

Abstract
The transcription factor Egr-1 functions as a key regulator in cellular growth, differentiation, and apoptosis. The loss of Egr-1 expression is closely associated with tumor development, although the molecular mechanism behind the suppression of Egr-1 is largely unknown. In this report, we show that growth factor-induced transcriptional activation of Egr-1 gene is downregulated by chronic expression of oncogenic H-Ras in NIH3T3 fibroblasts. Our results demonstrate that phosphoinositide 3-kinase (PI3K) signaling is necessary for oncogenic H-Ras-mediated reduction of Egr-1 gene expression. Aberrant activation of PI3K signaling by oncogenic Ras decreased the level of serum response factor (SRF) protein through the acceleration of proteolysis, which resulted in decreased SRF binding to the serum response element (SRE) sites within the Egr-1 promoter, leading to the suppression of Egr-1 transcription. Inhibition of PI3K signaling restored the downregulation of SRF and Egr-1 expression caused by oncogenic Ras. Our findings suggest a novel signaling mechanism by which prolonged activation of oncogenic H-Ras can trigger the loss of tumor suppressor Egr-1 through the PI3K pathway in NIH3T3 fibroblast model cell lines.
Keywords: Egr-1, oncogenic H-Ras, phosphoinositide 3-kinase, serum response element, serum response factor
Introduction
Transcription factor Egr-1, also known as NGFI-A, zif268, krox24, and Tis8, is an immediate-early response gene that is induced by stress, injury, mitogens, and differentiation factors (Sukhatme et al, 1988; Edwards et al, 1991; Liu et al, 1998). Egr-1 regulates the expression of genes that are involved in growth control and apoptosis by transactivation of p21, p53, PTEN, TGFβ1, fibronectin, and Gadd45 (Liu et al, 1998; Virolle et al, 2001; Krones-Herzig et al, 2003; Ragione et al, 2003; Baron et al, 2005; Thyss et al, 2005). Egr-1 is poorly expressed or not expressed at all in tumor cells, and the lack of Egr-1 expression is closely associated with tumor formation (Huang et al, 1997). The ectopic expression of the Egr-1 gene in tumor cells results in the attenuation of cell proliferation and tumorigenicity with increased cell attachment (de Belle et al, 1999; Liu et al, 1999, 2000). These results support the notion that functional loss of the Egr-1 gene contributes to tumorigenic potential.
The Ras proteins (H-Ras, K-Ras, and N-Ras) are small, GTP-binding proteins that initiate the activation of signaling networks that are involved in the regulation of cell growth and differentiation (Macara et al, 1996). Point mutations in the ras gene occur at a high frequency in approximately 30% of all human cancers (Bos, 1989). These mutant forms of Ras are constitutively activated in the absence of extracellular stimuli and play a central role in oncogenesis. The Egr-1 promoter contains the serum response element (SRE) cluster, which is implicated in the transcriptional activation of Egr-1 in response to various growth factors (Christy and Nathans, 1989; Clarkson et al, 1999; Tsai et al, 2000). Egr-1 SREs include both the CArG box (CC[A/T]6GG motif), which binds the serum response factor (SRF), and the Ets motif (GGA[A/T]), which binds a ternary complex factor (TCF) family member (Treisman, 1994). TCFs, which include Elk-1, Sap-1, and Sap-2/Net/Erp, can be phosphorylated by Erk MAPK (Price et al, 1995), and Elk-1 phosphorylation by the Ras–Raf–Erk MAPK cascade correlates with increased transcriptional activation of the Egr-1 gene (Hipskind et al, 1994; Watson et al, 1997; Hodge et al, 1998; Guha et al, 2001; Schratt et al, 2001). Previously, we, as well as others, had demonstrated that Egr-1 expression is downregulated in v-Ras-transformed NIH3T3 cells (Yu et al, 1993) and in HT1080 fibrosarcoma cells (Liu et al, 1999; Shin et al, 2001). Given that HT1080 cells contain an endogenous mutant allele of N-ras, which is critical for the transformation of these cells to the malignant status (Paterson et al, 1987), the suppression of Egr-1 expression in Ras-transformed cells seems paradoxical. In the present study, our aim was to understand the molecular mechanism underlying the suppression of Egr-1 expression by oncogenic Ras. We show that growth factor-induced transcriptional activation of Egr-1 gene is reduced by chronic expression of oncogenic H-Ras in NIH3T3 fibroblasts. The present report represents the first evidence that chronic expression of oncogenic H-Ras decreases the level of SRF protein through PI3K signaling, which results in the suppression of Egr-1 transcription. This suppression of Egr-1 expression in turn could reduce the induction of Egr-1 target genes, such as PTEN. Since Egr-1 and PTEN contribute significantly to human tumor development (Liu et al, 1998; Cantley and Neel, 1999), our findings have important implications for understanding the mechanisms involved in tumor progression caused by oncogenic Ras.
Results
Downregulation of PDGF-induced Egr-1 expression in H-RasV12-expressing NIH3T3 fibroblasts
To investigate whether Egr-1 is downregulated directly by Ras mutation or by secondary effects in the transformed state, we established a NIH3T3 cell line that expresses oncogenic H-RasV12 (NIH3T3/RasV12) (Figure 1A). Upon exposure to platelet-derived growth factor (PDGF) following serum deprivation, the levels of phosphorylated downstream effectors of Ras, such as Akt (Ser 473) and Erk1/2 (Thr 202/Tyr 204), were much increased in NIH3T3/RasV12 cells as compared to those of the empty vector-transfected control cells (NIH3T3/vec), which demonstrates that the overexpressed oncogenic H-RasV12 protein is functional. In NIH3T3/RasV12 cells, the PDGF-induced Egr-1 protein was barely detectable, compared with the NIH3T3/vec cells (Figure 1A). Northern blot analysis also demonstrated marked downregulation of Egr-1 mRNA expression in NIH3T3/RasV12 cells (Figure 1B). These results suggest that the overexpression of oncogenic H-RasV12 is sufficient to cause a decrease in the level of Egr-1 in response to PDGF stimulation.
Figure 1.
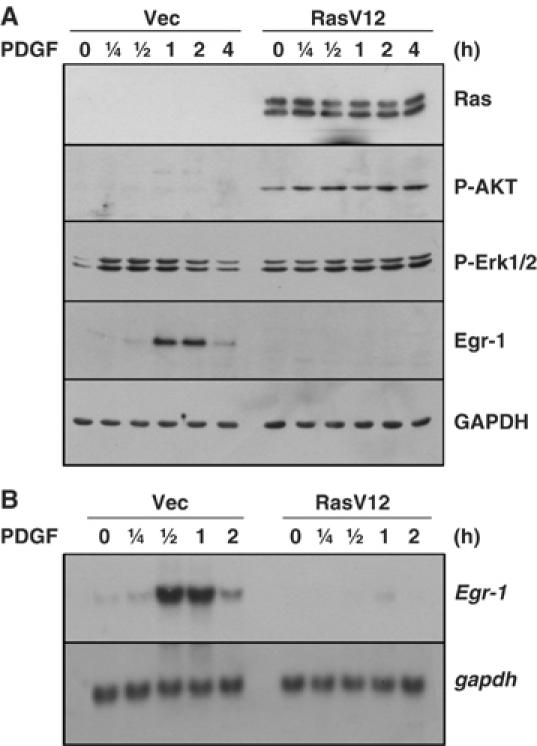
Suppression of Egr-1 expression in oncogenic H-RasV12-expressing NIH3T3 fibroblasts. (A) Serum-starved NIH3T3/vec and NIH3T3/RasV12 cells were treated with PDGF (50 ng/ml) for the indicated periods of time, and Western blot analysis was performed with antibodies against H-Ras, phospho-Erk1/2 (Thr 202/Tyr 204), phosphor-Akt (Ser 473), Egr-1, and GAPDH. (B) Serum-starved NIH3T3/vec and NIH3T3/RasV12 cells were treated with PDGF (50 ng/ml) for the indicated periods of time, and total RNA samples were isolated. Northern blot analyses were performed with the Egr-1 cDNA probe. The GAPDH probe was used as an internal control.
Suppression of Egr-1 expression by conditional expression of H-RasG12R in NIH3T3 fibroblasts
To determine whether the downregulation of Egr-1 was a direct consequence of oncogenic Ras, we used a NIH3T3 cell line that harbors a tetracycline-inducible expression vector that encodes a constitutively active mutant of H-Ras (NIH3T3tet-on/H-RasG12R). The H-RasG12R protein was detectable 24 h after the addition of doxycycline (2 μg/ml) and accumulated through 48 h (Figure 2A). The phosphorylation of Ras downstream effectors, which include MEK1 (Ser 217/221), Erk1/2 (Thr 202/Tyr 204), and Akt/PKB (Ser 473), was increased within 6 h of doxycycline treatment, which suggests that although the accumulation of H-RasG12R protein was not detected at earlier time points, its induction was sufficient to activate downstream effectors. Increased expression of cyclin D1 was also observed (Figure 8A). Furthermore, in agreement with the results on the induction of H-RasG12R, the cells exhibited morphological transformation between 2 and 3 days after doxycycline treatment (data not shown). Thus, the doxycycline-induced H-RasG12R protein is biologically functional.
Figure 2.
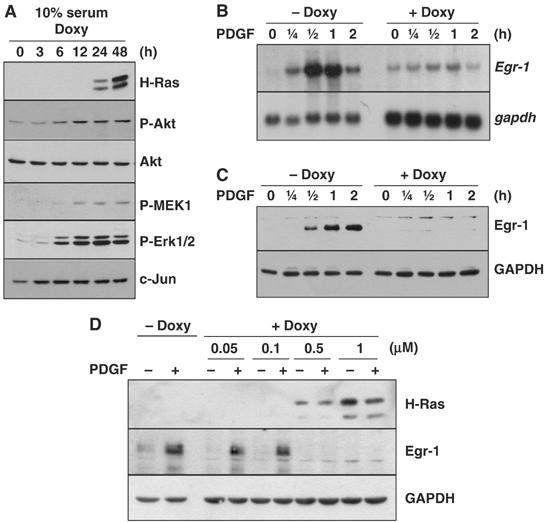
Suppression of Egr-1 expression by inducible expression of H-RasG12R in NIH3T3 cells. (A) Log-phase cultured cells were harvested at different time points after the addition of doxycycline (2 μg/ml). Protein lysates were prepared and subjected to Western blot analysis with antibodies against H-Ras, phosphor-Akt (Ser 473), phosphor-MEK1/2 (Ser 217/221), phospho-Erk1/2 (Thr 202/Tyr 204), and c-Jun. (B, C) NIH3T3tet-on/H-RasG12R cells were cultured in the absence (−Doxy) or presence (+Doxy) of doxycycline (B and C: 2 μg/ml; D: 0.05–1 μg/ml) for 24 h, serum-starved for an additional 24 h in the absence or presence of doxycycline, and then treated with PDGF (50 ng/ml) for various periods of time (B and C) or for 1 h (D). Egr-1 expression was analyzed in Northern (B) and Western (C and D) blots. GAPDH was used as an internal control.
Figure 8.
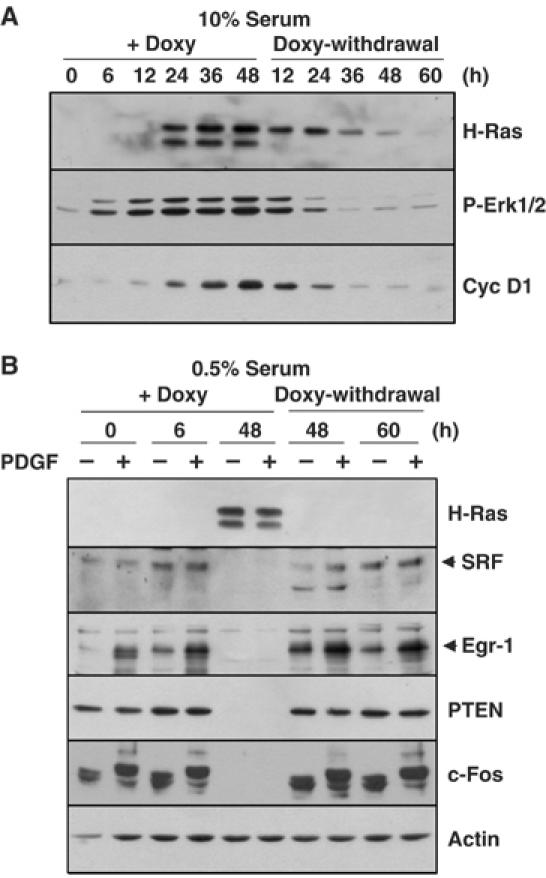
The suppression of SRF and Egr-1 expression by oncogenic H-Ras is reversible. (A) NIH3T3tet-on/H-RasG12R cells were cultured in the presence of doxycycline for 48 h, after which the doxycycline was removed by washing. At the indicated time points, total cell lysates were prepared and Western blot analysis was performed with antibodies directed against H-Ras, phospho-Erk1/2 (Thr 202/Tyr 204), and cyclin D1. (B) NIH3T3tet-on/H-RasG12R cells were serum starved for 24 h in medium that contained 0.5% FBS, and then stimulated with PDGF for 1 h. Doxycycline was added before, 6 h after, or 48 h after PDGF stimulation (+Doxy). Doxycycline was removed by washing out the medium after culturing the cells with doxycycline for 48 h. The cells were cultured in the absence of doxycycline for 48 or 60 h, and then stimulated with PDGF for 1 h (Doxy-withdrawal). Serum starvation was performed before 24 h of PDGF stimulation. Total protein extracts were prepared and used to detect the expression of H-Ras, SRF, Egr-1, PTEN, and c-Fos by Western blot analysis. GAPDH was used as a loading control.
To determine whether growth factor-induced Egr-1 expression is affected by prolonged expression of oncogenic H-Ras, NIH3T3tet-on/H-RasG12R cells were cultured for 48 h with 0.5% serum in the absence or presence of doxycycline. In the absence of H-RasG12R induction, the PDGF-induced increase in the level of Egr-1 mRNA was evident at 15 min, peaked at 30 min, and decreased gradually thereafter (Figure 2B). In contrast, in the NIH3T3tet-on/H-RasG12R cells cultured with doxycycline for 48 h, the PDGF-induced Egr-1 mRNA levels were much lower than those seen in the absence of doxycycline. Western blot analysis also demonstrated that PDGF-induced Egr-1 protein expression was suppressed by the addition of doxycycline in both time- and dose-dependent manners (Figure 2C and D).
The PI3K pathway participates in the suppression of Raf effector-mediated Egr-1 transcription
A number of Ras effector molecules, such as Raf, PI3K, and RalGDS, have been shown to bind preferentially to Ras in the GTP-bound state (Joneson and Bar-Sagi, 1997; Campbell et al, 1998). It has been well established that site-specific mutants of Ras can distinguish between the downstream effector pathways of Ras; RasV12G37 activates only Ral GDS, RasV12E38 activates only Raf, RasV12C40 activates only PI3K, and RasV12A38 activates none of these molecules (White et al, 1995; Joneson and Bar-Sagi, 1997; Rodriguez-Viciana et al, 1997). To analyze the role of the Ras effector pathways in the regulation of Egr-1 expression, we transiently transfected NIH3T3 cells with Ras effector mutants and analyzed Egr-1 promoter activity. The expression of either RasV12 or RasV12E38 led to a strong increase in reporter activity, while RasV12A38 and RasV12C40 had no effect (Figure 3A), which indicates that the induction of Egr-1 promoter activity is mediated through a Ras–Raf effector pathway. Interestingly, RasV12C40 partially inhibited the Egr-1 promoter activity induced by dominant-active MEK1 or RasE38 (Figure 3B). Expression of the constitutively active p110 subunit (p110-CAAX) led to partial inhibition of dominant-active MEK1-induced Egr-1 promoter activity, while the dominant-negative p85 regulatory subunit that lacks the SH2 domain (p85ΔSH2) or PTEN, which is a phosphatase that dephosphorylates the phosphatidylinositol 3,4,5-trisphosphate (PIP3) produced by PI3K, synergized with MEK1 to increase reporter activity (Figure 3C). These data suggest that the PI3K effector pathway functions to regulate, in a negative fashion, Raf-mediated Egr-1 transcription.
Figure 3.
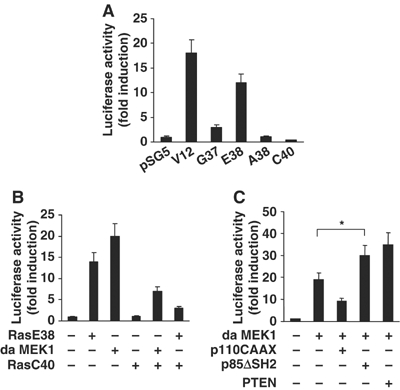
Role of the PI3K pathway in the suppression of Raf-mediated Egr-1 promoter activity. NIH3T3 cells were transfected with pGL2/Egr1-Luc reporter constructs and the Ras effector mutant constructs (A), pSG5/V12E38Ras, or pFC-MEK1 (dominant-active form of MEK1) in the absence or presence of pSG5/RasV12C40 (B), and pFC-MEK1 in the absence or presence of pSG5/p110-CAAX (constitutively active form of PI3K), pSG5/p85ΔSH2 (dominant-negative form of the p85 subunit of PI3K) or pcDNA/PTEN, as indicated (C). The pCMV/β-gal reporter vector was included as an internal control for the normalization of transfection efficiency. After 48 h of transfection, cell lysates were assayed for luciferase and β-galactosidase activities. Luciferase activity was normalized to β-galactosidase activity. The results are expressed as fold activation over control. Error bars represent the mean (±s.d.) of three independent experiments performed in triplicate. The statistical significance of the assay was evaluated using the Student's t-test (*, P<0.01 compared with the MEK1-transfected samples).
PI3K signaling acts downstream of Erk MAPK to suppress Egr-1 expression
To determine the role of the PI3K pathway in the suppression of Egr-1 expression, LY294002 was used to block the activation of the PI3K pathway in NIH3T3/RasV12 cells. The effect of LY294002 on the inhibition of PI3K was determined by measuring the level of phosphorylation of Akt, which is a downstream effector of PI3K (Figure 4A, second panel). Treatment with LY294002 alone or LY294002 plus PDGF increased the amounts of Egr-1 protein (Figure 4A, third panel) and mRNA (Figure 4B) in NIH3T3/RasV12 cells, but not in NIH3T3/vec cells, which suggests that activation of the PI3K pathway is involved in the suppression of Egr-1 expression in oncogenic Ras-expressing cells. Although LY294002 is also known to inhibit mTOR, treatment with rapamycin, which is an inhibitor of mTOR, had no effect on the restoration of Egr-1 expression (data not shown). It has been reported that Akt/PKB, which is a downstream effector of PI3K, downregulates the Ras–Raf–Erk pathway by reducing both the activity of Erk and the protein level of Elk-1, which leads to the inactivation of SRE-dependent transcription of c-fos in HEK 293 cells (Galetic et al, 2003). Since the Ras–Raf–MEK–Erk cascade is essential for growth factor-induced Egr-1 expression, we next determined whether the inhibition of PI3K signaling modulated Ras–Raf–MEK–Erk signaling. The activity of the Ras–Raf–MEK–Erk pathway was monitored according to the levels of phosphorylated Erk1/2 (Thr 202/Tyr 204). LY294002 had no effect on either the basal or EGF-stimulated phosphorylation of Erk (Figure 4A, fourth panel), which suggests that the PI3K pathway does not act directly on the Raf effector pathway. However, when MEK was inhibited by treatment with PD98059, LY294002-induced Egr-1 expression was strongly impaired (Figure 4C), which suggests that the inhibition of PI3K-induced Egr-1 expression requires MEK-Erk signaling. Thus, Raf-dependent stimulation of Egr-1 expression and PI3K-dependent inhibition may operate on different levels and it appears that, at least in oncogenic Ras-expressing cells, the latter becomes dominant.
Figure 4.
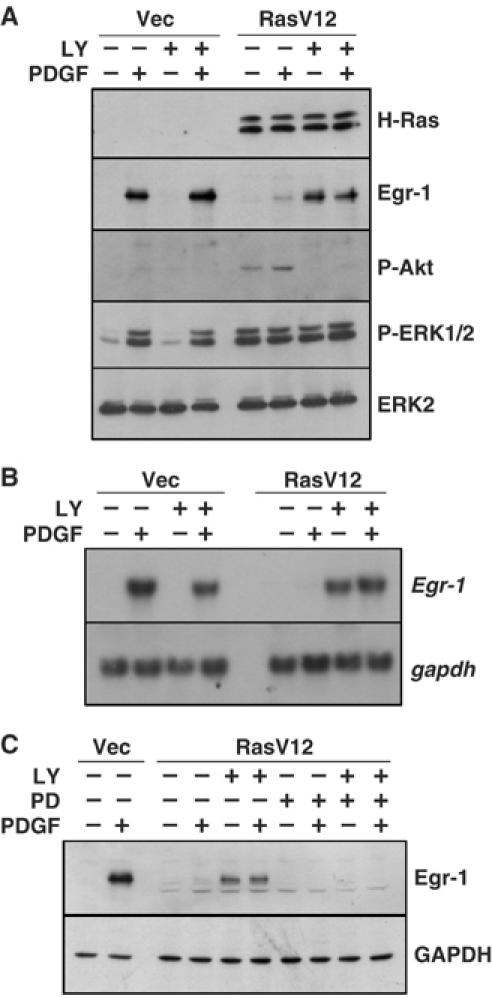
PI3K signaling acts downstream of Erk MAPK to suppress Egr-1 expression. Serum-starved NIH3T3/vec and NIH3T3/RasV12 cells were pretreated with LY294002 (20 μM), PD98059 (20 μM), or LY294002 plus PD98059, as indicated, for 1 h, and then either left untreated or treated with PDGF (50 ng/ml) for 1 h (A, C) or for 30 min (B). Cells were harvested and the Egr-1 expression was analyzed by Western blotting (A and B) or Northern blotting (C). GAPDH served as an internal control.
Oncogenic H-Ras reduces SRF binding to SRE regions
To identify PI3K signaling-responsive elements within the Egr-1 promoter, internal deletion mutants of the Egr-1 promoter (Aicher et al, 1999) were transiently cotransfected with the H-RasV12 expression plasmid into NIH3T3 cells. We found that SRE regions are necessary for LY294002-induced activation of Egr-1 promoter activity (Supplementary Figure S1). When NIH3T3tet-on/H-RasG12R cells were transfected with mutant reporter construct, which is deleted, the SRE 2-4 region (dSRE2-4), LY294002- or PDGF plus LY294002-induced Egr-1 promoter activity was markedly attenuated (Figure 5A), which suggests that SREs may be the potential target site of the PI3K effector pathway. The TCF forms a ternary complex with the SRF dimer on the SRE of the Egr-1 gene promoter and plays a crucial role in growth factor induction of Egr-1 transcription (Watson et al, 1997; Schratt et al, 2001). To investigate whether TCF trans-acting activity is altered by PI3K signaling, Elk-1-dependent trans-activation was assessed using the pFA2/Gal4-Elk1 and pFR-Luc plasmids. Elk-1-dependent trans-activation increased by ∼35-fold the inducible expression of oncogenic H-Ras. This activation was significantly reduced by cotransfection with dominant-negative H-RasN17, but not by p110-CAAX (Figure 5B). This result suggests that Elk-1 is not associated with oncogenic Ras-induced suppression of Egr-1 expression.
Figure 5.
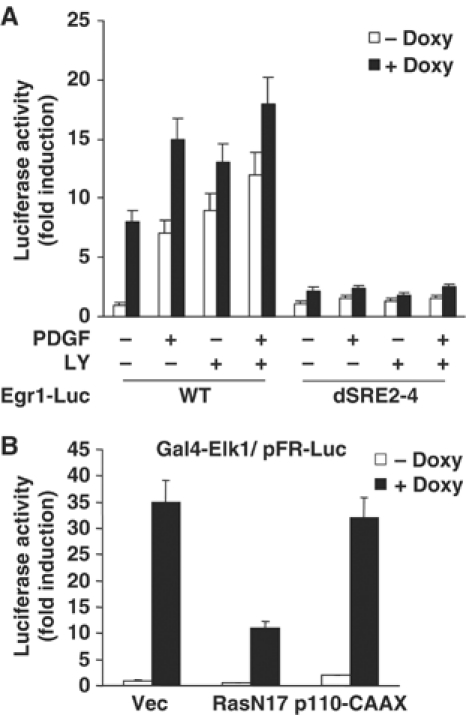
The SRE region is essential for Ras-mediated Egr-1 promoter activity. (A) NIH3T3tet-on/H-RasG12R cells were transfected with the wild-type or mutant Egr-1 promoter construct, which contained a deletion of the SRE 2-4 region (dSRE2-4). After 12 h of transfection, cells were cultured in the absence or presence of doxycycline (2 μg/ml) for 24 h in complete medium, serum starved for an additional 24 h in the absence or presence of doxycycline, and then treated with either PDGF (50 ng/ml), LY294002 (20 μM), or PDGF plus LY294002 for 12 h. Cell lysates were prepared and luciferase assay was performed. The results are expressed as fold increase +s.d. of three independent experiments performed in triplicate and normalized for transfection efficiency using pCMV/β-gal reporter. (B) PI3K signaling is not involved in Elk-1 trans-acting activity. NIH3T3 cells were transfected with the pFA2/Gal4-Elk1 and pFR-Luc plasmids along with the pSV/H-RasN17 or pSG5/p110-CAAX construct, as indicated. After 12 h of transfection, the cells were untreated or treated with doxycycline for 48 h, and then assayed for luciferase activity. The results are expressed as fold increase ±s.d. of three independent experiments performed in triplicate and normalized for transfection efficiency using the pCMV/β-gal reporter plasmid.
We also investigated whether the DNA-binding activity of SRF is involved in the suppression of Egr-1 transcription. An electrophoretic mobility shift assay (EMSA) was performed using the Egr-1 gene-derived SRE site (Figure 6A). In the absence of doxycycline, DNA–protein complexes were observed with the oligonucleotide probes that corresponded to the SRE2 and SRE3 regions of the Egr-1 gene (nucleotides −353 to −380) (Figure 6B, left panel). The addition of unlabeled oligonucleotides resulted in a reduction in DNA–protein binding, which indicates the specificity of this binding. This DNA–protein complex was supershifted when incubated with the anti-SRF antibody, but not with the anti-Elk-1 or anti-NFκB antibody, which demonstrates that the SRF protein is the main component of the complex. Notably, this SRF–DNA complex was barely detectable when the cells were treated with doxycycline for 48 h. Similar results were obtained when the consensus SRE probe was used (Figure 6B, right panel). These data strongly suggest that the loss of DNA-binding activity of SRF is associated with the suppression of Egr-1 transcription by oncogenic H-Ras. To examine the possible role of PI3K signaling in the loss of the SRF–DNA complex by oncogenic H-Ras, we next asked whether the inhibition of PI3K signaling affected the DNA-binding activity of SRF. Treatment with LY294002 abrogated oncogenic Ras-induced loss of the DNA-binding of SRF (Figure 6C), which suggests that PI3K signaling contributes to oncogenic Ras-induced suppression of the DNA-binding activity of SRF.
Figure 6.
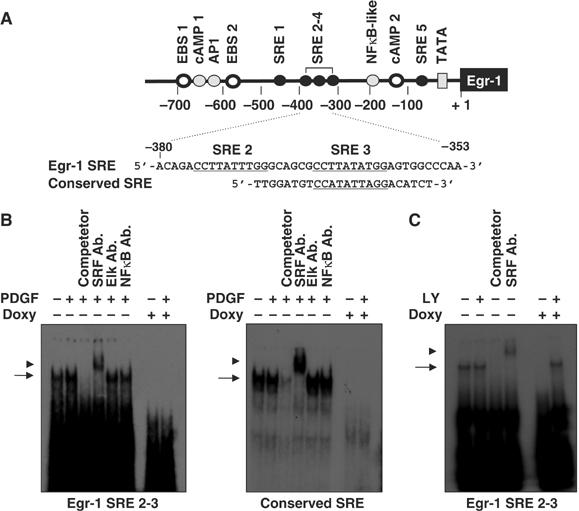
Binding of SRF to the Egr-1 promoter is decreased by the expression of oncogenic H-Ras. (A) Schematic diagram of the oligonucleotide probes used in the EMSA analysis. The SRE sites are underlined. (B) Suppression of the DNA-binding activity of SRF by oncogenic Ras. NIH3T3tet-on/H-RasG12R cells cultured in the absence or presence of doxycycline were serum starved for 24 h in medium that contained 0.5% FBS, and then stimulated with PDGF (50 ng/ml) for 30 min. Nuclear extracts (10 μg/lane) were prepared and subjected to EMSA analysis for the DNA-binding activity of SRF using oligonucleotides derived from the Egr-1 gene (left panel) or conserved SRE gene (right panel). Cold competitor (10-fold molar excess) or the anti-SRF, anti-Elk-1, and anti-NFκB antibodies were incubated for 15 min prior to the addition of 32P-labeled oligonucleotides. The arrow and arrowhead indicate the shift and supershift forms, respectively, corresponding to the position of the SRF protein–DNA complexes. (C) Effect of PI3K inhibition on the DNA-binding activity of SRF. NIH3T3tet-on/H-RasG12R cells were cultured for 24 h, serum starved with 0.5% FBS for an additional 24 h in the absence or presence of doxycycline (2 μg/ml) either with or without LY294002, as indicated. Nuclear extracts (10 μg/lane) were prepared and subjected to EMSA analysis for the DNA-binding activity of SRF, as described in (B).
The level of SRF protein is decreased by conditional expression of oncogenic H-Ras
Although SRF promoter activity has been shown to be stimulated by Ras signaling (Spencer et al, 1999), we hypothesized that hyperactivation of Ras might aberrantly deregulate the expression level of SRF. To test this possibility, we examined whether SRF expression could be altered by the inducible expression of oncogenic Ras. In concordance with the results of a previous study (Spencer et al, 1999), the inducible expression of H-RasG12R increased the amount of SRF mRNA, while the Elk-1 level remained relatively unchanged (Figure 7A). However, interestingly, the amount of SRF protein decreased dramatically 24 h after doxycycline treatment (Figure 7B). Furthermore, a decrease in the SRF protein level (Figure 7D), but not in the SRF mRNA level (Figure 7C), was observed in NIH3T3/RasV12 cells, as compared to NIH3T3/vec cells. In NIH3T3/RasV12 cells, PDGF-induced expression of the SRF-regulated genes, c-fos and Egr-1, was also severely reduced. To rule out the possibility that the loss of SRF protein was associated with changes in translocation, the subcellular localization of the SRF protein was determined by confocal microscopy. SRF immunoreactivity was concentrated in the perinuclear region in the absence of doxycycline, while it was enriched in the nucleus after 6 h of doxycycline treatment (Figure 7E). This nuclear accumulation was strongly reduced 48 h after the induction of H-RasG12R.
Figure 7.
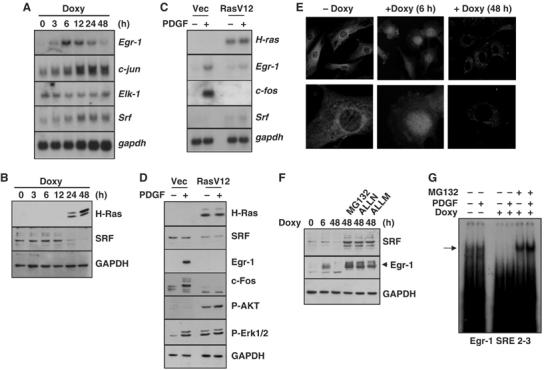
Loss of SRF protein following induced expression of oncogenic H-Ras. NIH3T3tet-on/H-RasG12R cells treated with doxycycline (2 μg/ml) were harvested at the indicated time points, and subjected to Northern blot (A) and Western blot (B) analyses. (C, D) Serum-starved NIH3T3/vec and NIH3T3/RasV12 cells were treated with PDGF for 30 min (C) or for 1 h (D), and subjected to Northern blot (C) or Western blot (D) analysis. (E) NIH3T3tet-on/H-RasG12R cells were cultured on glass slides and either left untreated (−Doxy) or treated (+Doxy) with doxycyline (2 μg/ml) for 6 or 48 h. The cells were fixed, stained with the anti-SRF antibody, and observed using the confocal microscope. (F) Effect of protease inhibitors on the SRF protein level. NIH3T3tet-on/H-RasG12R cells were cultured in the absence or presence of doxycyline (2 μg/ml) for 6 or 48 h, as indicated. Protease inhibitors, which included MG132 (20 μM), ALLN (20 μM), and ALLM (20 μM), were added for 1 h, and Western blot analysis was performed with the anti-SRF, anti-Egr-1, or anti-GAPDH antibody. (G) Effect of protease inhibition on the DNA-binding activity of SRF. NIH3T3tet-on/H-RasG12R cells were cultured for 24 h, serum starved for an additional 24 h in the absence or presence of doxycycline (2 μg/ml), and then treated with PDGF, either with or without MG132, for 1 h, as indicated. Nuclear extracts (10 μg/lane) were prepared and subjected to EMSA analysis for the DNA-binding activity of SRF, as described in Figure 6B.
We next asked whether the decrease in the level of SRF caused by oncogenic Ras was due to the acceleration of proteolysis. NIH3T3tet-on/H-RasG12R cells cultured in the presence of doxycycline for 48 h were treated with protease inhibitors. The proteasome inhibitor (N-carbobenzoxy-L-leucinyl-L-leucinyl-L-leucinal; MG132), calpain inhibitor I (N-acetyl-L-leucinyl-L-leucinyl-L-norleucinal; ALLN), and calpain inhibitor II (N-acetyl-L-leucinyl-L-leucinyl-L-methional; ALLM) prevented oncogenic Ras-induced loss of SRF protein (Figure 7F). Moreover, the DNA-binding activity of SRF was restored by MG132 treatment (Figure 7G). Together, these results demonstrate that oncogenic H-Ras accelerates the degradation of SRF protein via modulation of the proteolytic system, which results in reductions in the DNA-binding activity of SRF and SRE-mediated Egr-1 gene transcription.
Oncogenic Ras-induced reduction of the SRF protein level is reversible
To investigate whether the reduction in the level of SRF protein was caused by oncogenic H-Ras, the effect of doxycycline withdrawal was examined. NIH3T3tet-on/H-RasG12R cells were treated with doxycycline for 48 h followed by withdrawal of the drug. The amount of induced oncogenic H-Ras protein declined gradually after doxycycline withdrawal, which resulted in low-level expression of oncogenic H-Ras 60 h after withdrawal (Figure 8A). In parallel with the disappearance of H-Ras, the Erk1/2 phosphorylation and cyclin D1 levels also declined. Under these conditions, nuclear SRF protein was restored 48 h after the withdrawal of doxycycline (Figure 8B). Concomitant with the restoration of SRF expression, PDGF-induced expression of Egr-1 and c-Fos, which are targets of SRF, was also recovered. This result demonstrates that the reduction of the SRF protein level by oncogenic Ras is reversible.
PI3K signaling contributes to the reduction in the level of SRF protein caused by oncogenic H-Ras
Since oncogenic H-Ras-induced loss of SRF–DNA complex was recovered by inhibition of PI3K (Figure 6C), we investigated whether the PI3K pathway was involved in the reduction in the level of SRF protein by oncogenic Ras. We first examined the effect of a chemical inhibitor of PI3K on the abundance of SRF protein. Oncogenic Ras-induced suppression of the SRF protein level was abolished by treatment with LY294002, but not by the MEK inhibitor PD98059 (Figure 9A). To ascertain the contribution of PI3K signaling to the reduced SRF protein level, PTEN or dominant-active PI3K (p110-CAAX) was expressed using an adenovirus expression system in NIH3T3tet-on/H-RasG12R, and the levels of the SRF and Egr-1 proteins were examined. Transient expression of PTEN (Ax-PTEN) resulted in a decrease in the phosphorylation of Akt with concomitant increases in the amounts of SRF and Egr-1 proteins in response to PDGF stimulation (Figure 9B). In contrast, p110-CAAX recombinant adenovirus (Ad5-p110CAAX) infection of wild-type NIH3T3 cells decreased the levels of SRF and Egr-1 proteins, while it increased the phosphorylation of Akt (Figure 9C). Among the Ras effector mutants, RasV12C40 activates only PI3K (Rodriguez-Viciana et al, 1997). To further confirm the involvement of PI3K, we stably expressed the RasV12C40 mutant in NIH3T3 cells (NIH3T3/RasV12C40) and measured the levels of the SRF and Egr-1 proteins. Although the expression levels of the RasV12C40 mutant in the two selected clones were far lower than that of the RasV12 mutants, the expression levels of SRF and Egr-1 were comparable to those in RasV12-expressing cells (Figure 9D). This inhibitory effect of RasV12C40 was partially abrogated by the addition of LY294002 (Figure 9E), which lends further support to the role of PI3K effector signaling in the reduction of SRF protein levels by oncogenic Ras.
Figure 9.
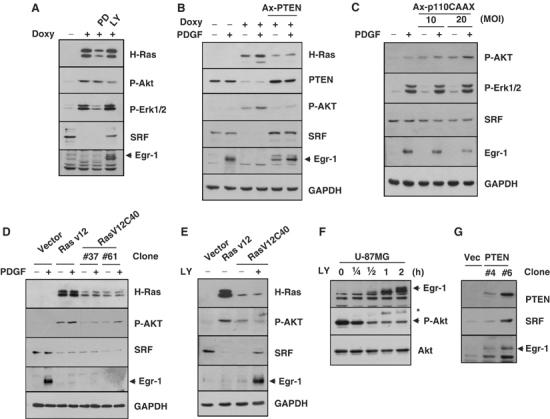
Inhibition of PI3K increases the levels of the SRF and Egr-1 proteins. (A) NIH3T3tet-on/H-RasG12R cells were cultured with doxycycline for 48 h in the absence or presence of PD98059 (20 μM), SB203580 (20 μM), or LY294002 (20 μM), and subjected to Western blot analysis with the anti-phospho-Erk1/2, anti-phospho-Akt, anti-SRF, or anti-Egr-1 antibody. (B) Effect of PTEN expression on the SRF protein level. NIH3T3tet-on/H-RasG12R cells were cultured with doxycycline for 24 h and serum starved for an additional 24 h in medium that contained 0.5% FBS medium and doxycycline in the absence or presence of recombinant PTEN-expressing adenovirus (Ax-PTEN). The serum-starved cells were treated with PDGF (50 ng/ml) for an additional hour. Total protein extracts were prepared and subjected to Western blot analysis to detect the expression of PTEN, SRF, and Egr-1. Actin was used as an internal control. (C) Effect of dominant-active PI3K expression on the SRF protein level. Wild-type NIH3T3 cells were infected with recombinant adenovirus that expressed p110-CAAX (Ad5-p110CAAX). Total protein extracts were prepared and subjected to Western blot analysis to detect the expression of SRF or Egr-1. GAPDH was used as an internal control. The expression of p110-CAAX was monitored by the detection of Akt phosphorylation. (D) Effect of RasV12C40 mutant expression on the SRF protein level. Serum-starved NIH3T3/vec, NIH3T3/RasV12, and NIH3T3/RasV12C40 cells were treated with PDGF (50 ng/ml) for 1 h. Total protein extracts were prepared and subjected to Western blot analysis to detect the expression of SRF or Egr-1. (E) Effect of LY294002 on the SRF protein level in RasV12C40-expressing cells. Serum-starved NIH3T3/vec, NIH3T3/RasV12, and NIH3T3/rasV12C40 cells were treated with LY294002 (20 μM) for 2 h. Total protein extracts were prepared and subjected to Western blot analysis to detect the expression of SRF and Egr-1. (F) Effect of LY294002 treatment on Egr-1 expression in PTEN-null U-87MG glioma cells. U-87MG cells were serum starved for 24 h, and then treated with LY294002 (20 μM) for the indicated periods of time. Total cell lysates were prepared and subjected to Western blot analysis to detect the expression of Egr-1. The same blot was stripped and reprobed with the anti-phospho-Akt (Ser 473) and anti-Akt antibodies. Asterisk indicates the undetached proteins stained with the anti-Egr-1 antibody. (G) Increased SRF and Egr-1 expression levels due to PTEN. U-87MG/vec and U-87MG/PTEN were lysed and subjected to Western blot analysis to detect the expression of PTEN, SRF, and Egr-1.
To extend these findings, we attempted to determine whether the activation of PI3K signaling modulates Egr-1 expression in PTEN-negative U-87MG glioma cells, in which PI3K signaling is constitutively activated. When U-87MG cells were treated with LY294002, the Egr-1 protein level increased in a time-dependent manner, which inversely correlated with the decrease in the level of phosphorylated Akt (Figure 9F). Furthermore, the forced expression of PTEN in U-87MG cells increased the steady-state levels of the SRF and Egr-1 proteins (Figure 9G). Thus, aberrant activation of PI3K effector signaling by oncogenic Ras seems to decrease in a specific manner the level of SRF protein via the acceleration of proteolysis, which results in the suppression of SRE-mediated Egr-1 gene transcription.
Discussion
Although Egr-1 was originally identified as a gene that is strongly induced by Ras signaling, the question as to how Egr-1 is downregulated in Ras-transformed cells has not been answered. In the present study, we have demonstrated that oncogenic H-Ras controls the transcription of the Egr-1 gene through differential effector pathways. The Raf pathway activates SRE-mediated Egr-1 transcription, while the PI3K pathway suppresses Raf pathway-mediated Egr-1 transcription. PI3K signaling prevents SRE-mediated Egr-1 transcriptional activity through promoting the degradation of the SRF protein. This event could lead to consequent suppression of Egr-1 target genes, such as PTEN, TGF-β1, p53, and fibronectin, which serve to maintain normal growth regulation (Baron et al, 2005).
We found that the protein level of SRF, but not the SRF mRNA level, was decreased by the expression of oncogenic H-Ras, which resulted in the suppression of the DNA-binding activity of SRF. Since Egr-1 and c-fos expression are severely impaired in SRF-null ES cells (Schratt et al, 2001), this loss of SRF protein is probably a critical factor in the suppression of Egr-1 expression. Our results demonstrate that the inhibition of PI3K activity by treatment with LY294002 or by adenovirus-mediated expression of PTEN prevents oncogenic Ras-induced reduction of the level of SRF protein. In addition, we have determined that stable expression of Ras effector mutant RasV12C40, which activates PI3K signaling only, or adenovirus-mediated expression of p110-CAAX, which is a dominant active form of PI3K, reduces the abundance of the SRF protein. Furthermore, stable expression of PTEN in PTEN-negative U87MG glioma cells resulted in increased levels of the SRF and Egr-1 proteins. Therefore, our results suggest that aberrantly activated PI3K signaling may play a critical role in reducing the SRF protein levels in oncogenic Ras-expressing cells.
Ras transformation may be qualitatively different from the Ras signaling that occurs during normal activation by growth factors; high levels of constitutively active Ras may engage effector proteins that are not activated by normal Ras (McCormick, 1999). Although the functional roles of Ras and its effector pathways can vary among cell types and species, even within the same cell type (Gire and Wynford-Thomas, 2000), multiple pathways are required for efficient Ras transformation (Campbell et al, 1998). Several lines of evidence suggest that the Raf and PI3K effector pathways are necessary and sufficient for Ras transformation of rodent fibroblasts (Cowley et al, 1994; Rodriguez-Viciana et al, 1997). Given that dominant-negative PI3K can block Ras-induced transformation of NIH3T3 cells (Rodriguez-Viciana et al, 1997), it is possible that PI3K pathway-mediated degradation of SRF protein may contribute to the development of tumor by oncogenic Ras through the downregulation of SRF-responsive genes (see Supplementary text).
Although the present study does not specifically address the mechanism by which chronic expression of oncogenic H-Ras causes degradation of the SRF protein, it is of particular interest that there are three potential PEST motifs at amino-acid residues 58–91 (score 6.6), 231–256 (score 8.59), and 369–402 (score 6.1) in the mouse SRF protein, as analyzed using a web-based algorithm maintained by EMBnet Austria (www.at.embnet.org/embnet/tools/bio/PESTfind/). PEST motifs, which are polypeptide sequences that are rich in proline (P), glutamate (E), serine (S), and threonine (T), have been proposed to induce rapid protein turnover by the 26S proteasome (Rechsteiner and Rogers, 1996; Spencer et al, 2004). Thus, it is possible that SRF is targeted for degradation by proteasome-dependent proteolytic pathway through the PEST sequences. We have found that treatment with the proteasome inhibitor MG-132 completely prevents the loss of both SRF and Egr-1 proteins caused by expression of oncogenic H-Ras, leading to the restoration of the DNA-binding activity of SRF. Alternatively, SRF degradation may be induced by a PEST motif-independent E3 ubiquitin ligase, which remains to be identified. In addition, we cannot rule out the possibility that multiple proteases are involved in oncogenic Ras-induced SRF proteolysis. It is known that the PEST sequence is often cleaved by a proline endopeptidase or by calpain (Rechsteiner and Rogers, 1996), and we have found that treatment with the calpain inhibitors ALLN and ALLM increases the amount of SRF protein in oncogenic Ras-expressing cells. Further characterizations of PI3K-induced signaling steps as well as the identification of specific protease(s) that target the SRF regulated in this manner will promise a deeper understanding of the molecular mechanisms that regulate tumor development due to oncogenic Ras. An understanding of the precise molecular mechanism of SRF proteolysis would be beneficial to the design of therapeutics for the future development of anticancer drugs.
PTEN is a phosphatase that dephosphorylates phosphatidylinositol 3,4,5-trisphosphate (PIP3), which is a product of PI3K, and focal adhesion kinase (Tamura et al, 1998; Cantley and Neel, 1999). PTEN functions as a tumor suppressor that plays an important role in the suppression of cell growth, focal adhesion formation, apoptosis, cell migration, and invasion (Tamura et al, 1998; Cantley and Neel, 1999; Parsons, 2004). In this study, we found that a decrease in PTEN expression was accompanied by suppression of Egr-1 expression by oncogenic H-Ras (Figures 8B and 9B, Supplementary Figure S3A). It seems likely that oncogenic Ras further facilitates the activation of the PI3K effector pathway through the negative regulation of PTEN. It has been reported that induction of Egr-1 expression by ultraviolet light upregulates the expression of PTEN mRNA and protein, and leads to apoptosis in wild-type but not in Egr-1-null mouse embryo fibroblasts (Virolle et al, 2001), which indicates that the PTEN gene is directly regulated by Egr-1. Indeed, we observed that PTEN expression was increased by PDGF stimulation in nontransformed NIH3T3 cells (Supplementary Figure S2), while transfection with small interfering RNA (siRNA) targeted to a specific sequence of Egr-1 partially blocked both basal and PDGF inducibility of PTEN protein expression (Supplementary Figure S3B). Further studies are required to elucidate the precise mechanism for the suppression of PTEN expression by oncogenic Ras, and whether the reduction of Egr-1 expression is directly linked to the downregulation of PTEN.
In this study, we have investigated the role of oncogenic H-Ras in the regulation of tumor suppressor Egr-1 expression. The major finding of our study is that oncogenic H-Ras controls Egr-1 gene expression by differential effector pathways. According to our model, H-Ras stimulates SRE-dependent transcription of the Egr-1 gene via the Raf effector pathway in nontransformed fibroblasts. However, if oncogenic H-Ras is expressed chronically at a high level, aberrant activation of the PI3K effector pathway decreases the nuclear level of SRF protein by accelerating proteolysis, which results in the suppression of Raf signaling-dependent SRE-mediated Egr-1 transcription. Since Egr-1 functions as a regulator of negative feed-back loop of PI3K signaling through the induction of PTEN, as well as of a regulation of cell cycle and apoptosis through induction of TGF-β1, p53, p21 Cip1, or Bax (Liu et al, 1998; Virolle et al, 2001; Krones-Herzig et al, 2003; Ragione et al, 2003; Baron et al, 2005; Thyss et al, 2005), loss of Egr-1 function could provide an excessive activation of PI3K signaling, which results in the inhibition of apoptosis and hyperactivation of cell proliferation, and thereby could promote a more aggressive malignant phenotype. It is noteworthy that the loss of PTEN function may occur due to oncogenic Ras without mutation or deletion of the PTEN gene. In summary, we suggest a novel signaling mechanism by which prolonged activation of oncogenic H-Ras can trigger the loss of tumor suppressor Egr-1 through the PI3K pathway in NIH3T3 fibroblasts.
Materials and methods
Cells and plasmid construction
NIH3T3 and the derivatives of NIH3T3 cell lines, including NIH3T3/RasV12, NIH3T3/RasV12C40, or NIH3T3tet-on/H-RasG12R, are described in the ‘Supplementary Materials and methods'. The Egr-1 mutant promoter constructs have been described elsewhere (Aicher et al, 1999). Full-length PTEN cDNA (a gift from Dr HJ Zhou, Ludwig Institute for Cancer Research, Melbourne, Australia) was subcloned into the EcoRI site of pcDNA3.1 (Invitrogen). The pSG5 Ras effector mutant constructs (pSG5/V12Ras, pSG5/V12A38Ras, pSG5/V12E38Ras, pSG5/V12G37Ras, and pSG5/V12C40Ras), pSG5/p110-CAAX, and pSG5/p85ΔSH2 (dominant-negative form of the p85 subunit of PI3K) plasmids have been described elsewhere (Marte et al, 1997; Wennstrom and Downward, 1999).
Northern blot analysis
Total RNA samples (10 μg) were separated by electrophoresis in a formaldehyde/agarose gel and transferred to a Hybond N+ nylon membrane (Amersham Pharmacia Biotech). Northern blotting was performed with the [γ-32P]dCTP-labeled (High Prime DNA Labeling Kit; Roche) Egr-1 cDNA probe, followed by hybridization with the GAPDH cDNA probe, as described previously (Shin et al, 2004).
Western blot analysis
Cells were lysed in buffer that contained 20 mM HEPES (pH 7.2), 1% Triton X-100, 10% glycerol, 400 mM NaCl, 10 μg/ml leupeptin, and 1 mM PMSF, and Western blot analysis was performed according to standard procedures using anti-Egr-1 (1:1000 dilution; Santa Cruz Biotechnology), anti-SRF (1:1000; Santa Cruz Biotechnology), anti-H-Ras (1:500; Oncogene), anti-phospho-MEK1 (1:1000; Cell Signaling), anti-phospho-Erk1/2 (Thr 202/Tyr 204; 1:1000, Cell signaling), anti-phospho-Akt (Ser 473; 1:1000; Cell Signaling), anti-Akt (1:1000; Cell Signaling), anti-Erk2 (1:5000; Santa Cruz Biotechnology), anti-GAPDH (1:2000; Santa Cruz Biotechnology), and anti-actin (1:5000; Santa Cruz Biotechnology) antibodies.
EMSA
After the serum-starved cells were treated with PDGF (50 ng/ml), nuclear extracts were prepared, and incubated with 25 000 cpm of 32P-labeled oligonucleotide probe (for details, see Supplementary Materials and methods) for 20 min at room temperature, as described previously (Shin et al, 2004). The resulting DNA–protein complexes were separated from the free probe by electrophoresis in a 6% nondenaturing polyacrylamide gel in 0.5 × Tris borate–EDTA buffer, and visualized by autoradiography.
Immunofluorescence staining
Cells seeded on cover glass were treated with doxycycline. After 6 or 48 h, the cells were fixed in 4% paraformaldehyde for 10 min, and permeabilized in 0.1% Triton X-100 and 2% BSA for 10 min. The samples were incubated with the anti-SRF antibody for 45 min, followed by incubation with rhodamine-labeled anti-rabbit IgG antibody for 30 min. The slides were then processed for indirect immunofluorescence microscopy.
Recombinant adenovirus
The preparation of recombinant adenovirus that expresses p110-CAAX (Ad5-p110CAAX) has been described elsewhere (Egawa et al, 1999). The recombinant adenovirus that expresses human PTEN1 under the control of the CAG promoter (AxCAhPTEN1) (Sakurada et al, 1999) was obtained from the RIKEN BioResource Center (Ibaraki, Japan). Preparation and infection of adenoviruses were described in Supplementary data.
Supplementary Material
Supplementary Information
Supplementary Figures
Acknowledgments
This research was supported in part by the Korea Science and Engineering Foundation (Grant no. R08-2004-000-10189-0 to SYS), the Korea Research Foundation (Grant no. KRF-2004-041-C00251 to SYS), and by the Brain Research Center of the 21st Century Frontier Research Program, funded by the Korean Ministry of Science and Technology (Grant no. M103KV010007-03K2201-00740 to YHL).
References
- Aicher WK, Sakamoto KM, Hack A, Eibel H (1999) Analysis of functional elements in the human Egr-1 gene promoter. Rheumatol Int 18: 207–214 [DOI] [PubMed] [Google Scholar]
- Baron V, Adamson ED, Calogero A, Ragona G, Mercola D (2005) The transcription factor Egr1 is a direct regulator of multiple tumor suppressors including TGFβ1, PTEN, p53, and fibronectin. Cancer Gene Ther; doi:10.1038/sj.cgt.7700896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos JL (1989) Ras oncogenes in human cancer: a review. Cancer Res 49: 4682–4689 [PubMed] [Google Scholar]
- Campbell SL, Khosravi-Far R, Rossman KL, Clark GJ, Der CJ (1998) Increasing complexity of Ras signaling. Oncogene 17: 1395–1413 [DOI] [PubMed] [Google Scholar]
- Cantley LC, Neel BG (1999) New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci USA 96: 4240–4245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christy B, Nathans D (1989) Functional serum response elements upstream of the growth factor-inducible gene zif268. Mol Cell Biol 9: 4889–4895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson RW, Shang CA, Levitt LK, Howard T, Waters MJ (1999) Ternary complex factors Elk-1 and Sap-1a mediate growth hormone-induced transcription of egr-1 (early growth response factor-1) in 3T3-F442A preadipocytes. Mol Endocrinol 13: 619–631 [DOI] [PubMed] [Google Scholar]
- Cowley S, Paterson H, Kemp P, Marshall CJ (1994) Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH3T3 cells. Cell 77: 841–852 [DOI] [PubMed] [Google Scholar]
- de Belle I, Huang RP, Fan Y, Liu C, Mercola D, Adamson ED (1999) p53 and Egr-1 additively suppress transformed growth in HT1080 cells but Egr-1 counteracts p53-dependent apoptosis. Oncogene 18: 3633–3642 [DOI] [PubMed] [Google Scholar]
- Edwards SA, Darland T, Sosnowski R, Samuels M, Adamson ED (1991) The transcription factor, Egr-1, is rapidly modulated in response to retinoic acid in P19 embryonal carcinoma cells. Dev Biol 148: 165–173 [DOI] [PubMed] [Google Scholar]
- Egawa K, Sharma PM, Nakashima N, Huang Y, Huver E, Boss GR, Olefsky JM (1999) Membrane-targeted phosphatidylinositol 3-kinase mimics insulin actions and induces a state of cellular insulin resistance. J Biol Chem 274: 14306–14314 [DOI] [PubMed] [Google Scholar]
- Galetic I, Maira SM, Andjelkovic M, Hemmings BA (2003) Negative regulation of ERK and Elk by protein kinase B modulates c-fos transcription. J Biol Chem 278: 4416–4423 [DOI] [PubMed] [Google Scholar]
- Gire V, Wynford-Thomas D (2000) RAS oncogene activation induces proliferation in normal human thyroid epithelial cells without loss of differentiation. Oncogene 19: 737–744 [DOI] [PubMed] [Google Scholar]
- Guha M, O'Connell MA, Pawlinski R, Hollis A, McGovern P, Yan SF, Stern D, Mackman N (2001) Lipopolysaccharide activation of the MEK–ERK1/2 pathway in human monocytic cells mediates tissue factor and tumor necrosis factor alpha expression by inducing Elk-1 phosphorylation and Egr-1 expression. Blood 98: 1429–1439 [DOI] [PubMed] [Google Scholar]
- Hipskind RA, Buscher D, Nordheim A, Baccarini M (1994) Ras/MAP kinase-dependent and -independent signaling pathways target distinct ternary complex factors. Genes Dev 8: 1803–1816 [DOI] [PubMed] [Google Scholar]
- Hodge C, Liao J, Stofega M, Guan K, Carter-Su C, Schwartz J (1998) Growth hormone stimulates phosphorylation and activation of elk-1 and expression of c-fos, egr-1, and junB through activation of extracellular signal-regulated kinases 1 and 2. J Biol Chem 273: 31327–31336 [DOI] [PubMed] [Google Scholar]
- Huang RP, Fan Y, de Belle I, Niemeyer C, Gottardis MM, Mercola D, Adamson ED (1997) Decreased Egr-1 expression in human, mouse and rat mammary cells and tissues correlates with tumor formation. Int J Cancer 72: 102–109 [DOI] [PubMed] [Google Scholar]
- Joneson T, Bar-Sagi D (1997) Ras effectors and their role in mitogenesis and oncogenesis. J Mol Med 75: 587–593 [DOI] [PubMed] [Google Scholar]
- Krones-Herzig A, Adamson E, Mercola D (2003) Early growth response 1 protein, an upstream gatekeeper of the p53 tumor suppressor, controls replicative senescence. Proc Natl Acad Sci USA 100: 3233–3238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Rangnekar VM, Adamson E, Mercola D (1998) Suppression of growth and transformation and induction of apoptosis by EGR-1. Cancer Gene Ther 5: 3–28 [PubMed] [Google Scholar]
- Liu C, Yao J, de Belle I, Huang RP, Adamson E, Mercola D (1999) The transcription factor EGR-1 suppresses transformation of human fibrosarcoma HT1080 cells by coordinated induction of transforming growth factor-beta1, fibronectin, and plasminogen activator inhibitor-1. J Biol Chem 274: 4400–4411 [DOI] [PubMed] [Google Scholar]
- Liu C, Yao J, Mercola D, Adamson E (2000) The transcription factor EGR-1 directly transactivates the fibronectin gene and enhances attachment of human glioblastoma cell line U251. J Biol Chem 275: 20315–20323 [DOI] [PubMed] [Google Scholar]
- Macara IG, Lounsbury KM, Richards SA, McKiernan C, Bar-Sagi D (1996) The Ras superfamily of GTPases. FASEB J 10: 625–630 [DOI] [PubMed] [Google Scholar]
- Marte BM, Rodriguez-Viciana P, Wennstrom S, Warne PH, Downward J (1997) R-Ras can activate the phosphoinositide 3-kinase but not the MAP kinase arm of the Ras effector pathways. Curr Biol 7: 63–70 [DOI] [PubMed] [Google Scholar]
- McCormick F (1999) Signalling networks that cause cancer. Trends Cell Biol 9: M53–M56 [PubMed] [Google Scholar]
- Parsons R (2004) Human cancer, PTEN and the PI-3 kinase pathway. Semin Cell Dev Biol 15: 171–176 [DOI] [PubMed] [Google Scholar]
- Paterson H, Reeves B, Brown R, Hall A, Furth M, Bos J, Jones P, Marshall C (1987) Activated N-ras controls the transformed phenotype of HT1080 human fibrosarcoma cells. Cell 51: 803–812 [DOI] [PubMed] [Google Scholar]
- Price MA, Rogers AE, Treisman R (1995) Comparative analysis of the ternary complex factors Elk-1, SAP-1a and SAP-2 (ERP/NET). EMBO J 14: 2589–2601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragione FD, Cucciolla V, Criniti V, Indaco S, Borriello A, Zappia V (2003) p21Cip1 gene expression is modulated by Egr1: a novel regulatory mechanism involved in the resveratrol antiproliferative effect. J Biol Chem 278: 23360–23368 [DOI] [PubMed] [Google Scholar]
- Rechsteiner M, Rogers SW (1996) PEST sequences and regulation by proteolysis. Trends Biochem Sci 21: 267–271 [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Warne PH, Khwaja A, Marte BM, Pappin D, Das P, Waterfield MD, Ridley A, Downward J (1997) Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell 89: 457–467 [DOI] [PubMed] [Google Scholar]
- Sakurada A, Hamada H, Fukushige S, Yokoyama T, Yoshinaga K, Furukawa T, Sato S, Yajima A, Sato M, Fujimura S, Horii A (1999) Adenovirus-mediated delivery of the PTEN gene inhibits cell growth by induction of apoptosis in endometrial cancer. Int J Oncol 15: 1069–1074 [DOI] [PubMed] [Google Scholar]
- Schratt G, Weinhold B, Lundberg AS, Schuck S, Berger J, Schwarz H, Weinberg RA, Ruther U, Nordheim A (2001) Serum response factor is required for immediate-early gene activation yet is dispensable for proliferation of embryonic stem cells. Mol Cell Biol 21: 2933–2943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin SY, Kim CG, Ko J, Min DS, Chang JS, Ohba M, Kuroki T, Choi YB, Kim YH, Na DS, Kim JW, Lee YH (2004) Transcriptional and post-transcriptional regulation of the PKC delta gene by etoposide in L1210 murine leukemia cells: implication of PKC delta autoregulation. J Mol Biol 340: 681–693 [DOI] [PubMed] [Google Scholar]
- Shin SY, Kim SY, Kim JH, Min DS, Ko J, Kang UG, Kim YS, Kwon TK, Han MY, Kim YH, Lee YH (2001) Induction of early growth response-1 gene expression by calmodulin antagonist trifluoperazine through the activation of Elk-1 in human fibrosarcoma HT1080 cells. J Biol Chem 276: 7797–7805 [DOI] [PubMed] [Google Scholar]
- Spencer JA, Major ML, Misra RP (1999) Basic fibroblast growth factor activates serum response factor gene expression by multiple distinct signaling mechanisms. Mol Cell Biol 19: 3977–3988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer ML, Theodosiou M, Noonan DJ (2004) NPDC-1, a novel regulator of neuronal proliferation, is degraded by the ubiquitin/proteasome system through a PEST degradation motif. J Biol Chem 279: 37069–37078 [DOI] [PubMed] [Google Scholar]
- Sukhatme VP, Cao XM, Chang LC, Tsai-Morris CH, Stamenkovich D, Ferreira PC, Cohen DR, Edwards SA, Shows TB, Curran T (1988) A zinc finger-encoding gene coregulated with c-fos during growth and differentiation, and after cellular depolarization. Cell 53: 37–43 [DOI] [PubMed] [Google Scholar]
- Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM (1998) Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science 280: 1614–1617 [DOI] [PubMed] [Google Scholar]
- Thyss R, Virolle V, Imbert V, Peyron JF, Aberdam D, Virolle T (2005) NF-kappaB/Egr-1/Gadd45 are sequentially activated upon UVB irradiation to mediate epidermal cell death. EMBO J 24: 128–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treisman R (1994) Ternary complex factors: growth factor regulated transcriptional activators. Curr Opin Genet Dev 4: 96–101 [DOI] [PubMed] [Google Scholar]
- Tsai JC, Liu L, Guan J, Aird WC (2000) The Egr-1 gene is induced by epidermal growth factor in ECV304 cells and primary endothelial cells. Am J Physiol 279: C1414–C1424 [DOI] [PubMed] [Google Scholar]
- Virolle T, Adamson ED, Baron V, Birle D, Mercola D, Mustelin T, de Belle I (2001) The Egr-1 transcription factor directly activates PTEN during irradiation-induced signalling. Nat Cell Biol 3: 1124–1128 [DOI] [PubMed] [Google Scholar]
- Watson DK, Robinson L, Hodge DR, Kola I, Papas TS, Seth A (1997) FLI1 and EWS-FLI1 function as ternary complex factors and ELK1 and SAP1a function as ternary and quaternary complex factors on the Egr1 promoter serum response elements. Oncogene 14: 213–221 [DOI] [PubMed] [Google Scholar]
- Wennstrom S, Downward J (1999) Role of phosphoinositide 3-kinase in activation of ras and mitogen-activated protein kinase by epidermal growth factor. Mol Cell Biol 19: 4279–4288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MA, Nicolette C, Minden A, Polverino A, Van Aelst L, Karin M, Wigler MH (1995) Multiple Ras functions can contribute to mammalian cell transformation. Cell 80: 533–541 [DOI] [PubMed] [Google Scholar]
- Yu C-L, Prochownik EV, Imperiale MJ, Jove R (1993) Attenuation of serum inducibility of immediate early genes by oncoprotein in tyrosine kinase signaling pathways. Mol Cell Biol 13: 2011–2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Supplementary Figures