

Abstract
Vibrio cholerae is a highly motile organism that secretes a Zn-dependent metalloprotease, hemagglutinin/protease (HapA). HapA has been shown to have mucinase activity and contribute to the reactogenicity of live vaccine candidates, but its role in cholera pathogenesis is not yet clear. The contribution of motility to pathogenesis is not fully understood, since conflicting results have been obtained with different strains, mutants, and animal models. The objective of this work was to determine the contribution of HapA and motility to the pathogenesis of El Tor biotype cholera. To this end we constructed isogenic motility (motY) and mucinase (hapA) single and double mutants of an El Tor biotype V. cholerae strain. Mutants were characterized for the expression of major virulence factors in vitro and in vivo. The motility mutant showed a remarkable increase in cholera toxin (CT), toxin coregulated pilus major subunit (TcpA), and HapA production in vitro. Increased TcpA and CT production could be explained by increased transcription of tcpA, ctxA, and toxT. No effect was detected on the transcription of hapA in the motility mutant. The sodium ionophore monensin diminished production of HapA in the parent but not in the motility mutant. Phenamil, a specific inhibitor of the flagellar motor, diminished CT production in the wild-type and motY strains. The hapA mutant showed increased binding to mucin. In contrast, the motY mutation diminished adherence to biotic and abiotic surfaces including mucin. Lack of HapA did not affect colonization in the suckling mouse model. The motility and mucinase defects did not prevent induction of ctxA and tcpA in the mouse intestine as measured by recombinase-based in vivo expression technology. Analysis of mutants in the rabbit ileal loop model showed that both V. cholerae motility and HapA were necessary for full expression of enterotoxicity.
Cholera is an acute diarrheal disease characterized by the passing of voluminous rice water stool. Vibrio cholerae of serogroups O1 and O139 continues to cause seasonal cholera outbreaks that affect highly populated regions in Asia, Africa, and Latin America. V. cholerae is a highly motile organism with a single sheathed polar flagellum. It colonizes the small intestine and expresses a variety of virulence determinants, such as the toxin-coregulated pilus (TCP), cholera toxin (CT), and other factors required to multiply and survive in the host.
V. cholerae produces a soluble Zn-metalloprotease, hemagglutinin/protease (HapA), encoded by hapA (18). HapA can proteolytically degrade several physiologically important host proteins including mucin (10). HapA perturbs the paracellular barrier of cultured intestinal epithelial cells (33, 46) by acting on tight junction-associated proteins (47). Inactivation of hapA increased adherence to mucin synthesized by HT29-18N2 cells (4), and expression of hapA was required for V. cholerae to penetrate a mucin-containing gel in vitro (42). Although analysis of hapA mutants in infant rabbits and suckling mice has not provided evidence that HapA is an essential virulence factor (11, 18, 36), HapA has been shown to contribute to reactogenicity of live attenuated cholera vaccine strains in humans (3, 13).
Expression of hapA requires transcriptional activation by HapR, RpoS, and the cyclic AMP receptor protein (22, 41). The regulators LuxO and HapR coordinate cell density-dependent expression of CT, TCP, HapA, and biofilm formation (44, 49). At low cell density, phospho-LuxO represses hapR, a condition conducive to expression of regulator AphA (26). AphA activates expression of TcpP/TcpH (25), which in concert with a second pair of transmembrane regulators (ToxR/ToxS) activates expression of the soluble regulator ToxT (31). ToxT then acts positively at the ctxA and tcpA promoters (48). At high cell density, LuxO is inactive and HapR is expressed to activate hapA and repress aphA (22, 26).
HapA and CT share the same extracellular protein secretion pathway which colocalizes with the site of the polar flagellum at the old pole of the cell (37, 39). Bile posttranscriptionally increases production of extracellular HapA (42). Monensin, an ionophore that collapses the transmembrane Na+ potential, diminishes HapA secretion to the medium (16).
It has been suggested that motility contributes to reactogenicity of live cholera vaccine candidates by promoting penetration of the protective mucus barrier (32). The role of motility in pathogenesis is not completely understood. Early studies using spontaneous or chemically induced nonmotile (NM) mutants provided conflicting results on the role of motility in adherence and enterotoxicity in the rabbit ileal loop model (1, 12, 35, 43). In one report, NM mutants of the classical biotype strain O395 were not found to be significantly defective in intestinal colonization in the suckling mouse model (14). In contrast, ΔflaA and ΔmotAB mutants of El Tor biotype V. cholerae exhibited significant reduction in colonization of the infant mouse intestine (29).
The V. cholerae polar flagellum is driven by sodium motive force (SMF) (24). Four genes have been identified to be required for flagellar rotation: pomA, pomB, motX, and motY. Inactivation of these genes by mutation abolishes motility but does not prevent flagellum assembly (30). Briefly, PomA and PomB translocate Na+ ions by forming the Na+ conducting channel. The MotX and MotY proteins are required for torque generation. The presence of an extended domain that could interact with peptidoglycan suggests that MotY constitutes the stator of the flagellar motor (30). A pomB mutant of the classical biotype strain O395 showed an increase in CT and TcpA production when the mutant was grown in Luria-Bertani (LB) medium at pH 8.5 (14). Inhibition of motility by addition of phenamil and monensin or by mutation slightly increased transcription of toxT in this biotype (17). These observations suggest a complex relationship between motility and the expression of virulence factors. The relationship between motility and expression of virulence factors in El Tor biotype strains is even less understood.
Bile has been reported to increase motility in V. cholerae (15). We observed that bile enhanced the ability of V. cholerae to penetrate a mucin column in vitro (42). Since this bile effect was not observed in a HapA-defective motile strain, we suggested that HapA is also required to penetrate mucus. The HapA mucinase activity could decrease the viscosity of the medium and allow motile V. cholerae to swim toward epithelial cells (42). In order to study the effect of HapA production and motility on cholera pathogenesis, we constructed flagellated NM, HapA-defective, and NM HapA-defective mutants of El Tor biotype V. cholerae. The mutants were characterized for the expression of major virulence factors in vitro and in vivo and for virulence in animal models of cholera. While only motility affected colonization in the suckling mouse model, both HapA and motility were required for full expression of enterotoxicity in the rabbit ileal loop model.
MATERIALS AND METHODS
Strains and media.
V. cholerae and Escherichia coli strains used in this study are shown in Table 1. For routine cloning, V. cholerae and E. coli were grown in LB broth at 37°C with agitation (250 rpm). For studies on the expression of CT and TcpA, V. cholerae strains were grown in AKI cultures as described elsewhere (21). For production of HapA, strains were grown in tryptic soy broth (TSB) at 37°C with shaking (250 rpm) or in tryptic soy agar (TSA) containing 1.5% skim milk. For motility determination, strains were stabbed in LB medium containing 0.3% agar (swarm agar) and incubated at 30°C for 16 h. Culture media were supplemented with ampicillin (Amp; 100 μg/ml), tetracycline (Tet; 1 μg/ml), kanamycin (Km; 50 μg/ml), 5-bromo-4-chloro-3-indolyl-ā-d-galactopyranoside (20 μg/ml); or isopropyl-ā-d-thiogalactopyranoside (20 μg/ml) as required. In some experiments, cultures were supplemented with monensin and phenamil (Sigma Chemical Co.) at final concentrations of 25 and 10 μM, respectively.
TABLE 1.
Strains, plasmids, and primers
| Strain, plasmid, or primer | Description | Source or reference |
|---|---|---|
| V. cholerae | ||
| AC-V66 | C6709-1, lacZ::res-tet-res | 28 |
| AJB31 | AC-V66, hapA::celA | This study |
| AJB32 | AC-V66, motY::Km | This study |
| AJB35 | AC-V66, hapA::celA motY::Km | This study |
| AC-V1745 | AC-V66 with tcpA::tnpR fusion | This study |
| AC-V1747 | AJB31 with tcpA::tnpR fusion | This study |
| AC-V1792 | AJB32 with tcpA::tnpR fusion | This study |
| AC-V1794 | AJB35 with tcpA::tnpR fusion | This study |
| AC-V1746 | AC-V66 with ctxA::tnpR fusion | This study |
| AC-V1748 | AJB31 with ctxA::tnpR fusion | This study |
| AC-V1793 | AJB32 with ctxA::tnpR fusion | This study |
| AC-V1795 | AJB35 with ctxA::tnpR fusion | This study |
| E. coli | ||
| TOP10 | F−mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80lacZΔM15 ΔlacX74 recA1 araD139 galU galKΔ(ara-leu)7697 rpsL (StrR) endA1 nupG | Invitrogen |
| SY327λPir | Δ(lac pro) argE(Am) rif nalA recA56 (λpirR6K) | 34 |
| SM10λPir | thi thr leu tonA lacY supE recA::RP4-2Tc::Mu (λpirR6K) | 34 |
| Plasmids | ||
| pGPH6 | Suicide vector pGP704 containing hapA::celA allele in BglII site (oriR6K, mobRP4, Ampr) | 36 |
| pCVD442 | Suicide vector derived from pGP704 (oriR6K, mobRP4, sacB, Ampr) | 8 |
| pUC4K | Source of the aminoglycoside 3′-phosphotransferase (Km) gene (Ampr, Kmr) | Amersham |
| PPUCMotY2 | 1,166-bp motY amplification product cloned in pUC19 as a SphI-SacI fragment | This study |
| pPUCMotYKm7 | PPUCMotY2 containing Km insertion in PstI site in motY | This study |
| pCVDMotYK1 | pCVD442 containing the motY::Km allele cloned as a SphI-SacI fragment | This study |
| Primers | ||
| RecA578 | 5′-GTGCTGTGGATGTCATCGTTGTTG | |
| RecA863 | 5′-CCACCACTTCTTCGCCTTCTTTGA | |
| Ctx120 | 5′-TATGCCAAGAAGACAGAGTGAGTAC | |
| Ctx529 | 5′-ACCTGCCAATCCATAACCATCTGC | |
| TcpA49 | 5′-CACGATAAGAAAACCGGTCAAGAGG | |
| TcpA488 | 5′-AGCGACAGCAGCGAAAGCACCTT | |
| Toxt189 | 5′-GCCCTCTATTCAGCGATTTTCTTTG | |
| ToxT400 | 5′-GCCCTCCATAGCATCAAGATCATC | |
| HapA1010 | 5′-GCACGGCGTTCAGTTATGCTTGTA | |
| HapA1551 | 5′-AGGTAAAACGCGCGGTTAAACACG | |
| MotY153 | 5′-GCATCATCTCCACCAAGTCTCGCA | |
| MotY1319 | 5′-GAAATCTAGATCCCTCAGTGCCATAA |
Construction of mucinase and motility mutants.
V. cholerae motY was amplified from strain C6709-1. Genomic DNA was purified as described by Ausubel et al. (2). The motY open reading frame (ORF) was amplified with primers MotY153 and MotY1319 (Table 1) and the Advantage PCR system (BD Biosciences). The 1,166-bp amplification product was cloned in pUC19 to create pPUCMotY2. A Km resistance gene from pUC4K (Amersham Biosciences) was then inserted at the unique PstI site located within the motY ORF to create pPUCMotYKm7. The motY::Km allele was subcloned in the suicide vector pCVD442 (8) to generate pCVDMotYK1. The motY::Km-containing suicide vector was constructed in SY327λpir, electroporated to strain SM10λpir, and transferred by conjugation to strain AC-V66. Integration in the motY locus was confirmed by Southern hybridization. Chromosomal DNA from exconjugants was digested with HindIII, and the fragments were separated in a 0.8% agarose gel and transferred to a nylon membrane. The membrane was hybridized with a digoxigenin-labeled 1,166-bp motY amplification product. Correct cointegrates were identified by the presence of HindIII junction fragments of the anticipated sizes. One cointegrate was allowed to segregate in LB broth and plated in LB agar without NaCl containing 5% sucrose. Twelve sucrose-resistant Amp-sensitive colonies were selected and characterized by Southern hybridization as described above. Strain AJB32 (motY) containing the motY::Km allele was selected for further studies. A hapA mutant of AC-V66 was constructed by conjugal transfer of pGPH6 essentially as described previously (36). The cointegrate was allowed to segregate in LB broth, and Amp-sensitive protease-negative colonies were selected in TSA containing 1.5% skim milk. Segregant AJB31 (hapA) was further confirmed by Southern hybridization and for production of the endoglucanase activity halo in carboxymethylcellulose agar plates as described previously (36). The same procedure was applied to strain AJB32 (motY) to obtain the double mutant AJB35 (hapA motY). In order to study the induction of ctxA and tcpA in mucinase and motility mutants in vivo, ctxA-tnpR and tcpA-tnpR resolvase fusions containing the tnpR135 allele were mated into each strain essentially as described by Lee et al. (28). The resulting strains containing ctxA-tnpR and tcpA-tnpR fusions were confirmed to resolve in AKI but not LB medium (data not shown).
Determination of HapA, cholera toxin, and toxin coregulated pilus.
The amount of HapA secreted to the medium was measured using an azocasein assay as described previously (41). One azocasein unit is the amount of enzyme that produces an increase of 0.01 optical density units in this assay. TcpA was determined in Western blots with a rabbit anti-TcpA serum kindly provided by R. K. Taylor (Dartmouth Medical School). For TcpA detection, the pellet corresponding to 1 optical density of cells at 600 nm was boiled in 100 μl sodium dodecyl sulfate-polyacrylamide gel electrophoresis loading buffer, and proteins were separated in a 12% polyacrylamide gel. Cholera toxin was determined by GM1 enzyme-linked immunosorbent assay using a standard curve of pure CT (Sigma Chemical Co.) as described previously (40).
Real-time reverse transcription PCR (RT-PCR).
Total RNA was isolated using the RNeasy kit (QIAGEN Laboratories). For RT-PCR the following primer combinations were used: Ctx120 and Ctx529 for ctxA mRNA; TcpA49 and TcpA488 for tcpA mRNA; HapA1010 and HapA1551 for hapA mRNA; ToxT189 and ToxT400 for toxT mRNA; and RecA578 and RecA863 for recA mRNA. An RT-negative control was performed for each reaction to exclude chromosomal DNA contamination. For quantitative comparisons RNA samples were analyzed by real-time RT-PCR using the iScript One-Step RT-PCR kit with SYBR Green (Bio-Rad Laboratories). Relative expression values (R) were calculated using the equation R = 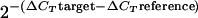 , where CT is the fractional threshold cycle. The recA mRNA was used as reference.
, where CT is the fractional threshold cycle. The recA mRNA was used as reference.
Adherence to abiotic and biotic surfaces.
V. cholerae was grown for 24 h in 96-well polystyrene microtiter plates at 30°C. Similarly, V. cholerae was grown in microtiter plates previously coated with porcine gastric mucin (1 mg/ml) (type III; Sigma Chemical Co.). Biofilm formation on these surfaces was measured by the crystal violet staining method (49). T84 cells were obtained from the American Type Culture Collection (Manassas, VA). Cells were grown in 24-well tissue culture plates in Dulbecco's modified Eagle medium (DMEM)-F12 medium at 37°C and 5% CO2 atmosphere to >95% confluence. Cells were infected with approximately 108 bacteria in DMEM-F12, and the plates were incubated for 30 min as described above. After the incubation period, each well was washed three times with phosphate-buffered saline (PBS) to remove unbound bacteria, and then adherent vibrios were released by addition of PBS plus 1% Triton X-100. The titer of adherent bacteria was determined by dilution plating in LB agar.
Suckling mice experiments.
Four- to 5-day-old CD-1 mice were starved for 6 h and inoculated orally through a 21-gauge feeding needle with 106 CFU of challenge strain in 50 μl or with a 1:1 mixture of strains in the same volume. Mice were maintained for 16 h in a Styrofoam box at 30°C. The animals were sacrificed by cervical dislocation, and the entire small intestine was removed, weighed, and homogenized in 5 ml of phosphate-buffered saline, pH 7.4 (PBS), in an ultraturrax T8 homogenizer. In competitive colonization assays the homogenate was plated in LB and LB supplemented with Km. The competitive index was calculated as the mutant-to-wild-type ratio after intraintestinal growth divided by the input ratio. For in vivo expression studies, mice were similarly inoculated with strains containing ctxA-tnpR and tcpA-tnpR fusions and sacrificed at 6 h postinoculation. Colonizing vibrios were plated in LB agar and replicated to agar plates containing tetracycline. Induction of ctxA and tcpA in this assay is estimated from the fraction of cells that become Tet sensitive (resolution) due to resolvase-mediated excision of the Tet resistance gene from the res-tet-res substrate located in the chromosome (28).
Rabbit ileal loop studies.
Rabbit ileal loop experiments were conducted as described by De and Chatterjee (7). New Zealand White male rabbits (1.5 to 2 kg) were fasted for 48 h prior to surgery and fed only water ad libitum. Rabbits were anesthetized by intramuscular administration of ketamine (35 mg/kg of body weight) and xylazine (5 mg/kg). A laparotomy was performed, and the ileum was washed and ligated into discrete loops of approximately 10 cm. Each loop was inoculated with 108 CFU of challenge strain in PBS. Pure CT (20 μg; Sigma Chemical Co.) and PBS were used as positive and negative controls, respectively. The intestine was returned to the peritoneum, and the animals were sutured and returned to their cages. After 9 h, rabbits were sacrificed by intravenous injection of Pentobarbital (150 mg/kg), and the loops were excised. Fluid volume and loop length were measured, and secretion was recorded as milliliters per centimeter. Results were analyzed by an analysis of variance of repeated measures followed by a posthoc Tukey's protected t test. In all cases, bacteria recovered from fluid were confirmed to be the challenge strain based on Km resistance and proteolytic activity in TSA containing 1.5% skim milk. The pellet of 1 ml fluid was collected by centrifugation and boiled in 100 μl of sodium dodecyl sulfate-polyacrylamide gel electrophoresis loading buffer for TcpA determination. The volume of extract applied per well was standardized by CFU. Fluids were cleared by high-speed centrifugation and saved for CT determination.
RESULTS
Effect of hapA, motY, and hapA motY mutations on the expression of virulence factors in vitro.
In order to study the relationships between mucinase production, motility, and cholera pathogenesis, we constructed isogenic motY and hapA single and double mutants. NM motY and hapA motY mutants were negative for motility in swarm agar but produced normal flagella detected by transmission electron microscopy (data not shown). The hapA and hapA motY mutants did not show proteolytic activity in milk agar plates (data not shown). The motY and hapA motY mutants produced significantly more CT than the wild-type strain and hapA mutant in AKI cultures (Fig. 1). The intensity of TcpA bands in the Western blot correlated with CT production (Fig. 1). A slightly stronger TcpA band was observed in the hapA mutant compared to the wild type, suggesting that HapA could act on TCP. The motY mutant also produced significantly more HapA than the wild type. To determine if the increase in CT, TcpA, and HapA production in the NM mutants was due to enhanced transcription, we performed quantitative real-time RT-PCR. The motY mutant produced more tcpA and toxT mRNA, respectively, compared to the wild type (Fig. 2). A smaller but statistically significant increase was observed for ctxA mRNA production at the same incubation time (Fig. 2). We did not observe a significant increase in hapA mRNA production in the motY mutant compared to the wild type. We conclude that increased TcpA and CT production in NM strains can be explained by elevated toxT transcription. The comparable levels of hapA transcription in the wild-type and motility-defective strains suggest participation of a posttranscriptional mechanism.
FIG. 1.

Production of CT, TcpA, and HapA. Strains AC-V66 (wild type), AJB31 (hapA), AJB32 (motY), and AJB35 (hapA motY) were grown in TSB for 16 h at 37°C with shaking to determine HapA production. For CT (supernatant) and cell-associated TcpA (pellet), strains were grown in AKI medium for 18 h at 30°C. Each value is the mean of six independent cultures. Error bars indicate the standard deviations. *, Significantly different from the wild type (t test, α = 0.05). **, Significantly different from the wild type (α = 0.01). OD600, optical density at 600 nm.
FIG. 2.

Relative expression of ctxA, tcpA, toxT, and hapA mRNA. Production of mRNA was measured by real-time RT-PCR with SYBR Green. The recA mRNA was used as a reference. For ctxA, tcpA, and toxT mRNA, AC-V66 (wild type) and AJB32 (motY) were grown in AKI medium for 8 h at 30°C. For hapA mRNA, strains were grown in TSB at 37°C to an optical density at 600 nm of 2. Each bar reflects the average of six independent cultures. Error bars indicate the standard deviations. *, Significantly different from the wild type (t test, α = 0.05). **, Significantly different from the wild type (α = 0.01).
It is known that the V. cholerae polar flagellum is driven by SMF (24). Monensin, which reduces SMF, diminishes secretion of HapA (16). We hypothesized that SMF not expended in flagellar rotation in the motY strain could be used to energize and enhance secretion of HapA. In this case, we would expect secretion of HapA to be less affected by monensin in the motY mutant. To this end, we examined the effect of monensin and phenamil (a specific Na+ channel inhibitor) on production of HapA. Because CT and HapA are secreted through the same pathway, we also tested the effect of these drugs on production of CT. In Fig. 3 we show that addition of monensin significantly diminished production of HapA in wild-type V. cholerae but not in the motY mutant. Monensin did not affect the relative expression of hapA in the wild-type strain measured by real-time RT-PCR (wild type, 2.3 ± 0.6; wild type plus monensin, 1.7 ± 0.5). Phenamil rather than monensin significantly reduced production of CT in the wild-type strain and the motY mutant, suggesting that Na+ translocation through the flagellar motor rather than SMF plays a predominant role in CT expression.
FIG. 3.

Effect of phenamil and monensin on production of HapA and CT. For HapA production (top panel), six cultures of AC-V66 (wild type, open bar) and AJB32 (motY, filled bar) were grown with and without phenamil or monensin in TSB for 18 h at 37°C. For CT production (lower panel), strains were grown with and without phenamil or monensin in AKI cultures for 18 h at 30°C. *, Significantly different from untreated control (t test, α = 0.05). **, Significantly different from untreated control (α = 0.01). OD600, optical density at 600 nm.
Adherence of hapA, motY, and hapA motY mutants to abiotic and biotic surfaces.
Motility has been shown to influence adherence of V. cholerae to abiotic surfaces (45), while HapA promotes detachment of vibrios from cultured cells and mucin (4, 11). We decided to test the joint effect of HapA and motility on adherence to several surfaces. As expected, motY and hapA motY strains produced a smooth colonial phenotype (data not shown) and were defective in biofilm formation (Fig. 4). The hapA mutant was not affected for biofilm formation. When plates were coated with mucin, the hapA mutant adhered significantly more compared to the wild type (Fig. 4). However, the motility defect prevailed over hapA inactivation for adherence to mucin (Fig. 4, hapA motY). Finally, NM motY and hapA motY mutants also adhered significantly less to monolayers of T84 cells (Fig. 4).
FIG. 4.

Adherence properties of the wild type, mucinase, and motility isogenic mutants. Strains AC-V66 (wild type), AJB31 (hapA), AJB32 (motY), and AJB35 (hapA motY) were grown in LB medium for 24 h at 30°C in 96-well polystyrene microtiter plates and in polystyrene microtiter plates coated with 1 mg/ml gastric porcine mucin in PBS. V. cholerae attachment to these surfaces is expressed as the optical density at 570 nm (OD570) (attached)/optical density at 600 nm (OD600) (growth) ratio. V. cholerae strains were grown in LB at 37°C for 16 h with shaking and approximately 108 vibrios in DMEM-F12 medium inoculated to T84 cells grown in 24-well tissue cultures plates to 95% confluence. Results are expressed as the fraction of V. cholerae cells bound to T84 cells. Each value is the mean of three independent cultures and six wells per culture. The error bars indicate the standard deviations. *, Significantly different from untreated control (t test, α = 0.05). **, Significantly different from untreated control (α = 0.01).
Analysis of NM and HapA-defective mutants in the suckling mouse model.
Inactivation of motY but not hapA diminished colonization in the suckling mouse model (Table 2). In a previous study we suggested that motility and mucinase activity could cooperate to achieve penetration of mucus (42). In order to test the effect of motY and hapA on intestinal colonization, we performed a competitive colonization assay. In a competition assay, the HapA secreted by the wild-type strain could be used by the mutant. To circumvent this difficulty, we tested the effect of mutating motY in hapA+ (wild type versus motY) and hapA mutant (hapA versus hapA motY) backgrounds. Hypothetically, if HapA and motility cooperate to overcome the mucus barrier, a double mutant lacking both functions should show a more severe colonization defect. However, competitive indices clearly indicated that the motility defect had a similarly negative effect on colonization in hapA+ and hapA vibrios (Table 2), suggesting that hapA does not influence colonization in this model either alone or in cooperation with motility.
TABLE 2.
Colonization of suckling mice by V. cholerae mucinase and motility mutant
| Strain and expt | Expt resultd |
|---|---|
| Single-strain infections (CFU per g tissuea) | |
| Wild type | 1.8 × 106 ± 0.8 × 106 |
| hapA | 2.5 × 106 ± 0.9 × 106 |
| motY | 0.4 × 106 ± 0.2 × 106* |
| hapA motY | 0.2 × 106 ± 0.1 × 106** |
| Mixed infections (competitive indexb) | |
| Wild type plus motY | 0.05 ± 0.02 |
| hapA plus hapA motY | 0.04 ± 0.01 |
| Induction of major virulence genes (tcpA-tnpB, ctxA-tnpR)c | |
| Wild type | 70 ± 17, 76 ± 18 |
| hapA | 57 ± 12, 93 ± 7 |
| motY | 61 ± 24, 72 ± 12 |
| hapA motY | 73 ± 20, 74 ± 12 |
Mean ± standard deviation for three mice per strain.
Mean competitive index ± standard deviation for five mice per mixture.
Percentage resolution of res-tet-res substrate ± standard deviation for three mice per strain.
*, Significantly different from wild type (t test, α = 0.05). **, Significantly different from wild type (t test, α = 0.01).
To allow for a qualitative test for virulence gene induction during infection, we introduced tcpA-tnpR and ctxA-tnpR fusions in the indicator strain AC-V66 and in derivatives of this strain containing the hapA, motY, and hapA motY mutations. Resolution was tested after growth in LB (nonpermissive for virulence gene expression), AKI (permissive), and the infant mouse intestine. As expected, little or no resolution was observed for any strain after growth in LB, whereas growth in AKI resulted in high levels of resolution for all strains (data not shown). As shown in Table 2, lack of HapA and motility did not prevent induction of tcpA and ctxA in this animal model.
Enterotoxicity of NM and HapA-defective mutants in adult rabbit ileal loops.
The above suckling mouse studies did not provide evidence that HapA alone or in concert with motility affects cholera pathogenesis. Therefore, we decided to test NM and HapA-defective mutants in the rabbit ileal loop model. As shown in Fig. 5A, inactivation of hapA and/or motY reduced significantly fluid accumulation (analysis of variance, P ≤ 0.01). The hapA motY double mutant showed the most severe effect on fluid accumulation (hapA motY versus hapA, P < 0.05; Fig. 5A). Less CT was detected in the fluid of hapA and hapA motY mutants compared to the wild type, although the former strains reached slightly higher titers in ileal loops that did not reach statistical significance. Additionally, less TcpA was detected in pellets recovered from loops inoculated with hapA and hapA motY mutants (Fig. 5B). We conclude that both motility and HapA are virulence factors that contribute to the pathogenesis of V. cholerae by enhancing enterotoxicity in the rabbit model.
FIG. 5.

Analysis of mucinase and motility mutants in the rabbit ileal loop model. A. Rabbit ileal loops were inoculated with 108 CFU of each strain AC-V66 (wild type), AJB31 (hapA), AJB32 (motY), and AJB35 (hapA motY) in PBS and incubated for 9 h. Results are expressed as fluid accumulation (FA) (in milliliters) per loop length (in centimeters). Shown are means ± standard deviations; n = 5. *, Different from the wild type (α = 0.01); **, different from hapA (α = 0.05) and motY (α = 0.01). B. Cell pellets and fluids recovered from three loops per strain were analyzed for production of TcpA and CT. ‡, Different from the wild type (α = 0.01).
DISCUSSION
Although production of mucinases and motility has been recognized as virulence factors in many pathogens, their role in cholera pathogenesis remains poorly understood. HapA has been shown to contribute to reactogenicity of live attenuated vaccines (3, 13), but its role in cholera pathogenesis has not been established. Motility also has been shown to contribute to reactogenicity (32) and to affect the expression of virulence factors (14). The motY and hapA motY mutants of El Tor biotype strain AC-V66 showed a remarkable increase in CT and TcpA production (Fig. 1) when grown in AKI cultures and of HapA in TSB (in the motY mutant). Classical biotype V. cholerae NM mutants produced more CT and variable levels of TcpA only under nonpermissive conditions (LB, pH 8.5) but did not overproduce protease (14). Furthermore, NM flagellated mutants of the classical biotype showed little effect in infant mouse colonization (14), while our motY and hapA motY mutants were significantly affected. These results suggest that the relationship between motility and expression of virulence factors is subject to important biotype-specific differences. The motY mutant showed a significant increase in tcpA, ctxA, and toxT mRNA (Fig. 2). This result is in agreement with the observation that a similar mutant of strain O395 showed a slight increase in the expression of a toxT-lacZ transcriptional fusion (17). The disparity between the increase in tcpA and ctxA in the motY strain could be due to differences in the timing of tcpA and ctxA induction during growth under AKI conditions or in the mechanism by which ToxT activates each promoter (31, 48). No differences in hapA mRNA were detected to explain the higher HapA production by the motY mutant. Since both flagellar rotation and HapA secretion require SMF (16, 24), we tested if production HapA in the motY mutant is sensitive to drugs that affect SMF and Na+ influx through the flagellar motor. As shown in Fig. 3, production of HapA is diminished by monensin in the wild-type but not in the motility-defective strain (motY). This result suggests that SMF not expended in flagellar rotation could be used to energize and enhance secretion of HapA. The finding that type II secretion does not require TonB-facilitated proton motive force suggested that ATP hydrolysis is the main source of energy for this system (5). It is known that under conditions of high SMF, H+-dependent FOF1-type ATPases can function as Na+-transporting ATP synthetase (19). Recently, it has been demonstrated that EpsE is an ATPase that could function to couple energy to the type II apparatus (6). However, a problem with the use of the above drugs is that it is difficult to uncouple translocation through the outer membrane from Sec-dependent translocation through the inner membrane, which also requires ATP hydrolysis. Production of CT in the wild-type and motY strains was sensitive to phenamil rather than monensin, indicating that Na+ flux through the flagellar motor is more important in CT expression. This result suggests that the sodium-conducting channel is still active in the motY mutant. This interpretation is supported by the finding that all phenamil-resistant mutations isolated in V. alginolyticus and V. parahaemolyticus reside within pomA and pomB and not motX or motY (30). Second, MotX and MotY do not copurify with the PomA/PomB complex (38). Furthermore, purified PomA/PomB reconstituted in proteoliposomes catalyze phenamil-sensitive Na+ transport in the absence of MotY (38). Recently, it has been shown that Na+ influx through the flagellar motor in El Tor biotype V. cholerae inhibited exopolysaccharide biosynthesis and biofilm formation (27). As proposed for exopolysaccharide biosynthesis, it is possible that Na+ influx could act through a signal transduction pathway to affect expression of CT. It is noteworthy that inhibition of motility by mutating motY and by poisoning the PomA/PomB Na+ conducting channel with phenamil had opposite effects on CT production. This could explain earlier conflicting data obtained with poorly characterized NM mutants. Our results contrast with those for classical biotype V. cholerae, in which blockage of the flagellar motor by mutating pomB or addition of phenamil increased production of CT (14, 17).
As expected, NM mutants were defective in biofilm formation (45). Here we show that motY mutants are also defective in adherence to mucin and T84 cells regardless of making HapA or not (Fig. 4). The finding that motY was epistatic to hapA for binding to mucin suggests that motility acts before HapA in the adherence process. This result supports the view that HapA diminishes adherence to mucin by promoting detachment of already bound vibrios. Inactivation of motY diminished colonization in the suckling mouse, while mutation of hapA had no effect (Table 2). The finding that motility and HapA are not required for induction of ctxA and tcpA expression in the recombinase-based in vivo expression technology assay (Table 2) suggests that vibrios do not need to overcome a mucus barrier to start making TCP and CT in this model. One should also consider that the suckling mouse might not have a fully developed protective mucus barrier (9). The thickness of the mucus protective barrier along the gastrointestinal tract has been difficult to measure, and estimates have been obtained recently in rats and humans (23).
The suckling mouse colonization model did not reveal a functional interaction between mucinase production and motility. However, analysis of mutants in the rabbit ileal loop model revealed a contrasting picture. Both motility and HapA played a significant role in this model (Fig. 5A). The hapA motY double mutant showed the most prominent effect (Fig. 5A). Our results are in agreement with the finding that pretreatment of ileal loops with purified V. cholerae HapA increased fluid accumulation in loops inoculated with live vibrios (20). The hapA and hapA motY mutants expressed less CT and TcpA (Fig. 5B) in rabbit ileal loops. Our results suggest that HapA could facilitate penetration of the mucus barrier and colonization of a region conducive to enhanced expression and delivery of virulence factors. In addition, the mucinolytic activity of HapA could release free amino acids and peptides that could enhance the expression of TCP and CT. Both interpretations would explain the predominant role of HapA in fluid accumulation in this animal model. We cannot rule out the possibility of HapA also promoting fluid secretion by acting on intestinal tight junctions (33, 46, 47). Elevated levels of CT were detected in loops inoculated with the motY mutant, but fluid accumulation was below wild type (Fig. 5). It is likely that most of this toxin remained in the fluid due to the motility and adherence defect of this strain. Based on the above results and volunteer studies with hapA mutants (3, 13), we propose that HapA is an important virulence factor that in concert with motility can strengthen the severity of cholera.
Acknowledgments
We are grateful to Andrew Shaw for assistance in growing T84 cells. We are grateful to Richard A. Finkelstein (University of Missouri School of Medicine) for helpful discussions.
A.J.S. and J.A.B. were supported by research grants S06GM08248 and RO1AI63187, respectively, from the National Institutes of Health.
Editor: V. J. DiRita
REFERENCES
- 1.Attridge, S. R., and D. Rowley. 1983. The role of the flagellum in the adherence of Vibrio cholerae. J. Infect. Dis. 147:864-872. [DOI] [PubMed] [Google Scholar]
- 2.Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl. 1995. Short protocols in molecular biology. John Wiley & Sons Inc., New York, N.Y.
- 3.Benitez, J. A., L. Garcia, A. J. Silva, H. Garcia, R. Fando, B. Cedre, A. Perez, J. Campos, B. L. Rodriguez, J. L. Perez, T. Valmaseda, O. Perez, A. Perez, M. Ramirez, T. Ledon, M. Diaz, M. Lastre, L. Bravo, and G. Sierra. 1999. Preliminary assessment of the safety and immunogenicity of a new CTXΦ-negative hemagglutinin/protease-defective El Tor strain as a cholera vaccine candidate. Infect. Immun. 67:539-545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benitez, J. A., R. G. Spelbrink, A. J. Silva, T. E. Phillips, C. M. Stanley, M. Boesman-Finkelstein, and R. A. Finkelstein. 1997. Adherence of Vibrio cholerae to cultured differentiated human intestinal cells: an in vitro colonization model. Infect. Immun. 65:3474-3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bose, N., S. M. Payne, and R. K. Taylor. 2002. Type 4 pilus biogenesis and type II-mediated protein secretion by Vibrio cholerae occurs independently of the TonB-facilitated proton motif force. J. Bacteriol. 184:2305-2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Camberg, J. L., and M. Sandkvist. 2005. Molecular analysis of the Vibrio cholerae type II secretion ATPase EpsE. J. Bacteriol. 187:249-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De, S. H., and D. N. Chatterjee. 1953. An experimental study of the mechanism of action of Vibrio cholerae on the intestinal mucous membrane. J. Pathol. Bacteriol. 46:559-562. [DOI] [PubMed] [Google Scholar]
- 8.Donnenberg, M. S., and J. B. Kaper. 1991. Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive-selection suicide vector. Infect. Immun. 59:4310-4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Faruque, S. M., G. Balakrish Nair, and J. J. Mekalanos. 2004. Genetics of stress adaptation and virulence in toxigenic Vibrio cholerae. DNA Cell Biol. 23:723-741. [DOI] [PubMed] [Google Scholar]
- 10.Finkelstein, R. A., M. Boesman-Finkelstein, and P. Holt. 1983. Vibrio cholerae hemagglutinin/lectin/protease hydrolyzes fibronectin and ovomucin: F.M. Burnet revisited. Proc. Natl. Acad. Sci. USA 80:1092-1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Finkelstein, R. A., M. Boesman-Finkelstein, Y. Chang, and C. C. Häse. 1992. Vibrio cholerae hemagglutinin/protease, colonial variation, virulence, and detachment. Infect. Immun. 60:472-478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Freter, R., and P. C. M. O'Brien. 1981. Role of chemotaxis in the association of motile bacteria with intestinal mucosa: chemotactic responses of Vibrio cholerae and description of motile nonchemotactic mutants. Infect. Immun. 34:215-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garcia, L., M. D. Jidy, H. Garcia, B. L. Rodriguez, R. Fernandez, G. Ano, B. Cedre, T. Valmaseda, E. Suzarte, M. Ramirez, Y. Pino, J. Campos, J. Menendez, R. Valera, D. Gonzalez, I. Gonzalez, O. Perez, T. Serrano, M. Lastre, F. Miralles, J. Del Campo, J. L. Maestre, J. L. Perez, A. Talavera, A. Perez, K. Marrero, T. Ledon, and R. Fando. 2005. The vaccine candidate Vibrio cholerae 638 is protective against cholera in healthy volunteers. Infect. Immun. 73:3018-3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gardel, C. L., and J. J. Mekalanos. 1996. Alterations in Vibrio cholerae motility phenotypes correlate with changes in virulence factor expression. Infect. Immun. 64:2246-2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gupta, S., and R. Chowdhury. 1997. Bile affects production of virulence factors and motility of Vibrio cholerae. Infect. Immun. 65:1131-1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hase, C. C. 2003. Ion motive force dependence of protease secretion and phage transduction in Vibrio cholerae and Pseudomonas aeruginosa. FEMS Microbiol. Lett. 227:65-71. [DOI] [PubMed] [Google Scholar]
- 17.Hase, C. C., and J. J. Mekalanos. 1999. Effects of changes in membrane sodium flux on virulence gene expression in Vibrio cholerae. Proc. Natl. Acad. Sci. USA 96:3183-3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Häse, C. C., and R. A. Finkelstein. 1991. Cloning and nucleotide sequence of the Vibrio cholerae hemagglutinin/protease (HA/protease) gene and construction of an HA/protease-negative strain. J. Bacteriol. 173:3311-3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hase, C. C., N. D. Fedorova, M. Y. Galperin, and P. A. Dibrov. 2001. Sodium ion cycle in bacterial pathogens: evidence from cross-genome comparisons. Microbiol. Mol. Biol. Rev. 65:353-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ichinose, Y., M. Ehara, T. Honda, and T. Miwatani. 1994. The effect on enterotoxicity of protease purified from Vibrio cholerae O1. FEMS Microbiol. Lett. 115:265-271. [DOI] [PubMed] [Google Scholar]
- 21.Iwanaga, M., K. Yamamoto, N. Higa, Y. Ichinose, N. Nakasone, and M. Tanabe. 1986. Culture conditions for stimulating cholera toxin production by Vibrio cholerae O1 El Tor. Microbiol. Immunol. 30:1075-1083. [DOI] [PubMed] [Google Scholar]
- 22.Jobling, M. G., and R. K. Holmes. 1997. Characterization of hapR, a positive regulator of the Vibrio cholerae HA/protease gene hap, and its identification as a functional homologue of the Vibrio harveyi luxR gene. Mol. Microbiol. 26:1023-1034. [DOI] [PubMed] [Google Scholar]
- 23.Jordan, N., J. Newton, J. Pearson, and A. Allen. 1998. A novel method for the visualization of the in situ mucus layer in rat and man. Clin. Sci. 95:97-106. [PubMed] [Google Scholar]
- 24.Kojima, S., K. Yamamoto, I. Kawagishi, and M. Homma. 1999. The flagellar motor of Vibrio cholerae is driven by a Na+ motive force. J. Bacteriol. 181:1927-1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kovacikova, G., and K. Skorupski. 2001. Overlapping binding sites for the virulence gene AphA, AphB and cAMP-CRP at the Vibrio cholerae tcpPH promoter. Mol. Microbiol. 41:393-407. [DOI] [PubMed] [Google Scholar]
- 26.Kovacikova, G., and K. Skorupski. 2002. Regulation of virulence gene expression in Vibrio cholerae by quorum sensing: HapR functions at the aphA promoter. Mol. Microbiol. 46:1135-1147. [DOI] [PubMed] [Google Scholar]
- 27.Lauriano, C. M., C. Ghosh, N. E. Correa, and K. E. Klose. 2004. The sodium-driven flagellar motor controls exopolysacharide expression in Vibrio cholerae. J. Bacteriol. 186:4864-4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee, S. H., D. L. Hava, M. K. Waldor, and A. Camilli. 1999. Regulation and temporal expression patterns of Vibrio cholerae virulence genes during infection. Cell 99:625-634. [DOI] [PubMed] [Google Scholar]
- 29.Lee, S. H., S. M. Butler, and A. Camilli. 2001. Selection for in vivo regulators of bacterial virulence. Proc. Natl. Acad. Sci. USA 98:6889-6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCarter, L. L. 2001. Polar flagellar motility of the Vibrionaceae. Microbiol. Mol. Biol. Rev. 65:445-462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Medrano, A. I., V. J. DiRita, G. Castillo, and J. Sanchez. 1999. Transient transcriptional activation of the Vibrio cholerae El Tor virulence regulator ToxT in response to culture conditions. Infect. Immun. 67:2178-2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mekalanos, J. J., M. K. Waldor, C. L. Gardel, T. S. Coster, J. Kenner, K. P. Killeen, D. T. Beattie, A. Trofa, D. N. Taylor, and J. C. Sadoff. 1995. Live cholera vaccines: perspectives on their construction and safety. Bull. Inst. Pasteur 93:255-262. [Google Scholar]
- 33.Mel, S. F., K. J. Fullner, S. Wimer-Mackin, W. I. Lencer, and J. J. Mekalanos. 2000. Association of protease activity in Vibrio cholerae vaccine strains with decrease in transcellular epithelial resistance of polarized T84 intestinal cells. Infect. Immun. 68:6487-6492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller, V. L., and J. J. Mekalanos. 1988. A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane protein and virulence determinants in Vibrio cholerae requires toxR. J. Bacteriol. 170:2575-2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Richardson, K. 1991. Roles of motility and flagellar structure in pathogenicity of Vibrio cholerae: analysis of motility mutants in three animal models. Infect. Immun. 59:2727-2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robert, A., A. J. Silva, J. A. Benitez, B. L. Rodriguez, R. Fando, J. Campos, D. K. Sengupta, M. Boesman-Finkelstein, and R. A. Finkelstein. 1996. Tagging a Vibrio cholerae El Tor candidate vaccine strain by disruption of its hemagglutinin/protease gene using a novel reporter enzyme: Clostridium thermocellum endoglucanase A. Vaccine 14:1517-1522. [DOI] [PubMed] [Google Scholar]
- 37.Sandkvist, M., L. O. Michel, L. P. Hough, V. M. Morales, M. Bagdasarian, M. Koomkey, V. J. DiRita, and M. Bagdasarian. 1997. General secretion pathway (eps) genes required for toxin secretion and outer membrane biogenesis in Vibrio cholerae. J. Bacteriol. 179:6994-7003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sato, K., and M. Homma. 2000. Functional reconstitution of the Na+-driven polar flagellar motor components of Vibrio alginolyticus. J. Biol. Chem. 275:5718-5722. [DOI] [PubMed] [Google Scholar]
- 39.Scott, M. E., Z. Y. Dossami, and M. Sandkvist. 2001. Directed polar secretion of protease from single cells of Vibrio cholerae via the type II secretion pathway. Proc. Natl. Acad. Sci. USA 98:13978-13983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Silva, A. J., R. Fando, and J. A. Benitez. 1998. Overexpression of a mutant B subunit in toxigenic Vibrio cholerae diminishes production of active cholera toxin in vivo. Curr. Microbiol. 37:231-235. [DOI] [PubMed] [Google Scholar]
- 41.Silva, A. J., and J. A. Benitez. 2004. Transcriptional regulation of Vibrio cholerae hemagglutinin/protease by the cyclic AMP receptor protein and RpoS. J. Bacteriol. 186:6374-6382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Silva, A. J., K. Pham, and J. A. Benitez. 2003. Hemaglutinnin/protease expression and mucin gel penetration in El Tor biotype Vibrio cholerae. Microbiology 149:1883-1891. [DOI] [PubMed] [Google Scholar]
- 43.Teppema, J. S., P. A. M. Guinee, A. A. Ibrahim, M. Paques, and E. J. Ruttenberg. 1987. In vivo adherence and colonization of Vibrio cholerae strains that differ in hemagglutinating activity and motility. Infect. Immun. 55:2093-2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vance, R. E., J. Zhu, and J. J. Mekalanos. 2003. A constitutively active variant of the quorum-sensing regulator LuxO affects protease production and biofilm formation in Vibrio cholerae. Infect. Immun. 71:2571-2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Watnick, P. I., C. M. Lauriano, K. E. Klose, L. Croal, and R. Kolter. 2001. The absence of a flagellum leads to altered colony morphology, biofilm development and virulence in Vibrio cholerae O139. Mol. Microbiol. 39:223-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu, Z., D. Milton, P. Nybon, A. Sjo, and K. E. Magnusson. 1996. Vibrio cholerae hemagglutinin/protease (HA/protease) causes morphological changes in cultured epithelial cells and perturbs their paracellular barrier function. Microb. Pathog. 21:111-123. [DOI] [PubMed] [Google Scholar]
- 47.Wu, Z., P. Nybom, and K. E. Magnusson. 2000. Distinct effects of Vibrio cholerae hemagglutinin/protease on the structure and localization of the tight junction-associated proteins occludin and ZO-1. Cell. Microbiol. 2:11-17. [DOI] [PubMed] [Google Scholar]
- 48.Yu, R. R., and V. J. DiRita. 2002. Regulation of gene expression in Vibrio cholerae by ToxT involves both anti-repression and RNA polymerase stimulation. Mol. Microbiol. 43:119-134. [DOI] [PubMed] [Google Scholar]
- 49.Zhu, J., M. B. Miller, R. E. Vance, M. Dziejman, B. L. Bassler, and J. J. Mekalanos. 2002. Quorum-sensing regulators control virulence gene expression in Vibrio cholerae. Proc. Natl. Acad. Sci. USA 99:3129-3134. [DOI] [PMC free article] [PubMed] [Google Scholar]