

Abstract
Two-pore-domain K+ channels provide neuronal background currents that establish resting membrane potential and input resistance; their modulation provides a prevalent mechanism for regulating cellular excitability. The so-called TASK channel subunits (TASK-1 and TASK-3) are widely expressed, and they are robustly inhibited by receptors that signal through Gαq family proteins. Here, we manipulated G protein expression and membrane phosphatidylinositol 4,5-bisphosphate (PIP2) levels in intact and cell-free systems to provide electrophysiological and biochemical evidence that inhibition of TASK channels by Gαq-linked receptors proceeds unabated in the absence of phospholipase C (PLC) activity, and instead involves association of activated Gαq subunits with the channels. Receptor-mediated inhibition of TASK channels was faster and less sensitive to a PLCβ1-ct minigene construct than inhibition of PIP2-sensitive Kir3.4(S143T) homomeric channels that is known to be dependent on PLC. TASK channels were strongly inhibited by constitutively active Gαq, even by a mutated version that is deficient in PLC activation. Receptor-mediated TASK channel inhibition required exogenous Gαq expression in fibroblasts derived from Gαq/11 knockout mice, but proceeded unabated in a cell line in which PIP2 levels were reduced by regulated overexpression of a lipid phosphatase. Direct application of activated Gαq, but not other G protein subunits, inhibited TASK channels in excised patches, and constitutively active Gαq subunits were selectively coimmunoprecipitated with TASK channels. These data indicate that receptor-mediated TASK channel inhibition is independent of PIP2 depletion, and they suggest a mechanism whereby channel modulation by Gαq occurs through direct interaction with the ion channel or a closely associated intermediary.
Keywords: G protein, KCNK, neuromodulation, phospholipase C, TASK
Receptors that couple specifically to Gαq-containing heterotrimeric G proteins regulate various types of ion channels in different settings. Included among these channels are the so-called TASK channel subunits (TASK-1, K2P3 and TASK-3, K2P9); these widely distributed members of the KCNK gene family generate pH- and anesthetic-sensitive background K+ currents and function to establish a negative resting membrane potential (1–3). Inhibition of TASK channels by various Gαq-coupled receptors is now established as an important mechanism for enhancing neuronal excitability and controlling hormone release (1–3).
Mechanisms engaged in receptor-mediated TASK channel inhibition have not been clearly established, in part because typical signaling intermediaries have not been implicated (2, 4). In this regard, an increasingly popular postulate has emerged for channel modulation by Gαq-coupled receptors that does not involve downstream mediators: Gαq-mediated activation of PLC depletes its substrate, phosphatidylinositol 4,5-bisphosphate (PIP2), which is a trace membrane phospholipid that is required for full activity of many channels (5, 6). This mechanism is implicated in receptor-mediated regulation of voltage-gated and inwardly rectifying K+ channels, voltage-dependent Ca2+ channels, and some TRP family cationic channels (5, 6). In the case of TASK channels, however, the evidence is less certain; although PIP2 can enhance recombinant TASK channel activity in heterologous expression systems (7, 8), Gαq-coupled receptors inhibit a native correlate of TASK channels in cerebellar neurons by a mechanism apparently independent of phospholipase C (PLC) (9).
In the studies reported here, we evaluated the PIP2 depletion hypothesis using mammalian heterologous expression systems in which receptor signaling to TASK channels was reconstituted and in which expression of G proteins and levels of PIP2 could be manipulated. Moreover, because our data were not easily reconciled with a mechanism involving PLC activation and PIP2 depletion, we also tested whether activated Gαq subunits associate with TASK channels in intact cells and inhibit TASK channel activity when applied directly to cell-free patches. These studies demonstrate a mechanism of ion channel modulation by Gαq-linked receptors that is independent of PLC-mediated PIP2 hydrolysis. Furthermore, they provide evidence that TASK channels or a closely associated intermediary may be direct effectors for activated Gαq subunits.
Results
Distinctly Different Characteristics of Receptor-Mediated TASK and Kir3.4* Channel Inhibition.
We reconstituted receptor-mediated TASK channel modulation in a mammalian heterologous expression system by transiently coexpressing either TASK-1 or TASK-3 in HEK293 cells that stably express a Gαq-linked TRHR1 receptor (Fig. 1A). In these cells, TASK channel inhibition by thyrotropin-releasing hormone (TRH) was robust (≈85% inhibition) and rapid (10.8 ± 1.8 s and 11.7 ± 2.1 s for 10–90% inhibition of TASK-1 and TASK-3). Receptor-mediated inhibition of TASK-1 and TASK-3 was unaffected by cotransfected βARKct, a Gβγ sink (10, 11) (Fig. 1B); inhibition was only slightly, although significantly, decreased by PLCβ1ct (Fig. 1B), a construct that interferes with PLC-mediated PIP2 hydrolysis (12) and with modulation of PIP2-regulated Ca2+ and K+ channels (11) by Gαq-coupled receptors.
Fig. 1.

Inhibition of TASK and Kir3.4* channels is distinctly different. (A) Whole cell conductance (slope conductance between −130 mV and −60 mV) in HEK293 cells stably expressing TRHR1 receptor and transiently transfected with TASK-1 (Left) or TASK-3 (Right). TRH (200 nM) inhibited pH-sensitive TASK channels. (B) Averaged (±SEM) TRH-mediated TASK inhibition (% control) in cells transfected with βARK-ct, a Gβγ sink, or PLCβ1-ct, a Gαq sink (∗, P < 0.05 by ANOVA, n ≥ 5 per group). (C) Conductance in cells expressing a homomeric mutant Kir channel [Kir3.4(S143T), Kir3.4*] in 25 mM extracellular KCl. TRH caused fast activation of Kir3.4* conductance followed by a delayed inhibition. (D) Averaged (±SEM) TRH-induced activation (Left) and inhibition (Right) of Kir3.4* (% of control) was reduced in cells transfected with βARK-ct or PLCβ1-ct (∗, P < 0.05 by ANOVA, n ≥ 11 per group).
For comparison, we examined the effect of TRH on a channel for which PIP2 depletion is the established mechanism of channel inhibition, specifically a pore-mutated Kir3.4(S143T) construct (Kir3.4*) that forms PIP2-sensitive homomeric inwardly rectifying K+ channels (13). TRH had a biphasic effect on currents from Kir3.4* channels (Fig. 1C). A rapid Kir3.4* activation was blocked by βARKct (Fig. 1D Left), as expected for a Gβγ-mediated action (14), whereas slow inhibition of Kir3.4* was substantially diminished by PLCβ1ct (Fig. 1D Right), as expected for PLC-mediated effects of Gαq (13, 15). In the presence of βARKct, to remove confounding effects of current activation, TRH inhibited ≈50% of Kir3.4* current with 10–90% inhibition requiring over a minute (73.2 ± 6.9 s). Thus, in the same test system, TASK channel inhibition by this Gαq-linked receptor is much faster and less sensitive to PLCβ1ct than inhibition of a Kir channel that is known to depend on PIP2 depletion (13, 15).
Receptor-Mediated TASK Channel Inhibition Requires Gαq.
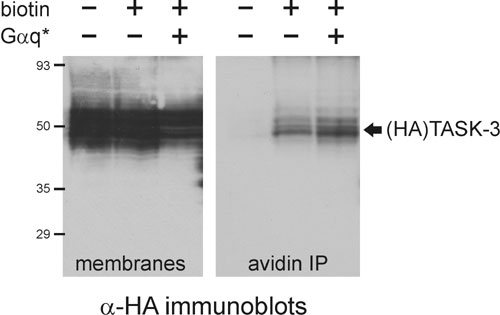
We tested whether constitutively active G protein subunits can modulate TASK channel activity independent of receptor activation. TASK channel currents were strongly inhibited in cells expressing GTPase-deficient forms of Gαq family proteins, Gαq* or Gα11*, as exemplified for TASK-3 (Fig. 2A Left); identical results were obtained with TASK-1 (data not shown). The decrease in current was not due to diminished cell-surface expression of the channels because we recovered similar levels of TASK-3 immunoreactivity in avidin precipitates from control and Gαq*-expressing cells after biotinylation of membrane proteins (see Fig. 6, which is published as supporting information on the PNAS web site). TASK currents were unaffected in cells transfected with other constitutively active Gα subunits, including Gαi2*, Gαs*, and Gα13*, or with Gβ1γ2 (Fig. 2A Left). We also examined effects of Gαq* on a TASK-3/TREK-1 chimeric channel in which six residues of TREK-1 were substituted into TASK channels at the C-terminal cytoplasmic-membrane interface; this chimera generates pH-sensitive currents that are not inhibited by Gαq-coupled receptors (4). Accordingly, we found that receptor-insensitive TASK-3/TREK-1 channel currents were unaffected by Gαq* (Fig. 2A Center). Notably, also, a mutated form of Gαq* that bears a C-terminal substitution rendering it deficient in PLC activation, Gαq*(AA) (16, 17), was as effective as the nonmutated form of Gαq* in mediating inhibition of WT TASK channels (Fig. 2A Right). These data suggest that inhibition of TASK currents by constitutively active Gαq/11 subunits does not require PLC and reflects a mechanism similar to that evoked by Gαq-coupled receptors.
Fig. 2.

TASK channel inhibition involves Gαq. (A) Normalized pH-sensitive conductance in cells transfected with TASK-3 and constitutively active G protein α subunits or Gβ1γ2. (Left) Only Gαq* or Gα11* inhibited TASK-3 currents. (Center) Gαq* did not decrease conductance in cells expressing a TASK-3/TREK chimeric channel that is not receptor modulated. (Right) The PLC-inactive Gαq*(AA) mutant was as effective as Gαq* at inhibiting TASK-3 (∗, P < 0.05 by ANOVA, n ≥ 6 per group). (B) The effect of bath acidification and TRH was determined in Fq/11 fibroblasts from Gαq/11 knockout mice transfected with the indicated constructs. (C) TRH inhibition of pH-sensitive TASK conductance in Fq/11 cells was rescued by expression of either Gαq or Gαq(AA) (∗, P < 0.05 by ANOVA, n ≥ 6 per group).
To demonstrate a role for Gαq in channel modulation by receptors, we reconstituted receptor-mediated TASK channel inhibition in fibroblasts derived from Gαq/11 knockout mice (Fig. 2 B and C) (18). In these Fq/11 cells transfected only with TASK-3 and TRHR1, the pH-sensitive TASK currents were unaffected by agonist application; in contrast, when Gαq was cotransfected along with TASK-3 and TRHR1, the signaling pathway was fully reconstituted and TRH strongly inhibited TASK currents (Fig. 2B). Likewise, TRH inhibition of TASK currents was rescued when Fq/11 cells were cotransfected with a Gαq subunit bearing the mutation that interferes with PLC activation [Gαq(AA), Fig. 2C]. These data indicate that Gαq is necessary for receptor-mediated TASK channel inhibition, but they also suggest that PLC activation is not required.
Receptor-Mediated TASK Channel Inhibition Is Independent of PLC Activity and PIP2 Depletion.
As an additional test of PLC and PIP2 involvement in TASK channel inhibition, we transfected TASK channels into a stable cell line in which expression of a lipid phosphatase, phosphoinositide-specific inositol polyphosphate 5-phosphatase IV (5ptase IV) is under tetracycline control (19). In these cells, induction of 5ptase IV causes ≈8- to 15-fold depletion of PIP2 after 10–12 h (19). Under these conditions, pH-sensitive TASK-3 currents were undiminished by 5ptase IV expression (Fig. 3A and B). Likewise, receptor-mediated channel inhibition was totally preserved in PIP2-depleted cells, whether induced by ATP via an endogenous P2Y receptor or by TRH via the coexpressed TRHR1 (Fig. 3 A and B). TASK-1 currents and modulation were also not different in control and PIP2-depleted cells (data not shown). Expression of 5ptase IV did not affect recovery of TASK-3 currents after receptor-mediated inhibition, either in magnitude or time course (Fig. 3A). Notably, recovery from TRH-mediated inhibition averaged 90.6 ± 4.5% in control cells and 83.2 ± 7.2% in PIP2-depleted cells, with 50% recovery occurring within 1.2 ± 0.2 and 1.3 ± 0.2 min (n = 5 cells each). By comparison with cells stably expressing TRHR1 (cf. Fig. 1), current recovery was faster and more complete in these transiently transfected cells, even after PIP2 depletion.
Fig. 3.

TASK channel inhibition is preserved in cells depleted of PIP2. (A) An HEK293 cell line stably expressing 5ptase IV under tetracycline control was transfected with TASK-3 and recorded without tetracycline exposure (control) or after 5ptase IV induction by tetracycline (tet) for 12–16 h (tetracycline). Bath acidification and alkalization revealed a robust pH-sensitive TASK conductance that was inhibited by P2Y receptor activation (ATP, 100 μM), even in tet-treated cells. (B) Averaged data (± SEM) illustrating that tet-induced expression of 5ptase IV was without effect on TASK-3 conductance (Left, n ≥ 15 per group) or channel inhibition by ATP or TRH (Right, n ≥ 6 per group). (C) In cells expressing PIP2-sensitive K+ channels (Kir3.4*, Kir2.1, and KV7.2/7.3 channels), tet-induced 5ptase IV expression decreased channel activity. Averaged normalized conductance (Kir3.4* or Kir2.1) or current density (at +20 mV; KV7.2/7.3) from control or tet-treated cells (∗, P < 0.05 by unpaired t test; n ≥ 14 per group). (D) Fura-2-imaging of ATP-induced calcium mobilization by endogenous P2Y receptors in control and tet-treated cells. Representative fields illustrate that calcium transients evoked by ATP (100 μM) in nearly all control cells were largely absent from tet-treated cells (Left). The histogram presents the percentage of control and PIP2-depleted cells capable of mounting a calcium response to ATP (Right; pooled data from six fields include >300 cells for each condition).
Control experiments demonstrated that 5ptase IV induction strongly reduced currents from PIP2-sensitive inwardly rectifying and voltage-dependent K+ channels, specifically Kir3.4*, Kir2.1, and KV7.2/7.3 (Fig. 3C). The results with Kir2.1 are particularly noteworthy. Those channels have an especially high affinity for PIP2 (13, 20, 21) so the inhibition of Kir2.1 currents after tetracycline treatment indicates substantial PIP2 depletion in these cells. Moreover, the high affinity of Kir2.1 channels for PIP2 is thought to account for their resistance to modulation by receptor-mediated PIP2 depletion (5, 13). If TASK channel activity requires PIP2, as suggested (7, 8), then the observation that TASK channels were totally unaffected under these same conditions implies that they have even greater affinity for PIP2 than Kir2.1 channels. Nevertheless, TASK channels remain robustly inhibited by activation of Gαq-linked receptors in 5ptase IV-expressing cells. These data from TASK channels, therefore, do not conform to results expected from the PIP2 depletion hypothesis of receptor modulation.
As an additional control for PIP2 depletion, we found that ATP-induced Ca2+ transients, which were universally observed in control cells, were abolished in tetracycline-treated, 5ptase IV-expressing cells (Fig. 3D). Ionomycin-induced Ca2+ transients were observed in all PIP2-depleted cells, indicating that intracellular Ca2+ stores were intact under these conditions (data not shown). So, even as these PIP2-depleted cells could no longer mount a PLC-dependent calcium response to P2Y receptor activation (Fig. 3D), ATP completely retained its ability to inhibit TASK channel currents (Fig. 3 A and B). Overall, these data indicate that TASK channels are relatively insensitive to changes in PIP2 levels and that receptor-mediated inhibition proceeds unabated in cells depleted of PIP2.
Activated Gαq Inhibits TASK Channels in Cell-Free Patches.
To this point, the data indicate that receptor-mediated inhibition of TASK channels involves Gαq but is independent of PLC. Therefore, we tested whether purified, recombinant Gαq can inhibit TASK channel activity when applied directly to excised inside-out macropatches (Fig. 4). TASK-3 macropatch currents from transfected cells were identified by their weakly rectifying current-voltage profile and activation by halothane (Fig. 4A). TASK-3 currents were strongly inhibited by GTPγS-loaded (i.e., activated) Gαq, but unaffected by the CHAPS (3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate) detergent used to solubilize the G proteins, by activation buffer alone, or by nonactivated Gαq-GDP (Fig. 4 A and D). Inhibition of TASK-3 macropatch currents was obtained with two separate preparations of purified Gαq. On the other hand, TASK channels were not inhibited by other classes of Gα subunits (Gαs-GTPγS and Gαi-GTPγS) or by Gβ1γ2 subunits (Fig. 4 B and D). The activity of all recombinant G protein subunits was verified in independent in vitro assays (i.e., all tested G protein subunits form heterotrimers, couple functionally to receptors, and activate relevant effectors) (22, 23). Because PLC activity is Ca2+-dependent (requiring ≈10–100 nM free Ca2+) (24) and our experiments were performed in a Ca2+-free bath (containing zero added Ca2+ and 5 mM EGTA), it is unlikely that channel inhibition resulted from Gαq-stimulation of any residual PLC retained in the patches. Moreover, recombinant Gαq-GTPγS was fully effective in patches pretreated with U73122, a PLC inhibitor (Fig. 4 C and D).
Fig. 4.

Activated Gαq subunits inhibit TASK-3 in excised inside-out macropatches. (A) Currents recorded from a representative multichannel macropatch excised from an HEK293 cell transfected with TASK-3. Time series of currents (at +60 mV) from patches exposed sequentially to halothane, G protein activation buffer, nonactivated Gαq-GDP, and Gαq preactivated with GTPγS. The boxed region is expanded in the Center, and ramp currents under the indicated conditions provided in the Right. (B) TASK-3 currents from a patch exposed to GTPγS-loaded Gαi before Gαq-GTPγS. (C) TASK-3 currents from a patch exposed to Gαq-GTPγS during prolonged treatment with the PLC inhibitor U73122 (5 μM). (D) Averaged inhibition (% of control, ± SEM) from macropatches treated with various G protein subunits. Of those tested, only Gαq-GTPγS inhibited TASK-3 currents.
Activated Gαq Coassociates with TASK Channels.
Because we observed TASK channel inhibition by Gαq in cell-free patches, we reasoned that the G protein might remain in close association with chronically inhibited channels observed in cells expressing constitutively active Gαq* subunits. To test this possibility, we cotransfected HEK293 cells with hemagglutinin (HA)-tagged TASK subunits along with either WT or constitutively active Gαq* and assayed HA immunoprecipitates for the presence of Gαq (Fig. 5). In accord with these expectations, we found that constitutively active Gαq* was selectively coimmunoprecipitated with either HA-tagged TASK-1 (HA-T1) or HA-tagged TASK-3 (HA-T3); WT Gαq was expressed at comparable levels but it did not associate with the channels. In addition, we were able to retrieve Gα11* from HA-immunoprecipitates when that highly homologous Gα subunit, which also caused TASK channel inhibition, was cotransfected with an HA-tagged TASK channel (see Fig. 7, which is published as supporting information on the PNAS web site). Thus, activated Gαq/11 subunits associate with TASK channels in cells under conditions when channel activity is strongly diminished.
Fig. 5.

Activated Gαq subunits coassociate with TASK channels. Cell lysates and HA-immunoprecipitates (HA-IP) were obtained from HEK293 cells cotransfected with the indicated HA-tagged TASK channel subunits (T1, TASK-1; T3, TASK-3) and either WT (q) or constitutively active Gαq (q*). After SDS/PAGE, immunoblotting for Gαq revealed only active Gαq* in the HA-IP (Left, arrow), but both Gαq and Gαq* in the cell lysate (Center); both HA-tagged TASK channels were obtained in the HA-IP (Right, arrowhead). TASK-3 channels expressed at higher levels than TASK-1 channels, as reflected in the HA blot. Blots are representative of at least four replicate experiments.
Discussion
Inhibition of background K+ channels by Gαq-coupled receptors is of widespread importance for regulation of cell excitability, but the relevant signaling mechanisms have proven elusive. We demonstrate that TASK channel subunits coassociate in intact cells with activated Gαq, the G protein subunit that recapitulates channel inhibition when applied directly to cell-free patches and which is necessary for receptor-mediated TASK channel inhibition in fibroblasts derived from Gαq/11 knockout mice. TASK channel inhibition by Gαq is independent of PLC because channel modulation can be reconstituted in cells expressing Gαq subunits deficient in PLC activation and in inside-out patches under conditions where PLC cannot be activated. Moreover, TASK channel currents are undiminished and receptor-mediated channel inhibition is completely unaffected in cells depleted of PIP2, the PLC substrate. The most parsimonious interpretation of the accumulated evidence is that TASK channels are themselves direct effectors for Gαq, although interaction with another closely associated intermediary has not been excluded.
PLC-Independent Inhibition of TASK Channels.
These studies identify a mechanism for channel inhibition by Gαq that is independent of PLC activation and PIP2 depletion. In this respect, the mechanism we propose for inhibition of TASK channels is distinctly different from that suggested earlier to explain TASK channel inhibition (7, 8), as well as other examples of channel modulation by Gαq-coupled receptors (e.g., Kv, Kir, TRP, and Ca2+ channels; for reviews see refs. 5 and 6).
According to the prevailing hypothesis, activation of PLC by Gαq-coupled receptors leads to a depletion of PIP2 in the membrane, thereby depriving the channels of this important channel-regulating lipid. The de facto standard of proof for implicating this mechanism includes primarily two types of observation: (i) PIP2 itself can activate the channels after membranes are depleted of PIP2 by using selective antibodies or polylysine; and (ii) pharmacological inhibition of PLC activation (with U73122) or of PIP2 replenishment (with wortmannin) disrupts receptor-mediated channel modulation or delays recovery of channel activity after receptor inhibition (5). To date, this minimum standard of proof represents the only evidence for PIP2 regulation of TASK channels (7, 8, 25), whereas a number of additional experimental tests, including channel mutagenesis and expression of minigene constructs that alter PIP2 levels, have been applied to validate the PIP2 hypothesis for other channels (15, 26, 27). For these other channels, such as the Kir and KV7.2/7.3 channels that we used as “positive controls,” our data also support their regulation by PIP2. However, for TASK channels that have not been so extensively scrutinized, we present multiple lines of evidence with alternative test systems that do not support the PIP2 depletion hypothesis.
We cannot fully explain the discrepant conclusions from our experiments and previous studies implicating PIP2 in TASK channel activation. It is possible that PIP2 application can activate TASK channel currents in patches after rundown but does not regulate constitutively active channels in intact cells. For example, it may be that other, more abundant phospholipids regulate TASK channel activity (as recently found for the related TREK-1 channels; see ref. 28). In this case, decreased channel activity in excised patches might reflect the ability of polylysine or high concentrations of PIP2 antibodies to disrupt interactions with other channel-regulating phospholipids; and addition of PIP2 after channel rundown may enhance TASK currents under those conditions even though PIP2 is not solely responsible for maintaining basal channel activity. Because our experiments provided little reason to suspect PIP2 as crucial for TASK channel activity or modulation, we did not attempt to replicate previous results with U73122 or wortmannin. We would point out that those pharmacological studies rely on use of a notoriously fickle blocker with many side effects to implicate PLC (i.e., U73122; see ref. 29 for excellent discussion) and, to provide evidence of a role for PIP2 depletion, they require high concentrations of wortmannin purposefully chosen to exploit nonselective effects of that drug (5). Instead, we established a simple experimental paradigm for testing the PIP2 hypothesis based on inducible expression of a lipid phosphatase in mammalian cells.
TASK Channels May Be Direct Effectors for Gαq.
Our functional data in excised patches, coupled with results from coimmunoprecipitation experiments, suggest a mechanism for TASK channel inhibition that involves direct actions of activated Gαq on the channel or on a closely associated intermediary. It is noteworthy that these conclusions are generally concordant with those from a recent study investigating modulation of Kir3.1/3.2 heteromeric channels by the Gαq-coupled substance P receptor (30). In that work, a role for PIP2 depletion in receptor-mediated channel modulation was discounted because mutations that enhanced apparent PIP2 affinity of the channels had no effect on the kinetics of receptor-mediated Kir channel inhibition. As with our results for TASK channels, Gαq was coimmunoprecipitated with the modulated Kir3.2 channels. These findings prompted the suggestion of a direct effect of Gαq on those Kir channels, although functional confirmation by application of purified Gαq to Kir channels in patches was not provided (30). Nevertheless, those data from Kir3.1/3.2 heteromeric channels suggest that additional or alternative actions of Gαq, independent of PLC activation and PIP2 depletion, may be a more general feature of signaling from Gαq-coupled receptors to ion channels than currently realized.
Aside from PLC, the only other direct effector described for Gαq to date is the protein tyrosine kinase Btk (31), although Btk is activated by other Gα subunits (e.g., Gα12) (32), and like PLC, also by Gβγ dimers (33). TASK channel modulation is not likely due to Btk because inhibition proceeds undiminished in mutated channels that remove identified tyrosine phosphorylation sites (4). There are other reports of PLC-independent effects of Gαq and Gαq-coupled receptors. For example, the PLC-inactive Gαq*(AA) inhibits phosphatidylinositol 3-kinase and activates glycogen synthase kinase (17, 34), and activation of the small G protein RhoA by Gαq seems to be independent of PLC (35). It is unlikely that TASK channel modulation is downstream of RhoA because channel activity was unaffected by cotransfected Gα13*, which activates RhoA, and because neither constitutively active or dominant negative RhoA constructs have any effect on receptor-mediated TASK channel inhibition (N.P., E.M.T., and D.A.B., unpublished observations). Finally, genetic evidence from Caenorhabditis elegans indicates that egl-30 (Gαq) functions largely independently of egl-8 (PLCβ) in serotonin-mediated egg-laying behavior (36), again consistent with alternative mediators for downstream effects of Gαq. The mechanisms underlying these other PLC-independent actions have not been determined, and additional direct effectors for Gαq subunits may yet be identified.
Materials and Methods
DNA Constructs.
Rat TASK channel constructs were as described (4, 37). Kir2.1 was obtained from L. Y. Jan (University of California, San Francisco), Kir3.4 from N. Davidson (California Institute of Technology, Pasadena), and KV7.2/7.3 from D. McKinnon (State University of New York, Stony Brook). We used PCR to generate a homomeric Kir3.4 channel bearing an S143T mutation (Kir3.4*) (38) and to introduce the PLC-disrupting mutation (R256T257→AA; see mutant no. 10 in ref. 16) into both WT Gαq and constitutively active (Q204L) Gαq construct (both from Guthrie cDNA Resource, University of Missouri-Rolla). The GFP-tagged PLCβ1-ct and βARK-ct were as described (15). All constructs were fully verified by sequence analysis.
Cell Culture and Transfection.
HEK 293 cells stably expressing TRHR1 receptor (E2 cells, from G. Milligan (39), tetracycline-inducible 5ptase IV cells, from P. Majerus (19), and embryonic fibroblasts from Gq/11 knockout mice [Fq/11 cells, from P. Hinkle (18)] were grown under conditions described by each investigator. Cells were transfected with indicated constructs, usually together with GFP (pGreenLantern) by using Lipofectamine 2000 (Invitrogen). Induction of 5ptase IV cells was achieved by treating with tetracycline (0.1 μg/ml for 12–16 h).
G Protein Purification and Activation.
Recombinant G protein α and βγ subunits were expressed in Sf9 cells and purified, as described (22, 23). G protein α subunits were activated in a buffer containing 10 mM Hepes (pH 7.2), 5 mM EGTA, 2 mM MgCl2, 10 NaCl, 130 mM KCl, 0.01% CHAPS, 0.2 mM DTT, and 1 mM GTPγS at 30°C for 30–45 min before addition to the excised inside-out patches at the indicated concentrations (25–50 nM). The activity of all G protein subunits was verified in independent in vitro assays before being applied to patches (22, 23).
Electrophysiology.
Whole-cell recordings were obtained at room temperature by using 3- to 5-MΩ patch pipettes and an Axopatch 200B amplifier (Axon Instruments, Union City, CA) in a bath solution composed of 140 mM NaCl, 3 mM KCl, 2 mM MgCl2, 2 mM CaCl2, 10 mM Hepes, and 10 mM glucose, with pH adjusted as indicated. For recording Kir channels, external KCl was raised to 25 mM, with corresponding reduction in NaCl. Internal solution contained 120 mM KCH4SO3, 4 mM NaCl2, 1 mM MgCl2, 0.5 mM CaCl2, 10 mM Hepes, 10 mM EGTA, 3 mM Mg-ATP, 0.3 mM GTP-Tris (pH 7.2). For excised inside-out macropatch recordings we used ≈1-MΩ patch pipettes containing 140 mM NaCl, 3 mM KCl, and 10 mM Hepes (pH 7.3) and a nominally Ca2+-free bath solution that contained 150 mM KCl, 5 mM EGTA, 1 mM MgCl2 and 10 mM Hepes (pH 7.3). Where indicated, bath KCl was substituted equimolar with CsCl or the bath solution was supplemented with 5 mM BaCl2 or halothane (by bubbling with a room air gas mixture through calibrated vaporizers).
Data Acquisition and Analysis.
Ramp voltage clamp commands were applied at 5-s intervals by using pclamp software and a Digidata 1322A digitizer (Axon Instruments). Whole-cell slope conductance was derived by linear fits to ramp currents (between −130 and −60 mV) and normalized to cell capacitance. The pH-sensitive conductance was taken as the difference in whole-cell conductance at pH 8.4 and pH 5.9. Patch current amplitudes were obtained at 60 mV. All data are expressed as means (± SEM); statistical tests included one-way analysis of variance (ANOVA) or Student’s t test, as indicated, with P < 0.05 considered statistically significant.
Calcium Imaging.
Cells were incubated with 5 μM Fura-2, AM (Molecular Probes) for 45 min at 37°C. Recordings were performed in a low-volume chamber continuously perfused at ≈2 ml·min−1 with solutions at 25°C. Fluorescence measurements were made with a Zeiss Axioskop FS upright microscope fitted with a ×20 water immersion objective. Fura-2 was excited at 340 nm and 380 nm with a Polychrome IV monochromator (TILL Photonics, Planegg, Germany) and emitted fluorescence filtered with a 510-nm longpass filter. Images were acquired by using an OrcaER charge-coupled device (CCD) camera (Hamamatsu). Acquisition and analysis were performed with metafluor software (Molecular Devices).
Immunoprecipitation and Western Blotting.
Cells were transfected with HA-tagged TASK-1 or TASK-3 (4) along with WT or constitutively active Gα subunits [Gαq*, a gift from J. H. Exton (Vanderbilt University, Nashville, TN); Gαq, a gift from W. F. Simonds (National Institutes of Health); and Gα11, a gift from M. Simon (California Institute of Technology)]. Cells were lysed in Tris-buffered saline containing 1% CHAPS and protease inhibitors. HA-tagged channels were immunoprecipitated by incubation first with mouse anti-HA (12CA5, gift from I. G. Macara, University of Virginia, Charlottesville) and then with protein G-agarose beads (Sigma-Aldrich). The precipitated complexes were separated by SDS/PAGE and immunoblotted by using a rabbit anti-Gαq antibody (Santa Cruz Biotechnology) or the 12CA5 anti-HA antibody directly conjugated to horseradish peroxidase. As a further control for nonspecific binding, in addition to transfection with WT Gαq, we found that neither the channel or Gαq subunits were precipitated from cells transfected with channels lacking the HA epitope (data not shown).
Supplementary Material
Acknowledgments
We thank Dr. J. C. Lawrence for help with immunoprecipitations and Laura Johnson for initial recordings from Kv7.2/7.3 channels. This work was supported by Spanish Ministry of Education and Science Grant SAF2004-01011 (to F.V.), the American Heart Association (E.M.T.), and National Institutes of Health Grants DK19552 (to J.C.G.) and NS33583 (to D.A.B.).
Abbreviations
- PIP2
phosphatidylinositol 4,5-bisphosphate
- PLC
phospholipase C
- TRH
thyrotropin-releasing hormone
- 5ptase IV
phosphoinositide-specific inositol polyphosphate 5-phosphatase IV
- HA
hemagglutinin.
Footnotes
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Goldstein S. A., Bockenhauer D., O’Kelly I., Zilberberg N. Nat. Rev. Neurosci. 2001;2:175–184. doi: 10.1038/35058574. [DOI] [PubMed] [Google Scholar]
- 2.Talley E. M., Sirois J. E., Lei Q., Bayliss D. A. Neuroscientist. 2003;9:46–56. doi: 10.1177/1073858402239590. [DOI] [PubMed] [Google Scholar]
- 3.Lesage F. Neuropharmacology. 2003;44:1–7. doi: 10.1016/s0028-3908(02)00339-8. [DOI] [PubMed] [Google Scholar]
- 4.Talley E. M., Bayliss D. A. J. Biol. Chem. 2002;277:17733–17742. doi: 10.1074/jbc.M200502200. [DOI] [PubMed] [Google Scholar]
- 5.Suh B. C., Hille B. Curr. Opin. Neurobiol. 2005;15:370–378. doi: 10.1016/j.conb.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 6.Hilgemann D. W., Feng S., Nasuhoglu C. Sci. STKE. 2001 Dec 4; doi: 10.1126/stke.2001.111.re19. [DOI] [PubMed] [Google Scholar]
- 7.Chemin J., Girard C., Duprat F., Lesage F., Romey G., Lazdunski M. EMBO J. 2003;22:5403–5411. doi: 10.1093/emboj/cdg528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lopes C. M., Rohacs T., Czirjak G., Balla T., Enyedi P., Logothetis D. E. J. Physiol. 2005;564:117–129. doi: 10.1113/jphysiol.2004.081935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boyd D. F., Millar J. A., Watkins C. S., Mathie A. J. Physiol. 2000;529:321–331. doi: 10.1111/j.1469-7793.2000.00321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koch W. J., Hawes B. E., Inglese J., Luttrell L. M., Lefkowitz R. J. J. Biol. Chem. 1994;269:6193–6197. [PubMed] [Google Scholar]
- 11.Kammermeier P. J., Ikeda S. R. Neuron. 1999;22:819–829. doi: 10.1016/s0896-6273(00)80740-0. [DOI] [PubMed] [Google Scholar]
- 12.Wu D., Jiang H., Katz A., Simon M. I. J. Biol. Chem. 1993;268:3704–3709. [PubMed] [Google Scholar]
- 13.Kobrinsky E., Mirshahi T., Zhang H., Jin T., Logothetis D. E. Nat. Cell Biol. 2000;2:507–514. doi: 10.1038/35019544. [DOI] [PubMed] [Google Scholar]
- 14.Wickman K., Clapham D. E. Physiol. Rev. 1995;75:865–885. doi: 10.1152/physrev.1995.75.4.865. [DOI] [PubMed] [Google Scholar]
- 15.Lei Q., Talley E. M., Bayliss D. A. J. Biol. Chem. 2001;276:16720–16730. doi: 10.1074/jbc.M100207200. [DOI] [PubMed] [Google Scholar]
- 16.Venkatakrishnan G., Exton J. H. J. Biol. Chem. 1996;271:5066–5072. doi: 10.1074/jbc.271.9.5066. [DOI] [PubMed] [Google Scholar]
- 17.Fan G., Ballou L. M., Lin R. Z. J. Biol. Chem. 2003;278:52432–52436. doi: 10.1074/jbc.M310982200. [DOI] [PubMed] [Google Scholar]
- 18.Yu R., Hinkle P. M. J. Biol. Chem. 1999;274:15745–15750. doi: 10.1074/jbc.274.22.15745. [DOI] [PubMed] [Google Scholar]
- 19.Kisseleva M. V., Cao L., Majerus P. W. J. Biol. Chem. 2002;277:6266–6272. doi: 10.1074/jbc.M105969200. [DOI] [PubMed] [Google Scholar]
- 20.Huang C. L., Feng S., Hilgemann D. W. Nature. 1998;391:803–806. doi: 10.1038/35882. [DOI] [PubMed] [Google Scholar]
- 21.Zhang H., He C., Yan X., Mirshahi T., Logothetis D. E. Nat. Cell Biol. 1999;1:183–188. doi: 10.1038/11103. [DOI] [PubMed] [Google Scholar]
- 22.Kerchner K. R., Clay R. L., McCleery G., Watson N., McIntire W. E., Myung C. S., Garrison J. C. J. Biol. Chem. 2004;279:44554–44562. doi: 10.1074/jbc.M406071200. [DOI] [PubMed] [Google Scholar]
- 23.McIntire W. E., MacCleery G., Garrison J. C. J. Biol. Chem. 2001;276:15801–15809. doi: 10.1074/jbc.M011233200. [DOI] [PubMed] [Google Scholar]
- 24.Rebecchi M. J., Pentyala S. N. Physiol. Rev. 2000;80:1291–1335. doi: 10.1152/physrev.2000.80.4.1291. [DOI] [PubMed] [Google Scholar]
- 25.Czirjak G., Petheo G. L., Spat A., Enyedi P. Am. J. Physiol. Cell Physiol. 2001;281:C700–C708. doi: 10.1152/ajpcell.2001.281.2.C700. [DOI] [PubMed] [Google Scholar]
- 26.Winks J. S., Hughes S., Filippov A. K., Tatulian L., Abogadie F. C., Brown D. A., Marsh S. J. J. Neurosci. 2005;25:3400–3413. doi: 10.1523/JNEUROSCI.3231-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y., Gamper N., Hilgemann D. W., Shapiro M. S. J. Neurosci. 2005;25:9825–9835. doi: 10.1523/JNEUROSCI.2597-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chemin J., Patel A. J., Duprat F., Lauritzen I., Lazdunski M., Honore E. EMBO J. 2005;24:44–53. doi: 10.1038/sj.emboj.7600494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Horowitz L. F., Hirdes W., Suh B. C., Hilgemann D. W., Mackie K., Hille B. J. Gen. Physiol. 2005;126:243–262. doi: 10.1085/jgp.200509309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koike-Tani M., Collins J. M., Kawano T., Zhao P., Zhao Q., Kozasa T., Nakajima S., Nakajima Y. J. Physiol. 2005;564:489–500. doi: 10.1113/jphysiol.2004.079285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bence K., Ma W., Kozasa T., Huang X. Y. Nature. 1997;389:296–299. doi: 10.1038/38520. [DOI] [PubMed] [Google Scholar]
- 32.Jiang Y., Ma W., Wan Y., Kozasa T., Hattori S., Huang X. Y. Nature. 1998;395:808–813. doi: 10.1038/27454. [DOI] [PubMed] [Google Scholar]
- 33.Langhans-Rajasekaran S. A., Wan Y., Huang X. Y. Proc. Natl. Acad. Sci. USA. 1995;92:8601–8605. doi: 10.1073/pnas.92.19.8601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ballou L. M., Lin H. Y., Fan G., Jiang Y. P., Lin R. Z. J. Biol. Chem. 2003;278:23472–23479. doi: 10.1074/jbc.M212232200. [DOI] [PubMed] [Google Scholar]
- 35.Vogt S., Grosse R., Schultz G., Offermanns S. J. Biol. Chem. 2003;278:28743–28749. doi: 10.1074/jbc.M304570200. [DOI] [PubMed] [Google Scholar]
- 36.Bastiani C. A., Gharib S., Simon M. I., Sternberg P. W. Genetics. 2003;165:1805–1822. doi: 10.1093/genetics/165.4.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berg A. P., Talley E. M., Manger J. P., Bayliss D. A. J. Neurosci. 2004;24:6693–6702. doi: 10.1523/JNEUROSCI.1408-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vivaudou M., Chan K. W., Sui J. L., Jan L. Y., Reuveny E., Logothetis D. E. J. Biol. Chem. 1997;272:31553–31560. doi: 10.1074/jbc.272.50.31553. [DOI] [PubMed] [Google Scholar]
- 39.Kim G. D., Carr I. C., Anderson L. A., Zabavnik J., Eidne K. A., Milligan G. J. Biol. Chem. 1994;269:19933–19940. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}