contains a 14-3-3 recognition site, but this involves phosphorylation of a threonine residue at position 32 (Brunet et al, 1999). Given these examples, it is clear that the recognition motif for 14-3-3 targets may be quite degenerate.
A role for DP-3 in checkpoint control
At a general level, the DNA damage response regulation of DP-3 may reflect a role in checkpoint control. This idea rests upon the observations described here, namely the regulation of the interaction between 14-3-3ɛ and DP-3, and the correlative induction of DP-3 upon DNA damage. Under normal conditions, 14-3-3ɛ appears to maintain DP-3 in a form that contributes to cell cycle progression and transcription. In contrast, under DNA damage the loss of 14-3-3ɛ binding may influence apoptosis (Figure 9). In turn, these results strengthen the evidence that the variety of E2F and DP subunits that constitute E2F activity endow E2F with distinct functional and biochemical properties. For the DP-3 subunit, our results suggest a role in the DNA damage response.
Materials and methods
Plasmids and expression vectors
The following plasmids have been described previously: pCMV-HA-E2F5 (Allen et al, 1997); pG-DP-3δ (de la Luna et al, 1996); pCMV-14-3-3ɛ-myc (Chaudhri et al, 2003); pG4-DBD, pG4-DBD-DP-3δN and pG4-DBD-DP1N (de la Luna et al, 1999); and pCyclinE-luc (Botz et al, 1996). The vector pG-DP-3 5S was constructed using the QuikChange Single Site-Directed Mutagenesis Kit (Stratagene). The pG-DP-3 5S mutant was obtained by successive rounds of individual mutations using pG-DP-3δ as the original template and the new mutant as the template thereafter. The vector pCMV-VP16-TAD-14-3-3ɛ-myc was constructed by subcloning full-length 14-3-3ɛ-myc into the vector pCMV-VP16-TAD. Both pGEX-DP-3δ and pGEX-DP-3 5S encoding bacterially expressed GST-DP-3δ and GST-DP-3 5S were constructed by subcloning full-length pG-DP-3δ and pG-DP-3 5S into the pGEX-KG vector.
Cell culture and transfection
COS7 cells were maintained in DMEM, supplemented with 10% (v/v) foetal calf serum (FCS). Cells were transfected using the calcium phosphate-DNA precipitate method and DNA levels were normalised by the addition of pcDNA3. Protein extracts were prepared 65 h after transfection by scraping the cells and re-suspending the cell pellet in 100 μl of micro-extraction (ME) buffer (20 mM HEPES, 100 mM NaCl, 25% glycerol, 50 mM NaF, 0.2 mM EDTA, 0.5 mM PMSF, 60 mM β-glycero-phosphate, 1 mM Na3VO4, 0.1% NP-40 and protease inhibitor cocktail) at 4°C for 30 min and then centrifuged at 13 000 r.p.m. for 20 min at 4°C. Supernatants were assayed for total protein using the Bradford method (Biorad).
Immunoblotting and immunoprecipitation
Immunoblotting was performed according to standard procedures using the polyclonal anti-DP3 antibody, 7.2, the monoclonal anti-Myc antibody, 9E10 (Santa Cruz), the monoclonal anti-HA antibody, HA11 (Babco), the monoclonal anti-DP-2 antibody, G12 (Santa Cruz), and the polyclonal anti-14-3-3ɛ antibody, T-16 (Santa Cruz). For immunoprecipitation, cells were lysed in ME buffer and incubated for 24 h at 4°C with the appropriate antibody bound to protein A–Sepharose beads. Proteins were released and resolved by SDS–PAGE, followed by immunoblotting.
In vitro binding
GST or GST-tagged protein (5 μg) bound to beads were washed in ME buffer. Either 1 mg total protein from extract or in vitro-translated protein was incubated with the beads for 1 h at 4°C. The beads were then washed in ME buffer and analysed by SDS–PAGE and immunoblotting.
Immunostaining
COS7 cells were seeded onto glass coverslips and transfected with the indicated plasmids. At 65 h post-transfection, the cells were washed twice with PBS and fixed with 4% paraformaldehyde, permeabilised with 0.1% Triton X-100 and blocked with PBS containing 10% FCS. The anti-DP3 and anti-Myc antibodies were incubated for 30 min at a dilution of 1:1000 in 1% FCS/PBS. Secondary anti-mouse Alexa 594 and anti-rabbit Alexa 488 antibodies were from Molecular Probes. Cells were counterstained with 4,6-diamidino-2-phenylindole (DAPI).
Flow cytometry
COS7 cells were transfected with the indicated plasmids together with 5 μg pCMV-CD20. At 65 h post-transfection, the cells were harvested in cell dissociation buffer (Sigma) and incubated with FITC-conjugated CD20 antibody (Becton-Dickinson) to identify the transfected cell population. The cells were then washed in PBS and fixed overnight at 4°C. Cells were sorted on a FACScan cell sorter (Becton-Dickinson) and analysed using the Cell Quest software package.
Luciferase assays
The pCyclinE-luc construct has been described previously (Botz et al, 1996). For reporter assays, COS7 cells were transfected with 0.5 μg of luciferase reporter plasmid, 1 μg of pCMV-β-galactosidase (β-gal) plasmid as an internal control and the indicated expression plasmids. Transfected cells were harvested by gentle scraping into 300 μl of luciferase assay lysis buffer (Promega). Lysates were transferred and centrifuged at 13 000 r.p.m. for 15 min at 4°C. The supernatants were used for luciferase and β-gal activity assays. All were performed in duplicate and were normalised to β-gal expression.
Cell proliferation (Brdu) assay
COS7 cells transfected with the indicated plasmids. At 65 h post-transfection, 10 μM Brdu (Roche) was added to the cells in culture and left for 1 h. The cells were washed twice in PBS and fixed for 45 min in ethanol (70%) in 50 mM glycine buffer, pH 2. The coverslips were washed twice in PBS and one coverslip was removed and used in an immunofluorescence assay to ensure efficient expression of the exogenous protein. The remaining coverslips were simultaneously incubated with 5 U/ml DNAse and the appropriate dilution of anti-Brdu-FLUOS antibody for 1 h at 37°C.
TUNEL assay
COS7 cells were transfected with the indicated plasmids. At 65 h post-transfection, the cells were fixed onto coverslips by the addition of 4% (w/v) paraformaldehyde for 1 h. One coverslip was removed and used in an immunofluorescence assay to ensure expression of the exogenous proteins. The remaining coverslips were washed twice in PBS and permeabilised on ice for 2 min by the addition of 1% (v/v) Triton X-100 in PBS. The coverslips were washed again in PBS and the TUNEL (TdT-mediated dUTP-X nick end labelling) reaction mixture (50% terminal transferase/50% fluorescein-dUTP) was added for 1 h at 37°C. The samples were washed twice in PBS and rinsed in DAPI stain to enable detection of the cells.
Cycloheximide treatment
COS7 cells were transfected with the indicated plasmids. After 48 h, 0.01 μg/μl cycloheximide (Promega) was added and harvested thereafter at the indicated time point. The cell extracts were lysed in ME buffer and the supernatant was removed after which the samples were subjected to SDS–PAGE and finally Western blotting.
Yeast two-hybrid assay
Saccharomyces cerevisiae CTY10.5d (MATa ade2 trp1-901 leu2-3,112 his3-200 gal80 URA3∷lexA op-lacZ) with integrated plasmid pSH18–34, which carries four binding site for LexA upstream of the transcription start site of a lacZ gene, was used. A 10.5 d.p.c. CD-1 mouse embryo library generated by random-primed cDNA synthesis and size selected to have insert sizes in the range 350–700 nucleotides cloned into vector pVP16, which caries the LEU2 gene, was screened against the DP-3δ bait carrying residues 1–110 cloned into vector pLexA that contains the HIS3 gene creating the DP-3 LexA-bait fusion protein.
Yeast were transformed simultaneously with bait and library plasmid using the lithium acetate method as described (de la Luna et al, 1999). Colonies were assayed for β-gal activity by filter assay. Blue colonies were isolated and plasmids conferring a Trp+ phenotype that gave a blue colony colour only in the presence of bait were selected and DNA sequenced.
Gel retardation assay
Gel retardation was carried out as described (Bandara et al, 1994) in 30 μl reactions containing 500 ng salmon sperm DNA, 500ng BSA, reaction buffer (40 mM HEPES, pH 7.9, 400 mM KCl, 4 mM EDTA, 16% Ficoll, 1 mM DTT, protease inhibitors) and in vitro-translated proteins. Where complexes were identified by antibody shifts, 2 μl of HA11 antibody (Babco) was added for 20 min, then radiolabelled probe was added and incubated for a further 20 min followed by electrophoresis on a 4% polyacrylamide gel run at 4°C. The E2F binding site probe contained the distal E2F site from the E2A promoter (Bandara et al, 1994). Proteins were in vitro-translated using the TNT-coupled reticulocyte lysate kit (Promega).
siRNA expression
The following siRNA oligonucleotides were used to interrupt the expression of endogenous 14-3-3ɛ and DP-3 proteins:
14-3-3ɛ
Oligonucleotide 1:
Forward sequence: 5′-GGGAGGAGAAGACAAGCUATT-3′
Reverse sequence: 5′-UAGCUUGUCUUCUCCUCCCTT-3′.
Oligonucleotide 2:
Forward sequence: 5′-GGUUAAUCACACUAUAGAGTT-3′
Reverse sequence: 5′-CUCUAUAGUGUGAUUAACCTC-3′.
Oligonucleotide 3:
Forward sequence: 5′-GGAGAAGACAAGCUAAAAATT-3′
Reverse sequence: 5′-UUUUUAGCUUGUCUUCUCCTC-3′.
DP-3
Oligonucleotide 1:
Forward sequence: 5′-GGCUUUAGAAGGUUAUAUCTT-3′
Reverse sequence: 5′-GAUAUAACCUUCUAAAGCCTT-3′.
Oligonucleotide 2:
Forward sequence: 5′-GGAGAUAAAAAUGGGAAAGTT-3′
Reverse sequence: 5′-CUUUCCCAUUUUUAUCUCCTT-3′.
Oligonucleotide 3:
Forward sequence: 5′-GGCUUGAGACACUUUUCAATT
Reverse sequence: 5′-UUGAAAAGUGUCUCAAGCCTT-3′.
Oligonucleotides were purchased from Ambion and were found to be functional in reducing endogenous 14-3-3ɛ and DP-3 protein expression in SiHa cervical carcinoma cells. The siRNA oligonucleotides against zyxin have been described previously (Griffith et al, 2005).
Chromatin immunoprecipitation
For ChIP, SiHa cells were treated with 1 μg/ml bleomycin (Sigma) for 6 h or left untreated (0 h). After crosslinking with 1% formaldehyde (Sigma) for 10 min, cells were washed twice with cold PBS containing 125 mM glycine and harvested. The nuclear pellet was resuspended in RIPA buffer (10 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1% NP-40, 1% deoxycholic acid 0.1% SDS, 1 mM EDTA, 1 mM PMSF and protease inhibitor cocktail (Roche)) and sonicated to produce soluble chromatin with average size between 300 and 1000 bp. The nuclear lysate was precleared by incubating with salmon sperm DNA/BSA-blocked Sepharose A/G beads for 4 h at 4°C. The lysate was used for immunoprecipitation with anti-Gal4-DNA-BD (control Ab), anti-14-3-3ɛ (T-16), anti-DP-2 (C-20) (all from SantaCruz Biotechnology) and without antibody (no antibody control) overnight at 4°C. The immune complex was collected by incubating with salmon sperm DNA/BSA-blocked Sepharose A/G beads for 4 h at 4°C. The beads were washed twice with buffer I (20 mM Tris–HCl, pH 8.1, 150 mM NaCl, 0.1% SDS, 2 mM EDTA, 1% Triton X-100), three times with buffer II (20 mM Tris–HCl, pH 8.1, 500 mM NaCl, 1% Triton X-100, 0.1% SDS, 2 mM EDTA), four times with buffer III (0.25 M LiCl, 1% NP-40, 1% doxycholic acid, 1 mM EDTA, 10 mM Tris–HCl, pH 8.1) and TE buffer. For de-crosslinking, the eluted complex was treated with 40 mM Tris–HCl, pH 6.5, 200 mM NaCl, 10 mM EDTA, 2 μl of 10 mg/ml proteanase K, 1 μl of 10 mg/ml RNase A and incubated at 55°C for 3 h and 65°C overnight. The de-crosslinked chromatin DNA was further purified by Qiaquick PCR purification kit (Qiagen) and eluted in 50 μl elution buffer supplied with the kit. A measure of 1 μl eluted DNA sample was used for each PCR reaction.
The primers used for amplification were as follows:
E2F-1:
Forward: 5′-AGGAACCGCCGCCGTTGTTCCCGT-3′,
Reverse: 5′-GCTGCCTGCAAAGTCCCGGCCACT-3′,
p73:
Forward: 5′-TGAGCCATGAAGATGTGCGAG-3′,
Reverse: 5′-GCTGCTTATGGTCTGATGCTTATG-3′
APAF-1:
Forward: 5′-CCGACTTCTTCCGGCTCTTCA-3′
Reverse: 5′-GAGCTGGCAGCTGAAAGACTC-3′
β-Actin:
Forward: 5′-CAAGGCGGCCAACGCCAAAACTCT-3′,
Reverse: 5′-GCCATAAAAGGCAACTTTCGGAACG-3′.
Acknowledgments
We thank Rosemary Williams for help in preparing the manuscript. We are grateful to AICR, LRF, MRC, EU and CRUK for supporting our research.
References
- Allen KE, de la Luna S, Kerkhoven RM, Bernards R, La Thangue NB (1997) Distinct mechanisms of nuclear accumulation regulate the functional consequence of E2F transcription factors. J Cell Sci 110: 2819–2831 [DOI] [PubMed] [Google Scholar]
- Bandara LR, Lam EWF, Sorensen TS, Zamanian M, Girling R, La Thangue NB (1994) DP-1-a cell-cycle-regulated and phosphorylated component of transcription factor DRTF1/E2F which is functionally important for recognition by pRB and the adenovirus E4-Orf-6/7 protein. EMBO J 13: 3104–3114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasina A, de Weyer IV, Laus MC, Luyten WH, Parker AE, McGowan CH (1999) A human homologue of the checkpoint kinase Cds1 directly inhibits Cdc25 phosphatase. Curr Biol 9: 1–10 [DOI] [PubMed] [Google Scholar]
- Blattner C, Sparks A, Lane D (1999) Transcription factor E2F1 is upregulated in response to DNA damage in a manner analogous to that of p53. Mol Cell Biol 19: 3704–3713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botz J, Zerfass-Thome K, Spitkovsky D, Delius H, Vogt B, Eilers M, Hatzigeorgiou A, Jansen-Durr P (1996) Cell cycle regulation of the murine cyclin E gene depends on an E2F binding site in the promoter. Mol Cell Biol 16: 3401–3409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96: 857–868 [DOI] [PubMed] [Google Scholar]
- Chaudhri M, Scarabel M, Aitken A (2003) Mammalian and yeast 14-3-3 isoforms form distinct patterns of dimers in vivo. Biochem Biophys Res Commun 300: 679–685 [DOI] [PubMed] [Google Scholar]
- Chehab I, Malikzay A, Appel M, Halazonetis TD (2000) Chk2/hCds1 functions as a DNA damage checkpoint in G(1) by stabilising p53. Genes Dev 14: 278–288 [PMC free article] [PubMed] [Google Scholar]
- de la Luna S, Allen KE, Mason SL, La Thangue NB (1999) Integration of a growth-suppressing BTB/POZ domain protein with the DP component of the E2F transcription factor. EMBO J 18: 212–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Luna S, Burden MJ, Lee CW, La Thangue NB (1996) Nuclear accumulation of the E2F heterodimer regulated by subunit composition and alternative splicing of a nuclear localization signal. J Cell Sci 109: 2443–2452 [DOI] [PubMed] [Google Scholar]
- Demonacos C, Kristic-Demonacos M, Smith L, Xu D, O'Connor DP, Jansson M, La Thangue NB (2004) A new effector pathway links ATM kinase with the DNA damage response. Nat Cell Biol 6: 968–976 [DOI] [PubMed] [Google Scholar]
- Field SJ, Tsai FY, Kuo F, Zubiaga AM, Kaelin WG Jr, Livingston DM, Orkin SH, Greenberg ME (1996) E2F1 functions in mice to promote apoptosis and suppress proliferation. Cell 85: 549–561 [DOI] [PubMed] [Google Scholar]
- Fu H, Subramanian RR, Masters SC (2000) 14-3-3 proteins: structure, function, and regulation. Annu Rev Pharmacol Toxicol 40: 617–647 [DOI] [PubMed] [Google Scholar]
- Girling R, Partridge JF, Bandara LR, Burden N, Totty NF, Hsuan JJ, La Thangue NB (1993) A new component of the transcription factor DRTF1/E2F. Nature 362: 83–87 [DOI] [PubMed] [Google Scholar]
- Griffith E, Coutts AS, Black DM (2005) RNAi knockdown of the focal adhesion protein TES reveals its role in actin stress fibre organisation. Cell Motil Cytoskeleton 60: 140–152 [DOI] [PubMed] [Google Scholar]
- Grozinger CM, Schreiber SL (2000) Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14-3-3 dependent cellular localisation. Proc Natl Acad Sci USA 97: 7835–7840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermeking H, Lengauer C, Polyak K, He TC, Zhang L, Thiagalingam S, Kinzler KW, Vogelstein B (1997) 14-3-3 sigma is a p53-regulated inhibitor of G2/M progression. Mol Cell 1: 3–11 [DOI] [PubMed] [Google Scholar]
- Hofferer M, Wirbelauer C, Humar B, Krek W (1999) Increased levels of E2F1-dependent DNA binding activity after UV- or γ-irradiation. Nuc Acid Res 27: 491–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DG, Schwarz JK, Cress WD, Nevins JR (1993) Expression of transcription factor E2F1 induces quiescent cells to enter S phase. Nature 365: 349–352 [DOI] [PubMed] [Google Scholar]
- Kohn MJ, Bronson RT, Harlow E, Dyson NJ, Yamasaki L (2003) Dp1 is required for extra-embryonic development. Development 130: 1295–1305 [DOI] [PubMed] [Google Scholar]
- Ku NO, Liao J, Omary MB (1998) Phosphorylation of human keratin 18 serine 33 regulates binding to 14-3-3 proteins. EMBO J 17: 1892–1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai A, Dunphy WG (1999) Binding of 14-3-3 proteins and nuclear export control the intracellular localisation of the mitotic inducer Cdc25C. Genes Dev 13: 1067–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Thangue NB (2003) The yin and yang of E2F-1: balancing life and death. Nat Cell Biol 5: 587–589 [DOI] [PubMed] [Google Scholar]
- Liu YC, Liu Y, Elly C, Yoshida H, Lipkowitz S, Altman A (1997) Serine phosphorylation of Cbl induced by phorbol ester enhances its association with 14-3-3 proteins in T cells via a novel serine-rich 14-3-3 binding motif. J Biol Chem 272: 9979–9985 [DOI] [PubMed] [Google Scholar]
- Muslin AJ, Tanner JW, Allen PM, Shaw AS (1996) Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell 84: 889–897 [DOI] [PubMed] [Google Scholar]
- Muslin AJ, Xing H (2000) 14-3-3 proteins: regulation of subcellular localization by molecular interference. Cell Signal 12: 703–709 [DOI] [PubMed] [Google Scholar]
- O'Connor DJ, Lu X (2000) Stress signals induce transcriptionally inactive E2F1 independently of p53 and Rb. Oncogene 19: 2369–2376 [DOI] [PubMed] [Google Scholar]
- Ormondroyd E, de la Luna S, La Thangue NB (1995) A new member of the DP family, DP-3, with distinct protein products suggests a regulatory role for alternative splicing in the cell cycle transcription factor DRTF1/E2F. Oncogene 11: 1437–1446 [PubMed] [Google Scholar]
- Peng CY, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica-Worms H (1997) Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science 277: 1501–1505 [DOI] [PubMed] [Google Scholar]
- Qin XQ, Livingston DM, Kaelin WG Jr, Adams PD (1994) Deregulated transcription factor E2F-1 expression leads to S-phase entry and p53-mediated apoptosis. Proc Natl Acad Sci USA 91: 10918–10922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittinger K, Budman J, Xu J, Volinia S, Cantley LC, Smerdon SJ, Gamblin SJ, Yaffe MB (1999) Structural analysis of 14-3-3 phosphopeptide complexes identifies a dual role for the nuclear export signal of 14-3-3 in ligand binding. Mol Cell 4: 153–166 [DOI] [PubMed] [Google Scholar]
- Rogers KT, Higgins PDR, Milla MM, Phillips RS Horowitz JM (1996) DP-2, a heterodimeric partner of E2F: Identification and characterisation of DP-2 proteins expressed in vivo. Proc Natl Acad Sci USA 93: 7594–7599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roseboom PH, Weller JL, Babila T, Aitken A, Sellers LA, Moffett JR, Namboodiri MA, Klein DC (1994) Cloning and characterisation of the epsilon and zeta isoforms of the 14-3-3 proteins. DNA Cell Biol 13: 629–640 [DOI] [PubMed] [Google Scholar]
- Stevens C, La Thangue NB (2003) E2F and cell cycle control: a double-edged sword. Arch Biochem Biophys 412: 157–169 [DOI] [PubMed] [Google Scholar]
- Stevens C, Smith L, La Thangue NB (2003) Chk2 activates E2F1 in response to DNA damage. Nat Cell Biol 5: 401–409 [DOI] [PubMed] [Google Scholar]
- Trimarchi JM, Lees JA (2002) Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol 3: 11–20 [DOI] [PubMed] [Google Scholar]
- Tsai KY, Hu Y, Macleod KF, Crowley D, Yamasaki L, Jacks T (1998) Mutation of E2F1 suppresses apoptosis and inappropriate S phase entry and extends survival of RB-deficient mouse embryos. Mol Cell 2: 293–304 [DOI] [PubMed] [Google Scholar]
- Tzivion G, Shen YH, Zhu J (2001) 14-3-3 proteins; bringing new definitions to scaffolding. Oncogene 20: 6331–6338 [DOI] [PubMed] [Google Scholar]
- van Hemert MJ, Steensma HY, van Heusden GP (2001) 14-3-3 proteins: key regulators of cell division, signalling and apoptosis. Bioessays 23: 936–946 [DOI] [PubMed] [Google Scholar]
- Waterman MJ, Stavridi ES, Waterman JL, Halazonetis TD (1998) ATM-dependent activation of p53 involves dephosphorylation and association with 14-3-3 proteins. Nat Genet 19: 175–178 [DOI] [PubMed] [Google Scholar]
- Xing H, Zhang S, Weinheimer C, Kovacs A, Muslin AJ (2000) 14-3-3 proteins block apoptosis and differentially regulate MAPK cascades. EMBO J 19: 349–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe MB, Rittinger K, Volinia S, Caron PR, Aitken A, Leffers H, Gamblin SJ, Smerdon SJ, Cantley LC (1997) The structural basis for 14-3-3: phosphopeptide binding specificity. Cell 91: 961–971 [DOI] [PubMed] [Google Scholar]
- Yamasaki L, Bronson R, Williams BO, Dyson N, Harlow E, Jacks T (1998) Loss of E2F1 reduces tumorigenesis and extends the lifespan of RB+/− mice. Nat Genet 18: 360–364 [DOI] [PubMed] [Google Scholar]
- Yamasaki L, Jacks T, Bronson R, Goillot E, Harlow E, Dyson NJ (1996) Tumor induction and tissue atrophy in mice lacking E2F-1. Cell 85: 537–548 [DOI] [PubMed] [Google Scholar]
- Zeng Y, Piwnica-Worms H (1999) DNA damage and replication checkpoints in fission yeast require nuclear exclusion of the Cdc25 phosphatase via 14-3-3 binding. Mol Cell Biol 19: 7410–7419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chellappan SP (1995) Cloning and characterisation of human DP-2, a novel dimerisation partner of E2F. Oncogene 10: 2085–2093 [PubMed] [Google Scholar]