

Abstract
Bacteria have long been thought of as little more than sacks of homogeneously distributed enzymes. However, recent cytological studies indicate that bacteria are compartmentalized with proteins involved in processes such as cell division, motility, chemotaxis, and development located at distinct sites. We have used the green fluorescent protein as a reporter to determine the cellular distribution of the extracellular protein secretion (eps)-encoded type II secretion complex responsible for extracellular secretion of cholera toxin and hemagglutinin/protease in Vibrio cholerae. Real-time monitoring of green fluorescent protein fused to EpsM in living cells indicated that, like the single polar flagellum, the Eps complex is located at the old pole after cell division. Eps-dependent protease secretion was also visualized in single cells by fluorescence microscopy by using intramolecularly quenched casein. This analysis demonstrated that active protease secretion is focused at the poles and colocalizes with the site of the polar Eps apparatus. These results suggest that the type II secretion complex is responsible for directed delivery of virulence factors during cholera pathogenesis.
Cholera is a major cause of life-threatening diarrheal disease endemic to southern Asia and parts of Africa and Latin America, where seasonal outbreaks are common (1). Cholera infection occurs through ingestion of water or food contaminated with toxigenic Vibrio cholerae, a motile Gram-negative rod-shaped bacterium. V. cholerae expresses a number of virulence and colonization factors, including the coordinately expressed cholera toxin (CT) and the toxin coregulated pilus (TCP), to cause disease (2). The primary virulence factor CT, which is largely responsible for the symptoms of cholera, is a hexameric protein complex composed of five B subunits and a single A subunit (3–5). The B-subunit pentamer is responsible for binding the toxin to its receptor, GM1-ganglioside (5). After endocytosis and retrograde transport, the A subunit activates adenylate cyclase, which increases the production of cAMP (5–7), leading to massive chloride and water secretion from the cell with diarrhea as a consequence (5).
A critical step in the pathogenesis of V. cholerae is its ability to actively secrete CT to the extracellular environment. CT is transported in a two-step process, which first involves the translocation of the individual subunits across the cytoplasmic membrane via the Sec pathway (8). In the periplasmic compartment, the subunits assemble into the hexameric AB5 complex, which is subsequently translocated across the outer membrane via the type II secretion pathway, encoded by the extracellular protein secretion genes (epsC-epsN) and vcpD (pilD) (9–13). This pathway is also responsible for extracellular secretion of other potential virulence factors that include hemagglutinin/protease (HA/protease), chitinase, neuraminidase, and lipase (11, 14).
The eps genes belong to a large and widespread family of homologous genes, which encode components that are required for outer membrane translocation of a wide range of proteins in species belonging to the proteobacteria family (15). The secreted proteins, which include hydrolytic enzymes and toxins, display different structures and exhibit diverse functions; several are known to play a crucial role in the pathogenesis of their hosts (15). The unique ability to transport these apparently unrelated proteins across the outer membrane in their fully or nearly folded forms distinguishes the type II pathway from most other membrane transport systems (16–18). The type II secretion apparatus is composed of at least 13 different proteins and, despite their role as mediators of outer membrane translocation, several of these components are localized to the cytoplasmic membrane (16, 18, 19). It is believed that they interact with components in the outer membrane, including a putative gated pore to form a multiprotein secretion complex that spans the Gram-negative cell envelope (18, 20–23). The number of assembled secretion complexes per bacterium is thought to be relatively low. As few as 50–100 complexes were estimated to exist during logarithmic growth in Pseudomonas aeruginosa (22). However, it was not known whether these complexes were localized to a specific region or were uniformly distributed in the cell envelope. In this paper, we have determined the relative distribution of the type II secretion apparatus in V. cholerae and found that it is primarily localized to one of the cell poles.
Materials and Methods
Bacterial Strains and Plasmids.
The following strains were used: V. cholerae strains TRH7000 [wild type for Eps-dependent secretion (24)]; Mut8 [(epsL mutant (25)]; PU3 [(epsM mutant (26)]; HAP-1 [(hap mutant (27)]; Escherichia coli strain MC1061 [(F −lac−(28)].
To construct fusion proteins that contained green fluorescent protein (GFP) at the N terminus in frame with either EpsL or EpsM at the C terminus a gfpmut2 (29), fragment was PCR-amplified with primers 5′CGAATTCGATTTAAGAAGGAGATATAC3′ and 5′TGGATCCTTTGTATAGTTCATCCA3′ and plasmid pTM111 (gift of T. Merkel, U.S. Food and Drug Administration), a derivative of pKEN-gfpM2 (29). The pGFP-EpsL plasmid was constructed by cloning the gfp fragment into the low-copy isopropyl β-d-thiogalactoside (IPTG)-inducible vector pMMB66 (30) that contained the epsL gene. Amplification of epsM was obtained with primer pair 5′GAGATCTAAAGAATTATTGGCTCCTG3′ and 5′TCTGCAGATATCAGCCTCCACGCTT3′. The gfp-epsM fusion was constructed by stepwise cloning of the gfp and epsM fragments into pMMB66 to yield pGFP-EpsM. The gfp-epsM construct was also subcloned into the arabinose-inducible vector pAR3 (31) to yield pGFP-EpsM-ara for colocalization of the Eps apparatus and the site of protease secretion. Native epsM was subcloned into the arabinose-inducible vector pBAD33 (32) to yield pBAD-EpsM for coexpression of epsM and gfp-epsL from pGFP-EpsL in E. coli. The complete gfp sequence was subcloned from pTM111 into pMMB66 to yield pGFP. The Gene Releaser Kit (Bio-Ventures Group, Portland, ME) was used to isolate chromosomal DNA from V. cholerae strain TRH7000, and the hap gene was PCR amplified with primers 5′CTGCAGCTCTAGGATTGAGAAATG3′ and 5′AGGATCCAAGGAAGTTAGTCCAAG3′ and then cloned into pMMB66 to yield pHAP.
Growth Conditions.
V. cholerae or E. coli were grown to logarithmic phase under conditions optimal for Eps-mediated secretion at 37°C in M9 growth medium (M9GM) supplemented with 4% casamino acids/0.4% glucose/100 μg/ml of ampicillin, and expression of different GFP constructs was induced with IPTG (10 μM) for 1 h. The cells were placed on a microscope slide that contained a thin 1.5% agarose layer (M9GM supplemented with 4% casamino acids and 0.4% glucose). Over 800 bacteria that expressed the GFP-EpsM fusion were counted, and 91% of these bacteria were found to express active GFP at the poles under these conditions. When GFP-EpsL and EpsM were coexpressed from pGFP-EpsL and pBAD-EpsM, respectively, glucose was replaced with 0.5% glycerol, and 0.1 mM arabinose was added to induce the expression of EpsM. Agarose slides for time-lapse photography of V. cholerae PU3/pGFP-EpsM were prepared as above and kept on the microscope stage at 20°C for the duration of the experiment. The agarose layer was supplemented with 100 μg/ml of ampicillin and 10 μM IPTG. Images were captured every 30–40 min for a total of 8 h.
Immunofluorescence.
The procedure for immunofluorescence microscopy was based on a published method (33). Cells of V. cholerae were fixed with 1% paraformaldehyde/0.1% glutaraldehyde/30 mM sodium phosphate, washed in PBS (pH 7.4), and resuspended in 50 mM glucose/20 mM Tris⋅HCl (pH 7.5)/1 mM EDTA. Lysozyme (2 mg/ml) was added, and the cells were attached to slides. Slides were dried and treated with cold methanol followed by cold acetone. The polar flagellum of V. cholerae was detected after growth overnight on solid M9GM that contained 1.5% agarose. Colonies were resuspended into M9GM and applied to slides pretreated with 0.1% (wt/vol) poly-L-lysine (Sigma). Attached bacteria were fixed with 2% glutaraldehyde in PBS, washed with Tris buffer, and permeabilized with cold methanol. The immobilized fixed cells were incubated with blocking buffer (10–20% goat sera in PBS) and then polyclonal antibodies directed to EpsL (34), EpsG (M. Bagdasarian, Michigan State University) or Vibrio paraheamolyticus flagellin (35) in blocking buffer containing 0.05% Tween-20. Slides were washed and incubated with Alexa flour 488 F(ab′)2 goat anti-rabbit IgG (Molecular Probes) for rabbit anti-Eps IgG detection or Alexa fluor 546 goat anti-rabbit IgG (Molecular Probes) for antiflagellin detection. Slides were washed and mounted in antifade mounting medium (Molecular Probes).
Visualization of Protease Secreted from Single Cells.
Cells of V. cholerae were embedded in 1.5% agarose that contained M9GM supplemented with 4% casamino acids, 0.4% glucose, 0.3 μg/ml of BODIPY TR-X casein (Molecular Probes), 0.1 mM IPTG (and 100 μg/ml of ampicillin when the cells contained the pHAP plasmid) to detect extracellular HA/protease activity. Colocalization of protease and GFP-EpsM in cells of wild-type V. cholerae TRH7000 containing pHAP and pGFP-EpsM-ara was done under the same conditions, except that glucose was omitted from the agarose, and 0.2 mM arabinose was added to induce GFP-EpsM expression.
Microscopy.
Microscopy and photography were done with a Nikon Eclipse E800 fluorescence microscope equipped with a Nikon PlanApo100 × 1.4 N.A. oil-immersion objective and a Spot RT slider digital camera with Kodak KAI-2092 cooled color charge-coupled device chip. An excitation cube unit B-2E/C FITC with a 465- to 495-nm excitation filter and a 515- to 555-nm barrier filter was used for GFP fluorescence and visualization of Alexa fluor 488-stained cells, whereas BODIPY TR-X casein hydrolysis (EnzCheck protease assay kit), and the Alexa fluor 546-stained samples were observed with an excitation cube unit G-2E/C tetramethyl/rhodamine isothiocyanate (TRITC) with a 528- to 553-nm excitation filter and a 600- to 660-nm barrier filter. No spectral crossover fluorescence was observed with these filter sets. Images were overlaid when indicated with the use of Adobe photoshop 5.0 (Adobe Systems, Mountain View, CA).
Results
Polar Distribution of Eps Components.
The fluorescent reporter protein GFP was used for localization of the Eps apparatus within the cell envelope of V. cholerae. GFP was fused to the cytoplasmically located N terminus of EpsL to keep the GFP portion of the fusion protein in the cytoplasm to ensure the formation of active GFP (36, 37). GFP-EpsL complemented the secretion defect in a V. cholerae epsL mutant, suggesting that the EpsL portion of this chimera must interact with the rest of the secretion apparatus (not shown). Fluorescence microscopy of living cells revealed that the GFP-EpsL fusion protein localized predominantly to one of the poles of the V. cholerae cell (Fig. 1A). GFP was also fused to the N terminus of the cytoplasmic membrane protein EpsM, which has previously been shown to interact with EpsL (34), to determine whether the polar location is common to other Eps proteins. Like GFP-EpsL, this fusion protein was also active and could restore extracellular secretion in a V. cholerae epsM mutant (not shown). When examined by fluorescence microscopy, the location of GFP-EpsM was found to be confined to the poles of the V. cholerae cell as well (Fig. 1B). A control that expressed GFP without a fusion partner gave only diffuse cytoplasmic staining, thus confirming that GFP itself is not directed to the poles (Fig. 1C and ref. 36). An additional control, in which GFP was fused to a protein encoded by the hsp15 gene, located immediately upstream up the eps gene cluster, was also cytoplasmic (not shown), demonstrating that not every GFP fusion protein is localized to the poles of V. cholerae.
Figure 1.

Polar distribution of GFP fused to either EpsL or EpsM. The location of different GFP constructs was determined by fluorescent microscopy by using FITC filter. (A) GFP-EpsL expression in a V. cholerae epsL mutant. (B) GFP-EpsM expression in a V. cholerae epsM mutant. (C) GFP expression in the epsL mutant. (D) GFP-EpsL expression in E. coli MC1061. (E) GFP-EpsM expression in E. coli MC1061. (F) GFP-EpsL and EpsM coexpression in E. coli MC1061. The arrow indicates the brighter pole in a cell with bipolar fluorescence.
Next, the locations of EpsL and EpsG were determined by immunofluorescence to confirm the polar distribution of native Eps proteins. EpsL, as noted above, is anchored in the cytoplasmic membrane, and EpsG is detected both in the cytoplasmic and outer membrane after cell fractionation. Areas of bright fluorescence at one end of the wild-type V. cholerae cells were observed with both anti-Eps antibodies (Fig. 2 A and B). Polar distribution of the putative outer membrane pore was also observed with anti-EpsD antibodies (not shown). No staining was obtained with secondary antibody alone (Fig. 2C). Hence, the immunofluorescence data supported the GFP-protein fusion data, which indicated that the Eps machinery is primarily localized to one of the poles.
Figure 2.

Polar localization of native Eps proteins. Cells of wild-type V. cholerae were fixed with paraformaldehyde, treated with lysozyme, and subjected to immunofluorescence by using anti-EpsG (A) or anti-EpsL (B) antibodies and detected with Alexa fluor 488-conjugated F(ab′)2 goat anti-rabbit IgG. C shows result observed with secondary antibody only.
Polar Localization of EpsM in E. coli.
The GFP-EpsL fusion appeared to be confined to the poles of V. cholerae in a species-specific manner, because expression of this fusion protein in the absence of other Eps proteins in E. coli resulted in only weak cytoplasmic and membrane staining (Fig. 1D). GFP-EpsM, on the other hand, was restricted to the poles in E. coli, suggesting that EpsM carries all of the information necessary for polar localization (Fig. 1E). When GFP-EpsL and native EpsM were coexpressed from two different plasmids in the same E. coli cells, the level of GFP-EpsL fluorescence was increased (Fig. 1F), thus confirming the previous finding that showed that EpsM stabilizes EpsL and prevents its degradation (34). Furthermore, in the presence of EpsM, GFP-EpsL was detected at the cell poles in E. coli (Fig. 1F).
The Eps Complex Is Confined to the “Old” Pole.
Although the Eps apparatus is predominantly localized to one pole, a significant proportion (22%) of cells carried GFP-EpsM or GFP-EpsL at both poles. However, one end of the bacteria very often was brighter than the other (Fig. 1B, arrow). Moreover, some of the cells in which fluorescence was detected at both poles appeared to be longer than the average cell, and a few of them had a constricted middle, suggesting that cell division had been initiated. The hypothesis that the Eps apparatus is preferentially localized to the old pole was tested by time-lapse fluorescence microscopy that followed the location of GFP-EpsM throughout the growth cycle (Fig. 3). Cells of the V. cholerae epsM mutant expressing GFP-EpsM were placed on agarose containing M9GM supplemented with amino acids and glucose on a glass slide, a coverslip was positioned on top and the slide was sealed. The slide was then placed on the microscope stand at ambient temperature (20°C), and images were retrieved at ≈30-min intervals. Under these slow growth conditions, growth of individual cells and the location of the Eps apparatus could be followed in real time through several division cycles. A time-lapse experiment in which a cell with one bright fluorescent pole grows and subsequently ends up with two fluorescent poles before cell division is shown in Fig. 3. These results confirm that the Eps apparatus is confined to the old pole. Interestingly, when bacteria were incubated for extended periods (i.e., overnight) under these conditions, the number of bacteria with bipolar fluorescence increased to ≈65%.
Figure 3.

The Eps apparatus is localized to the “old” pole. Cells of V. cholerae epsM mutant that express GFP-EpsM were placed on a thin film of agarose-M9GM on a slide and subjected to time-lapse fluorescence and phase-contrast microscopy. The images were overlaid with the use of Adobe photoshop 5.0. The numbers indicate time in minutes. The white arrows indicate the appearance of new fluorescent poles. The black arrow shows the cell division site.
The Eps Apparatus and the Single Flagellum Are Located at the Same Pole.
The mechanism by which the Eps apparatus is targeted to the old pole is not known; however, it may be similar to that which directs the unipolar localization of the V. cholerae flagellum. The location of the Eps apparatus relative to the polar flagellum was determined by immunofluorescence microscopy of the V. cholerae epsM mutant that expressed the GFP-EpsM fusion by using anti-V. paraheamolyticus flagellin antibodies. The results indicated that the flagellum and the Eps apparatus assemble at the same pole (Fig. 4) and suggest that the flagellum is also confined to the old pole of V. cholerae.
Figure 4.
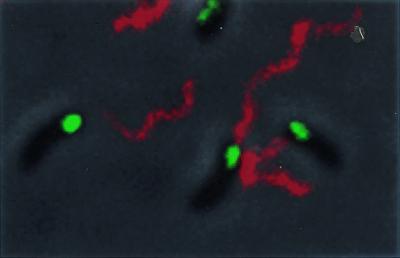
The Eps apparatus and flagellum assemble at the same pole. Fixed cells of V. cholerae epsM mutant that express GFP-EpsM were subjected to immunofluorescence by using antiflagellin antibodies and detected with Alexa fluor 546 goat anti-rabbit IgG. FITC and TRITC filters were used for visualization of GFP-EpsM and the single flagellum, respectively. The bacteria were visualized by phase-contrast microscopy. The images were overlaid with the use of Adobe photoshop 5.0.
Identification of the Site of Active Protease Secretion.
The polar location of the type II apparatus inferred that secretion would also be polar. Therefore, we developed an assay to determine the site of extracellular secretion in single cells. The site of HA/protease secretion was visualized by proteolysis of intramolecularly quenched BODIPY TR-X casein, which releases highly fluorescent fragments upon cleavage by a protease. Cells of V. cholerae, to prevent motility, were embedded in agarose-M9GM, supplemented with glucose, amino acids, and BODIPY TR-X casein, then incubated at 37°C overnight to allow for HA/protease secretion and accumulation of fluorescent casein fragments. No fluorescence was detected with a HA/protease mutant, HAP-1 (27), whereas introduction of plasmid pHAP that expressed HA/protease into HAP-1 resulted in bright areas of fluorescence predominantly at the poles (not shown). The expression level of endogenous HA/protease in the wild-type strain TRH7000 was too low to be visualized (Fig. 5B), but bright fluorescence was observed at the poles of TRH7000 that contained the pHAP plasmid (Fig. 5A). Although many of the cells appeared to secrete protease at both poles, a significant number of cells (26%) showed casein hydrolysis at only one pole. Even in the presence of high levels of HA/protease expression from the pHAP plasmid, the secretion-deficient epsM mutant PU3 did not display any fluorescence (Fig. 5C). This indicates that the site of Eps-dependent protease secretion is, like the Eps apparatus itself, confined to the poles of the V. cholerae cell.
Figure 5.
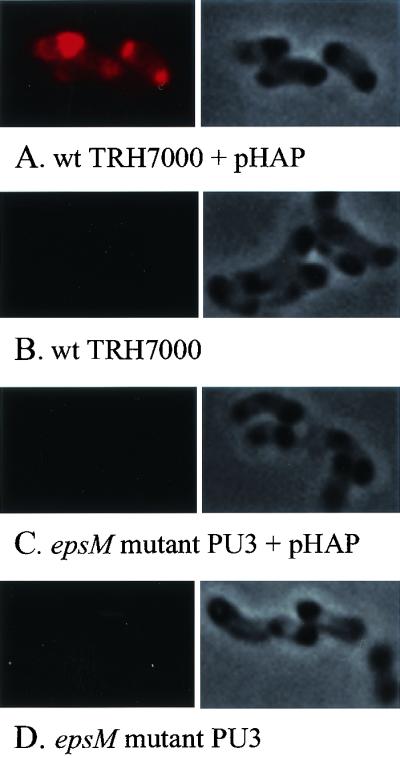
Polar Eps-dependent HA/protease secretion in single cells. Cells of V. cholerae were embedded in agarose containing M9 salts, amino acids, BODIPY TR-X casein, and IPTG to induce HA/protease expression and grown overnight at 37°C. The slides were subjected to fluorescence microscopy by using a TRITC filter. (A) Wild-type V. cholerae TRH7000 expressing HA/protease from pHAP. (B) TRH7000. (C) epsM mutant PU3 expressing HA/protease from pHAP. (D) PU3. The corresponding phase-contrast images are shown next to the fluorescent images to indicate the presence of bacteria on each slide.
Colocalization of the Eps Apparatus and the Site of Eps-Dependent Protease Secretion.
The site of protease secretion and the location of GFP-EpsM were identified in the same cells to determine whether the GFP-EpsM fusion protein represents functional Eps complexes that actively secrete protease. The assay was performed as described above, and the site of secreted protease and location of the GFP-EpsM were visualized independently as red and green fluorescence, respectively (Fig. 6 A and B). The results indicated that the site of active protease secretion and the location of the GFP-EpsM fusion colocalize to the same pole in living cells (Fig. 6C). When GFP-EpsM was detected at both poles, hydrolysis of the quenched fluorescent substrate was also observed at both poles. In contrast, when GFP-EpsM was detected at only one of the poles, only this pole had HA/protease activity (Fig. 6, arrow).
Figure 6.

Colocalization of the Eps apparatus and the site of Eps-dependent protease secretion. Cells of wild-type V. cholerae that contained plasmids pHAP and pGFP-EPSM-ara were embedded in agarose containing M9 salts, amino acids, BODIPY TR-X casein, and IPTG and arabinose to induce HA/protease and GFP-EpsM expression, respectively. The slide was incubated at 37°C overnight, and fluorescent images were retrieved by using a TRITC filter to detect casein hydrolysis (A) or an FITC filter to detect GFP-EpsM (B). The images were imported into Adobe photoshop and layered for the purpose of colocalization, which is shown as yellow (C). A cell in which GFP-EpsM is detected at only one pole and this pole alone secretes HA/protease is indicated by an arrow.
Discussion
The biological advantage to the restriction of the secretion apparatus to the pole is not known. However, it may be a way to concentrate the relatively few secretion complexes to obtain a critical amount of secreted material at one site. Furthermore, it is possible that, under certain environmental conditions, if secretion occurred all over the cell surface a significant portion of secreted material could be lost and wasted. A polar apparatus, on the other hand, would provide directed secretion to a discrete location, thus conserving both secreted material and energy. For example, directed secretion may be important for chitinase secretion during nutrient acquisition, when V. cholerae is attached to chitinous particles in the aquatic milieu. Similarly, if the Eps apparatus were localized to the same pole that is responsible for attaching V. cholerae to the epithelial cell surface during colonization of the small intestine, this would provide for the directed delivery of virulence factors such as CT. Thus, the inference is that the polar confinement of the secretion apparatus may play an important role in the pathogenesis of this organism. The type IV pilus TCP is required for intestinal colonization of V. cholerae (2, 38). Although the colonization process is likely to be multifactorial, and a specific receptor for adhesion has not yet been identified, one role for TCP may be to function as a ligand for cell-surface attachment. A previous study has shown that during V. cholerae colonization in infant mice, expression of the tcp genes is required for and proceeds the expression of the genes for CT, suggesting that the expression and secretion of CT occurs after initiation of colonization (39). It is not known to which end of the bacterium TCP are attached. However, by analogy with Caulobacter and Myxococcus (40, 41), we suggest that in V. cholerae, the polar type IV pilus TCP is localized to the same pole as the flagellum. If this is the case, TCP should also be present at the same pole as the Eps apparatus, and provide localized secretion of CT directly at the site of cell surface attachment. Finally, directed protease secretion may be beneficial in the dissemination process. Finkelstein and colleagues have suggested that HA/protease may be responsible for detachment of V. cholerae from the epithelial cell surface by digestion of several putative receptors (42). Delivery of the “detachase” activity, specifically where bacterial cells are attached, would likely result in rapid detachment and dissemination.
The mechanism used to target the Eps apparatus to the pole and maintain it there is not known. Several hypotheses for localization of other polar complexes, such as flagella and the cell division apparatus, have been proposed (43, 44). These include the possibility that, in comparison to the lateral wall, the presence or absence of specific components at the poles may mark these domains for delivery of polar complexes. The protein as well as lipid and peptidoglycan content may be different at the poles (45, 46). In addition, during peptidoglycan synthesis, new glycan strands are incorporated into the lateral wall and the septum, but not into the old pole, which is inert (45). Thus, the stable environment of the old pole may provide a mechanism by which complexes are maintained at this location (43) but may not fully explain how the polar complexes initially reach this locale. The results of our time-lapse fluorescence microscopy experiments showed that the Eps apparatus is assembled at the old pole. However, as the cells aged and stopped dividing, there was an apparent shift from unipolar to bipolar distribution of the Eps machinery. It is possible that when the cells stop growing, the “new” poles gradually mature and develop properties typical of the old pole. This may signal the Eps components to assemble at this pole too.
Our preliminary finding that EpsM can localize to the pole in the absence of other Eps components in E. coli, whereas detection of EpsL at the pole requires EpsM expression suggests that not all Eps proteins contain information required for polar targeting. It is possible that EpsM alone, or EpsM in combination with a subset of Eps proteins, serves as a polar nucleation or retention factor for the other Eps components. Future experiments are aimed at understanding the mechanism of polar secretion and identifying the species-specific properties of V. cholerae that are responsible for polar confinement of the Eps apparatus. Another level of complexity to the polar Eps-dependent secretion exists. Namely, not only must there be a mechanism to localize the type II secretion apparatus to the pole, but the proteins that are secreted via this apparatus must also be directed to the pole before outer membrane translocation. The secreted proteins are initially transported via the Sec pathway to the periplasmic compartment. Are there specialized Sec-machineries located at the pole that preferentially transport these proteins, or are the secreted proteins randomly transported across the cytoplasmic membrane and uniformly distributed in the periplasmic compartment, and then targeted to the pole?
Acknowledgments
We thank M. Bagdasarian, K. C. Ingham, D. A. Lawrence, and M. Russel for helpful discussions and comments on the manuscript. We also thank M. Bagdasarian for providing anti-EpsG antiserum, L. L. McCarter for antiflagellin antiserum, R.A. Finkelstein for the HAP-1 mutant, and T. Merkel for plasmid pTM111. The American Red Cross supported this work.
Abbreviations
- CT
cholera toxin
- TCP
toxin coregulated pilus
- GFP
green fluorescent protein
- M9GM
M9 growth medium
- IPTG
isopropyl β-d-thiogalactoside
- HA
hemagglutinin
- TRITC
tetramethyl/rhodamine isothiocyanate
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Faruque S M, Albert M J, Mekalanos J J. Microbiol Mol Biol Rev. 1998;62:1301–1314. doi: 10.1128/mmbr.62.4.1301-1314.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Herrington D A, Hall R H, Losonsky G, Mekalanos J J, Taylor R K, Levine M M. J Exp Med. 1988;168:1487–1492. doi: 10.1084/jem.168.4.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang R G, Scott D L, Westbrook M L, Nance S, Spangler B D, Shipley G G, Westbrook E M. J Mol Biol. 1995;251:563–573. doi: 10.1006/jmbi.1995.0456. [DOI] [PubMed] [Google Scholar]
- 4.Sixma T K, Kalk K H, van Zanten B A, Dauter Z, Kingma J, Witholt B, Hol W G. J Mol Biol. 1993;230:890–918. doi: 10.1006/jmbi.1993.1209. [DOI] [PubMed] [Google Scholar]
- 5.Spangler B D. Microbiol Rev. 1992;56:622–647. doi: 10.1128/mr.56.4.622-647.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lencer W I, Constable C, Moe S, Jobling M G, Webb H M, Ruston S, Madara J L, Hirst T R, Holmes R K. J Cell Biol. 1995;131:951–962. doi: 10.1083/jcb.131.4.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsai B, Rodighiero C, Lencer W I, Rapoport T A. Cell. 2001;104:937–948. doi: 10.1016/s0092-8674(01)00289-6. [DOI] [PubMed] [Google Scholar]
- 8.Hofstra H, Witholt B. J Biol Chem. 1984;259:15182–15187. [PubMed] [Google Scholar]
- 9.Hirst T R, Holmgren J. Proc Natl Acad Sci USA. 1987;84:7418–7422. doi: 10.1073/pnas.84.21.7418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirst T R, Holmgren J. J Bacteriol. 1987;169:1037–1045. doi: 10.1128/jb.169.3.1037-1045.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sandkvist M, Michel L O, Hough L P, Morales V M, Bagdasarian M, Koomey M, DiRita V J. J Bacteriol. 1997;179:6994–7003. doi: 10.1128/jb.179.22.6994-7003.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marsh J W, Taylor R K. Mol Microbiol. 1998;29:1481–1492. doi: 10.1046/j.1365-2958.1998.01031.x. [DOI] [PubMed] [Google Scholar]
- 13.Fullner K J, Mekalanos J J. Infect Immun. 1999;67:1393–1404. doi: 10.1128/iai.67.3.1393-1404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Connell T D, Metzger D J, Lynch J, Folster J P. J Bacteriol. 1998;180:5591–5600. doi: 10.1128/jb.180.21.5591-5600.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sandkvist M. Infect Immun. 2001;69:3523–3535. doi: 10.1128/IAI.69.6.3523-3535.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Russel M. J Mol Biol. 1998;279:485–499. doi: 10.1006/jmbi.1998.1791. [DOI] [PubMed] [Google Scholar]
- 17.Lory S. Curr Opin Microbiol. 1998;1:27–35. doi: 10.1016/s1369-5274(98)80139-2. [DOI] [PubMed] [Google Scholar]
- 18.Sandkvist M. Mol Microbiol. 2001;40:271–283. doi: 10.1046/j.1365-2958.2001.02403.x. [DOI] [PubMed] [Google Scholar]
- 19.Filloux A, Michel G, Bally M. FEMS Microbiol Rev. 1998;22:177–198. doi: 10.1111/j.1574-6976.1998.tb00366.x. [DOI] [PubMed] [Google Scholar]
- 20.Nouwen N, Ranson N, Saibil H, Wolpensinger B, Engel A, Ghazi A, Pugsley A P. Proc Natl Acad Sci USA. 1999;96:8173–8177. doi: 10.1073/pnas.96.14.8173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sauvonnet N, Vignon G, Pugsley A P, Gounon P. EMBO J. 2000;19:2221–2228. doi: 10.1093/emboj/19.10.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brok R, Van Gelder P, Winterhalter M, Ziese U, Koster A J, De Cock H, Koster M, Tommassen J, Bitter W. J Mol Biol. 1999;294:1169–1179. doi: 10.1006/jmbi.1999.3340. [DOI] [PubMed] [Google Scholar]
- 23.Nunn D. Trends Cell Biol. 1999;9:402–408. doi: 10.1016/s0962-8924(99)01634-7. [DOI] [PubMed] [Google Scholar]
- 24.Hirst T R, Sanchez J, Kaper J B, Hardy S J, Holmgren J. Proc Natl Acad Sci USA. 1984;81:7752–7756. doi: 10.1073/pnas.81.24.7752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sandkvist M, Bagdasarian M, Howard S P, DiRita V J. EMBO J. 1995;14:1664–1673. doi: 10.1002/j.1460-2075.1995.tb07155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Overbye L J, Sandkvist M, Bagdasarian M. Gene. 1993;132:101–106. doi: 10.1016/0378-1119(93)90520-d. [DOI] [PubMed] [Google Scholar]
- 27.Hase C C, Finkelstein R A. J Bacteriol. 1991;173:3311–3317. doi: 10.1128/jb.173.11.3311-3317.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Casadaban M J, Cohen S N. J Mol Biol. 1980;138:179–207. doi: 10.1016/0022-2836(80)90283-1. [DOI] [PubMed] [Google Scholar]
- 29.Cormack B P, Valdivia R H, Falkow S. Gene. 1996;173:33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- 30.Furste J P, Pansegrau W, Frank R, Blocker H, Scholz P, Bagdasarian M, Lanka E. Gene. 1986;48:119–131. doi: 10.1016/0378-1119(86)90358-6. [DOI] [PubMed] [Google Scholar]
- 31.Perez-Perez J, Gutierrez J. Gene. 1995;158:141–142. doi: 10.1016/0378-1119(95)00127-r. [DOI] [PubMed] [Google Scholar]
- 32.Guzman L M, Belin D, Carson M J, Beckwith J. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harry E J, Pogliano K, Losick R. J Bacteriol. 1995;177:3386–3393. doi: 10.1128/jb.177.12.3386-3393.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sandkvist M, Hough L P, Bagdasarian M M, Bagdasarian M. J Bacteriol. 1999;181:3129–3135. doi: 10.1128/jb.181.10.3129-3135.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCarter L L. J Bacteriol. 1995;177:1595–1609. doi: 10.1128/jb.177.6.1595-1609.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Margolin W. Methods. 2000;20:62–72. doi: 10.1006/meth.1999.0906. [DOI] [PubMed] [Google Scholar]
- 37.Sandkvist M, Keith J M, Bagdasarian M, Howard S P. J Bacteriol. 2000;182:742–748. doi: 10.1128/jb.182.3.742-748.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thelin K H, Taylor R K. Infect Immun. 1996;64:2853–2856. doi: 10.1128/iai.64.7.2853-2856.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee S H, Hava D L, Waldor M K, Camilli A. Cell. 1999;99:625–634. doi: 10.1016/s0092-8674(00)81551-2. [DOI] [PubMed] [Google Scholar]
- 40.Skerker J M, Shapiro L. EMBO J. 2000;19:3223–3234. doi: 10.1093/emboj/19.13.3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wall D, Kaiser D. Mol Microbiol. 1999;32:1–10. doi: 10.1046/j.1365-2958.1999.01339.x. [DOI] [PubMed] [Google Scholar]
- 42.Finkelstein R A, Boesman-Finkelstein M, Chang Y, Hase C C. Infect Immun. 1992;60:472–478. doi: 10.1128/iai.60.2.472-478.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lybarger S R, Maddock J R. J Bacteriol. 2001;183:3261–3267. doi: 10.1128/JB.183.11.3261-3267.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shapiro L, Losick R. Cell. 2000;100:89–98. doi: 10.1016/s0092-8674(00)81686-4. [DOI] [PubMed] [Google Scholar]
- 45.de Pedro M A, Quintela J C, Holtje J V, Schwarz H. J Bacteriol. 1997;179:2823–2834. doi: 10.1128/jb.179.9.2823-2834.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mileykovskaya E, Dowhan W. J Bacteriol. 2000;182:1172–1175. doi: 10.1128/jb.182.4.1172-1175.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]