

Abstract
Increased production of mitochondrial reactive oxygen species (ROS) by hyperglycemia is recognized as a major cause of the clinical complications associated with diabetes and obesity [Brownlee, M. (2001) Nature 414, 813–820]. We observed that dynamic changes in mitochondrial morphology are associated with high glucose-induced overproduction of ROS. Mitochondria undergo rapid fragmentation with a concomitant increase in ROS formation after exposure to high glucose concentrations. Neither ROS increase nor mitochondrial fragmentation was observed after incubation of cells with the nonmetabolizable stereoisomer l-glucose. However, inhibition of mitochondrial pyruvate uptake that blocked ROS increase did not prevent mitochondrial fragmentation in high glucose conditions. Importantly, we found that mitochondrial fragmentation mediated by the fission process is a necessary component for high glucose-induced respiration increase and ROS overproduction. Extended exposure to high glucose conditions, which may mimic untreated diabetic conditions, provoked a periodic and prolonged increase in ROS production concomitant with mitochondrial morphology change. Inhibition of mitochondrial fission prevented periodic fluctuation of ROS production during high glucose exposure. These results indicate that the dynamic change of mitochondrial morphology in high glucose conditions contributes to ROS overproduction and that mitochondrial fission/fusion machinery can be a previously unrecognized target to control acute and chronic production of ROS in hyperglycemia-associated disorders.
Keywords: DLP1/Drp1, mitochondrial fission, dynamin, diabetes, obesity
The vascular and multiorgan complications in diabetes and obesity are causally associated with hyperglycemia-induced overproduction of reactive oxygen species (ROS) (1–4). In addition, surgical and intensive care patients often exhibit high blood glucose levels, which contributes to multiorgan injury (5, 6). Hyperglycemic conditions increase input of metabolic substrate into mitochondria and overwhelm the electron transport system, resulting in ROS overproduction (2).
Mitochondria are dynamic organelles, frequently changing shape and distribution (7). Defined sets of proteins that mediate mitochondrial fission and fusion constitute the critical regulatory components controlling mitochondrial dynamics (8–14). The basic morphology of mitochondria in cells is tubular, and balanced frequency of fission and fusion determines mitochondrial morphology (15). Growing evidence indicates that maintaining correct mitochondrial morphology is critical for cell function (16). Recent findings that mutations in genes encoding fission and fusion proteins cause human diseases (17–20) signify the importance of mitochondrial dynamics.
Mitochondrial function is reflected in their structure and morphology (13, 14, 21–24). A frequently observed characteristic in tissues of hyperglycemic patients and animals is mitochondrial deformation, most notably mitochondrial swelling or accumulation of small mitochondria (25–27). In this study, we found that dynamic change of mitochondrial morphology is an important factor contributing to ROS overproduction in high glucose (HG) conditions. Mitochondria become rapidly fragmented in HG concentrations with a concomitant increase of ROS. Whereas glucose metabolism is necessary for both mitochondrial fragmentation and ROS increase, mitochondrial pyruvate uptake is not required for mitochondrial fragmentation in HG conditions. We demonstrated that mitochondrial fragmentation is necessary for HG-induced respiration increase and ROS overproduction. We also found that ROS levels fluctuate in prolonged HG conditions, which is prevented by inhibiting mitochondrial fission. These findings suggest that mitochondrial dynamics can be a previously undescribed target to control ROS overproduction in diabetes, obesity, and other hyperglycemia-associated disorders.
Results
HG-Induced ROS Increase Coincides with Morphological Change of Mitochondria.
We examined cellular ROS levels in Clone 9 rat liver cell line or H9c2 rat myoblasts. Upon shifting to HG concentrations (25 or 50 mM), cells displayed increased ROS levels. Dichlorofluorescein (DCF) fluorescence resulting from increased ROS showed a perinuclear distribution (Fig. 1A), consistent with the ROS increase in mitochondria during hyperglycemia (4, 27, 28). Time course experiments revealed that ROS increased within 15 min and decreased to the normal level in 60 min (Fig. 1B), indicating that HG incubation produces a transient increase in ROS. The results observed by using DCF fluorescence were confirmed with another ROS probe, dihydroethidium (DHE). Ethidium produced by DHE oxidation accumulates as red fluorescence in the nucleus (Fig. 6, which is published as supporting information on the PNAS web site).
Fig. 1.

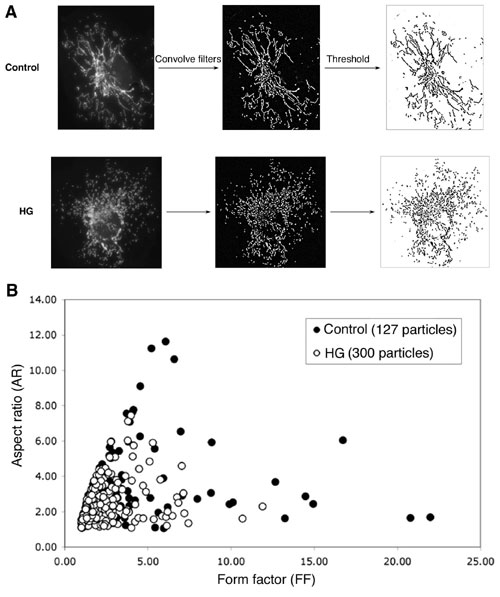
HG-induced ROS increase coincides with mitochondrial fragmentation. (A and B) HG incubation transiently increased ROS production. ROS was measured by oxidation of carboxy-dichlorodihydrofluorescein diacetate (Carboxy-DCF) at different times after HG incubation. Experiments were repeated more than five times and produced consistent results. Representative fluorescence quantification is shown in B. (C–F) Mitochondrial fragmentation in HG incubation. Clone 9 cells, whose mitochondria were labeled with red fluorescent protein, were incubated in 25 mM glucose. (C) Fragmented mitochondria were prevalent after 15 and 30 min in HG and normal tubular mitochondria after 60 min. (D) Cell counting is shown. (E) Analyses of mitochondrial morphology by form factor and aspect ratio. (E Left) Graphs plotted for form factor and aspect ratio of individual mitochondria in one cell are shown. (E Right) Enlarged images of a part of the corresponding cell used for the analysis, showing detailed mitochondrial morphology. (F) Average values of form factor and aspect ratio, as well as number of mitochondria, from five cells at each time point show transient fragmentation of mitochondria after incubation in HG conditions.
The HG-induced ROS production was accompanied by a marked change in mitochondrial morphology. Normal tubular mitochondria became short and small within 15 min, indicating mitochondrial fragmentation (Fig. 1C). Importantly, this morphological change was transient, similar to the temporal profile of ROS levels. The number of cells containing fragmented mitochondria increased to ≈70% in 15 min and decreased to control levels in 60-min HG incubation (Fig. 1D). We analyzed mitochondrial morphologies by computer-assisted morphometric analyses that calculate form factor (FF) and aspect ratio (AR) (refs. 29 and 30; Fig. 7, which is published as supporting information on the PNAS web site). Both parameters have a minimal value of 1, which represents a perfect circle, and the values increase as mitochondria elongate. Plotting AR against FF showed that most mitochondria from cells in HG for 15 and 30 min had lower values of FF and AR compared to the control, 60-, and 120-min incubations (Fig. 1E), indicating transient fragmentation and recovery of mitochondria in HG incubation. Average values of FF and AR, as well as the number of mitochondria, at each time point showed transient fragmentation of mitochondria in HG conditions (Fig. 1F).
The Mitochondrial Fission Protein DLP1 Mediates HG-Induced Mitochondrial Fragmentation.
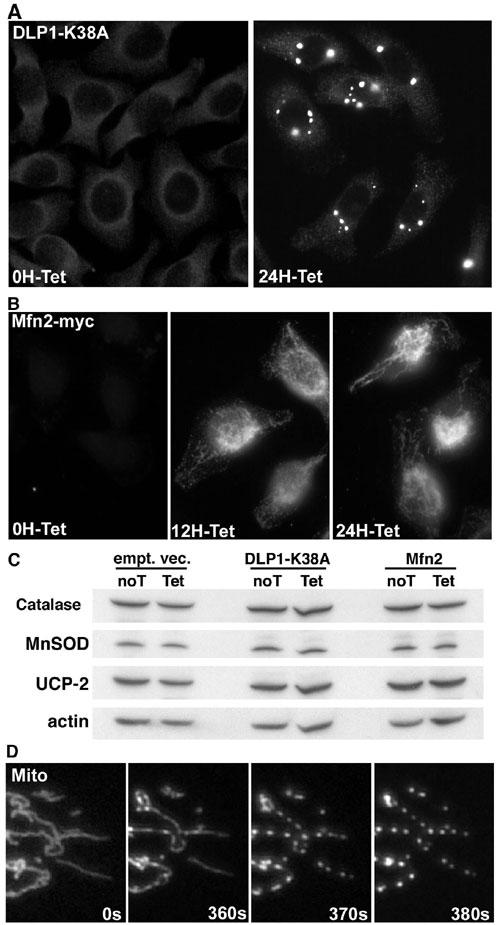
The dynamin-like GTPase DLP1/Drp1 mediates mitochondrial fission (8–10, 31). A dominant-negative mutant form of DLP1, DLP1-K38A (a point mutation at Lys-38 to Ala), inhibits fission, forming elongated mitochondrial tubules (8–10). To test whether the mitochondrial fragmentation observed in HG was mediated by mitochondrial fission, we examined mitochondrial morphology in cells transfected with DLP1-K38A. Mitochondrial tubules were elongated, entangled, and often collapsed around the nucleus in cells overexpressing DLP1-K38A (Fig. 2A, 0 min, “T”). Upon shifting to HG, mitochondria in untransfected cells became fragmented (Fig. 2A, 15 min and 30 min, “U”), whereas cells overexpressing DLP1-K38A maintained elongated mitochondrial tubules (Fig. 2A, 15 min and 30 min, T). These results show that mitochondrial fragmentation in HG conditions requires mitochondrial fission machinery.
Fig. 2.

Roles of DLP1, glucose metabolism, and pyruvate transport in HG-induced ROS increase and mitochrondrial fragmentation. (A) Mitochondrial fragmentation in HG requires mitochondrial fission machinery. Cells transfected with DLP1-K38A (T) maintained elongated tubular mitochondria in HG media, whereas mitochondria in untransfected cells (U) were short and fragmented. (B–D) d-glucose, but not l-glucose, causes ROS increase and mitochondrial fragmentation. (B and C) DCF fluorescence increased in cells incubated in d-glucose, but not in l-glucose. (D) Both form factor and aspect ratio of mitochondria did not change in cells from l-glucose incubation. (E and F) Inhibiting mitochondrial pyruvate uptake prevents ROS increase in HG conditions. Preincubation with 0.1 mM CHC blocked ROS increase (E) but did not affect mitochondrial fragmentation (F).
HG-Induced Mitochondrial Fragmentation Requires Glucose Metabolism.
Increased glucose metabolism in hyperglycemia leads to ROS overproduction (2, 4). To test whether glucose metabolism is also necessary for HG-induced mitochondrial fragmentation, we used the stereoisomer l-glucose that cannot be transported and metabolized by cells. Neither increased ROS nor mitochondrial fragmentation was observed in the presence of l-glucose. ROS increase was negligible in cells incubated in 25 or 50 mM l-glucose (Fig. 2 B and C). Morphometric analyses showed no change of mitochondrial length in cells incubated in l-glucose (Fig. 2D). These results indicate that both mitochondrial morphology change and ROS increase in HG conditions require glucose metabolism.
Mitochondrial Pyruvate Uptake Is Necessary for HG-Induced ROS Increase but Not for Mitochondrial Fragmentation.
Metabolism of pyruvate taken into mitochondria provides electron donors to the electron transport system through the tricarboxylic acid cycle. We tested whether mitochondrial pyruvate uptake is necessary for ROS increase and mitochondrial fragmentation in HG conditions by using an inhibitor of mitochondrial pyruvate transport, α-cyano-4-hydroxycinnamate (CHC) (4, 32, 33). Preincubating cells with 0.1 mM CHC blocked ROS increase in HG (Fig. 2E). Under the same conditions, however, CHC-treated cells displayed the transient mitochondrial fragmentation in HG conditions (Fig. 2F). These results indicate that mitochondrial pyruvate uptake is necessary for HG-induced ROS increase but not for mitochondrial fragmentation.
Inhibition of Mitochondrial Fission Prevents HG-Induced ROS Increase.
The temporal profiles of changes in mitochondria morphology and ROS levels indicate a correlation between these two events. We first tested whether increased ROS is required for mitochondrial fragmentation in HG conditions. Because mitochondrial hyperpolarization in hyperglycemia is known to cause the ROS increase (2, 4, 34, 35), we blocked the ROS increase by reducing the mitochondrial membrane potential and examined mitochondrial morphology. Mild uncoupling of mitochondria with 100 nM carbonyl cyanide p-(trifluoromethoxy)phenylhydrazone (FCCP) prevented ROS increase upon HG exposure (Fig. 3A). However, mitochondrial fragmentation still occurred in the absence of the ROS increase (Fig. 3B), indicating that increased ROS is not the cause of mitochondrial fragmentation in HG.
Fig. 3.

Mitochrondrial fragmentation is necessary for HG-induced ROS overproduction and respiration increase. (A and B) ROS increase is not required for mitochondrial fragmentation in HG incubation. Cells pretreated with 100 nM carbonyl cyanide p-(trifluoromethoxy)phenylhydrazone (FCCP) blocked ROS increase (A) but mitochondria were still fragmented upon HG exposure (B). (C and D) Inhibition of mitochondrial fission prevented HG-induced ROS production. (C) Cells transfected with GFP-DLP1-K38A showed bright aggregates of GFP signals (Left, arrows and arrowheads). Under normal glucose conditions, ethidium fluorescence of transfected cells (Upper Right, arrows) was similar to that in untransfected cells. At 30-min exposure to 25 mM glucose, ethidium fluorescence remained low in transfected cells (Lower Right, arrowheads) whereas more intense ethidium fluorescence was observed in untransfected cells. (D) Quantification of fluorescence intensity is shown. (E and F) Inhibition of mitochondrial fission prevents respiration increase in HG conditions. (E) Oxygen consumption profiles of cells in normal and HG conditions. DLP1-K38A overexpression was induced by tetracycline (Tet-induced). (F) Respiration rates show that DLP1-K38A overexpression (Tet-induced) blocked HG-induced respiration increase (arrowheads).
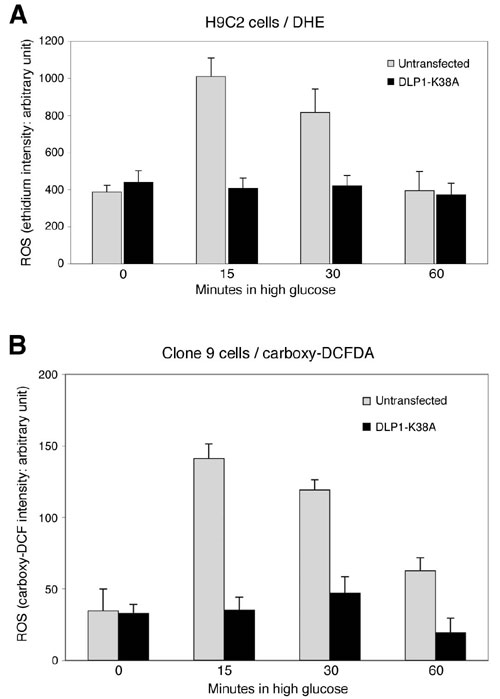
To test whether mitochondrial fragmentation by HG is a cause of the ROS increase, we blocked mitochondrial fragmentation by DLP1-K38A and measured ROS levels. GFP-tagged DLP1-K38A (GFP-DLP1-K38A) was used to identify cells expressing the mutant DLP1 in live experimental conditions. Overexpressed GFP-DLP1-K38A forms small aggregates in the cytoplasm (refs. 9 and 31; Fig. 3C). An increase of ethidium fluorescence in nuclei was observed in untransfected cells in HG conditions. Remarkably, in cells expressing GFP-DLP1-K38A, the ethidium fluorescence remained low in HG (Fig. 3 C and D). Experiments with H9c2 cells or with H2DCFDA as an ROS indicator gave similar results (Fig. 8, which is published as supporting information on the PNAS web site). These results show that the mitochondrial fragmentation observed in cells exposed to HG concentrations is a causal factor leading to increased ROS production.
Inhibition of Mitochondrial Fission Prevents HG-Induced Increase of Respiration.
To explore the role of mitochondrial morphology change in HG-induced ROS increase, we measured oxygen consumption in control and DLP1-K38A-overexpressing cells under normal and HG conditions. Because oxygen consumption is measured with populations of cells, we used a cell line in which DLP1-K38A is overexpressed in a tetracycline-inducible manner (Fig. 9, which is published as supporting information on the PNAS web site). Oxygen consumption profiles indicated that HG exposure accelerated respiration in control cells (Fig. 3E). Respiration rates were increased by 1.6- and 1.5-fold in steady-state and maximal respiration, respectively, in HG incubations (Fig. 3F). In contrast, we found that cells overexpressing DLP1-K38A did not increase the respiration rate in HG. Both steady-state and maximal respirations of DLP1-K38A-overexpressing cells in HG incubation were similar to those measured in normal glucose conditions (Fig. 3F, arrowheads). These results indicate that mitochondrial fragmentation is a necessary component for increased respiration in HG incubation.
Promoting Mitochondrial Fusion Prevents HG-Induced ROS Increase.
Mitochondrial morphology is maintained by balanced fission and fusion (15). Shifting this balance changes mitochondrial morphology to either a fragmented or an elongated shape. Because mitochondrial fragmentation can occur by increased fission, decreased fusion, or both, we tested whether increasing fusion can prevent ROS overproduction in HG conditions. We used a cell line that overexpresses the mitochondrial fusion protein Mfn2 in a tetracycline-inducible manner. Cells treated with tetracycline for 24 h formed elongated tubular mitochondria, indicating increased mitochondrial fusion (Fig. 9). Cells overexpressing Mfn2 showed little ROS increase in HG incubation, whereas control cells exhibited ROS increases (Fig. 4A and B). This result indicates that not only inhibiting fission but also increasing fusion of mitochondria prevents HG-induced ROS increase. Low ROS levels in HG conditions by overexpression of Mfn2 or DLP1-K38A were not due to the increased levels of antioxidant enzymes or mitochondrial uncoupling protein as judged by immunoblot (Fig. 9C).
Fig. 4.

Fission/fusion dynamics of mitochondria in HG-induced ROS increase. (A and B) Promoting mitochondrial fusion prevented HG-induced ROS increase. Whereas the ethidium fluorescence increased without tetracycline induction (A Upper and Control in B), Mfn2 overexpression by tetracycline greatly reduced ROS levels in HG (A Lower and Mfn2 in B). (C) Time-lapse images of mitochondria upon HG exposure. Tubular mitochondria (Upper Left) became fragmented in HG (Upper Right). Concurrent fissions occurred within tubules (Lower). Contraction and condensation of mitochondria are apparent (arrows).
It is unclear whether HG conditions induce increased fission or decreased fusion to fragment mitochondria. Time-lapse imaging of cells exposed to HG displayed multiple concurrent fissions within mitochondrial tubules along with contractions of short tubules to generate a fragmented phenotype (Figs. 4C and 9D), which shows a greatly increased rate of fission but no fusion regardless of the actual cause of mitochondrial fragmentation. It is clear, however, that functional mitochondrial fission machinery is required for HG-induced mitochondrial fragmentation (Fig. 2A).
ROS Levels Fluctuate During Prolonged Exposure to HG Conditions.
Because we observed a transient short burst of ROS in HG incubation, we investigated why continuous hyperglycemic conditions elicit a more harmful effect than insulin-controlled glucose concentrations (5, 25, 36–38). ROS levels measured during extended HG incubation revealed that after the initial transient, ROS increased again for a prolonged period and then decreased, suggesting that ROS levels cyclically fluctuate with continuous exposure to HG. In ROS measurements obtained every 2 h during continuous HG conditions, ROS increased a second time around the 10-h time and remained high until approximately the 18-h time of incubation before decreasing to the basal level (Fig. 5A). The extent of the ROS increase during the second burst was similar to the initial increase, but the duration was vastly increased (8–10 h) (Fig. 5A).
Fig. 5.

ROS levels fluctuate during prolonged exposure to HG conditions. (A) ROS levels were measured in Clone 9 cells with DHE every 2 h after incubation with 50 mM glucose. Fluorescence intensity was normalized against the intensity at time 0 (F/F0). The second phase of ROS increase was observed at 10-h incubation, and the ROS levels remained high until the 18-h time point, then decreased thereafter. (B and C) Fragmented mitochondria were prevalent in cells incubated for 10–18 h, and mitochondria returned to tubular morphology at 22–24 h in HG (gray bars in C). Mitochondria from cells incubated for 16 and 22 h are shown in B. Cells overexpressing DLP1-K38A maintained elongated tubular mitochondria during prolonged incubation with HG concentrations (C, solid bars). (D) Cells overexpressing GFP-DLP1-K38A (arrows in D) showed little change in ROS levels, which remained low throughout the extended incubation in HG (black bars) whereas ROS levels in untransfected cells (gray bars) increased and decreased.
Inhibition of DLP1 Function Blocks HG-Induced ROS Fluctuation.
As observed during the early period of HG incubation, cells containing fragmented mitochondria were prevalent at the time points showing high ROS levels during the extended incubation in HG conditions (Fig. 5 B and C), indicating that the temporal correlation between mitochondrial morphology and ROS increase persisted during the extended HG incubation. Again, the HG-induced mitochondrial fragmentation was prevented by DLP1-K38A (Fig. 5C). More importantly, inhibiting mitochondrial fission prevented the prolonged second ROS increase. ROS levels in cells overexpressing DLP1-K38A remained consistently low (Fig. 5D), indicating that mitochondrial fission participates in HG-induced ROS fluctuation.
Discussion
In this study, we observed a change of mitochondrial morphology that was accompanied by a ROS increase upon exposing cells to HG conditions. Whereas mitochondrial deformations that have been observed in diabetic patients and animals are likely terminal phenotypes caused by ROS-induced injury in prolonged hyperglycemic conditions, we identified a rapid and reversible change of mitochondrial morphology in HG conditions. Our study indicates that the early dynamic change of mitochondrial morphology is a causal factor necessary for HG-induced ROS increase. While our study was ongoing, mitochondrial fragmentation was also reported in cultured endothelial cells exposed to HG conditions (39). This independent observation is consistent with our findings and suggests that HG-induced change of mitochondrial morphology is a common event occurring in a variety of tissues.
Respiration Increase, Hyperpolarization, and ROS Overproduction in HG Conditions.
HG conditions increase the pyruvate production that subsequently enhances the input of reducing equivalents into the mitochondrial electron transport chain and leads to ROS production via mitochondrial hyperpolarization (2). Decreasing the mitochondrial membrane potential by uncoupling agents or overexpression of uncoupling protein prevents ROS production in hyperglycemic conditions (4, 34, 35). We also showed that mild mitochondrial uncoupling prevented the ROS increase in HG conditions (Fig. 3A), suggesting that mitochondrial hyperpolarization caused ROS increase in our experimental system.
Our data indicate that respiration increases in HG conditions (Fig. 3 E and F). Increased respiration was also observed along with increased mitochondrial membrane potential at the onset of hyperglycemia in animal studies (40). The respiratory chain pumps protons across the inner mitochondrial membrane to generate the proton-motive force. This proton circuit links the respiratory chain and the ATP synthase. The respiratory chain sets an upper limit on the rate of electron transport (respiration) and on the membrane potential. Under well coupled conditions, rapidly phosphorylating mitochondria have slightly lower membrane potential and, thus, generate less ROS (41). In HG conditions, however, more ROS are generated by hyperpolarization with increased respiration, suggesting a possible loss of coupling and a modification in the regulatory capacity of the respiratory chain. It is possible that the morphological change of mitochondria that we observed in HG may contribute to this process. Perhaps, mitochondrial fragmentation may affect structural and spatial organization of the respiratory chain and ATP synthase in such a way that alters electron transport and coupling, causing mitochondrial hyperpolarization and ROS increase in HG conditions.
Morphological Change of Mitochondria in HG Conditions.
Our data indicate that the mitochondrial morphology change in HG is required for the respiration increase and ROS overproduction (Fig. 3). Mitochondrial fragmentation still occurred under the mild mitochondrial uncoupling that prevented the ROS increase in HG conditions (Fig. 3B), suggesting that mitochondrial fragmentation precedes the hyperpolarization as well. In support of this notion, our preliminary data suggested that inhibition of mitochondrial fission prevented HG-induced mitochondrial hyperpolarization, as judged by JC-1 staining (results not shown). Further evaluation of these data are necessary because of many possible pitfalls in measuring the membrane potential in intact cells by using fluorescent probes (42). Our data also showed that inhibition of mitochondrial pyruvate uptake prevented HG-induced ROS increase, whereas mitochondrial fragmentation persisted (Fig. 2 E and F), which places the change of mitochondrial morphology upstream to the mitochondrial pyruvate uptake in the HG-induced sequence of events. This series of experimental data indicate that morphological change of mitochondria in HG incubation occurs as an early event that is necessary for respiration increase, mitochondrial hyperpolarization, and ROS overproduction.
What causes mitochondrial fragmentation in HG conditions is unknown. Our data showed that l-glucose did not cause mitochondrial fragmentation (Fig. 2D), whereas mitochondria still fragmented when mitochondrial pyruvate uptake was inhibited in HG conditions (Fig. 2F). These results suggest that mitochondrial fragmentation is likely induced by a signal generated between the cellular uptake of glucose and pyruvate production. Studies are underway to identify the signal that induces mitochondrial fragmentation in HG conditions.
Our data indicate that either inhibiting fission or promoting fusion prevents ROS increase in HG conditions (Figs. 3 and 4). Time-lapse imaging showed fission predominating over fusion (Fig. 4C). In some occasions, extremely rapid and concurrent fission events were observed within a 10-sec period (Fig. 9D). However, it is unknown whether HG-induced mitochondrial fragmentation is caused by increased fission, decreased fusion, or both. The aforementioned undefined signal generated from the glucose metabolism is likely to modulate the fission/fusion balance to induce fragmented mitochondria. Detecting specific molecular changes in fission or fusion proteins upon HG exposure would provide insight into not only determining whether fission increases or fusion decreases but also aid in identifying the signal that mediates HG-induced mitochondrial fragmentation.
Potential Role of HG-Induced Mitochondrial Fragmentation.
It has been hypothesized that mitochondrial activity is reflected in ultrastructural changes of mitochondria. Active mitochondria were proposed to be more condensed and to have an electron dense matrix (21). It is possible that small spherical mitochondria formed in HG conditions may represent condensed, metabolically active mitochondria. Time-lapse imaging showed that, after fissions, short tubular mitochondria became contracted to form spherical mitochondria, suggestive of possible condensation via the fragmentation process. Because our data show that the early morphological change of mitochondria is necessary for increased respiration in HG conditions (Fig. 3), mitochondrial fragmentation by HG exposure likely generates more active mitochondria. The morphological change leading to increased respiration, along with the reversibility of fragmentation in HG conditions, suggests that HG-induced mitochondrial fragmentation is a physiological event, as opposed to pathological fragmentation of mitochondria often occurring in apoptosis. Possibly, HG-induced morphological change of mitochondria is a cellular response to increased metabolic substrate to facilitate metabolic input into mitochondria. Rapid fragmentation of mitochondria, concomitantly increasing total mitochondrial surface area, may increase accessibility of metabolic substrate (e.g., pyruvate) to carrier proteins. Our data suggests that a signal generated during excess glucose metabolism induces mitochondrial fragmentation that leads to ROS overproduction via enhanced respiration and mitochondrial hyperpolarization.
Mitochondrial Fission/Fusion Machinery as a Therapeutic Target for ROS Injury in Hyperglycemia.
Why the early ROS increase is transient in HG is not known. Although insulin can decrease ROS by lowering glucose levels in the body, the rapid ROS transient we observed in cultured cells is an insulin-independent event. It is possible that initial ROS increase activates cellular ROS defense mechanism. Our observations that ROS levels fluctuate with prolonged increases under continuous exposure to HG may implicate diminished ability of cells to control ROS levels, possibly due to less-effective antioxidant mechanism in the later burst of ROS. These observations may account for the significantly increased morbidity associated with untreated hyperglycemia (5, 36, 37). Most importantly, we were able to prevent HG-induced ROS increase and subsequent fluctuation by inhibiting mitochondrial fragmentation. Our results indicate that mitochondrial dynamics can be a previously unrecognized therapeutic target to prevent the pathological effects of increased ROS production in not only diabetes and obesity, but also other disease states associated with hyperglycemia.
Materials and Methods
Cell Culture.
The cell lines Clone 9 [American Type Culture Collection (ATCC) CRL-1439] and H9c2 (ATCC CRL-1446) were maintained in Ham’s F-12K and DMEM plus 10% FBS. Transfections were performed by using Lipofectamine (Invitrogen). Cells (T-REx-HeLa; Invitrogen) overexpressing DLP1-K38A or Mfn2 in a tetracycline-inducible manner were maintained in MEM in the presence of Zeocin (200 μg/ml) and Blasticidin (5 μg/ml). DLP1-K38A or Mfn2 were overexpressed by incubating with 1 μg/ml tetracycline for 24 h.
ROS Measurements.
ROS production was detected by using the fluorescent probe carboxy-dichlorodihydrofluorescein diacetate (Molecular Probes) or DHE (Molecular Probes). Cells were preloaded with 25 μM carboxy-dichlorodihydrofluorescein diacetate or 5 μM DHE at 37°C for 30 min before shifting to a HG concentration. For experiments with longer incubation periods in a HG concentration, cells were loaded with the probe at each time point for 30 min.
Respiration Assay.
Oxygen consumption rates were measured in whole cells by using a Clark-type O2 electrode. Briefly, cells were suspended at concentration of 2 × 106 cell/ml at 37°C in a magnetically stirred 1.2-ml chamber. The decreases of the O2 concentration in the sealed chamber were measured as cellular oxygen consumption by an O2 meter connected to the electrode. At the end of each experiment, 5 μM rotenone was added to the chamber to confirm that decreases of the O2 concentration originated from mitochondrial respiration, and maximal mitochondrial respiration was obtained by adding 1 μM carbonyl cyanide p-(trifluoromethoxy)phenylhydrazone.
Fluorescence Microscopy and Image Analyses.
Images were acquired with a Evolution QEi camera (Media Cybernetics, Silver Spring, MD) driven by IPLab imaging software (Scanalytics, Billerica, MA) and fluorescence intensity was measured by using the iplab software. For morphological analyses of mitochondria, acquired images were analyzed as described in Fig. 7 by using imagej (National Institutes of Health).
Supplementary Material
Acknowledgments
We thank P. S. Brookes, S. Nadtochiy, and R. Fox for technical assistance and T. Gunter for his valuable insight. This study was supported by National Institutes of Health Grants DK073858 and DK061991 (to Y.Y.).
Abbreviations
- DCF
dichlorofluorescein
- DHE
dihydroethidium
- GFP-DLP1-K38A
GFP-tagged DLP1-K38A
- HG
high glucose
- ROS
reactive oxygen species.
Footnotes
Conflict of interest statement: No conflicts declared.
References
- 1.Baynes J. W., Thorpe S. R. Diabetes. 1999;48:1–9. doi: 10.2337/diabetes.48.1.1. [DOI] [PubMed] [Google Scholar]
- 2.Brownlee M. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 3.Green K., Brand M. D., Murphy M. P. Diabetes. 2004;53(Suppl. 1):S110–S118. doi: 10.2337/diabetes.53.2007.s110. [DOI] [PubMed] [Google Scholar]
- 4.Nishikawa T., Edelstein D., Du X. L., Yamagishi S., Matsumura T., Kaneda Y., Yorek M. A., Beebe D., Oates P. J., Hammes H. P., et al. Nature. 2000;404:787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 5.Van den Berghe G. J. Clin. Invest. 2004;114:1187–1195. doi: 10.1172/JCI23506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ouattara A., Lecomte P., Le Manach Y., Landi M., Jacqueminet S., Platonov I., Bonnet N., Riou B., Coriat P. Anesthesiology. 2005;103:687–694. doi: 10.1097/00000542-200510000-00006. [DOI] [PubMed] [Google Scholar]
- 7.Bereiter-Hahn J., Voth M. Microsc. Res. Tech. 1994;27:198–219. doi: 10.1002/jemt.1070270303. [DOI] [PubMed] [Google Scholar]
- 8.Smirnova E., Shurland D. L., Ryazantsev S. N., van der Bliek A. M. J. Cell Biol. 1998;143:351–358. doi: 10.1083/jcb.143.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pitts K. R., Yoon Y., Krueger E. W., McNiven M. A. Mol. Biol. Cell. 1999;10:4403–4417. doi: 10.1091/mbc.10.12.4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smirnova E., Griparic L., Shurland D. L., van der Bliek A. M. Mol. Biol. Cell. 2001;12:2245–2256. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoon Y., Krueger E. W., Oswald B. J., McNiven M. A. Mol. Cell. Biol. 2003;23:5409–5420. doi: 10.1128/MCB.23.15.5409-5420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen H., Chan D. C. Curr. Top. Dev. Biol. 2004;59:119–144. doi: 10.1016/S0070-2153(04)59005-1. [DOI] [PubMed] [Google Scholar]
- 13.Yoon Y. Cell Biochem. Biophys. 2004;41:193–205. doi: 10.1385/CBB:41:2:193. [DOI] [PubMed] [Google Scholar]
- 14.Yoon Y. Sci. STKE. 2005;2005(280):pe18. doi: 10.1126/stke.2802005pe18. [DOI] [PubMed] [Google Scholar]
- 15.Sesaki H., Jensen R. E. J. Cell Biol. 1999;147:699–706. doi: 10.1083/jcb.147.4.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen H., Chan D. C. Hum. Mol. Genet. 2005;14:R283–R289. doi: 10.1093/hmg/ddi270. [DOI] [PubMed] [Google Scholar]
- 17.Niemann A., Ruegg M., La Padula V., Schenone A., Suter U. J. Cell Biol. 2005;170:1067–1078. doi: 10.1083/jcb.200507087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alexander C., Votruba M., Pesch U. E., Thiselton D. L., Mayer S., Moore A., Rodriguez M., Kellner U., Leo-Kottler B., Auburger G., et al. Nat. Genet. 2000;26:211–215. doi: 10.1038/79944. [DOI] [PubMed] [Google Scholar]
- 19.Delettre C., Lenaers G., Griffoin J. M., Gigarel N., Lorenzo C., Belenguer P., Pelloquin L., Grosgeorge J., Turc-Carel C., Perret E., et al. Nat. Genet. 2000;26:207–210. doi: 10.1038/79936. [DOI] [PubMed] [Google Scholar]
- 20.Zuchner S., Mersiyanova I. V., Muglia M., Bissar-Tadmouri N., Rochelle J., Dadali E. L., Zappia M., Nelis E., Patitucci A., Senderek J., et al. Nat. Genet. 2004;36:449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
- 21.Hackenbrock C. R. J. Cell Biol. 1966;30:269–297. doi: 10.1083/jcb.30.2.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Riva A., Tandler B., Loffredo F., Vazquez E., Hoppel C. Am. J. Physiol. 2005;289:H868–H872. doi: 10.1152/ajpheart.00866.2004. [DOI] [PubMed] [Google Scholar]
- 23.Skulachev V. P. Trends Biochem. Sci. 2001;26:23–29. doi: 10.1016/s0968-0004(00)01735-7. [DOI] [PubMed] [Google Scholar]
- 24.Westermann B. EMBO Rep. 2002;3:527–531. doi: 10.1093/embo-reports/kvf113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vanhorebeek I., De Vos R., Mesotten D., Wouters P. J., De Wolf-Peeters C., Van den Berghe G. Lancet. 2005;365:53–59. doi: 10.1016/S0140-6736(04)17665-4. [DOI] [PubMed] [Google Scholar]
- 26.Kelley D. E., He J., Menshikova E. V., Ritov V. B. Diabetes. 2002;51:2944–2950. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- 27.Vincent A. M., Brownlee M., Russell J. W. Ann. N.Y. Acad. Sci. 2002;959:368–383. doi: 10.1111/j.1749-6632.2002.tb02108.x. [DOI] [PubMed] [Google Scholar]
- 28.Kiritoshi S., Nishikawa T., Sonoda K., Kukidome D., Senokuchi T., Matsuo T., Matsumura T., Tokunaga H., Brownlee M., Araki E. Diabetes. 2003;52:2570–2577. doi: 10.2337/diabetes.52.10.2570. [DOI] [PubMed] [Google Scholar]
- 29.De Vos K. J., Allan V. J., Grierson A. J., Sheetz M. P. Curr. Biol. 2005;15:678–683. doi: 10.1016/j.cub.2005.02.064. [DOI] [PubMed] [Google Scholar]
- 30.Koopman W. J., Verkaart S., Visch H. J., van der Westhuizen F. H., Murphy M. P., van den Heuvel L. W., Smeitink J. A., Willems P. H. Am. J. Physiol. 2005;288:C1440–C1450. doi: 10.1152/ajpcell.00607.2004. [DOI] [PubMed] [Google Scholar]
- 31.Yoon Y., Pitts K. R., McNiven M. A. Mol. Biol. Cell. 2001;12:2894–2905. doi: 10.1091/mbc.12.9.2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Halestrap A. P., Denton R. M. Biochem. J. 1974;138:313–316. doi: 10.1042/bj1380313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Halestrap A. P. Biochem. J. 1975;148:85–96. doi: 10.1042/bj1480085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Du X. L., Edelstein D., Dimmeler S., Ju Q., Sui C., Brownlee M. J. Clin. Invest. 2001;108:1341–1348. doi: 10.1172/JCI11235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin Y., Berg A. H., Iyengar P., Lam T. K., Giacca A., Combs T. P., Rajala M. W., Du X., Rollman B., Li W., et al. J. Biol. Chem. 2005;280:4617–4626. doi: 10.1074/jbc.M411863200. [DOI] [PubMed] [Google Scholar]
- 36.U.K. Prospective Diabetes Study Group Lancet. 1998;352:837–853. [PubMed] [Google Scholar]
- 37.Brown J. B., Nichols G. A., Perry A. Diabetes Care. 2004;27:1535–1540. doi: 10.2337/diacare.27.7.1535. [DOI] [PubMed] [Google Scholar]
- 38.Langouche L., Vanhorebeek I., Vlasselaers D., Vander Perre S., Wouters P. J., Skogstrand K., Hansen T. K., Van den Berghe G. J. Clin. Invest. 2005;115:2277–2286. [Google Scholar]
- 39.Paltauf-Doburzynska J., Malli R., Graier W. F. J. Cardiovasc. Pharmacol. 2004;44:423–436. doi: 10.1097/01.fjc.0000139449.64337.1b. [DOI] [PubMed] [Google Scholar]
- 40.Ferreira F. M., Palmeira C. M., Seica R., Moreno A. J., Santos M. S. J. Biochem. Mol. Toxicol. 2003;17:214–222. doi: 10.1002/jbt.10081. [DOI] [PubMed] [Google Scholar]
- 41.Nicholls D. G. Aging Cell. 2004;3:35–40. doi: 10.1111/j.1474-9728.2003.00079.x. [DOI] [PubMed] [Google Scholar]
- 42.Nicholls D. G., Ward M. W. Trends Neurosci. 2000;23:166–174. doi: 10.1016/s0166-2236(99)01534-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}