

Abstract
Here we investigate the involvement of PKCɛ in apical actin remodeling in carbachol-stimulated exocytosis in reconstituted rabbit lacrimal acinar cells. Lacrimal acinar PKCɛ co-sedimented with actin filaments in an actin filament binding assay. Stimulation of acini with carbachol (100 μM, 2–15 min) significantly (p ≤ 0.05) increased PKCɛ recovery with actin filaments in two distinct biochemical assays, while confocal fluorescence microscopy showed a significant increase in PKCɛ association with apical actin in stimulated acini as evidenced by quantitative colocalization analysis. Overexpression of dominant negative (DN) PKCɛ in lacrimal acini using replication-defective adenovirus (Ad) resulted in profound alterations in apical and basolateral actin filaments while significantly inhibiting carbachol-stimulated secretion of bulk protein and β-hexosaminidase. The chemical inhibitor GF 109203X (10 μM, 3 hrs) which inhibits PKCα, β, δ and ɛ, also elicited more potent inhibition of carbachol-stimulated secretion relative to Gö 6976 (10 μM, 3 hrs) which inhibits only PKCα and β. Transduction of lacrimal acini with Ad encoding syncollin-GFP resulted in labeling of secretory vesicles that were discharged in response to carbachol stimulation while co-transduction of acini with Ad-DN-PKCɛ significantly inhibited carbachol-stimulated release of syncollin-GFP. Carbachol also increased the recovery of secretory component in culture medium while Ad-DN-PKCɛ transduction suppressed its carbachol-stimulated release. We propose that DN-PKCɛ alters lacrimal acinar apical actin remodeling, leading to inhibition of stimulated exocytosis and transcytosis.
Keywords: actin, PKCɛ, exocytosis, lacrimal gland, acinar epithelial cell
Introduction
Production and release of ocular fluid of appropriate composition is essential for maintenance of corneal health. The lacrimal acinar cells of the lacrimal gland are the major source of tear proteins released into ocular fluid. Most tear protein stores are released at the apical plasma membrane (APM)1 of lacrimal acini from mature secretory vesicles (SVs) that are coated with the small GTPase, rab3D (27, 41). These SVs fuse with the APM rapidly upon secretagogue stimulation (41). Additional tear proteins such as the extracellular domain of the polymeric immunoglobulin receptor (pIgR), either alone (secretory component or SC) or complexed to dimeric IgA (secretory IgA) can be released at the APM through the transcytotic pathway. The area under the APM in lacrimal acini is enriched in a dense network of actin filaments which appear to form a barrier preventing uncontrolled SV release in unstimulated acini. Several studies have shown that extensive remodeling of the underlying actin network accompanies stimulated secretion in acinar epithelial cells from pancreas (24, 38, 39), parotid gland (29) and lacrimal gland (Galina Jerdeva and Sarah Hamm-Alvarez, unpublished observations). This remodeling includes increased actin filament turnover as well as transient formation of actin coated invaginations thought to represent SV fusion intermediates. Stabilization of actin coated structures is associated with inhibition of exocytosis (39). Little is known about the effectors that regulate actin filament remodeling in acinar exocytosis.
In this study, we have investigated the contribution of the novel protein kinase C (PKC) isoform, PKCɛ, to actin filament remodeling in apical exocytosis. Three subfamilies of PKC are distinguished based on their requirements for activation: 1) the Ca2+-dependent or conventional PKCs; 2) the Ca2+-independent or novel PKCs; and 3) the phorbol ester-insensitive or atypical PKCs (reviewed in 12, 26). All PKCs consist of a single polypeptide chain containing regulatory N-terminal and catalytic C-terminal domains. While conventional isoforms have domains for binding to diacylglycerol and Ca2+, novel PKC isoforms lack the Ca2+-binding domain, making them maximally responsive to diacylglycerol or phorbol esters. Uniquely, PKCɛ has an actin-binding site spanning amino acids 223–228 (30, 31, reviewed in 2). Association of PKCɛ with actin filaments is triggered by binding of diacylglycerol or phorbol esters; conversely, association of PKCɛ with actin filaments maintains the kinase in the active state. It is thought that the phosphorylation of specific substrate proteins by PKCɛ is regulated by its targeting to actin filaments or its other partner, Golgi beta’-COP, which brings the activated kinase into close proximity to target molecules.
Secretagogue-mediated activation of diacylglycerol and Ca2+-dependent pathways in the lacrimal gland and consequent activation of Ca2+- and phospholipid-dependent PKCs has long been associated with stimulation of exocytosis (11). At least 5 PKC isoforms are present in lacrimal gland acini: α, ɛ, δ, μ and ι/λ, each exhibiting unique locations and translocation patterns in response to activation (47). In rat lacrimal acini, the muscarinic agonist, carbachol (CCH), evokes a secretory response by activation of PKCα and PKCɛ (46). In contrast, the secretory response evoked by α1-adrenergic agonists such as phenylephrine (PE) in rat lacrimal acini occurs primarily through activation of PKCɛ (46). PKCɛ facilitates synaptic vesicle exocytosis (31) as well as prolactin secretion from pituitary cells (1). Given the important role for actin filament remodeling in lacrimal acinar exocytosis and the established link between PKCɛ and exocytosis in other systems, we hypothesized that PKCɛ participated in actin filament remodeling in lacrimal acinar exocytosis. Here we have utilized biochemical strategies, confocal fluorescence microscopy and functional assays to demonstrate in lacrimal acini that 1) PKCɛ is an actin binding protein; 2) PKCɛ association with actin is responsive to secretagogue stimulation; 3) introduction of dominant negative (DN) PKCɛ using replication-defective adenovirus (Ad) elicits profound alterations in actin cytoskeleton including accumulation of actin-coated invaginations and 4) expression of DN-PKCɛ significantly (p ≤ 0.05) inhibits CCH-stimulated release of bulk protein, β-hexosaminidase, co-transduced syncollin-GFP and SC. We propose that, in stimulated acini, activated PKCɛ is targeted to apical actin where it phosphorylates actin-associated proteins, leading to the changes in actin organization required for exocytosis. We further suggest that the impaired CCH-stimulated exocytosis at the APM of acini transduced with Ad-DN-PKCɛ is a consequence of the changes in apical actin remodeling exerted by DN-PKCɛ.
Materials and Methods
Reagents
CCH, PE, rhodamine-phalloidin and goat anti-rabbit secondary antibody conjugated to FITC were obtained from Sigma Chemical Co (St. Louis, MO). GF 109203X and Gö 6976 were obtained from EMD Biosciences (San Diego, CA). _Sheep anti-rabbit antibody to SC was generated against purified SC from rabbit bile (Pel-Freeze, Rogers, AK) by preparative gel electrophoresis and used to produce sheep anti-rabbit SC polyclonal antiserum (Capralogics, Hardwick, MA). The antiserum was of sufficient titer to use diluted for Western blotting. For immunofluorescence, antibodies were purified from antiserum using protein G-Sepharose (Amersham-Pharmacia). Mouse monoclonal anti-syncollin antibody was kindly provided by Dr. Michael Edwardson (Cambridge University). Rabbit polyclonal antibody to PKCɛ was obtained from Santa Cruz Biotechnology, Inc (Santa Cruz, CA) and used for Western blotting and immunofluorescence labeling. Mouse monoclonal antibody to PKCɛ was purchased from BD Transduction Laboratories (Lexington, KY) and used for immunofluorescence labeling in acini transduced with Ad-DN-PKCɛ. Rabbit ProLong antifade mounting medium, 4',6-diamidino-2-phenylindole, dihydrochloride (DAPI), goat anti-rabbit secondary antibody conjugated to Alexa Fluor-568 or Alexa Fluor-647 and Alexa Fluor-647-phalloidin were from Molecular Probes (Eugene, OR). Mouse monoclonal antibody to actin was obtained from Roche Diagnostics Inc (Indianapolis, IN). Goat anti-mouse, anti-rabbit and donkey anti-sheep IRDye™800-conjugated secondary antibodies were purchased from Rockland (Gilbertsville, PA). Cell culture reagents were from Life-Technologies.
Cell isolation, culture and treatments
Isolation of lacrimal acini from female New Zealand white rabbits (1.8–2.2 kg) obtained from Irish Farms (Norco, CA) was in accordance with the Guiding Principles for Use of Animals in Research. Lacrimal acini were isolated as described (9, 12) and cultured for 2–3 days. Cells prepared in this way aggregate into acinus-like structures while individual cells within these structures display distinct apical and basolateral domains and maintain a robust secretory response (9, 10, 41). CCH and PE were used at 100 μM.
Ad amplification and transduction
Replication-defective Ad constructs used in this study include Ad expressing DN-PKCɛ and GFP separately (Ad-DN-PKCɛ), Ad encoding GFP alone (Ad-GFP) and Ad encoding a syncollin-GFP fusion protein (Ad-syncollin-GFP). QB1 cells were infected with Ad constructs and grown at 37°C and 5% CO2 in DMEM (high glucose) containing 10% fetal bovine serum for 66 hrs until cells were completely detached from the flask surface. The virus was purified using cesium chloride ultracentrifugation as described (41). Viral titers were determined using the tissue culture infectious dose50 assay on 293 cells. Transduction with Ad constructs involved exposure for 1–3 hrs on day 2 of culture at a MOI of 5, followed by washing well in Dulbecco’s phosphate buffered saline (DPBS) and incubation in fresh culture medium for 18–20 hrs at 37°C and 5% CO2. Cells were analyzed on day 3 of culture. Transduction efficiency was maintained ~80%, consistent with previous reports (41). For co-transduction studies, an MOI of 5 was used for each Ad construct. Dual transduction efficiency was difficult to quantify using flow cytometry since the constructs of interest both expressed GFP (Ad-DN-PKCɛ co-expressing GFP and Ad-syncollin-GFP). However, analysis by confocal fluorescence microscopy in fixed acini showed co-expression of cytosolic PKCɛ and large syncollin-containing vesicles in ~70% of lacrimal acini, consistent with efficiencies seen for co-transduction by other constructs in lacrimal acini.
Confocal fluorescence microscopy
For analysis of effector proteins and actin filaments in fixed cells, reconstituted rabbit lacrimal acini cultured on Matrigel-coated coverslips were fixed and processed as described (9, 10, 41). Acini were incubated with appropriate primary and fluorophore-conjugated secondary antibodies and rhodamine- or Alexa Fluor-647-phalloidin. Most confocal images were obtained with a Zeiss LSM 510 Meta NLO imaging system equipped with Argon, red HeNe and green HeNe lasers mounted on a vibration-free table and attached to an incubation chamber controlling temperature, humidity and CO2. The ability of this system to acquire fluorescence emission signals resolved within narrow ranges in multitrack mode, and the use of singly-labeled control samples ensured the validity of co-localization studies. Panels were compiled in Adobe Photoshop 7.0 (Adobe Systems Inc, Mountain View, CA).
For live cell imaging of acini expressing syncollin-GFP, rabbit lacrimal acini seeded on Matrigel-covered glass-bottomed round 35 mm dishes (MatTek, Ashland MA) at a density of 4 x 106 cells/dish for 2 days were transduced with Ad-syncollin-GFP as described and cultured in fresh medium for 18–24 hrs. On day 3, lacrimal acini were analyzed by time-lapse confocal fluorescence and DIC microscopy using Zeiss Multiple Time Series V3.2 and Physiology V3.2 software modules. Live cell analyses were performed at 37°C. DIC images and GFP fluorescence were acquired simultaneously using the 488 line of the Argon Laser.
For analysis of transduction efficiency, rabbit lacrimal acini transduced with Ad-DN-PKCɛ co-expressing GFP as above were fixed in 4% paraformaldehyde for 15 min followed by permeabilization with 0.5% Triton X-100. These acini were then incubated with appropriate primary and secondary antibodies to visualize PKCɛ, as well as Alexa-647 phalloidin and DAPI to visualize actin filaments and nuclei, respectively. Most of the expressed GFP fluorescence was preserved using this fixation method. To evaluate transduction efficiency, 7–10 random fields per slide were chosen from each of 4 different preparations and all reconstituted acini in the field were subjected to quantitation of transduction efficiency. The cells in the acinus expressing DN-PKCɛ were easily determined based on the increased intensity of PKCɛ labeling of apical and basolateral membranes in parallel with the extended process formation. The cells in the acinus expressing GFP were also easily determined. The number of nuclei within the reconstituted acini were used for normalization to cell number (the small numbers of single cells were not counted since they did not represent reconstituted and functionally competent acini).
Analysis of the extent of colocalization between apical actin and PKCɛ in confocal microscopy images was done using the Enhanced Colocalization tool available with the Zeiss LSM510 software. Fluorescence intensities shown in red (actin filaments labeled with rhodamine phalloidin) were recorded with the 543 nm laser at 100% transmission. Fluorescence intensities shown in green (PKCɛ labeled with appropriate primary and FITC-labeled secondary antibodies) were measured with the 488 nm laser at 2% transmission. Narrow bandpass filters were utilized to avoid bleeding through the channels while the palette function of the software was used to eliminate intensity saturation. For measurement of colocalization, a region of interest was selected around each lumenal area which extended beneath it by ~1 μm; the lumenal area was defined by accumulation of actin filaments as previously described (9). The thresholding function was used to establish the background intensity. The colocalization coefficients obtained were calculated as follows and they reflect the total number of colocalizing pixels within the region of interest for each marker divided by the total pixels in that channel:
Biochemical analysis of PKCɛ-actin filament binding
For actin filament binding assays, the non-muscle Actin Binding Protein Biochemistry Kit from Cytoskeleton, Inc (Denver, CO) was utilized. Cell lysates were prepared by lysing acinar cells (unstimulated or exposed to CCH for 2, 5 or 15 min at 100 μM) in A buffer [5 mM Tris-HCl, pH 8.0, containing 0.2 mM CaCl2 and protease inhibitor cocktail (12)] through a 21 gauge needle on ice and clarified by low speed centrifugation as described above. The supernatant fraction was concentrated in Amicon 3000 filters before addition of polymerized actin and sedimentation according to the manufacturer’s protocol. PKCɛ and actin contents in supernatant and pellet fractions were measured by Western blotting.
Sequential detergent extraction was performed as described (41). The distribution of PKCɛ and actin across each of the pools was determined by SDS-PAGE and Western blotting. Western blots were processed utilizing appropriate primary antibodies and either goat anti-mouse, anti-rabbit or anti-sheep secondary antibodies conjugated to IRDye™800. Blots were quantified using an Odyssey Scanning Infrared Fluorescence Imaging System (Li-Cor, Lincoln Nebraska). For display, fluorescent signals were converted digitally to black and white.
Secretion assays
Lacrimal acini seeded in Matrigel-coated 24-well plates and either transduced with Ad-GFP or Ad-DN-PKCɛ or treated with GF 109203X and Gö 6976 for 3 hours as previously described in pancreatic acini (34) were treated without or with CCH (100 μM, 30 minutes) or PE (100 μM, 30 minutes, transduced acini only) and processed to evaluate release of bulk protein and β-hexosaminidase as described (9, 12, 32, 41). In each assay, values were calculated from 5–6 replicate wells/treatment and normalized to total cellular protein. For analysis of SC or syncollin-GFP release into culture medium, culture medium from resting and CCH-stimulated acini treated or transduced with constructs as described above was collected, concentrated on Centricon 10 filters, equal volumes resolved by SDS-PAGE, and proteins of interest detected by Western blotting. Signal intensity was normalized to pellet protein in each sample and expressed as fluorescence intensity/mg protein before normalization to control and comparison across treatments. Differences in experimental groups in all secretion assays were determined using a paired t-test with p ≤ 0.05.
Results
Lacrimal acinar PKCɛ associates with actin filaments
To demonstrate that lacrimal acinar PKCɛ associates with actin, we performed several in vitro and intact cell assays. Actin binding proteins can be identified by their co-sedimentation with polymerized non-muscle actin. During establishment of the parameters of the actin sedimentation assays, control experiments revealed that added bovine serum albumin did not pellet with the polymerized actin but remained in the supernatant fraction while added α-actinin, a known actin binding protein, did partition with polymerized actin filaments in the pellet (Figure 1A). A weaker protein signal showing a the major band with a MW ~95kDa that likely represented PKCɛ could also be detected by Coomassie blue staining of the gel in the Lysate lane in the Pellet fraction when non-muscle actin was added to the reaction. As shown in Figure 1B, analysis of the actin pellet by Western blotting revealed that PKCɛ was co-sedimented in unstimulated or control (CON, + actin) acini, while equivalent sedimentation in the absence of exogenous polymerized actin (CON, - actin) did not result in sedimentation of PKCɛ. Analysis of PKCɛ in the supernatant remaining after sedimentation of actin also revealed depletion of the cytosolic pool (data not shown).
Figure 1. PKCɛ is an actin binding protein in lacrimal acini.
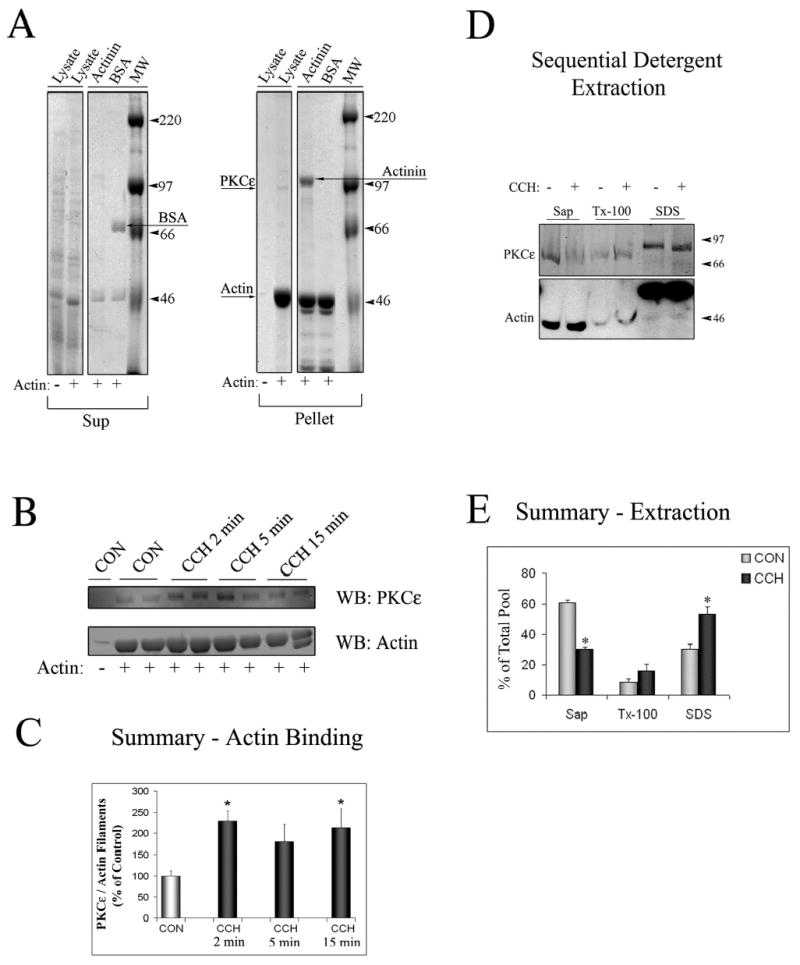
A. SDS-PAGE gel stained with Coomassie blue staining depicting the Supernatant (Sup) and Pellet fractions from actin-binding assays. Lysates from lacrimal acini (Lysate) were incubated without (−) or with (+) non-muscle actin and filaments were pelleted by centrifugation as described in Methods. α-Actinin (an actin-binding protein) and bovine serum albumin (BSA) were used as positive and negative controls, respectively. α-Actinin was pelleted with actin filaments (Arrow labeled Actinin showing its position in the Pellet fraction) while BSA was not (Arrow labeled BSA showing its position in the Sup fraction). A weaker protein signal showing a the major band with a MW corresponding to PKCɛ ~95kDa could also be detected in the Lysate lane in the Pellet fraction when non-muscle actin was added to the reaction (Arrow labeled PKCe). B. Western Blot analysis of the Pellet fraction from a representative actin-binding assay, when lysates (from equal amounts of cells) from acini stimulated without (CON) or with CCH for the indicated periods of time (2–15 min, 100 μM) were used for the actin binding assay and Pellet fractions were blotted for PKCɛ and actin as indicated. Duplicate samples were run in each assay as shown. C. Summary graph of actin-binding experiments in B. obtained from three independent preparations; *, significant at p ≤ 0.05. D. Acini without or with CCH (100 μM, 15 min) lacrimal acini were subjected to sequential detergent extraction to isolate soluble (Sol), membrane (Mem) and cytoskeletal (Cyt) pools as described in Methods. Equal volumes of each of the fractions were resolved by SDS-PAGE and the sample content of PKCɛ and actin determined by Western blotting. E. Composite values reflecting PKCɛ enrichment within soluble, membrane and cytoskeletal pools from acini without or with CCH as described in C. and expressed as a percentage of total cellular PKCɛ. Stimulation did not affect the recovery of marker proteins in the three fractions (data not shown). Results are from n=3 experiments; error bars represent sem; *, p ≤ 0.05.
Since PKCɛ is activated by secretagogue exposure in lacrimal acini (46) including agents acting through M3 muscarinic receptors, we investigated whether the interaction between PKCɛ and actin was increased in lysates from CCH-stimulated lacrimal acini. When actin filament binding assays were performed with lysates from acini exposed to CCH (100 μM) for 2, 5 or 15 min, the amount of PKCɛ recovered with polymerized actin in the pellet was increased significantly at 2 min and 15 min of CCH stimulation, as evidenced by an increase in the ratio of PKCɛ to actin signal on Western blots (Figures 1B and 1C). Although the increase in PKCɛ/actin signal was not statistically significant at 5 min of CCH stimulation, a trend towards an increase was evident at this time, consistent with the other time points chosen for analysis and with confocal fluorescence microscopy findings (see below).
To further verify the apparent increased association of PKCɛ with actin filaments associated with CCH stimulation, we employed an additional protocol based on isolation of subcellular protein pools with sequential detergent extraction to analyze PKCɛ partitioning between soluble (saponin-soluble), membrane (Triton X-100 soluble) and cytoskeletal (SDS-soluble) pools isolated from unstimulated and CCH-stimulated lacrimal acini. A representative blot of PKCɛ and actin contents of each pool from unstimulated and CCH-stimulated acini (100 μM, 15 min) is shown in Figure 1D while Figure 1E shows summary data from several preparations. It should be noted that PKCɛ and actin in the cytoskeletal pool migrate more slowly, due to the presence of high amounts of detergent in this fraction necessary to solubilize filaments. In unstimulated acini, most PKCɛ (~60%) was recovered in the cytosolic pool with only a trace amount in the membrane pool and the remainder (~30%) enriched in the cytoskeletal pool. In CCH-stimulated acini, a significant (p ≤ 0.05) shift in the partitioning of PKCɛ was noted, with only ~30% recovered in the cytosolic fraction and ~55% detected in the cytoskeletal fraction containing most of the cellular actin. This finding, as well as the findings from actin sedimentation assays in Figures 1B–C, strongly suggested that PKCɛ translocation to actin filaments occurred in response to CCH stimulation. The α1-adrenergic agonist, phenylephrine (PE) showed only a modest trend towards an increase in PKCɛ association with actin in these assays (data not shown), consistent with previous findings that this agent is a weak secretagogue in rabbit lacrimal acini (32).
The cellular localization of PKCɛ in unstimulated and CCH-stimulated acini was investigated in parallel. Figure 2 shows results from a representative preparation with actin filament labeling in red and PKCɛ in green. Cytoskeletal organization in reconstituted lacrimal acini has been characterized previously (9, 10). Briefly, the apical or lumenal regions in lacrimal acini can be distinguished by the more intense actin filament labeling detected in roughly circular regions (marked by *) associated with actin filament enrichment beneath the APM and within microvilli. As shown in the images in Figure 2A, apical actin filaments in unstimulated acini appear continuous, without the appearance of multiple invaginations. Fainter actin filament labeling can also be detected beneath basolateral membranes. In unstimulated acini, some PKCɛ was co-localized with apical actin and also in the cytosol in punctuate structures. CCH stimulation (100 μM, 5 min) appeared to increase the co-localization of PKCɛ with apical actin at the lumen and also within transient actin-coated structures, seen as apparent invaginations of the apical actin array in the lumenal region. The intensity profile in Figure 2B depicts the fluorescence in each channel across the region marked in each image by the line in Figure 2A, confirming, by the increase in coincident peaks of fluorescence pixel intensity, the increased co-localization. Co-localization of these two markers beneath the lumen was detected as early as 2 min of CCH stimulation and was sustained to 15 min (data not shown). To quantify the co-localization of apical PKCɛ and actin across multiple samples, we measured the extent of co-localization of lumenal PKCɛ pixels with actin pixels as a percentage of total PKCɛ pixels, and the co-localization of lumenal actin pixels with PKCɛ pixels as a percentage of total actin pixels (Figure 2C). These measurements were conducted in resting (CON) acini as well as in acini exposed to CCH (100 μM) for 5 min, within the time interval associated with microscopic and biochemical colocalization and well within the initial secretory burst from 0–10 min (9). While some co-localization was seen in resting acini, CCH caused a significant increase in co-localization of each marker with the other. PE treatment caused a modest increase in co-localization of PKCɛ with apical actin (data not shown). Biochemical and confocal fluorescence microscopy data in Figures 1 and 2 collectively suggested a strong interaction between lacrimal acinar PKCɛ and actin filaments that was increased in acini exposed to CCH.
Figure 2. Stimulation with carbachol increases PKCe colocalization with apical actin in lacrimal acini.
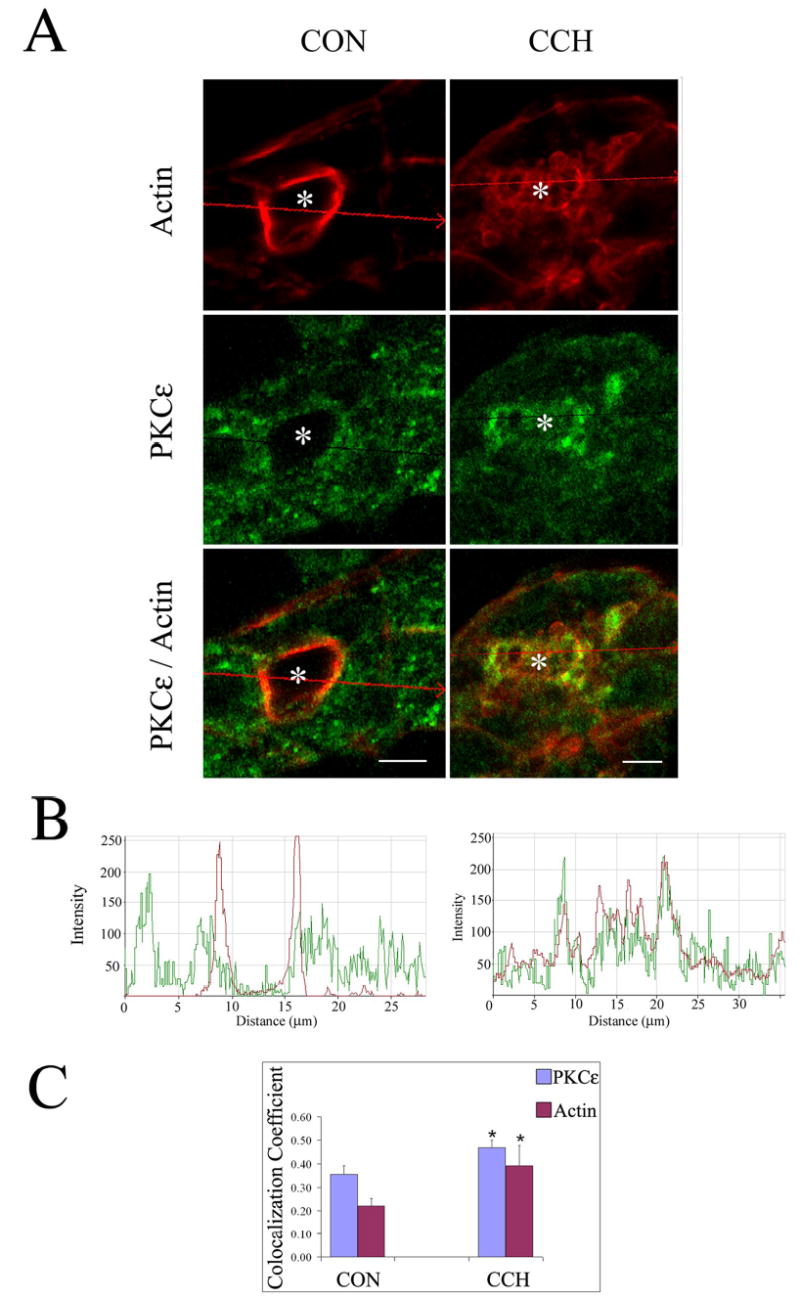
A. Confocal micrographs of control (CON) and CCH-stimulated (CCH; 100 μM, 5 min) acini processed as described in Methods to label PKCe (green; rabbit polyclonal antibody to PKCe, secondary antibody- goat anti-rabbit-FITC) and actin (red; rhodamine phalloidin) which are displayed as separate signals and also as overlaid images. Red arrows across the images indicate the linear region of focus and the direction of the displayed signals from left to right for the intensity plots in B. *, lumenal regions and bars, 5 μm. B. Intensity plots showing the relative intensity of green (PKCe) and red (actin) fluorescence in the regions indicated from the images in A. Regions exhibiting coincident peaks are increased by CCH stimulation, suggestive of increased colocalization. C. Colocalization coefficients which express, for each marker, the number of colocalizing pixels over the number of total pixels, were calculated as described in Methods from unstimulated acini (CON) or acini exposed to CCH (5 min, 100 μM). The relative number of colocalizing pixels in channels 1 or 2, respectively, are expressed as compared to the total number of pixels above threshold. The value range is from 0 – 1 where 0 indicates no colocalization and 1 indicates that all pixels colocalize. All pixels above background count irrespective of their intensity. Results were obtained from 45 (CON) and 47 (CCH) lumena acquired randomly over n=4 separate preparations (7–20 lumena per preparation), *, significant at p ≤ 0.05.
Introduction of DN-PKCɛ alters acinar actin organization
DN mutations in PKC isoforms utilize a point mutation in the ATP binding domain that renders the enzyme inactive (35). Such a mutation in PKCɛ has been generated (K437R) and inserted into an Ad expression vector (17). We transduced lacrimal acini with Ad-DN-PKCɛ co-expressing GFP and examined the efficiency of transduction. Although expression of GFP was clearly high as evidenced by fluorescence microscopy (data not shown), GFP is the second gene inserted into the Ad expression vector and would not be expected to show as much expression as DN-PKCɛ, the first gene expressed by the Ad expression vector. We therefore conducted additional evaluations of transduction efficiency based on confocal fluorescence microscopy. Using acini fixed and processed to label PKCɛ, actin filaments, nuclei and GFP as shown in Figure 3A, we quantified DN-PKCɛ and GFP overexpression in multiple fields chosen randomly from each of several preparations. All reconstituted acini in the field were subjected to quantitation of transduction efficiency. The number of nuclei within the reconstituted acini were used for normalization. The percentage of cells within the reconstituted acini overexpressing DN-PKCɛ was 85 ± 2% and the percentage expressing GFP was 62 ± 2% (n=4). Therefore, in excess of 80% of reconstituted acini were transduced with Ad-DN-PKCɛ at an MOI of 5. Analysis of PKCɛ content in lysates of acini transduced at different MOIs revealed considerable overexpression of the DN-PKCɛ at an MOI of 5, with little additional expression elicited above that dose (Figure 3B). Subsequent experiments utilized a MOI of 5 for transduction with Ad-DN-PKCɛ or Ad-GFP (as a control). The increased PKCɛ immunofluorescence detected in transduced acini was extensively co-localized with apical and basolateral actin filaments while organization of actin at these domains was markedly different (Figure 3C and D). At the APM, actin accumulation was evident in acini overexpressing DN-PKCɛ, and lumena had a compressed appearance. Additionally, actin-coated invaginations were abundant (Figure 3D, arrowheads) that were similar to the actin-coated structures detected transiently in CCH-stimulated acini (Figure 2A). However, the actin coats in acini expressing DN-PKCɛ were always detected. Transduced acinar cells also exhibited increased attachment and spreading at the basolateral domains, resulting in an extended or elongated appearance (Figure 3D, arrows) relative to their normally globular shape (Figure 2). In some cases, small processes could be detected extending from the basolateral surface. Transduction with Ad-GFP alone elicited no change in actin cytoskeleton (data not shown), as previously published (41).
Figure 3. High efficiency transduction of lacrimal acini with Ad-DN-PKCɛ results in co-localization of overexpressed DN-PKCɛ with actin filaments.
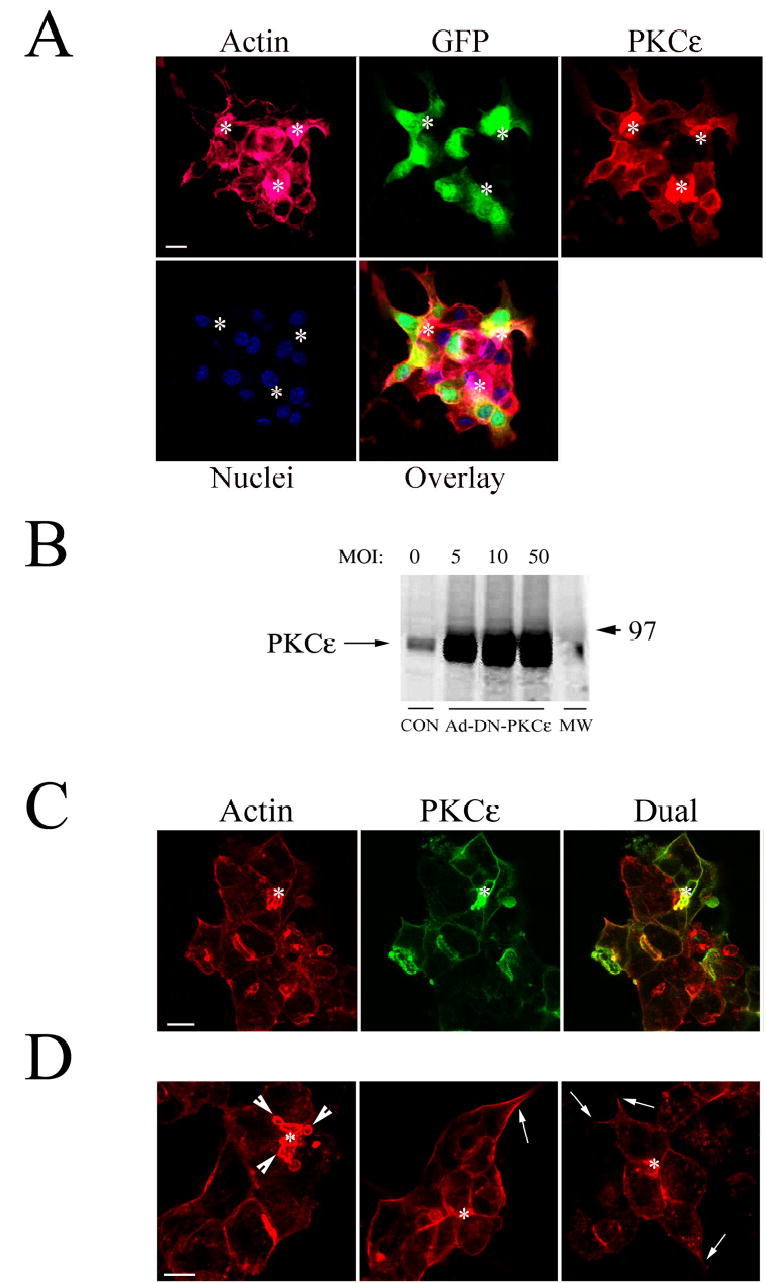
A. PKCɛ (red), actin filaments (pink), nuclei (blue) and GFP (green) in acini transduced with Ad-DN-PKCɛ co-expressing GFP at an MOI of 5 and fixed and processed for quantitation of transduction efficiency as described in Methods. B. Western blot showing the expression of PKCɛ in lysates of lacrimal acini transduced with Ad-DN-PKCɛ at the indicated MOI. Equal amounts of protein were loaded in each lane. C. Confocal fluorescence micrographs of lacrimal acini transduced with Ad-DN-PKCɛ and fixed and processed as described in Methods to label actin filaments (red) and PKCɛ (green). Note, cytosolic GFP fluorescence co-expressed by the Ad-DN-PKCɛ construct is destroyed during fixation. D. Confocal fluorescence micrographs showing actin filament organization in lacrimal acini transduced with Ad-DN-PKCɛ. Arrowheads in C. and D. indicate accumulation of actin-coated structures at the APM; arrows point to basolateral actin filaments associated with areas of cell spreading and process formation; *, lumenal regions; bars, 5 μm in all panels.
Control experiments revealed that overexpression of DN-PKCɛ did not alter the normal recruitment of the conventional PKC isoform, PKCα, to basolateral membranes in response to CCH stimulation for 15 min (data not shown) as has been reported (47). Moreover, the normal binding of the actin binding protein, ezrin, to apical actin filaments in acini was not disrupted by overexpression of DN-PKCɛ (data not shown). These control studies suggested that the overall cellular signaling pathways involving diacylglycerol were not disrupted by DN-PKCɛ overexpression, and furthermore that the apical actin filaments were not decorated with DN-PKCɛ to such an extent that other actin-binding proteins were unable to bind.
To better visualize the morphological changes associated with overexpression of DN-PKCɛ in the lacrimal acini, we performed 3-dimensional reconstruction of serial sections acquired in lacrimal acini. Figure 4 shows the 3D reconstructed shapes of non-transduced acini and acini overexpressing DN-PKCɛ (Ad-DN-PKCɛ) rotated at different angles for better visualization of the features induced by DN-PKCe [available online as movie 1 (CON) and movie 2 (Ad-DN-PKCɛ)]. The straight arrows indicate elongated neuritic-like projections extending from the basolateral surface that are commonly detected in acini transduced with DN-PKCɛ but not in non-transduced acini. While clearly revealing the more convoluted lumena associated with accumulation of actin-coated invaginations in acini transduced with DN-PKCɛ, these and additional projections did not reveal systematic evidence for alterations in the apparent accessibility of the lumena to the culture medium associated with DN-PKCɛ overexpression.
Figure 4. Transduction of lacrimal acini with Ad-DN-PKCɛ is associated with changes in acinar shape.
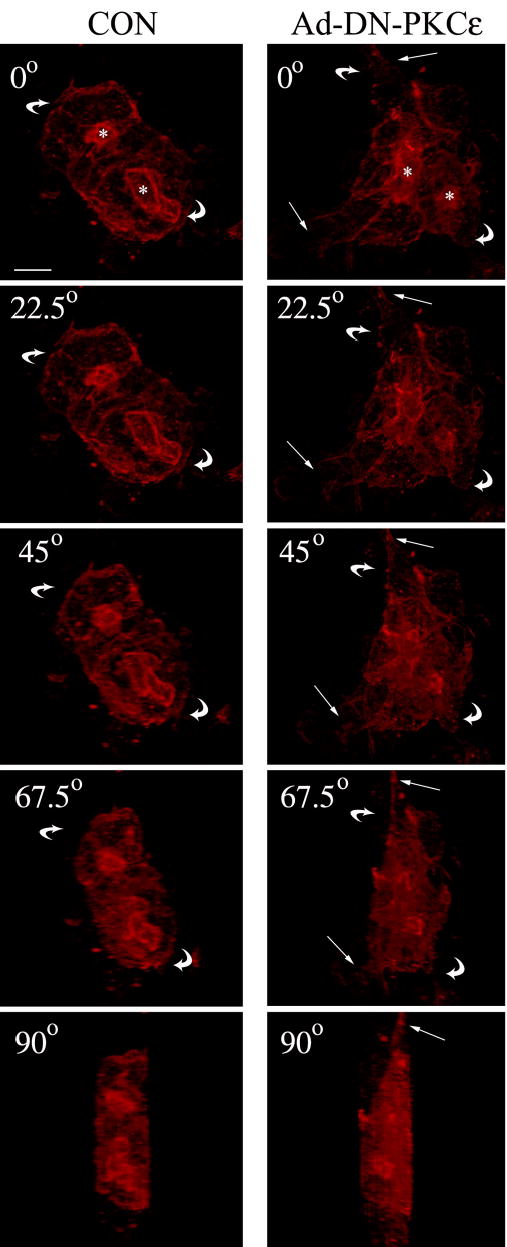
3D reconstruction of the actin filament network labeled with Alexa Fluor 647-phalloidin in non-transduced acini (CON, left panel) or acini transduced with Ad-DN-PKCɛ (right panel) as described in Methods. Images were acquired at Z intervals of 0.5 μm and were reconstructed into a 3D movie file which could be rotated to demonstrate the structure viewed at different angles. Selected frames are presented at 0°, 22.5 °, 45 °, 67.5 °, and 90 °. Straight arrows indicate projections at the basolateral side of acini; curved arrows indicate the direction of the rotation of the acinar projection; *, lumenal regions; bar, 5 μm.
The effects of DN-PKCɛ overexpression on basal and stimulated secretion of bulk protein and the secretory product, β-hexosaminidase are shown in Figure 5A. Many lumena in reconstituted acini are open to the culture medium, so that secretion can be measured by collecting culture medium, measuring the marker of interest, and normalizing the signal to pellet protein as previously described (9, 10, 41). CCH is a robust secretogogue in reconstituted rabbit lacrimal acini, eliciting ~3.5-fold increase in both bulk protein and β-hexosaminidase release. GFP overexpression minimally affected this pattern of release. In contrast, DN-PKCɛ caused a significant (p ≤ 0.05) increase in basal protein and β-hexosaminidase release that was significant relative to Ad-GFP-treated acini, with a concomitant significant decrease in both total and stimulated components of the release. We tried to evaluate the effects of DN-PKCɛ on secretion evoked by PE in parallel. Consistent with previous results (32), PE only weakly increased secretory functions, promoting only a 0.5x increase in bulk protein release and minimally affecting β-hexosaminidase release. A trend towards increased basal release relative to Ad-GFP transduced acini and a complete inhibition of PE-stimulated release were noted in these assays, although the magnitude of the effect made it difficult to distinguish actual changes from the normal variation inherent in the assay.
Figure 5. PKCɛ inhibition impairs secretagogue-stimulated release of protein and β-hexosaminidase in lacrimal acini.
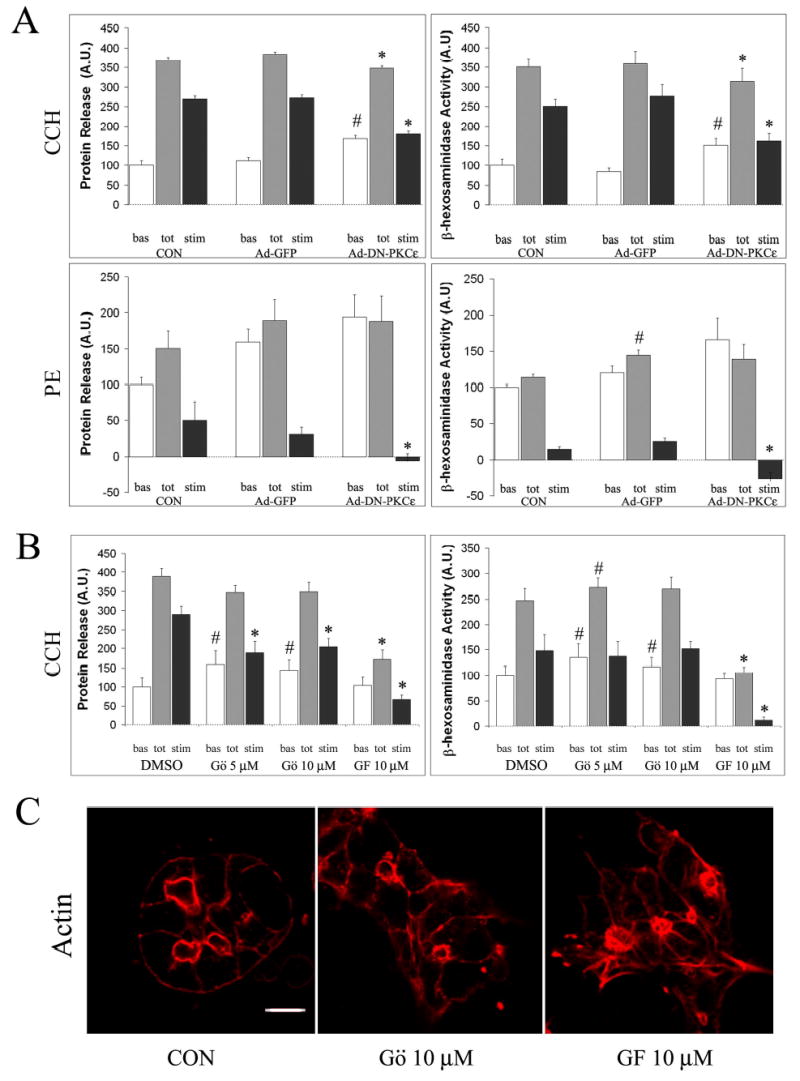
A. Lacrimal acini grown on Matrigel-coated dishes were transduced with Ad-DN-PKCɛ or Ad-GFP on day 2 of culture as described in Methods and analyzed on day 3 for secretion. Bulk protein secretion and β-hexosaminidase activity released into culture medium from transduced acini exposed without or with CCH (100 μM, 30 min) or PE (100 μM, 30 min) is shown. B. Lacrimal acini grown on Matrigel-coated dishes were treated with Gö 6976 (Gö, 5 or 10 μM) or GF 109203X (GF, 10 μM) for 3 hours prior to stimulation with CCH (100 μM, 30 min). Bulk protein and β-hexosaminidase activity released into culture medium is shown. In A. and B., basal (unstimulated) release is shown in white bars; total release (basal plus stimulated) is shown in grey bars; the stimulated component (total minus basal) is shown in black bars. Values were normalized to cell protein before comparison across samples. n=7 separate preparations for CCH stimulation and 4 separate preparations for PE stimulation in acini transduced with Ad-DN-PKCe or Ad-GFP; n=5 separate preparations for acini treated with Gö 6976 or GF 109203X; error bars represent sem; *, significant decrease at p ≤ 0.05 from paired control; #, significant increase at p ≤ 0.05 from paired control; &, significant decrease at p ≤ 0.05 from Gö 6976-treated, CCH-stimulated acini. C. Confocal fluorescence micrographs of lacrimal acini without (Control or CON) or with treatment with Gö 6976 (10 μM, 3 hrs) or GF 109203X (10 μM, 3 hrs) fixed and processed for labeling of actin filaments with rhodamine phalloidin as described in Methods. Bar, 5 μm.
To confirm that this inhibition of CCH-stimulated protein and β-hexosaminidase secretion was caused specifically by the inhibition of cellular PKCɛ activity caused by introduction of the DN variant, we also utilized chemical inhibitors of PKC isoforms in these assays. The PKC inhibitor, Gö 6976, specifically inhibits only the Ca2+-dependent PKC isoforms, α and β, without exerting any effects on PKCδ, ɛ or ζ isoforms (21). In contrast, the PKC inhibitor, GF 109203X inhibits PKC α, β, δ, and ɛ isoforms with equimolar potency (21). As shown in Figure 5B, pretreatment of lacrimal acini with Gö 6976 elicited a small but significant increase in basal release of protein and β-hexosaminidase, but caused no suppression of total release. CCH-stimulated release was significantly reduced. In contrast, pretreatment of lacrimal acini with GF 109203X elicited no effects on basal release of protein or β-hexosaminidase but promoted a significant inhibition of total and CCH-stimulated release. The CCH-stimulated decrease in protein release promoted by GF 109203X was also significantly reduced relative to the decrease promoted by Gö 6976. Relative to the spectrum of PKC isoforms expressed in lacrimal acini (α,ɛ, δ, μ and ι/λ), GF 109203X should elicit additional inhibition of PKCδ and PKCɛ, relative to Gö 6976 at these doses, suggesting that the impaired secretion was due to effects on one or both of these kinases. Since comparable effects on CCH-stimulated protein and β-hexosaminidase secretion were noted in acini transduced with Ad-DN-PKCɛ, it was reasonable to conclude that PKCɛ was at least a major target associated with the inhibition of secretion by GF 109203X.
Investigation of actin filament organization in lacrimal acini exposed to these broad spectrum inhibitors revealed that pretreatment of lacrimal acini with GF 109203X also caused changes similar to those elicited by DN-PKCɛ transduction including accumulation and bundling of apical actin filaments and elongation of cellular actin-enriched processes from the basolateral membrane (Figure 5C). In contrast, actin filament organization in acini pretreated with Gö 6976 showed some evidence for increased apical actin but not to the same extent as that caused by GF 109203X or DN-PKCɛ; no significant changes in basolateral actin or process formation were noted (Figure 5C).
These results suggested that the normal pathways of CCH-stimulated secretion were inhibited by introduction of DN-PKCɛ. However, because bulk protein is a relatively non-specific marker of secretion, it was difficult to discern whether the inhibition was exclusively due to alterations in apical targeting and release or whether it was complicated by changes in the basolateral release profile. Although β-hexosaminidase is a secretory protein, it is also present in basolateral membrane compartments which may, under some conditions, contribute to basolateral exocytosis. We therefore investigated whether DN-PKCɛ could modulate the CCH-stimulated release of two apically-targeted proteins, syncollin-GFP and SC, into culture medium. PE treatment was not further pursued because of its limited efficacy as a secretagogue.
CCH-stimulated release of syncollin-GFP is inhibited by DN-PKCɛ
Studies with a syncollin-GFP fusion protein have shown labeling of large protein-enriched SVs in diverse systems including zymogen granules in pancreatic acini (15), large dense core vesicles in AT-20 cells (15), insulin granules in pancreatic β-cells (20), but not synaptic-like or smaller SVs. These findings suggested syncollin-GFP as an excellent candidate for labeling of protein-enriched mature lacrimal acinar SVs. Figure 6A shows a 3D reconstructed image of the subapical region and interior cytoplasm from a live reconstituted acinus transduced with syncollin-GFP [available online as movie 3 (Syncollin-GFP)]. A number of large spherical vesicles delineated by syncollin-GFP fluorescence were detected throughout the acinus, with a particular enrichment beneath the APM (*). Some of these vesicles appeared to have “tails”, due to saltatory movement during image acquisition. The diameter of the vesicles ranged from 0.2–1 μM with most vesicles (60%) sized from 300–500 nm in diameter. The 1 μM vesicles, representing ~30% of the total, were more apically-localized. This fluorescence pattern suggested specific labeling of a subpopulation of mature SVs, consistent with previous work (15, 20).
Figure 6. CCH stimulation of lacrimal acini transduced with Ad-syncollin-GFP depletes subapical syncollin-GFP fluorescence.
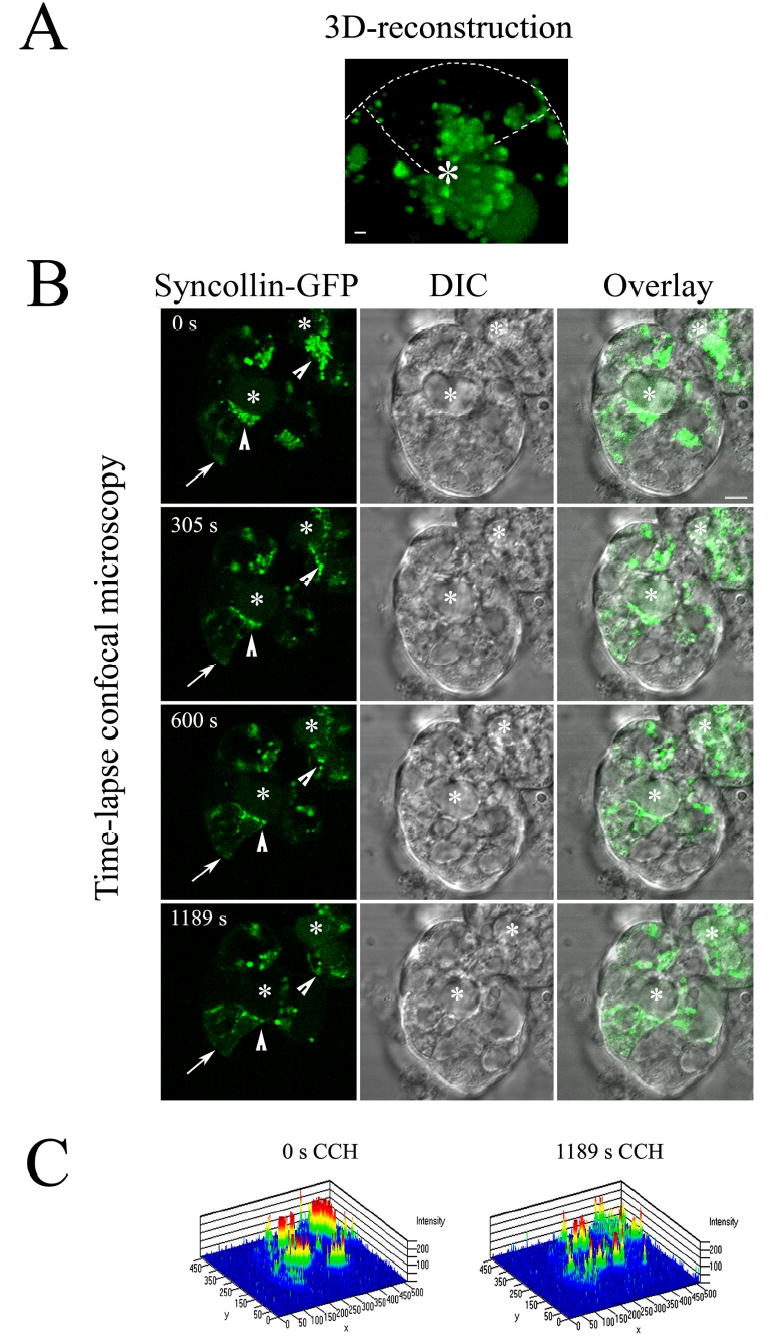
Lacrimal acini grown on Matrigel-covered glass-bottomed round 35 mm dishes were transduced with Ad-syncollin-GFP on day 2 of culture as described in Methods and imaged on day 3 of culture. A. 3D reconstruction at high magnification of the interior regions of a reconstituted acinus formed by three lacrimal acinar cells organized around a central lumen (*), each expressing syncollin-GFP. The reconstruction was obtained by compression of XY images acquired at Z intervals of 0.5 μm. Bar, 1 μm; dashed line, boundary of uppermost cell relative to the other two, deduced by comparison to DIC image. B. Live acini were imaged in the presence of CCH (100 μM) at the indicated times by time-lapse confocal fluorescence microscopy. Arrowheads indicate regions of major loss of syncollin-GFP intensity surrounding lumenal regions (*); bar, 5 μm. No major changes in syncollin-GFP intensity were observed when acini were imaged without CCH. C. 2.5 D graphical reconstruction of the overall intensity profile of the imaged areas presented in A. at 0 and 1189 sec of stimulation with CCH, illustrating individual intensities per pixel utilizing the rainbow scale. The resolution is ~10 pixels per micron.
Figure 6B shows selected images from live rabbit lacrimal acini transduced with Ad-syncollin-GFP and acquired by time-lapse microscopy over a 20 min interval within a single focal plane. As in the 3D reconstruction, syncollin-GFP was detected in large apparent vesicles throughout the acini which were particularly abundant (arrowheads) beneath the lumenal regions (*) detected in DIC micrographs acquired in parallel. Fainter, more diffuse syncollin-GFP labeling was detected above the basolateral membrane adjacent to the nucleus (arrow), likely reflecting enrichment in biosynthetic membrane compartments located in these regions. Upon CCH stimulation, a depletion of the subapical stores of syncollin-GFP was detected over time (arrowheads). This graph in Figure 6C plots the fluorescence intensity of the entire region at 0 sec and at 1189 sec. The subapical syncollin-GFP localized adjacent to the lumena in the untreated acinus was clearly the most intense (red labeling). At 1189 sec of CCH sec, this intensity was clearly diminished.
We were unable to analyze lacrimal acini dually transduced with Ad-syncollin-GFP and Ad-DN-PKCɛ (which co-expresses GFP) by live cell confocal fluorescence microscopy because syncollin-GFP-enriched vesicles could not be resolved above the strong cytosolic GFP fluorescence. Syncollin-GFP can be visualized in fixed, transduced acini under fixation conditions associated with quenching of GFP and using appropriate primary and fluorescently-labeled secondary antibodies in parallel with other markers. Figure 7A shows syncollin-GFP (green) detected by immunofluorescence in lacrimal acini co-transduced with Ad-syncollin-GFP and Ad-DN-PKCɛ (red) relative to Ad-syncollin-GFP alone. These images show that Ad-DN-PKCɛ overexpressing-acini (detected easily by the intense PKCɛ immunofluorescence associated with apical and basolateral actin) does not affect the abundance of syncollin-GFP-enriched vesicles or their apical enrichment in the same acinus. Interestingly, some co-localization of syncollin-GFP was detected with the actin-coated structures induced by overexpression of DN-PKCɛ (arrows, green and purple labels), consistent with our proposal that these actin-enriched structures were SV fusion intermediates.
Figure 7. DN-PKCɛ inhibits CCH-stimulated release of syncollin-GFP into culture medium in co-transduced lacrimal acini.
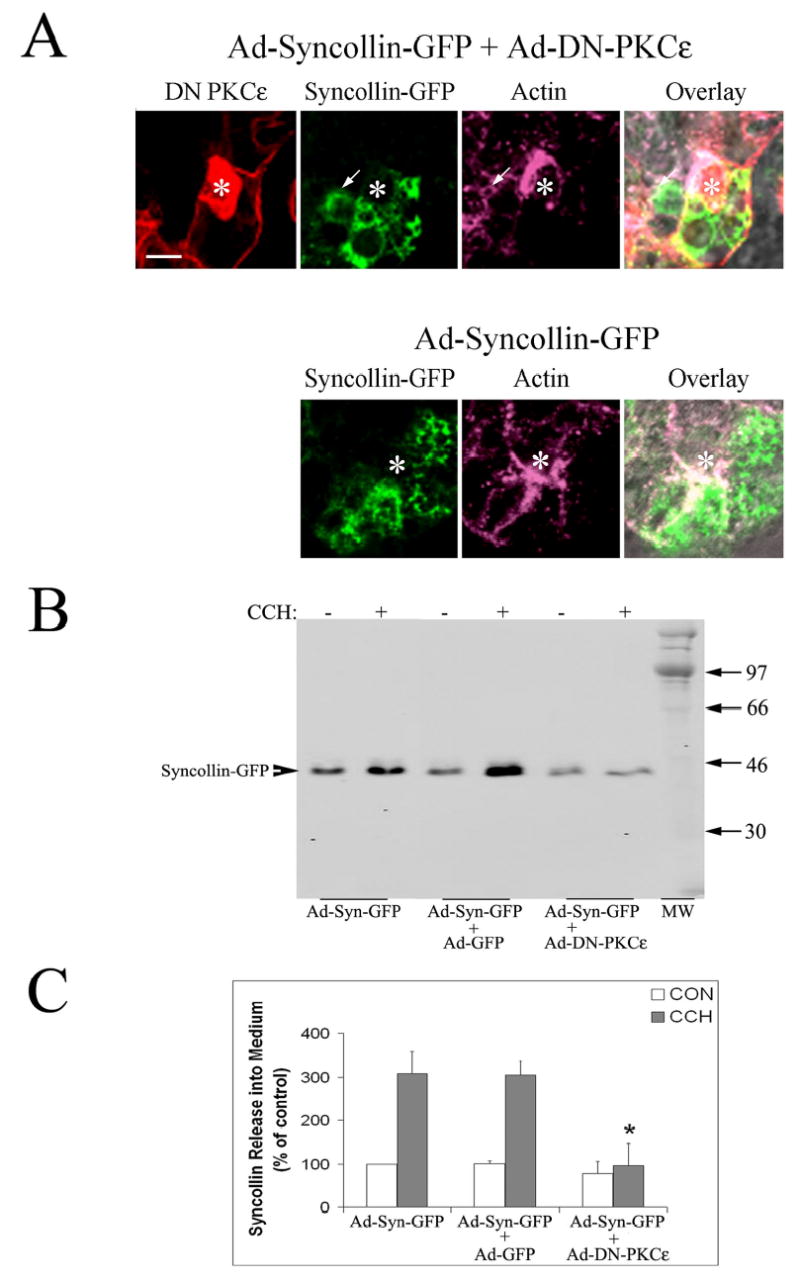
A. High magnification view of the APM region of acini co-transduced with Ad-syncollin-GFP and Ad-DN-PKCɛ, or Ad-syncollin-GFP alone. Transduced acini were fixed and labeled as described in Methods to detect syncollin (green), PKCɛ (red), and actin filaments (purple). Overlay shows all three fluorescence labels as well as the paired DIC image. Fixation quenches the intrinsic GFP fluorescence present in cytosol and on syncollin in these transduced acini. *, lumenal region; arrow, co-localization of syncollin-GFP with an actin-coated invagination; bar, 5 μm. B. Western blots showing syncollin-GFP release into culture medium in the absence (−) and presence (+) of 100 μM CCH for 30 min in lacrimal acini transduced with Ad-syncollin-GFP without or with Ad-GFP or Ad-DN-PKCɛ. Syncollin release was detected with an anti-syncollin antibody combined with an appropriate IRDye™800 conjugated secondary antibody. C. Syncollin-GFP release under each experimental condition was quantified as shown in A., normalized to cell protein in the pellet, and compared across treatments. White bars show basal (CON) release while grey bars show CCH-stimulated release. N=3 separate preparations; error bars show sem and *, significant at p ≤ 0.05 from samples co-transduced with Ad-GFP.
Since we were unable to use live cell confocal microscopy to investigate the effect of DN-PKCɛ on exocytosis of syncollin-GFP in real time, we utilized Western blotting to measure syncollin-GFP release into culture medium under different experimental conditions. As shown in Figure 7B, CCH stimulation caused a 3-fold increase in the recovery of syncollin-GFP in the culture medium of Ad-syncollin-GFP transduced acini which paralleled the loss of subapical fluorescence seen in live acini exposed to CCH (Figure 6). Co-transduction of acini with Ad-DN-PKCɛ but not Ad-GFP significantly inhibited CCH-stimulated release of syncollin-GFP into culture medium (Figure 7B and C). Co-transduction of acini with Ad-DN-PKCɛ and Ad-syncollin-GFP moderately reduced syncollin-GFP expression relative to co-transduction with GFP and syncollin-GFP (data not shown), an effect that was likely due to non-specific changes associated with overexpression of three exogenous proteins in co-transduced acini (GFP and PKCɛ by Ad-DN-PKCɛ and syncollin-GFP by Ad-syncollin-GFP). The percentage of total cellular syncollin-GFP released in response to CCH under each experimental condition represented ~20–30% of total cellular syncollin-GFP, suggesting that sufficient cellular stores were available for exocytosis under all conditions (data not shown). Abundant syncollin-GFP immunofluorescence could also be detected in acini co-transduced with Ad-DN-PKCɛ (Figure 7A). Finally, acini dually transduced with Ad-syncollin-GFP and Ad-DN-PKCɛ or Ad-GFP exhibited the same general profiles of β-hexosaminidase release reported for acini transduced with Ad-DN-PKCɛ or Ad-GFP alone in Figure 5 (Table 1). These findings indicated that the overall acinar secretory pathway was intact in acini expressing syncollin-GFP, and that DN-PKCɛ had the same magnitude of inhibitory effect on β-hexosaminidase release regardless of syncollin-GFP co-expression. We also determined that GF 109203X but not Gö 6976 was able exert the same significant inhibition of CCH-stimulated syncollin-GFP release, as additional evidence that this change was reliant on the reduction in PKCɛ activity (Table 2). From these data, we concluded that DN-PKCɛ significantly and specifically inhibited apical exocytosis of syncollin-GFP in lacrimal acini.
Table 1.
Effects of Ad-syncollin-GFP on lacrimal acinar secretion of β-hexosaminidase.
| Cell Treatment | Experimental Treatment | β-hexosaminidase release (% of non-transduced control) |
|---|---|---|
| No Ad | Basal | 100% + 8% |
| Total (in presence of CCH) | 174% + 34% | |
| Stimulated (difference) | 74% + 34% | |
| Ad-syncollin-GFP + Ad-GFP | Basal | 97% + 2% |
| Total (in presence of CCH) | 159% + 29% | |
| Stimulated (difference) | 62% + 27% (84% of the value elicited in non- transduced) | |
| Ad-syncollin-GFP + Ad-DN-PKCɛ | Basal | 106% + 6% |
| Total (in presence of CCH) | 149% + 25% | |
| Stimulated (difference) | 43% + 21% (58% of the value elicited in non- transduced) |
Results are averaged from n=3 separate preparations and errors indicate sem. CCH stimulation was for 30 min at 100 μM.
, significant at p
Table 2.
Effects of Gö 6976 and GF 109203X on lacrimal acinar secretion of syncollin-GFP.
| Cell Treatments | Resting (% of control) | CCH-stimulated (% of resting control) |
|---|---|---|
| Control | 100 ± 49% | 183 ± 26% |
| DMSO-treated | 100 ± 8 % | 176 ± 4% |
| Gö 6976 | 111 ± 22% | 156 ± 15% |
| GF 109203X | 73 ± 8% | 109 ± 19%* |
Results are averaged from n=3 separate preparations and errors indicate sem. CCH-stimulation was for 30 min at 100 μM.
, significant reduction from DMSO/CCH at p ≤ 0.05.
CCH-stimulated apical release of SC from pIgR is inhibited by DN-PKCɛ
Free SC and its dimeric IgA bound counterpart, secretory IgA, are enriched in ocular fluid (13, 40), presumably derived by proteolytic cleavage of transcytosed pIgR, either free or bound to dimeric IgA, respectively, at the APM. We evaluated the intracellular localization of pIgR in unstimulated and CCH-stimulated lacrimal acini by confocal fluorescence microscopy using an antibody against the extracellular domain of the pIgR. This antibody therefore recognizes both intact pIgR and free SC. As shown in Figure 8A, pIgR immunofluorescence is recovered in punctuate spots throughout the cytoplasm, and at basolateral (arrows) and apical (arrowheads) domains. Figure 8B shows a higher magnification of the subapical pIgR immunofluorescence in acini also labeled to detect endogenous PKCɛ. As shown in the triple overlay, there was considerable co-localization of pIgR, PKCɛ and apical actin (arrows). Co-localization of pIgR, PKCɛ and apical actin was also detected in CCH-stimulated acini; furthermore, the intensity of PKCɛ and pIgR immunofluorescence was increased. Also, recruitment of pIgR and PKCɛ immunofluorescence to actin-coated structures was evident (Figure 8C, arrowheads). These findings suggested that traffic of pIgR to the actin-enriched region beneath the APM was enhanced by CCH, and further that PKCɛ associated with apical actin filaments was enriched in areas actively undergoing pIgR trafficking.
Figure 8. pIgR is enriched with actin and PKCɛ at the APM in lacrimal acini.
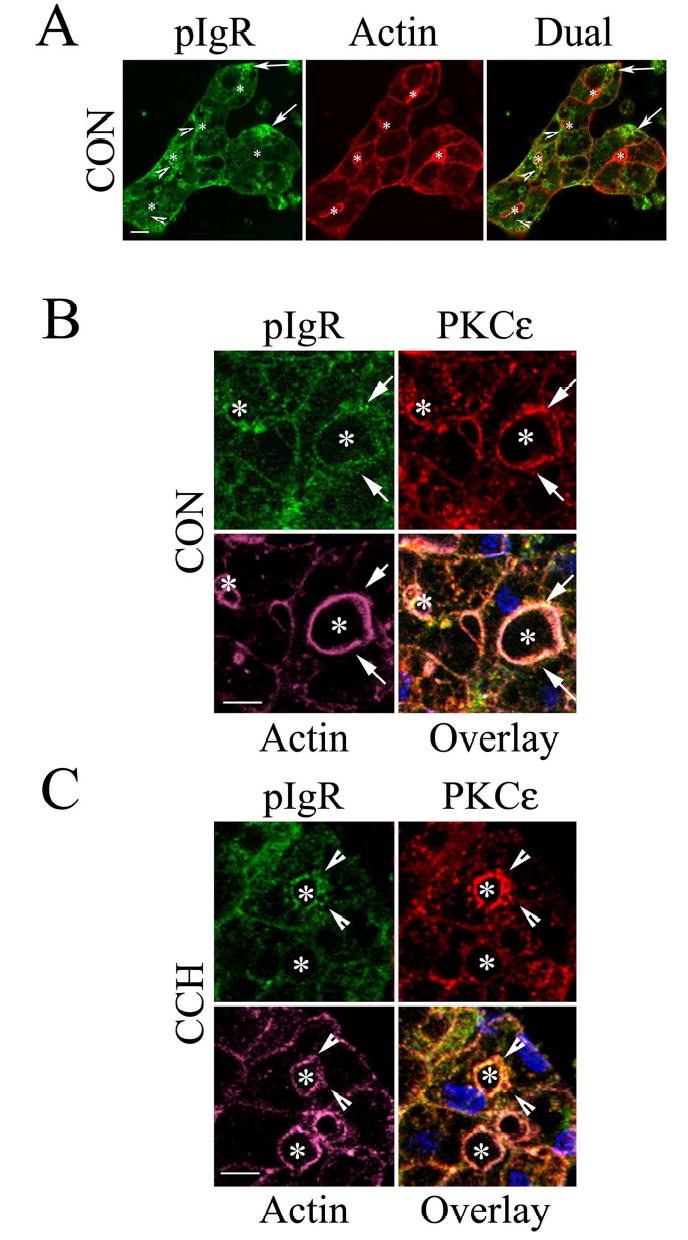
A. Lacrimal acini were fixed and processed as described in Methods to label pIgR (green) and actin filaments (red) for detection by confocal fluorescence microscopy. Arrows, basolateral labeling; arrowheads labeling around lumenal regions. *, lumenal regions. B. High magnification view of the APM region of control (CON) lacrimal acini labeled as described in Methods to detect pIgR (green), PKCɛ (red), and actin filaments (purple). The triple overlay reveals extensive co-localization of all three constituents together at the APM (arrows) and *, lumenal region. C. High magnification view of the APM region of CCH-stimulated (100 μM, 5 min) acini fixed and processed as in B. The triple overlay shows actin-coated invaginations co-localized with pIgR and PKCɛ (arrowheads) and *, lumenal region. Bars, 5 μm.
The cleaved extracellular domain of the pIgR, SC, can be detected in culture medium from rabbit lacrimal acini. Remarkably, CCH stimulated release of SC from lacrimal acini by 3.5-fold (Figure 9). Transduction of lacrimal acini with Ad-GFP did not affect the CCH-stimulated release of free SC but transduction with Ad-DN-PKCɛ significantly inhibited CCH-stimulated SC release. Examination of lacrimal acini transduced with Ad-DN-PKCɛ revealed abundant pIgR immunoreactivity detected at and beneath the apical actin array of both unstimulated and CCH-stimulated acini, similar to that seen in the CCH-stimulated acinus in Figure 8, and consistent with accumulation of pIgR at the APM or subapical stores (data not shown). Finally, we determined that GF 109203X but not Gö 6976 was able to significantly reduce CCH-stimulated SC release, as additional evidence that this change was reliant on changes in PKCɛ activity (Table 3).
Figure 9. Transduction with Ad-DN-PKCɛ inhibits CCH-stimulated release of SC from lacrimal acini.
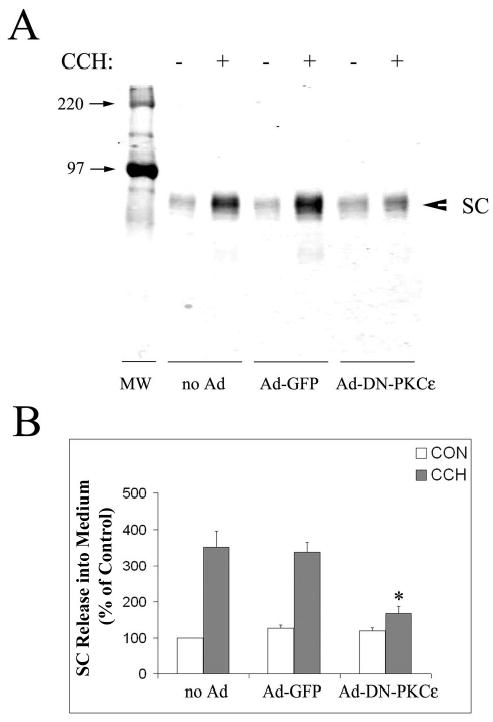
A. Western blots showing SC release into culture medium in the absence (−) and presence (+) of 100 μM CCH for 30 min in non-transduced acini (no Ad), or acini transduced with Ad-GFP or Ad-DN-PKCɛ. SC release was detected with an antibody to the extracellular, cleaved domain of pIgR combined with an appropriate IRDye™800 conjugated secondary antibody. B. SC release under each condition was quantified as shown in A., normalized to cell protein in the pellet, and compared across treatments. White bars show control (CON) release while grey bars show CCH-stimulated release. N=3 separate preparations; error bars show sem and *, significant at p ≤ 0.05 from samples co-transduced with Ad-GFP.
Table 3.
Effects of Gö 6976 and GF 109203X on lacrimal acinar secretion of SC.
| Cell Treatments | Resting (% of control) | CCH-stimulated (% of resting control) |
|---|---|---|
| Control | 100 ± 57% | 223 ± 26% |
| DMSO | 109 ± 14% | 217 ± 32% |
| Gö 6976 | 113 ± 27% | 257 ± 43% |
| GF 109203X | 63 ± 29% | 120 ± 19%* |
Results are averaged from n=3 separate preparations and errors indicate sem. CCH-stimulation was for 30 min at 100 μM.
, significant reduction from DMSO/CCH at p ≤ 0.05.
Discussion
Inspired by the fact that PKCɛ is the only PKC with an actin-binding site (2), and by observations that it is activated both by muscarinic and α1-adrenergic receptor agonists in lacrimal gland (46), we investigated whether PKCɛ participated in regulation of apical actin and/or apical exocytosis. We found that the association of PKCɛ with apical actin filaments and actin-coated structures was increased after CCH stimulation in lacrimal acini. To probe its functional role in exocytosis, we needed to selectively inhibit its activity. We chose a DN strategy for inhibition of PKCɛ activity in acini, using replication-defective Ad to introduce the DN-PKCɛ. Lacrimal acini were readily transduced at high efficiency with Ad constructs, enabling us to express the DN-PKCɛ in essentially all cells and to then identify individual transduced cells by immunofluorescence for examination of other features. Overexpression of DN-PKCɛ resulted in profound changes in apical and basolateral actin filament organization in parallel with inhibition of the CCH-stimulated secretion of protein and β-hexosaminidase. Independent verification of the ability of PKCɛ inhibition to affect CCH-stimulated secretion and actin filament organization was obtained by our findings comparing the effects of the chemical PKC inhibitors, Gö 6976 and GF 109203X, in acini. Although CCH-stimulated protein secretion was significantly reduced by both of these inhibitors, the decrease promoted by GF 109203X was significantly reduced relative to that elicited by Gö 6976. These studies showed an inhibitory effect of GF 109203X but not Gö 6976 on CCH-stimulated β-hexosaminidase secretion in parallel with changes in apical and basolateral actin filament organization elicited by GF 109203X. The additional effects of GF 109203X are attributable to the ability of this inhibitor to inhibit PKCɛ and δ in addition to PKCα and β, which are also inhibited by Gö 6976. The greater inhibitory effect of GF109203X on CCH-stimulated protein and β-hexosaminidase secretion relative to DN-PKCɛ suggests the additional involvement of PKCδ in facilitating an aspect of exocytosis.
After confirmation that the effects on actin filament organization and impaired exocytosis could be elicited through inhibition of PKCɛ by either introduction of DN-PKCɛ or by treatment with GF109203X, we continued to explore the cellular effects of DN-PKCɛ overexpression. Acini transduced with DN-PKCɛ exhibited inhibition of the CCH-stimulated release of two markers of apical secretion newly established in this study, syncollin-GFP and SC. These effects could likewise be mimicked by GF 109203X but not Gö 6976. We propose a direct relationship between the alterations in apical actin filaments and the inhibition of exocytotic and transcytotic traffic.
Early work on PKCɛ implicated its increased expression in the transition to a transformed cell phenotype. The oncogenic activity of PKCɛ has been reported in several fibroblast and colonic as well as prostate epithelial cell lines (2, 23, 28, 42). Epidermis-specific transgenic overexpression of wild type PKCɛ caused development of metastatic carcinomas in mice (16). Convergence of PKCɛ signaling events on the actin cytoskeleton are likely to explain many of the oncogenic effects reported for this kinase.
More recent studies on the cellular roles of PKCɛ have focused on its role in actin filament remodeling associated with a variety of intracellular events. PKCɛ was first identified in association with actin as an effector of exocytosis in hippocampal neurons (31). It has subsequently been shown to also have a role in actin filament remodeling in endocytosis in epithelial cells (36). Cell spreading is dependent upon actin cytoskeleton reorganization while integrins play an important role in linking the extracellular matrix with the cytoskeleton and signaling machinery of the cell. PKCɛ is known to influence cell adhesion and motility through its interactions with β1 integrin through RACK1 (receptor for activated C kinase 1) and F actin (2, 6, 37). Co-expression of PKCɛ can restore cell spreading inhibited by tac-β1, a DN inhibitor of integrin function in normal human fibroblasts and CHO cells; an intact kinase domain of PKCɛ was required for this process (5). Finally, PKCɛ has recently been implicated as a signaling mediator in the Toll-like signaling receptor pathway and to mediate macrophage and dendritic cell activation in response to lipopolysaccharide (3).
Our studies add to the body of literature implicating PKCɛ in actin filament remodeling in membrane trafficking, specifically exocytosis. In our working model, release of second messengers triggered by CCH stimulation normally results in PKCɛ activation followed by its translocation to apical actin filaments. The activated PKCɛ acts by phosphorylating key targets associated with actin filaments that result in the remodeling of apical actin in ways that facilitate SV exocytosis, including the transient formation of actin-coated structures. We and others have shown that actin filament remodeling is an integral part of apical exocytosis in acinar epithelial cells (24, 29, 38, 39, Galina Jerdeva and Sarah Hamm-Alvarez, unpublished observations). Further, stabilization of actin-coated structures, thought to represent fusion intermediates, was correlated with inhibition of acinar exocytosis (39, Galina Jerdeva and Sarah Hamm-Alvarez, unpublished observations).
In acini transduced with Ad-DN-PKCɛ, overexpressed PKCɛ was detected with actin filaments of altered organization even in unstimulated acini. The changes caused by DN-PKCɛ included accumulation of actin at the APM in the underlying filament network as well as accumulation of actin-coated structures representing prospective fusion intermediates. We hypothesize that the inhibitory effects of DN-PKCɛ on apical exocytosis were caused by stabilization of apical actin, either in the underlying actin filament network or in actin-coated structures, either of which could impair SV exocytosis. These effects are likely due to the absence of catalytic activity in the DN-PKCɛ which, when recruited to apical actin, is unable to appropriately phosphorylate actin-associated proteins that are normally required for its remodeling. The inhibitory effect on DN-PKCɛ on SC exocytosis suggested a comparable role for actin filament remodeling in the terminal events associated with its release (through transcytotic or exocytotic pathways), a hypothesis supported by the co-localization of pIgR and PKCɛ with actin-coated structures in CCH-stimulated acini. Our studies implicating PKCɛ as a major effector of apical actin remodeling in exocytosis in lacrimal acini are consistent with findings from other systems. Prekeris et al. (31) found that PKCɛ was necessary for exocytosis of synaptic vesicles in hippocampal neurons. DN-PKCɛ also significantly reduced glucose-stimulated exocytosis of insulin in insulin-secreting INS-1E cells (22). The latter study also demonstrated glucose-induced association of endogenous PKCɛ with insulin granules, confirming a specific translocation to vesicles involved with exocytosis. Although its function in exocytosis was not specifically investigated, PKCɛ was also detected at the APM of pancreatic acini (4).
Our previous investigations on the mechanisms of exocytosis in lacrimal acini have been hampered by our inability to track specific secretory products released exclusively at the APM. Syncollin-GFP, previously established as a marker of large dense core vesicles and zymogen granules in related systems (15, 20), was enriched in large, 1 μM diameter, spherical structures beneath the APM, consistent with its incorporation into lacrimal acinar mature SVs. Additional syncollin-GFP was also present in smaller vesicles ranging from 300–500 nm in diameter; these vesicles were detected in the cytoplasm as well as beneath the APM. This pattern suggested specific labeling of a subpopulation of mature 1 μM SVs that might be generated from the smaller, more abundant 300–500 nm SVs representing ~60% of the syncollin-GFP population. The fluorescence associated with these subapical vesicles was substantially diminished after CCH stimulation in parallel with the increased recovery of syncollin-GFP in the culture medium, confirming that the syncollin-GFP-enriched vesicles were in fact SVs. The inhibition of syncollin-GFP release in acini co-transduced with Ad-DN-PKCɛ but not Ad-GFP enabled us to conclude that apical exocytosis, in particular, was affected.
Little is known about the cellular mechanisms underlying the release of SC and secretory IgA at the APM of lacrimal acinar cells, although common wisdom suggests that some stores are transported via the transcytotic pathway as in other epithelial cells. One previous study investigated the release of SC from cultured lacrimal acini over 4–7 days, demonstrating that CCH modestly but significantly inhibited its release (18). The inhibitory effect of CCH in this previous study might have been influenced by the duration of the exposure, which could have influenced the expression of signaling effectors or affected other acinar functions. Our study reports for the first time that CCH stimulated the acute (30 min) release of SC from pIgR at the APM of lacrimal acini. Moreover, DN-PKCɛ significantly inhibited the CCH-stimulated release of SC from lacrimal acini, suggesting a general role for PKCɛ in regulation of exocytotic and transcytotic pathways. Our laboratory is currently investigating whether these pathways function independently in acini, or whether they converge at a common apical intermediate.
Some of our findings obtained with DN-PKCɛ overexpression in acini remain perplexing. A significant amount of overexpressed DN-PKCɛ was associated with actin filaments even in the absence of stimulation. In fact, the intensity of immunofluorescence labeling was so great in unstimulated acini that the increased association of DN-PKCɛ triggered by CCH stimulation could not be distinguished by confocal fluorescence microscopy. PKCɛ content in supernatant fractions from cell lysates from CCH-stimulated acini transduced with DN-PKCɛ was markedly decreased relative to its content in untreated acini (data not shown), suggestive of increased sedimentation with actin filaments and also consistent with the existence of a pool of DN-PKCɛ that was still recruited to actin by CCH stimulation. Acini may maintain a certain percentage of total PKCɛ in association with the actin cytoskeleton even in the resting state in order to maintain unique structures or morphology. The substitution of DN-PKCɛ for wild type PKCɛ in transduced acini may underlie some of the remarkable changes in apical actin morphology seen even in the resting state. However, it is clear that the amount of DN-PKCɛ associated with actin cytoskeleton in resting transduced acini is far greater than the amount of endogenous PKCɛ associated with actin cytoskeleton in resting acini.
PKCɛ was not detected with basolateral actin in resting or CCH-stimulated non-transduced acini. However, DN-PKCɛ was extensively co-localized with basolateral actin in parallel with a profound change in the organization of basolateral actin filaments into neuritic like-extensions. As discussed above, PKCɛ overexpression is well established as a contributor to cell transformation and cell metastasis (2), processes which involve extensive actin filament remodeling, branching and extension. However, recent work on the role of PKCɛ in neurite outgrowth has reported that overexpression of PKCɛ or overexpression of the regulatory domain of PKCɛ (containing the actin-binding but not catalytic domains) exerted the same effects on outgrowth (43, 44). It is possible that association of the regulatory domain present in the DN-PKCɛ with the acinar actin cytoskeleton may be able to modulate actin filament reorganization in the absence of catalytic activity, influencing both basolateral and apical actin filament dynamics.
Although a complete consideration of the mechanisms underlying the increased association of DN-PKCɛ with basolateral actin is beyond the scope of this study, it should be noted that such changes could influence apical exocytosis. Interactions between lacrimal acini and extracellular matrix can regulate exocytosis, while integrins, in concert with actin cytoskeleton are thought to modulate some of these interactions (7). Previous work has established that β1 integrin interacts with PKCɛ and actin filaments through its receptor, RACK1, and that these interactions regulate cell migration and adhesion (2, 6, 8, 37). Recent work has also suggested that the regulatory (actin-binding) domain of PKCɛ can inactivate RhoA, and that this mechanism is involved in the switch from an adherent (stress fiber-enriched) to a motile (neuritic process) state in neuronal cells (19). RhoA has recently been implicated as a positive effector in the formation of actin-coated fusion intermediates in pancreatic acini (25).
Interestingly, DN-PKCɛ overexpression also elevated basal release of β-hexosaminidase. We have observed a similar effect by overexpression of dynamitin, an effector of the cytoplasmic dynein complex, in lacrimal acini using using an Ad-expression system (41). Dynamitin overexpression inhibits cytoplasmic dynein activity which is essential for apical exocytosis, but it also elevated basal release of β-hexosaminidase. This suggests that β-hexosaminidase already present in basolateral membrane compartments (33) may be released at an increased rate when the secretory pathway is inhibited, even under resting conditions. The altered actin filament organization noted at basolateral membranes may contribute to this phenomenon. This scenario is consistent with the observation that basal release of syncollin-GFP, which is exclusively present in secretory vesicles, was not influenced by Ad-DN-PKCɛ. This latter alternative is also consistent with previous observations that the endosomal and lysosomal compartments in acini are highly enriched in β-hexosaminidase. It is interesting that the inhibition of PKCɛ (and δ) associated with GF 109203X shown in Figure 5B does not cause increased basal release of β-hexosaminidase in parallel with the decreased CCH-stimulated release. This phenomenon of altered basolateral release of β-hexosaminidase seems therefore to occur primarily in acini transduced with Ad vectors expressing proteins that impact on the secretory pathway, suggesting that this phenomenon may be triggered by alterations in the secretory pathway as well as cellular stress elicited by protein overexpression driven by the Ad vectors.
We have established here that PKCɛ is an actin-binding protein recruited transiently to apical actin filaments and actin-coated structures, possibly representing fusion intermediates, in CCH-stimulated lacrimal acini. We have further established that its inhibition, through overexpression of DN-PKCɛ and by use of chemical inhibitors, stabilized actin-coated structures in parallel with inhibition of stimulated exocytosis of a variety of secretory products at the APM. Future work will focus on the identification of actin-associated proteins that are likely to facilitate apical actin remodeling and that serve as targets of PKCɛ.
Supplementary Material
Acknowledgments
This work was supported by NIH grants EY-11386 to SHA and EY-06177 to DAD. Additional salary support to SHA was from NIH grants EY-13949, EY-05081, NS-38246, DK-56040 and GM-59297. We thank Dr. George King, Joslin Diabetes Center, Harvard Medical School, for the kind gift of the Ad-DN-PKCɛ.
Footnotes
APM, apical plasma membrane; SV, mature secretory vesicles; pIgR, polymeric immunoglobulin receptor; SC, secretory component; PKC, protein kinase C; CCH, carbachol; PE, phenylephrine; DAPI, 4',6-diamidino-2-phenylindole, dihydrochloride; Ad, replication-defective adenovirus serotype 5; DN, dominant negative
References
- 1.Akita Y, Ohno S, Yajima Y, Konno Y, Saido TC, Mizuno K. Overproduction of a Ca2+-independent protein kinase C isozyme, nPKC epsilon, increases the secretion of prolactin from thyrotropin-releasing hormone-stimulated rat pituitary GH4C1 cells. J Biol Chem. 1994;269:4653–3660. [PubMed] [Google Scholar]
- 2.Akita Y. Protein kinase C-ɛ (PKC-ɛ): its unique structure and function. J Biochem. 2002;132:847–852. doi: 10.1093/oxfordjournals.jbchem.a003296. [DOI] [PubMed] [Google Scholar]
- 3.Aksoy E, Goldman M, Willems F. Protein kinase C epsilon: a new target to control inflammation and immune-mediated disorders. Int J Biochem Cell Biol. 2003;36:183–188. doi: 10.1016/s1357-2725(03)00210-3. [DOI] [PubMed] [Google Scholar]
- 4.Bastani B, Yang L, Baldassare JJ, Pollo DA, Gardner JD. Cellular distribution of isoforms of protein kinase C (PKC) in pancreatic acini. Biochim Biophys Acta. 1995;1269:307–15. doi: 10.1016/0167-4889(95)00120-0. [DOI] [PubMed] [Google Scholar]
- 5.Berrier AL, Mastrangelo AM, Downward J, Ginsberg M, LaFlamme SE. Activated R-Ras, Rac1, PI 3-kinase and PKCɛ can each restore cell spreading inhibited by isolated integrin β1 cytoplasmic domains. J Cell Biol. 2000;151:1549–1560. doi: 10.1083/jcb.151.7.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Besson A, Wilson TL, Yong VW. The anchoring protein RACK1 links protein kinase C epsilon to integrin beta chains. Requirements for adhesion and motility. J Biol Chem. 2002;277:22073–22084. doi: 10.1074/jbc.M111644200. [DOI] [PubMed] [Google Scholar]
- 7.Chen L, Glass JD, Walton SC, Laurie GW. Role of laminin-1, collagen IV, and an autocrine factor(s) in regulated secretion by lacrimal acinar cells. Am J Physiol (Cell Physiol) 1998;275:C278–C284. doi: 10.1152/ajpcell.1998.275.1.C278. [DOI] [PubMed] [Google Scholar]
- 8.Chun JS, Ha MJ, Jacobson BS. Differential translocation of protein kinase Ce during HeLa cell adhesion to a gelatin substratum. J Biol Chem. 1996;271:13008–13012. doi: 10.1074/jbc.271.22.13008. [DOI] [PubMed] [Google Scholar]
- 9.da Costa SR, Yarber FA, Zhang L, Sonee M, Hamm-Alvarez SF. Microtubules facilitate the stimulated secretion of β-hexosaminidase in lacrimal acinar cells. J Cell Sci. 1998;111:1267–1276. doi: 10.1242/jcs.111.9.1267. [DOI] [PubMed] [Google Scholar]
- 10.da Costa SR, Sou E, Yarber FA, Okamoto CT, Pidgeon M, Kessels MM, Mircheff AK, Schechter J, Qualmann B, Hamm-Alvarez SF. Impairing actin filament or syndapin functions promotes accumulation of clathrin-coated vesicles at the apical plasma membrane of polarized cells. Mol Biol Cell. 2003;14:4397–4413. doi: 10.1091/mbc.E03-05-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dartt DA. Signal transduction and activation of the lacrimal gland. In Principles and Practice of Ophthalmology. (Eds Albert DM and Jacobiec FA), Saunders, Philadelphia, 2ndEdition, pp 458–465, 1994.
- 12.Dekker LV, Parker PJ. Protein kinase C – a question of specificity. Trends in Biol Sci. 1994;19:73–77. doi: 10.1016/0968-0004(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 13.Gundmundsson OG, Sullivan DA, Bloch KJ, Allansmith MR. The ocular secretory immune system of the rat. Exp Eye Res. 1985;40:231–238. doi: 10.1016/0014-4835(85)90008-9. [DOI] [PubMed] [Google Scholar]
- 14.Hamm-Alvarez SF, da Costa S, Yang T, Wei X, Gierow JP, Mircheff AK. Cholinergic stimulation of lacrimal acinar cells promotes redistribution of membrane-associated kinesin and the secretory protein, β-hexosaminidase, and activation of soluble kinesin. Exp Eye Res. 1997;64:141–156. doi: 10.1006/exer.1996.0198. [DOI] [PubMed] [Google Scholar]
- 15.Hodel A, Edwardson JM. Targeting of the zymogen-granule protein syncollin in AR42J and AT-20 cells. Biochem J. 2000;350:637–643. [PMC free article] [PubMed] [Google Scholar]
- 16.Jansen AP, Verwiebe EG, Dreckschmidt NE, Wheeler DL, Oberley TD, Verma AK. Protein kinase C-epsilon transgenic mice: a unique model for metastatic squamous cell carcinoma. Cancer Res. 2001;6:808–12. [PubMed] [Google Scholar]
- 17.Kaneto H, Suzuma K, Sharma A, Bonner-Weir S, King GL, Weir GC. Involvement of protein kinase C β2 in c-myc induction by high glucose in pancreatic β-cells. J Biol Chem. 2002;277:3680–3685. doi: 10.1074/jbc.M109647200. [DOI] [PubMed] [Google Scholar]
- 18.Kelleher RS, Hann LE, Edwards JA, Sullivan DA. Endocrine, neural and immune control of secretory component output by lacrimal gland acinar cells. J Immunol. 1991;146:3405–3412. [PubMed] [Google Scholar]
- 19.Ling M, Troller U, Zeidman R, Lundberg C, Larsson C. Induction of neurites by the regulatory domains of PKCδ and ɛ is counteracted by PKC catalytic activity and by the Rho A pathway. Exp Cell Res. 2004;292:135–150. doi: 10.1016/j.yexcr.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 20.Ma L, Bindokas VP, Kuznetsov A, Rhodes C, Hays L, Edwardson JM, Ueda K, Steiner DF, Philpson LH. Direct imaging shows that insulin granule exocytosis occurs by complete vesicle fusion. Proc Natl Acad Sci USA. 2004;101:9266–9271. doi: 10.1073/pnas.0403201101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martiny-Baron G, Kazanietz M, Mischak H, Blumberg P, Kochs G, Hug H, Marme D, Schachtele C. Selective inhibition of protein kinase C isozymes by the indolocarazole Gö6976. J Biol Chem. 1993;268:9194–9197. [PubMed] [Google Scholar]
- 22.Mendez CF, Leibiger IB, Leibiger B, Hoy M, Gromada J, Berggren P, Bertorello AM. Rapid association of protein kinase C-ɛ with insulin granules is essential for insulin exocytosis. J Biol Chem. 2003;278:44753–44757. doi: 10.1074/jbc.M308664200. [DOI] [PubMed] [Google Scholar]
- 23.Mischak H, Goodnight JA, Kolch W, Martiny-Baron G, Schaechtle C, Kazanietz MG, Blumberg PM, Pierce JH, Mushinski JF. Overexpression of protein kinase C-delta and –epsilon in NIH 3T3 cells induces opposite effects on growth, morphology, anchorage dependence, and tumorigenicity. J Biol Chem. 1993;268:6090–6096. [PubMed] [Google Scholar]
- 24.Muallem S, Kwiatkowska K, Xu X, Yin HL. Actin filament disassembly is a sufficient final trigger for exocytosis in nonexcitable cells. J Cell Biol. 1995;128:589–598. doi: 10.1083/jcb.128.4.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nemoto T, Kojima T, Oshima A, Bito H, Kasai H. Stabilization of exocytosis by dynamic F-actin coating of zymogen granules in pancreatic acini. J Biol Chem. 2004;279:37544–37550. doi: 10.1074/jbc.M403976200. [DOI] [PubMed] [Google Scholar]
- 26.Newton AC. Interaction of proteins with lipid head groups: lessons from protein kinase C. Annu Rev Biophys Biomol Struct. 1993;22:1–25. doi: 10.1146/annurev.bb.22.060193.000245. [DOI] [PubMed] [Google Scholar]
- 27.Ohnishi H, Ernst SA, Wys N, McNiven M, Williams JA. Rab3D localizes to zymogen granules in rat pancreatic acini and other exocrine glands. Am J Physiol (Gastro Liv Physiol) 1996;271:G531–G538. doi: 10.1152/ajpgi.1996.271.3.G531. [DOI] [PubMed] [Google Scholar]
- 28.Perletti GP, Concari P, Brusaferri S, Marras E, Piccinini F, Tashjian AH. Protein kinase C epsilon is oncogenic in colon epithelial cells by interaction with the ras signal transduction pathway. Oncogene. 1998;16:3345–3348. doi: 10.1038/sj.onc.1201871. [DOI] [PubMed] [Google Scholar]
- 29.Perrin D, Möller K, Hanke K, Söling HD. cAMP and Ca2+-mediated secretion in parotid acinar cells is associated with reversible changes in the organization of the cytoskeleton. J Cell Biol. 1992;116:127–133. doi: 10.1083/jcb.116.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prekeris R, Klumperman J, Chen YA, Scheller RH. Syntaxin 13 Mediates Cycling of Plasma Membrane Proteins via Tubulovesicular Recycling Endosomes. J Cell Biol. 1998;143:957–971. doi: 10.1083/jcb.143.4.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prekeris R, Mayhew MW, Cooper JB, Terrian DM. Identification and localization of an actin-binding motif that is unique to the epsilon isoform of protein kinase C and participates in the regulation of synaptic function. J Cell Biol. 1996;132:77–90. doi: 10.1083/jcb.132.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qian L, Wang W, Xie J, Rose CM, Yang T, Nakamura T, Sandberg M, Zeng H, Schechter JE, Chow RH-P, Hamm-Alvarez SF, Mircheff AK. Biochemical changes contributing to functional quiescence in lacrimal gland acinar cells after chronic ex vivo exposure to a muscarinic agonist. Scand J Immunol. 2003;58:550–565. doi: 10.1046/j.1365-3083.2003.01343.x. [DOI] [PubMed] [Google Scholar]
- 33.Rose CM, Qian L, Hakim L, Wang Y, Jerdeva GY, Marchelletta R, Nakamura T, Hamm-Alvarez SF, Mircheff AK. Accumulation of catalytically active cathepsins in lacrimal gland acinar cell endosomes during chronic ex vivo muscarinic receptor stimulation. Scand J Immunol. 2005;61:36–50. doi: 10.1111/j.0300-9475.2005.01527.x. [DOI] [PubMed] [Google Scholar]
- 34.Satoh A, Gukovskaya AS, Nieto JM, Cheng JH, Gukovsky I, Reeve JR, Shimosegawa T, Pandol SJ. PKC-δ and -e regulate NF-κB activation induced by cholecystokinin and TNF-α in pancreatic acinar cells. Am J Physiol (Gastrointest Liver Physiol) 2004;287:G582–G591. doi: 10.1152/ajpgi.00087.2004. [DOI] [PubMed] [Google Scholar]
- 35.Soh J, Lee EH, Prywes R, Weinstein IB. Novel roles of specific isoforms of protein kinase C in activation of the c-fos serum response element. Mol Cell Biol. 1999;19:1313–1324. doi: 10.1128/mcb.19.2.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Song JC, Rangachari PK, Matthews JB. Opposing effects of PKCα and PKCɛ on basolateral membrane dynamics in intestinal epithelia. Am J Physiol (Cell Physiol) 2002;283:C1548–C1556. doi: 10.1152/ajpcell.00105.2002. [DOI] [PubMed] [Google Scholar]
- 37.Tachado SD, Mayhew MW, Wescott GG, Foreman TL, Goodwin CD, McJilton MA, Terrian DM. Regulation of tumor invasion and metastasis in protein kinase C epsilon-transformed NIH3T3 fibroblasts. J Cell Biochem. 2002;85:785–97. doi: 10.1002/jcb.10164. [DOI] [PubMed] [Google Scholar]
- 38.Valentijn KM, Gumkowski FD, Jamieson JD. The subapical actin cytoskeleton regulates secretion and membrane retrieval in pancreatic acinar cells. J Cell Sci. 1999;112:81–96. doi: 10.1242/jcs.112.1.81. [DOI] [PubMed] [Google Scholar]
- 39.Valentijn JA, Valentijn K, Pastore LM, Jamieson JD. Actin coating of secretory granules during regulated exocytosis correlates with the release of rab3D. Proc Natl Acad Sci USA. 2000;97:1091–1095. doi: 10.1073/pnas.97.3.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Haeringen NJ. Clinical biochemistry of tears. Curr Eye Res. 1981;26:84–96. doi: 10.1016/0039-6257(81)90145-4. [DOI] [PubMed] [Google Scholar]
- 41.Wang Y, Jerdeva G, Yarber FA, da Costa SR, Xie J, Qian L, Rose CM, Mazurek C, Kasahara N, Mircheff AK, Hamm-Alvarez SF. Cytoplasmic dynein participates in apically targeted stimulated secretory traffic in primary rabbit lacrimal acinar epithelial cells. J Cell Science. 2003;116:2051–2065. doi: 10.1242/jcs.00398. [DOI] [PubMed] [Google Scholar]
- 42.Wu D, Foreman TL, Gregory CW, McJilton MA, Wescott GG, Ford OH, Alvey RF, Mohler JL, Terrian DM. Protein kinase C epsilon has the potential to advance the recurrence of human prostate cancer. Cancer Res. 2002;62:2423–2429. [PubMed] [Google Scholar]
- 43.Zeidman R, Löfgren B, Påhlman S, Larsson C. PKCɛ, via its regulatory domain and independently of its catalytic domain, induces neurite-like processes in neuroblastoma cells. J Cell Biol. 1999;145:713–726. doi: 10.1083/jcb.145.4.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zeidman R, Trollér U, Raghunath A, Påhlman S, Larsson C. Protein kinase Ce actin-binding site is important for neurite outgrowth during neuronal differentiation. Mol Biol Cell. 2002;13:132–14. doi: 10.1091/mbc.01-04-0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zoukhri D, Hodges RR, Willert S, Dartt DA. Immunolocalization of lacrimal gland PKC isoforms. Effect of phorbol esters and cholinergic agonists on their cellular distribution. J Memb Biol. 1997;157:169–75. doi: 10.1007/s002329900226. [DOI] [PubMed] [Google Scholar]
- 46.Zoukhri D, Hodges RR, Sergheraert C, Toker A, Dartt DA. Lacrimal gland PKC isoforms are differentially involved in agonist-induced protein secretion. Am J Physiol (Cell Physiol) 1997;272:C263–C269. doi: 10.1152/ajpcell.1997.272.1.C263. [DOI] [PubMed] [Google Scholar]
- 47.Zoukhri D, Hodges RR, Sergheraert C, Dartt DA. Lacrimal gland functions are differentially controlled by protein kinase C isoforms. Ann N Y Acad Sci. 1998;842:217–220. doi: 10.1111/j.1749-6632.1998.tb09653.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.