

Abstract
Reelin is a glycoprotein that is essential for the correct cytoarchitectonic organization of the developing CNS. Its function in the adult brain is less understood, although it has been proposed that Reelin is involved in signaling pathways linked to neurodegeneration. Here we analyzed Reelin expression in brains and cerebrospinal fluid (CSF) from Alzheimer’s disease (AD) patients and nondemented controls. We found a 40% increase in the Reelin protein levels in the cortex of AD patients compared with controls. Similar increases were detected at the Reelin mRNA transcriptional level. This expression correlates with parallel increases in CSF but not in plasma samples. Next, we examined whether CSF Reelin levels were also altered in neurological diseases, including frontotemporal dementia, progressive supranuclear palsy, and Parkinson’s disease. The Reelin 180-kDa band increased in all of the neurodegenerative disorders analyzed. Moreover, the 180-kDa Reelin levels correlated positively with Tau protein in CSF. Finally, we studied the pattern of Reelin glycosylation by using several lectins and the anti-HNK-1 antibody. Glycosylation differed in plasma and CSF. Furthermore, the pattern of Reelin lectin binding differed between the CSF of controls and in AD. Our results show that Reelin is up-regulated in the brain and CSF in several neurodegenerative diseases and that CSF and plasma Reelin have distinct cellular origins, thereby supporting that Reelin is involved in the pathogenesis of a number of neurodegenerative diseases.
Keywords: HNK-1, neurodegeneration, cerebrospinal fluid, blood, biomarker
Reelin is an extracellular 420-kDa glycoprotein that binds to the transmembrane receptors apolipoprotein receptor 2 and very-low-density lipoprotein receptor (1, 2), which transduce the Reelin signal through the intracellular adapter disabled-1 (3–5). Reelin signaling triggers a disabled-1-dependent signaling cascade involving several kinases, which ultimately controls proper neuronal migration and positioning during CNS development (for review see ref. 6).
The complex pattern of Reelin expression is consistent with evidence that this protein has multiple roles in brain development and adult brain function (7–9). In the adult mammalian brain, Reelin has been proposed to influence synaptogenesis and neural plasticity and to favor memory formation (8–12). Reelin is also expressed in peripheral tissues, including the liver, and is detected in blood (10, 13). However, whether brain and other tissues contribute to the pool of Reelin in blood remains to be elucidated. In this context, we recently reported the presence of detectable levels of Reelin in adult cerebrospinal fluid (CSF) (14).
Furthermore, the involvement of the Reelin signaling pathway in neurodegeneration has also been proposed (1, 6, 9, 15–17). First, Reelin binds to apolipoprotein E (ApoE) receptors, and some ApoE gene polymorphisms are considered risk factors for Alzheimer’s disease (AD). Moreover, the lack of Reelin is associated with increased phosphorylation of Tau (1, 2, 18), whose hyperphosphorylation leads to intracellular tangles and neuronal degeneration (19). Disabled-1 binds to β-amyloid precursor protein (20, 21) family members, whose proteolytic processing leads to the Aβ-peptide-forming amyloid plaques. Recent data also indicate that Reelin is present in β-amyloid plaques in a transgenic mouse model of AD (22).
Reelin is cleaved in vivo at two sites, which results in the production of several fragments whose relative abundance differs in distinct tissues (13, 23). We previously demonstrated the presence of the three Reelin bands (full-length 420-kDa, 310-kDa, and 180-kDa fragments) in human CSF and a significant increase in the 180-kDa band in a cohort of AD patients as compared with control individuals (14). Here we further corroborate these findings and show that increased Reelin levels in CSF of AD patients correlate with augmented brain expression of Reelin at mRNA and protein levels. We also show that Reelin levels in CSF are increased in other neurological disorders including frontotemporal dementia, progressive supranuclear palsy, and Parkinson’s disease (PD). These findings indicate that Reelin may be a reliable molecular marker for neurodegenerative diseases. Finally, we show that the pattern of Reelin glycosylation in CSF and plasma differ, which indicates a distinct cellular origin.
Results
Western Blot Analysis of CSF Reelin in AD.
To confirm whether Reelin levels are altered in AD CSF, we analyzed Reelin expression in 19 AD patients and 11 nondemented controls (NDC) (Fig. 1A). Because methodological handling and storage conditions may influence the processing of Reelin (see ref. 24 and Fig. 6, which is published as supporting information on the PNAS web site), all analyses were performed in samples frozen at −80°C, thawing-freezing cycles were avoided, and heating before electrophoresis was limited to 3 min. Three typical Reelin-immunoreactive bands were observed in all CSF analyses by using the 142 antibody, in agreement with previous studies (14, 25, 26). In all cases, the 180-kDa fragment was more abundant than the 310-kDa and 420-kDa bands, the latter corresponding to full-length Reelin. A significant ≈40% increase in the 180-kDa Reelin fragment in AD cases was detected, compared with NDC subjects (P < 0.001; Fig. 1B). Moreover, 15 of 19 AD cases had values >0.62 (arbitrary densitometric units) for this band, whereas only 2 of 11 controls were above this value (Fig. 1B).
Fig. 1.

Immunodetection of CSF Reelin. (A) Representative blot of Reelin in CSF samples from AD and NDC. (B) Scatter plots for 180-kDa Reelin. The dashed line represents an arbitrary cutoff (0.62 arbitrary units). The data represent the means ± SE (determinations by duplicate). MMSE, MiniMental State Examination (see Supporting Text). ∗, Significantly different (P < 0.05) from the NDC group as assessed by Student’s t test. Immunoreactive bands are also shown. (C) Scatter plots for the full-length 420-kDa and 310-kDa Reelin fragments. Because the predominant 180-kDa band displayed greater immunoreactivity than the 410-kDa and 320-kDa fragments, semiquantitative analysis of all the fragments was performed over a range of exposure times to avoid loss of linearity during the long exposure times required for the detection of the large fragments. Accumulative immunoreactivity from the sum of higher-molecular-mass Reelin bands is also shown.
Increased 180-kDa Reelin immunoreactivity may not necessarily indicate increased full-length Reelin abundance, but increased Reelin proteolytic processing of Reelin. To determine whether the increased 180-kDa band reflected increased protein processing, we compared the concentrations of all three Reelin fragments in CSF. Our analyses revealed that the levels of the 420-kDa and 310-kDa bands were similar in AD and NDC cases (Fig. 1C). These observations confirm our previous findings and indicate that total Reelin levels are increased in the CSF of AD patients and mostly correspond to augmented intensity of the 180-kDa band.
Expression of Reelin in Brains of AD and Control Subjects.
To determine whether changes in CSF Reelin reflect differences in brain levels of this protein, we examined the expression of Reelin in tissue samples from the frontal cortex and cerebellum of AD and NDC cases. In agreement with previous reports (27, 28), the Western blot of cortical extracts revealed the typical three Reelin bands, similar to CSF samples. The major 180-kDa Reelin fragment was detected in all samples, with the 420-kDa and 310-kDa bands being faintly stained in most cases (Fig. 2A). Quantitative analyses showed a significant increase in both the 180-kDa band (P = 0.001; a 33% increase) and total Reelin content (the sum of the three bands; P = 0.001; a 40% increase) in the frontal cortex of AD patients compared with NDC subjects (Fig. 2C). In contrast, we found similar Reelin levels in the cerebellum of AD and NDC subjects (Fig. 2 B and D). These results show that Reelin levels in CSF parallel protein expression in brain areas targeted by AD.
Fig. 2.

Concentrations and gel mobility for Reelin fragments in AD and NDC brain. (A and B) Three Reelin bands at 420, 310, and 180 kDa in frontal cortex (A) and cerebellum extracts (B). In each determination (made in triplicate) protein was adjusted to ≈20 μg by lane, and α-tubulin (1:1,000; Sigma) immunoreactive intensity was used as a control of blotting efficiency (Lower). (C and D) Reelin immunoreactivity from the 180-kDa fragment, and accumulative from the three Reelin bands, for NDC and AD subjects in frontal cortex (C) and cerebellum (D). In one NDC sample, no higher-molecular-mass Reelin fragments were detected and estimated in frontal cortex. The data represent the means ± SE (determinations by duplicate). ∗, Significantly different (P < 0.05) from the NDC group.
A semiquantitative PCR assay was further designed to determine whether changes in Reelin protein corresponded to alterations in mRNA expression. mRNA was purified from the frontal cortex and cerebellum of the same cases described above, retrotranscribed, and PCR-amplified by using 33P-dATP as a tracer and a low number of cycles to guarantee a scalar cDNA amplification. Identical findings were observed in three independent assays. The gels shown in Fig. 3A exhibit an apparent increase in Reelin cDNA content in the AD frontal cortex compared with controls. Reelin mRNA levels showed a significant (64%) increase in AD patients (levels normalized with respect to GADPH mRNA; Fig. 3C). In contrast, Reelin mRNA expression was not altered in samples from the cerebellum of the same AD and control cases (Fig. 3 B and D). Together, these data indicate a transcription alteration of the Reelin gene in cortical regions affected by AD.
Fig. 3.

RNA expression in AD cerebral tissue. (A and B) RT-PCR-amplified cDNAs of Reelin and GAPDH from frontal cortex (A) and cerebellum (B) extracts were mixed in a single tube and electrophoretically separated in 4% acrylamide gels. The amplified bands were identifed according to their sizes (513 bp for Reelin cDNA and 444 bp for GAPDH cDNA) and developed by Typhoon 8600 scanning. (C and D) Densitometric quantitation of the Reelin/GAPDH ratio of RNA expression from frontal cortex (C) and cerebellar extracts (D). ∗, Significantly different (P < 0.05) from the NDC group.
Western Blot Analysis of Plasma Reelin in Subjects with AD.
To determine whether the Reelin increase in CSF of AD cases is also detected in other biopsy fluids, we analyzed the levels of this protein in the plasma of 9 AD patients, 12 subjects with mild cognitive impairment, and 44 NDC subjects (Fig. 7, which is published as supporting information on the PNAS web site). Similar to previous reports (13, 24), the relative abundance of Reelin bands differed in plasma and CSF. Neither the intensity of the individual bands nor their relative banding pattern was altered in plasma from AD and mild cognitive impairment patients compared with NDC cases. These findings show that the increased Reelin expression in brain tissue and CSF samples of AD patients does not correlate with plasma levels of this protein.
Western Blot Analysis of CSF Reelin in Other Neurological Disorders.
In a previous report we found that the 180-kDa Reelin band was also increased in the CSF of frontotemporal dementia patients compared with controls (14). To assess whether Reelin levels in CSF are increased in other pathological disorders, we examined the levels of this protein in CSF samples from several neurological diseases (Fig. 8, which is published as supporting information on the PNAS web site). Significant increases in Reelin were detected in the 180-kDa band in samples from frontotemporal dementia, progressive supranuclear palsy, and PD patients, similar to those observed in AD cases. Once again, the concentrations of the full-length 420-kDa Reelin and 310-kDa bands did not differ significantly from controls (data not shown). We thus conclude that the 180-kDa Reelin band increases significantly in the CSF of all of the neurodegenerative diseases analyzed.
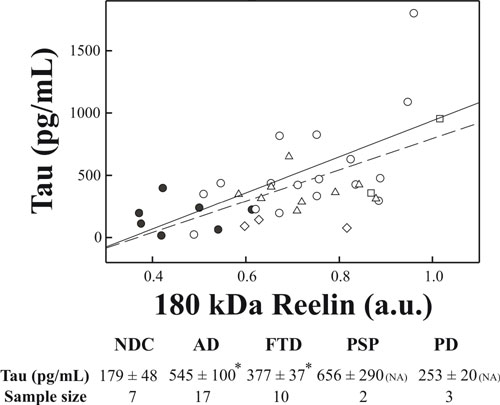
Because Tau protein is a well established CSF marker of many neurodegenerative disorders, we next compared Tau and Reelin levels in CSF samples (Fig. 9, which is published as supporting information on the PNAS web site). Interestingly, there was a positive correlation between Tau levels and the intensity of the 180-kDa band in AD patients and NDC cases (r = 0.66 and P = 0.006, linear regression), which was maintained when all samples were analyzed together (r = 0.55 and P < 0.001).
Lectin Binding and HNK-1 Immunoreactivity of CSF and Plasma Indicate Distinct Reelin Origins.
The above data indicated that CSF and plasma Reelin might originate in distinct tissues. To compare the pattern of Reelin glycosylation in CSF and plasma, samples from five NDC cases were incubated with several immobilized lectins [Canavalia ensiformis lectin (Con A), Lens culinaris agglutinin (LCA), Ricinus communis agglutinin (RCA120), and Triticum vulgaris agglutinin (wheat germ, WGA)] (Fig. 4A) that recognize distinct sugar sequences (29). Plasma Reelin was bound strongly to Con A, RCA, and WGA lectins and more weakly to LCA. All three Reelin fragments exhibited this pattern of labeling. In contrast, 420-kDa Reelin in CSF was strongly recognized by Con A and WGA, but also by LCA, whereas binding was weaker for RCA. Indeed, in contrast to CSF samples, RCA binding was conspicuous for the 180-kDa band in plasma, whereas this band was poorly recognized by LCA and WGA. These differences in the binding properties of plasma and CSF Reelin to lectins indicate a distinct pattern of glycosylation and thus a different cellular origin of Reelin protein in these two fluids.
Fig. 4.

Glycosylation of Reelin in CSF and plasma. (A) Five nonpathological CSF and plasma samples were incubated with immobilized lectins Con A, LCA, WGA, and RCA. Attempts to assay the Reelin bound to each lectin by resuspending and boiling the resin showed only 60–80% of recovery of the glycoprotein. The unbound Reelin, assayed by Western blotting, in the supernatant fraction was therefore used to compare differences in lectin binding between groups. The data represent the percentages of bound Reelin calculated after subtraction of unbound immunoreactivity for each band. We grouped values into four categories: +++, 75–100% of Reelin binding to lectin; ++, 50–74% binding; +, 25–49% binding; −, 0–24% binding. (B) Lectin binding of 180-kDa Reelin in gender- and age-matched CSF samples for 9 NDC and 11 AD subjects. (C) Scatter plots for the 180-kDa Reelin LCA/Con A ratio (% 180-kDa Reelin unbound to LCA/% 180-kDa Reelin unbound to Con A). The dashed line represents an arbitrary cutoff that maximally discriminated between AD and NDC groups (≈35 arbitrary units). The data represent the means ± SE. ∗, Significantly different (P < 0.05) from the NDC group as assessed by Student’s t test.
To study whether Reelin glycosylation is altered in AD, we further analyzed the pattern of glycosylation in CSF from NDC and AD subjects (Fig. 4B). There was a small but significant difference in the binding of the 180-kDa Reelin to LCA (P = 0.02) between the AD group and controls and a nonstatistically significant increase in the proportion of 180-kDa Reelin that does not bind to Con A (P = 0.06) in AD samples compared with the NDCs. We defined a quotient LCA/Con A as the 180-kDa Reelin unbound to LCA divided by the 180-kDa Reelin unbound to Con A for each sample, which provided greater discrimination between the two groups (P = 0.008; Fig. 4C). With this analysis, 10 of 11 samples from cases with AD were below an arbitrary cutoff point, whereas seven of the nine CSF samples from control cases were above this cutoff.
The HNK-1 glycoepitope, an unusual 3′-sulfated glucuronic acid sequence characteristic of neural recognition molecules, serves as a ligand in cell interactions (30). To determine whether Reelin contains HNK-1, we processed for Western blot G10-immunoprecipitated Reelin using the anti-HNK-1 antibody. Immunoprecipitation was confirmed by Western blotting by using the anti-Reelin 142 antibody (Fig. 5). The 180-kDa band contained the HNK-1 epitope in samples from the frontal cortex. We also identified a faint HNK-1 band in the 180-kDa Reelin band of CSF samples but not in plasma samples. These results further indicate that brain/CSF and plasma Reelin have essentially distinct cellular origins. Moreover, the presence of the HNK-1 epitope in brain Reelin opens up the possibility that this sequence may participate in Reelin functions in the brain.
Fig. 5.

Reelin carries the HNK-1 carbohydrate. Reelin from frontal cortex (lane 1), CSF (lane 3), and plasma samples (lane 5) was immunoprecipitated by using the G10 antibody (lanes 2, 4, and 6). The experiments were repeated three times. Western blot analyses were performed by using 142 (anti-Reelin; A) and Ab2 (anti-HNK-1; B) antibodies. B shows the immunoreactivity of the 180-kDa cortical frontex (lane 2) for HNK-1 and more weakly for CSF-Reelin (lane 4). In contrast, no HNK-1 band was detected in Reelin immunoprecipitated from plasma (lane 6).
Discussion
Reelin Levels Are Increased in the CSF and Brain, but Not Plasma, of AD Patients.
Several studies have reported decreased Reelin mRNA and protein levels in brains in several psychiatric disorders, including schizophrenia, bipolar disorder, and autism, which were correlated with decreased plasma protein levels (27, 28, 31). Recent evidence indicates that this is mainly caused by alterations in the methylation of the Reelin gene, which may lead to altered Reelin gene expression throughout the body (32). Our previous study showed detectable levels of Reelin in CSF and increased levels of the 180-kDa fragment in AD patients (14). Detection of Reelin in CSF has also been reported in another study (26). Here we used a cohort of AD samples, analyzed with the 142 anti-Reelin antibody, which exhibits a high affinity for human Reelin (13, 33). We detected the 420-kDa, 310-kDa, and 180-kDa Reelin bands, confirming a marked increase in the shorter fragment in the CSF and brains of AD. Although a differential proteolytic processing of Reelin in AD may also contribute to the increase in the 180-kDa band, the observation that the higher-molecular-mass Reelin bands are constant in AD and NDC indicates a net increase in Reelin protein abundance in AD.
We also found that the pattern of Reelin-lectin binding is altered for two mannose-specific lectins, LCA and Con A. Several neuropathological disorders, including AD, cause characteristic changes in the glycosylation pattern of proteins (34), which may be due to alterations in the contribution of different cell types to the protein pool, imbalance of protein glycoforms, or altered glycosylation mechanism. The cellular origin of the abnormally glycosylated Reelin and whether the altered glycosylation pattern in AD is a direct consequence of altered metabolism or reflects changes in differentiation state warrant further study.
In a recent report Ignatova et al. (26) failed to confirm altered levels of the 180-kDa Reelin fragment in AD samples. We believe that either the reduced sample size or the handling of the samples may have contributed to the divergent results. Here we report consistent evidence not only of increased Reelin levels in CSF and affected brain areas of AD patients but also of increased transcriptional activity of the Reelin gene. Moreover, our data show that CSF storage and heating conditions are key factors in determining Reelin protein levels, in agreement with another study (24). Our covariance analyses further show that these differences are not attributable to age or gender (see Supporting Text and Table 1, which are published as supporting information on the PNAS web site).
Reelin as a Marker of Neurodegenerative Diseases.
To examine whether Reelin could be used as a tool in the differential diagnosis of AD and other dementias and neurological disorders, we also analyzed CSF Reelin levels in samples from frontotemporal dementia, progressive supranuclear palsy, and PD subjects. We found a significant increase in the levels of this protein in the CSF of all these neurodegenerative diseases when compared with controls. Furthermore, Reelin levels were not substantially different among the distinct diseases, indicating that increased expression of this protein in this fluid is not disease-specific. Our findings indicate that increased Reelin levels in CSF could be considered a general marker for neurodegenerative diseases. Whether this reflects the participation of Reelin in the pathogenesis of these diseases or is secondary to the degenerative process itself remains to be elucidated.
CSF and Plasma Reelin Are Likely to Be Synthesized in Distinct Tissues.
Because of the lack of efficient biomarkers for neurodegenerative disorders in blood, here we studied whether plasma Reelin levels correlate with AD. We did not detect major changes in Reelin protein levels in plasma, in contrast to the data in CSF and brain. These results imply that Reelin detection in plasma is not an efficient prognostic marker for CNS neurodegenerative disorders and indicate that plasma and CSF Reelin may have distinct cell origins.
In addition to the CNS, Reelin is expressed in other organs including kidney, liver, and adrenal and pituitary glands (10, 13). We found that Reelin is differentially glycosylated in CSF and plasma, as detected with several lectins. Moreover, only brain and CSF Reelin reacts with the Ab-2 antibody recognizing the glycoepitope HNK-1. Because it is believed that different cell types add distinct carbohydrate moieties to the same glycoproteins, our results indicate that Reelin in these two fluids mainly reflects their expression and secretion by distinct cell types. The observations that Reelin levels in CSF correlate with those in brain and that Reelin is probably not expressed in human choroid plexus cells (unpublished observations) indicate that CSF Reelin is largely originated in brain. However, we cannot discard a small contribution of brain Reelin to plasma Reelin levels, because the Reelin receptor apolipoprotein receptor 2 is expressed by choroid plexus cells (35, 36).
Reelin is likely to play a role in long-term synaptic plasticity in adulthood by activating signaling cascades similar to those acting in neural development (37). The expression of the recognition molecule-associated carbohydrate HNK-1 in Reelin raises the possibility that this epitope may contribute to Reelin function. HNK-1 expression in molecules such as integrins, proteoglycans, and glycolipids is believed to be involved in the functional fine-tuning of these molecules in processes such as migration, cell aggregation, and differentiation (30, 38), processes in which Reelin has been implicated.
Reelin and Neurodegeneration in AD.
The observation of marked increases in Reelin protein levels in brains from AD patients, accompanied by a similar increase in Reelin transcripts, further supports the notion that the levels of this protein are intrinsically altered in this pathology. Furthermore, Reelin is overexpressed in cortical regions targeted by AD but not in the cerebella of the same patients. These observations, together with the close correlation of the 180-kDa Reelin band levels and Tau protein in CSF, indicate that altered expression of Reelin and abnormal Reelin signaling may participate in the pathogenesis of AD. The molecular mechanisms by which Reelin contributes to AD are not known. In this context, recent data have shown the presence of Reelin associated with amyloid plaques in a transgenic AD mouse model (22). Although alterations in Reelin-positive Cajal–Retzius cells in AD appear to be conflictive (39, 40), the expression of Reelin in cortical interneurons has not been investigated. Moreover, Reelin binds to ApoE receptors, and several downstream targets of the Reelin pathway are mediators of Tau hyperphosphorylation (2). In this context, it is interesting to note that Reelin signaling is believed to control both cyclin-dependent kinase 5 and glycogen synthase kinase 3, two key kinases that regulate Tau phosphorylation in AD (41–43). Furthermore, the in vitro interaction of Reelin with lipoprotein receptors is inhibited in the presence of ApoE3 and ApoE4 alleles (1), the latter being genetically associated with late-onset AD (44). The presence of ApoE also reduces Tau protein phosphorylation protein kinase activity in Reelin-deficient mice (17).
In summary, the present findings show that the expression of the Reelin gene and Reelin protein is increased in brains of AD patients. This expression correlates with similar increases in CSF but not in plasma samples, indicating that Reelin has distinct cell origins in these two tissue fluids. Finally, our data indicate that Reelin overexpression in CSF may be a general biomarker for several neurodegenerative diseases, although the precise mechanisms through which Reelin participates in the pathogenesis of these disorders require further study.
Materials and Methods
Preparation of Samples.
For diagnosis criteria see Supporting Text. CSF and plasma aliquot samples were stored at −80°C until their use. Pieces (0.2 g) of human frontal cortex and cerebellum (same cases) stored at −80°C were thawed slowly at 4°C and homogenized (10% wt/vol) in 50 mM Tris-HCl, pH 7.4/150 mM NaCl/0.5% Triton X-100/0.5% Nonidet P-40 and a mixture of proteinase inhibitors (34). The homogenates were sonicated and centrifuged at 20,000 × g at 4°C for 20 min, and the supernatant was collected and frozen at −80°C until assays.
Western Blot Analysis of Reelin.
Reelin was determined essentially as described (24). Briefly, CSF (50 μl), plasma (1.2 μl), and brain (20 μg) samples were analyzed on 6% SDS resolving gels. The sample boiling time was minimized to 3 min, and electrophoresis was allowed to proceed at a voltage that prevented excessive heat. Proteins were blotted onto nitrocellulose membranes (Schleicher & Schuell). Blots were blocked and incubated in monoclonal mouse anti-Reelin antibody 142 (1:200 dilution; Chemicon International; see ref. 33). Reelin was detected by ECL by using the ECL-Plus kit (Amersham Pharmacia). Two control samples were used to normalize immunoreactive Reelin signal. For semiquantitative studies, the intensity of Reelin bands was measured by densitometry by using science lab image gauge v4.0 software provided by Fuji Photo Film.
Semiquantitative PCR Assay for Reelin RNA.
Total RNA from brain tissue was purified by using an SV Total RNA purification kit (Promega), and 2 μg of RNA was retrotranscribed by adding the antisense oligonucleotides to be used in the PCR assays instead of oligo(dT). This assay consisted essentially of the same methodology reported by Álvarez-Dolado et al. (45) for the quantification of Reelin mRNA (see Supporting Text).
Lectin Binding Analysis of Reelin.
Aliquots (100 μl) of CSF and plasma (diluted 1:20 in PBS) were mixed with 40 μl of immobilized lectins (Sigma) and incubated overnight at 4°C. Unbound Reelin was separated by centrifugation and examined by Western blotting.
Reelin Immunoprecipitation and HNK-1 Western Blot Analysis.
Brain (frontal cortex), CSF, and plasma (diluted 1:2 and 1:20, respectively, in RIPA buffer) samples from NDC subjects were preadsorbed to protein A Sepharose (Amersham Pharmacia Biosciences) and incubated with the anti-Reelin G10 antibody (Chemicon International; see ref. 33). Immune complexes were precipitated by adding protein A Sepharose. The supernatant was collected, and the resin was washed, resuspended, and boiled in SDS/PAGE sample buffer. Reelin was detected by using the 142 antibody, and HNK-1 was detected by using the mouse Ab-2 antibody (1:500 dilution; Neomarkers Lab Vision).
Supplementary Material
Acknowledgments
We thank Dr. A. M. Goffinet (University of Louvain Medical School, Brussels) for the generous gift of Reelin antibodies used during preliminary experiments. J.S.-V. was supported by grants from La Caixa Foundation and the Fondo de Investigaciones Sanitarias Grant 03/0038; E.S. and R.B. were supported by La Caixa Foundation, the Pfizer Foundation, La Marató TV3, and Ministerio de Ciencia y Tecnología Grant SAF94-07929; J.M.U. was supported by Fondo de Investigaciones Sanitarias Grant 04/2280; and J.A.D.R. was supported by Ministerio de Ciencia y Tecnología Grant BFI2003-03594.
Abbreviations
- AD
Alzheimer’s disease
- ApoE
apolipoprotein E
- CSF
cerebrospinal fluid
- PD
Parkinson’s disease
- Con A
Canavalia ensiformis lectin
- LCA
Lens culinaris agglutinin
- RCA
Ricinus communis agglutinin
- WGA
Triticum vulgaris agglutinin
- NDC
nondemented control.
Footnotes
Conflict of interest statement: No conflicts declared.
References
- 1.D’Arcangelo G., Homayouni R., Keshvara L., Rice D. S., Sheldon M., Curran T. Neuron. 1999;24:471–479. doi: 10.1016/s0896-6273(00)80860-0. [DOI] [PubMed] [Google Scholar]
- 2.Hiesberger T., Trommsdorff M., Howell B. W., Goffinet A., Mumby M. C., Cooper J. A., Herz J. Neuron. 1999;24:481–489. doi: 10.1016/s0896-6273(00)80861-2. [DOI] [PubMed] [Google Scholar]
- 3.Bar I., Goffinet A. M. Nature. 1999;399:645–646. doi: 10.1038/21340. [DOI] [PubMed] [Google Scholar]
- 4.Cooper J. A., Howell B. W. Cell. 1999;97:671–674. doi: 10.1016/s0092-8674(00)80778-3. [DOI] [PubMed] [Google Scholar]
- 5.Trommsdorff M., Gotthardt M., Hiesberger T., Shelton J., Stockinger W., Nimpf J., Hammer R. E., Richardson J. A., Herz J. Cell. 1999;97:689–701. doi: 10.1016/s0092-8674(00)80782-5. [DOI] [PubMed] [Google Scholar]
- 6.Rice D. S., Curran T. Annu. Rev. Neurosci. 2001;24:1005–1039. doi: 10.1146/annurev.neuro.24.1.1005. [DOI] [PubMed] [Google Scholar]
- 7.Alcántara S., Ruiz M., D’Arcangelo G., Ezan F., de Lecea L., Curran T., Sotelo C., Soriano E. J. Neurosci. 1998;18:7779–7799. doi: 10.1523/JNEUROSCI.18-19-07779.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martínez-Cerdeño V., Galazo M. J., Cavada C., Clasca F. Cereb. Cortex. 2002;12:1298–1311. doi: 10.1093/cercor/12.12.1298. [DOI] [PubMed] [Google Scholar]
- 9.Weeber E. J., Beffert U., Jones C., Christian J. M., Forster E., Sweatt J. D., Herz J. J. Biol. Chem. 2002;277:39944–39952. doi: 10.1074/jbc.M205147200. [DOI] [PubMed] [Google Scholar]
- 10.Ikeda Y., Terashima T. Dev. Dyn. 1997;210:157–172. doi: 10.1002/(SICI)1097-0177(199710)210:2<157::AID-AJA8>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 11.Niu S., Renfro A., Quattrocchi C. C., Sheldon M., D’Arcangelo G. Neuron. 2004;41:71–84. doi: 10.1016/s0896-6273(03)00819-5. [DOI] [PubMed] [Google Scholar]
- 12.Roberts R. C., Xu L., Roche J. K., Kirkpatrick B. J. Comp. Neurol. 2005;482:294–308. doi: 10.1002/cne.20408. [DOI] [PubMed] [Google Scholar]
- 13.Smalheiser N. R., Costa E., Guidotti A., Impagnatiello F., Auta J., Lacor P., Kriho V., Pappas G. D. Proc. Natl. Acad. Sci. USA. 2000;97:1281–1286. doi: 10.1073/pnas.97.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sáez-Valero J., Costell M., Sjögren M., Andreasen N., Blennnow K., Luque J. M. J. Neurosci. Res. 2003;72:132–136. doi: 10.1002/jnr.10554. [DOI] [PubMed] [Google Scholar]
- 15.Bothwell M., Giniger E. Cell. 2000;102:271–273. doi: 10.1016/s0092-8674(00)00032-5. [DOI] [PubMed] [Google Scholar]
- 16.Herz J., Beffert U. Nat. Rev. Neurosci. 2000;1:51–58. doi: 10.1038/35036221. [DOI] [PubMed] [Google Scholar]
- 17.Ohkubo N., Lee Y. D., Morishima A., Terashima T., Kikkawa S., Tohyama M., Sakanaka M., Tanaka J., Maeda N., Vitek M. P., et al. FASEB J. 2003;17:295–297. doi: 10.1096/fj.02-0434fje. [DOI] [PubMed] [Google Scholar]
- 18.Tueting P., Costa E., Dwivedi Y., Guidotti A., Impagnatiello F., Manev R., Pesold C. NeuroReport. 1999;10:1329–1334. doi: 10.1097/00001756-199904260-00032. [DOI] [PubMed] [Google Scholar]
- 19.Iqbal K., Grundke-Iqbal I., Zaidi T., Merz P. A., Wen G. Y., Shaikh S. S., Johnson G., Moore S. W. Int. J. Dev. Neurosci. 2001;19:439–445. [Google Scholar]
- 20.Howell B. W., Lanier L. M., Frank R., Gertler F. B., Cooper J. A. Mol. Cell. Biol. 1999;19:5179–5188. doi: 10.1128/mcb.19.7.5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Homayouni R., Rice D. S., Sheldon M., Curran T. J. Neurosci. 1999;19:7507–7515. doi: 10.1523/JNEUROSCI.19-17-07507.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wirths O., Multhaup G., Czech C., Blanchard V., Tremp G., Pradier L., Beyreuther K., Bayer T. A. Neurosci. Lett. 2001;316:145–148. doi: 10.1016/s0304-3940(01)02399-0. [DOI] [PubMed] [Google Scholar]
- 23.Jossin Y., Ignatova N., Hiesberger T., Herz J., Lambert de Rouvroit C., Goffinet A. M. J. Neurosci. 2004;24:514–521. doi: 10.1523/JNEUROSCI.3408-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lugli G., Krueger J. M., Davis J. M., Persico A. M., Keller F., Smalheiser N. R. BMC Biochem. 2003;4:9. doi: 10.1186/1471-2091-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lacor P. N., Grayson D. R., Auta J., Sugaya I., Costa E., Guidotti A. Proc. Natl. Acad. Sci. USA. 2000;97:3556–3561. doi: 10.1073/pnas.050589597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ignatova N., Sindic C. J., Goffinet A. M. Neurobiol. Dis. 2004;15:326–330. doi: 10.1016/j.nbd.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 27.Impagnatiello F., Guidotti A. R., Pesold C., Dwivedi Y., Caruncho H., Pisu M. G., Uzunov D. P., Smalheiser N. R., Davis J. M., Pandey G. N., et al. Proc. Natl. Acad. Sci. USA. 1998;95:15718–15723. doi: 10.1073/pnas.95.26.15718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fatemi S. H., Kroll J. L., Stary J. M. NeuroReport. 2001;12:3209–3215. doi: 10.1097/00001756-200110290-00014. [DOI] [PubMed] [Google Scholar]
- 29.Lis H., Sharon N. Annu. Rev. Biochem. 1986;55:35–67. doi: 10.1146/annurev.bi.55.070186.000343. [DOI] [PubMed] [Google Scholar]
- 30.Schachner M., Martini R. Trends Neurosci. 1995;18:183–191. doi: 10.1016/0166-2236(95)93899-9. [DOI] [PubMed] [Google Scholar]
- 31.Persico A. M., D’Agruma L., Maiorano N., Totaro A., Militerni R., Bravaccio C., Wassink T. H., Schneider C., Melmed R., Trillo S., et al. Mol. Psychiatry. 2001;6:150–159. doi: 10.1038/sj.mp.4000850. [DOI] [PubMed] [Google Scholar]
- 32.Grayson D. R., Jia X., Chen Y., Sharma R. P., Mitchell C. P., Guidotti A., Costa E. Proc. Natl. Acad. Sci. USA. 2005;102:9341–9346. doi: 10.1073/pnas.0503736102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Bergeyck V., Naerhuyzen B., Goffinet A. M., Lambert de Rouvroit C. J. Neurosci. Methods. 1998;82:17–24. doi: 10.1016/s0165-0270(98)00024-7. [DOI] [PubMed] [Google Scholar]
- 34.Sáez-Valero J., Sberna G., McLean C., Small D. H. J. Neurochem. 1999;72:1600–1608. doi: 10.1046/j.1471-4159.1999.721600.x. [DOI] [PubMed] [Google Scholar]
- 35.Kim D. H., Iijima H., Got K., Sakai J., Ishii H., Kim H. J., Suzuki H., Kondo H., Saeki S., Yamamoto T. J. Biol. Chem. 1996;271:8373–8380. doi: 10.1074/jbc.271.14.8373. [DOI] [PubMed] [Google Scholar]
- 36.Stockinger W., Hengstschlager-Ottnad E., Novak S., Matus A., Hüttinger M., Bauer J., Lassmann H., Schneider W. J., Nimpf J. J. Biol. Chem. 1998;273:32213–32221. doi: 10.1074/jbc.273.48.32213. [DOI] [PubMed] [Google Scholar]
- 37.Chen Y., Beffert U., Ertunc M., Tang T. S., Kavalali E. T., Bezprozvanny I., Herz J. J. Neurosci. 2005;25:8209–8216. doi: 10.1523/JNEUROSCI.1951-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bronner-Fraser M. Dev. Biol. 1987;123:321–331. doi: 10.1016/0012-1606(87)90390-3. [DOI] [PubMed] [Google Scholar]
- 39.Riedel A., Miettinen R., Stieler J., Mikkonen M., Alafuzoff I., Soininen H., Arendt T. Acta Neuropathol. 2003;106:291–302. doi: 10.1007/s00401-003-0729-7. [DOI] [PubMed] [Google Scholar]
- 40.Baloyannis S. J. Int. J. Neurosci. 2005;115:965–980. doi: 10.1080/00207450590901396. [DOI] [PubMed] [Google Scholar]
- 41.Patrick G. N., Zukerberg L., Nikolic M., de la Monte S., Dikkes P., Tsai L. H. Nature. 1999;402:615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- 42.Beffert U., Morfini G., Bock H. H., Reyna H., Brady S. T., Herz J. J. Biol. Chem. 2002;277:49958–49964. doi: 10.1074/jbc.M209205200. [DOI] [PubMed] [Google Scholar]
- 43.González-Billault C., Del Río J. A., Ureña J. M., Jiménez-Mateos E. M., Barallobre M. J., Pascual M., Pujadas L., Simó S., Torre A. L., Gavín R., et al. Cereb. Cortex. 2005;15:1134–1145. doi: 10.1093/cercor/bhh213. [DOI] [PubMed] [Google Scholar]
- 44.Schmechel D. E., Saunders A. M., Strittmatter W. J., Crain B. J., Hulette C. M., Joo S. H., Pericak-Vance M. A., Goldgaber D., Roses A. D. Proc. Natl. Acad. Sci. USA. 1993;90:9649–9653. doi: 10.1073/pnas.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Álvarez-Dolado M., Ruiz M., del Río J. A., Alcántara S., Burgaya F., Sheldon M., Nakajima K., Bernal J., Howell B. W., Curran T., et al. J. Neurosci. 1999;19:6979–6993. doi: 10.1523/JNEUROSCI.19-16-06979.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}