

Abstract
Tertiary RNA structures from thermophilic bacteria generally are more stable than their mesophilic homologs. To understand the structural basis of the increase in stability, we investigated equilibrium folding of the specificity domain (S-domain) of RNase P RNA from a mesophilic (Escherichia coli) and a thermophilic (Thermus thermophilus) bacterium. Equilibrium folding of both S-domains is described by a minimal, three-state folding scheme, U-to-I-to-N. In the I-to-N transition of the thermophilic S-domain, more structure forms and protections are stronger against T1 nuclease and hydroxyl radical reactions. Phylogenetic comparison in the context of the native structure reveals that among 39 nucleotide differences between these S-domains, 12 likely contribute to higher stability. These residues participate in extensive networks of hydrogen bonding, stacking, and metal ion coordination throughout the molecule. The thermophilic S-domain achieves higher stability by mutating strategic base pairs to G-C, decreasing surface accessibility of the native state, and increasing the amount of structure formation in the native folding transition. An E. coli S-domain mutant containing these 12 nt has the same stability and folding cooperativity as the T. thermophilus S-domain. E. coli S-domain mutants containing a subset of 4 or 6 nt have the same stability as the T. thermophilus S-domain but the same folding cooperativity as the E. coli S-domain. These results show that increasing stability can be accomplished by mutations within a local structure, but increasing folding cooperativity needs concerted changes among multiple structural units.
Keywords: folding, stability, structure, thermophilic
INTRODUCTION
The optimal growth temperatures for the mesophilic bacterium Escherichia coli and the thermophilic bacterium Thermus thermophilus are 37°C and 75°C, respectively (Hartmann and Erdmann 1991; Brown et al. 1993). The essential tRNA processing ribozyme, ribonuclease P, in both bacteria has the same secondary and tertiary structure (Frank and Pace 1998; Altman and Kirsebom 1999). How then, does the T. thermophilus RNase P RNA maintain its stability at high temperatures?
One possible mechanism to elevate stability of the thermophilic RNase P is to significantly increase the stability of its protein subunit (Kazantsev et al. 2003). In fact, substituting the E. coli RNase P protein with that from the thermophile Thermotoga maritima allowed the E. coli RNase P to cleave its substrates at higher temperatures in vitro (Paul et al. 2001). The contribution from the protein subunit, however, is insufficient to fully account for the entire increase in stability. Previous folding studies have shown that the thermophilic RNA is more stable than the homologous RNase P RNA and its domains in the absence of the protein subunit (Brown et al. 1993; Fang et al. 2001). Hence, features of the thermophilic RNA must make significant contributions to the stability of this ribonucleoprotein enzyme.
The increased stability of a thermophilic RNA can be derived from multiple factors. More than a decade ago, Pace and coworkers (Brown et al. 1993) suggested that increasing number of G-C pairs and eliminating mismatched base pairs in the thermophilic RNA are important. Studies on thermostable group I introns revealed that increased stability can be attributed to improved packing in the interior of its structure (Guo and Cech 2002; Guo et al. 2004). Differential folding properties also played a role in achieving higher stability for a thermophilic RNA (Fang et al. 2001). The dynamics or rigidity of the native structure may also influence RNA stability (Brion and Westhof 1997). In summary, multiple factors—secondary structure improvements, tertiary packing, variations in the folding pathway, and dynamic properties of the native structure—are utilized to increase stability. But, their quantitative contribution likely depends on the specific RNA structure.
What has been missing to date is an explicit structural description of the relevant residues and their links to folding properties for increased stability in thermophilic RNA. These issues are explored here by studying the equilibrium folding of two homologous specificity domains of RNase P RNA (Loria and Pan 1996; Massire et al. 1998), one from a mesophile, E. coli, and the other from a thermophile, T. thermophilus (Fig. 1 ▶). These two ~160 nucleotide domains have 39 sequence differences and bind the T–stem–loop portion of a pre-tRNA substrate (Qin et al. 2001). The crystal structure of the T. thermophilus S-domain has been solved recently at 2.9 Å resolution (Krasilnikov et al. 2004). Our folding studies, together with phylogenetic comparison of 28 homologous thermophilic and mesophilic S-domains, suggest a structural basis on how this thermophilic RNA attains higher stability.
FIGURE 1.

Sequence and structure of S-domains with nucleotide differences highlighted in red. (A) E. coli secondary structure arranged according to the crystal structure of T. thermophilus S-domain. (B) T. thermophilus secondary structure arranged according to the crystal structure (Krasilnikov et al. 2004). Nucleotide insertions relative to E. coli S-domain are circled. (C) Three-dimensional structure of T. thermophilus S-domain (1U96).
RESULTS AND DISCUSSION
Folding monitored by CD and absorbance spectroscopy
Folding of the S-domains is monitored with circular dichroism (CD) and UV absorbance over a range of Mg2+ concentrations (Fig. 2 ▶). CD signals at 287 nm exhibit a definitive I-to-N transition occurring at 0.08–0.16 mM Mg2+ (Table 1 ▶). The absorbance signal at this wavelength displays another transition occurring at lower Mg2+ concentration than that observed by CD. This folding behavior is similar to those for other tertiary RNAs, wherein the equilibrium folding pathway was described by a minimal three-state scheme, U-to-I-to-N (Sosnick and Pan 2003). At 287 nm, UV absorbance is primarily sensitive to the U-to-I transition, whereas CD primarily reports on the I-to-N transition. For these S-domains, CD and absorbance signals at 260 and 278 nm also can track one or both transitions (data not shown).
FIGURE 2.

Folding of E. coli (□) and T. thermophilus (▪) S-domains monitored by CD (top panel) and absorbance (bottom panel) at 287 nm (0.3 μM RNA, 37°C, 20 mM Tris at pH 8.1). The inset shows a plot for titrations performed at various [urea] to determine the m-value. Changes in absorbance and CD track the U-to-I transition and the I-to-N transition, respectively.
TABLE 1.
Folding parameters for the I-to-N transition obtained by CD
| S-domain | No. of nt changes | KMg (mM) | n | m (kcal mol−1 m−1) |
| E. coli | 0 | 0.15 ± 0.01 | 3.9 ± 0.5 | 1.5 ± 0.1 |
| T. thermophilus | 39 | 0.08 ± 0.01 | 6.2 ± 0.6 | 2.4 ± 0.2 |
| Mutants | ||||
| P12 | 2 | 0.17 ± 0.01 | 3.0 ± 0.6 | |
| P9 + P11 | 4 | 0.07 ± 0.02 | 3.8 ± 1.7 | |
| J11/12 + P14 | 6 | 0.09 ± 0.01 | 2.9 ± 0.7 | |
| All 12 | 12 | 0.09 ± 0.01 | 6.4 ± 1.6 | |
20 mM TrisHCl (pH 8.1), 37°C.
Mg2+ titration curves are fit according to a cooperative Hill-type analysis to obtain the Mg2+-midpoint, KMg, and the Hill coefficient, n (Equation 1; Table 1 ▶). The denaturant-sensitive surface burial, m, for the I-to-N transition is obtained from variations in KMg upon the addition of urea (Shelton et al. 1999). The thermophilic S-domain folds at lower Mg2+ concentration (lower KMg) with increased co-operativity (larger n and m values), indicating that more structure forms in this transition. The decreased [Mg2+] requirement and higher folding cooperativity are similar to the folding properties of another homologous mesophilic/thermophilic RNA pair, the catalytic domain of B-type RNase P RNA (Fang et al. 2001).
Folding monitored by partial nuclease T1 digestion
Folding of the S-domains at varying Mg2+ concentrations also is monitored by partial nuclease T1 digestion (Fig. 3 ▶) to obtain residue-specific information during folding. Nuclease T1 cleaves unpaired or exposed guanosines, and the amount of cleavage decreases upon structure formation.
FIGURE 3.

(A) Partial T1 nuclease digestion at an [Mg2+] where I and N are highly populated. T. thermophilus S-domain exhibits strongly contrasting patterns in the I and the N states. Some protection factors (PF = amount of cleavage product in I state divided by amount of cleavage product in N state normalized to the total amount of RNA in each lane) are indicated for comparison (number following semicolon). Numbering for both RNAs is according to the T. thermophilus sequence. (B) T1 cleavage at 0.05 or 0.1 mM Mg2+, where the I state predominates. (C) T1 cleavage at 1.6 mM Mg2+, where the N state predominates.
The nuclease T1 cleavage patterns of these S-domains have noticeable differences (Fig. 3 ▶). The pattern of the thermophilic S-domain has an abrupt change between the I and the N states. Most cleavage bands are intense in the I state (0.05 mM Mg2+), but they dramatically diminish in intensity in the N state. In contrast, the pattern of the mesophilic S-domain has a much smaller change between the I (0.1 mM Mg2+) and the N states. This result can also be presented quantitatively as protection factors, defined as the ratio of the amount of cleavage in the I state to the amount in the N state, normalized to the amount of radioactivity in each lane (Fig. 3A ▶).
The dramatic difference in the protection factors between the I and the N states can be explained in at least two ways. In the first scenario, both S-domains have similar native structures, while the I state of the thermophilic S-domain is less structured. In the second scenario, both S-domains have similar I states, while the N state of the thermophilic S-domain is more structured, e.g., residues are better packed or more buried in the interior of its structure. Comparing the amount of cleavage products for both states (Fig. 3B,C ▶) indicates that cleavage in the I state of both S-domains is similar, whereas cleavage in the N state is markedly reduced in the thermophilic S-domain. This result is consistent with a less solvent-exposed structure for the native thermophilic S-domain that reduces the accessibility to T1 cleavage.
In order to relate sequence changes to folding properties, specific features in the crystal structure of the T. thermophilus S-domain are compared with quantitative differences in the hydroxyl radical protection and nuclease T1 cleavage to explore the link between the native structure and the I-to-N folding transition (Fig. 4 ▶). A standard correlation of this kind is between the extent of hydroxyl radical protection and the surface burial of C4′ of each nucleotide (Cate et al. 1996). Protection factors for T. thermophilus S-domain correlate well with the buried surface area of C4′ (colored red in Fig. 4A ▶). Poor correlations (colored blue) are generally found in surface loops.
FIGURE 4.

Comparisons of surface burial calculated from the T. thermophilus S-domain structure and structural mapping, which records the difference between the I and the N states. (A) Plot of C4′ burial and normalized hydroxyl radical protection factors (normalized PF = PF − 1; red and blue squares). Red nucleotides have a better correlation than blue nucleotides. Hydroxyl radical data are from Krasilnikov et al. (2004) and Loria and Pan (1996). (B) Plot of whole nucleotide burial and normalized T1 nuclease protection factors (red squares). (C) Plot of normalized hydroxyl radical protection factors for T. thermophilus (red) and E. coli (black) S-domains. Regions of significant differences are indicated by red bars and are illustrated within the structure of the T. thermophilus S-domain in D.
The nuclease T1 protection factors can be compared with the accessible surface area of the whole nucleotide (Leontis and Westhof 2003; Fig. 4B ▶). Here, a correlation holds for nearly all detectable positions, suggesting that protection from nuclease cleavage is largely due to surface burial difference in both states. Several nucleotides on the surface of the molecule, G93 in P8, and G115 and G117 in P11, have largely accessible ribose-phosphate backbones but inaccessible bases and are therefore protected from nuclease T1 cleavage.
There are many similarities and significant differences between the extent of hydroxyl radical protection among the mesophilic and the thermophilic S-domains (Fig. 4C ▶). Protection factors for residues in the intertwined loop structure (J11/12–J12/13) and the P12/L13 peripheral structure have similar magnitudes. On the other hand, protection factors for residues in the P10 region in the core and the P14/P8 peripheral structure (red bars in Fig. 4C ▶) are markedly higher in the thermophilic S-domain. This result suggests that reinforcing the interactions in the P14/P8 peripheral structure, as discussed in detail below, reduces the “breathing” of the native structure, thus increasing the rigidity of the thermophilic S-domain.
Phylogenetic differences in the context of the S-domain structure
There are 39 sequence differences between E. coli and T. thermophilus S-domain, including five nucleotide insertions (from the RNase P database) (Brown 1999; Fig. 1 ▶). Six are base-pair switches (residue numbering in T. thermophilus: 144–155, 146–153, 180–190) and five are in loop regions (residues 101, 148, 149, 150, 151). These residues are not involved in tertiary interactions and are considered to be unimportant in altering stability; they are likely to stabilize I and N to a similar extent. Of the 28 remaining sequence changes, phylogenetic comparison shows that 16 residues (110, 111, 112, 229 in P10; 127, 130 in J11/12; 137, 139, 147, 152, 159, 161 in P12; 178 in P13, 198, 206, 215 in P14) are <50% conserved among the thermophilic S-domains and therefore are not considered further. However, 12 positions (96, 107, 119, 128, 138, 164, 175, 197, 201, 212, 216, 224) are >50% conserved (Figs. 5 ▶, 6 ▶) and are described in detail below.
FIGURE 5.

Phylogenetic comparison of 10 thermophilic and 18 mesophilic S-domains. Base-paired nucleotides are connected by lines. The thermophilic S-domain sequences are from T. thermophilus, T. album, T. aquaticus, T. commune, T. luteus, T. tumescens, T. maritima, T. neapolitana, T. pallidum, and T. thioparus. The mesophilic S-domain sequences are from E. coli, S. typhimurium, N. gonnorhoeae, Synechococcus, V. cholera, C. vinosum, Anabaena, C. pneumoniae, H. pylori, M. luteus, C. trachomatis, Wolbachia, H. influenza, B. burgdorferi, Synechocystis, M. leprae, D. radiodurans, and M. tuberculosis.
FIGURE 6.

Locations and structural networks of the 12 residues in T. thermophilus S-domain proposed to affect its stability. They are phylogenetically conserved among thermophilic S-domains. (•) Ba2+ sites found in the crystal structure.
C119 is completely conserved in all thermophilic S-domains. C119 in T. thermophilus S-domain forms a Watson-Crick base pair with G222 and a tertiary hydrogen bond with the phosphate backbone of A127 in J11/12 (Fig. 6A ▶). This tertiary interaction is missing in the E. coli S-domain due to geometric constraints imposed by its mismatched A129·G229 pair.
An extra base pair in P9 (C96–G107) is present in all thermophilic S-domains, with 80% being C–G (Fig. 6D ▶). Although this base pair is not involved in tertiary interactions, it may provide additional stability to the P9 helix. In particular, it may improve the stacking interaction of the bulged A108 onto A226 of P11.
An extra base pair in P14 (C201–G212) is present in nine of the 10 thermophilic S-domains, eight of which are G–C. This base pair is not involved in tertiary interactions, but it likely adjusts the pitch of the P14 helix such that L14 can make new or modified interactions with P8 (Fig. 6C ▶).
Another base pair in P14 (C197–G216) is 100% Watson-Crick paired in thermophilic S-domains, eight of which are C–G. In mesophilic S-domains, however, only 56% are Watson-Crick paired and mostly not C–G. The O2 of C197 forms a hydrogen bond with the N6 of A220 (Fig. 6B ▶). This interaction orients and locks P14 in position, resulting in a strengthening of the peripheral L14–P8 interaction.
U → C224 substitution in P11 is in 70% of thermophilic S-domains, forming the C224–G117 base pair. Each nucleotide in this base pair contributes a ligand to a bound metal ion (Fig. 6A ▶).
Position 128 in J11/12 is a G in six of 10 thermophilic S-domains, as compared with just one of 18 mesophilic S-domains, where it is mostly a C. N2 of G128 is involved in a hydrogen bond with the phosphate backbone of C221 in J14/11 (Fig. 6E ▶). Position 175 in J11/12 is a pyrimidine in 80% of thermophilic S-domains, but a purine in 78% of mesophilic S-domains. O2 of C/U175 is a ligand for a bound metal ion near J12/13 (Fig. 6E ▶). This same metal ion can also be ligated by G128 and A129. Hence, these interactions strengthen the structure formed by the intertwined J11/12 and J12/13 loops.
Positions 134 and 164 in P12 form a G–C base pair in 70% of the thermophilic S-domains, but only 17% of mesophilic S-domains even have a Watson-Crick base pair at this position. O2 of C164 is involved in a hydrogen bond with N6 of A166 and 2′OH of C164 forms a hydrogen bond with 2′OH of G186 (Fig. 6F ▶).
The bulge at position 138 in P12 is primarily a G in thermophilic S-domains, but not in mesophilic RNAs. O6 of G138 forms a hydrogen bond with N2 of G162 and stacks between A184 of L13 and C139 of P12 (Fig. 6G ▶). These interactions strengthen the peripheral structure between L13 and P12.
Converting the E. coli S-domain to a thermophilic RNA
The 12 sequence changes in the thermophilic S-domain suggested by phylogeny to be pertinent in achieving higher stability, are located in three regions.
Two are in the P12 receptor, which forms a peripheral interaction with the L13 loop (G138, C164 in T. thermophilus S-domain). These changes may modulate the strength of this peripheral interaction.
Four are in the P11 portion of the core, including the additional base pair in P9 (C96–G107, C119, C224). All residues in P11 form Watson-Crick base pairs in the thermophilic S-domain. Such pairings reduce wobbles and mismatches and introduce new tertiary interactions and metal ion-binding ligands in the core. The increased number of interactions may lead to a more rigid core structure.
Two are in the structure made of J11/12 and J12/13 loops (G128, C175); two others are in P14 (C197–G216) and form a base triple with A220. These changes in the thermophilic S-domain lead to new cross-strand hydrogen bonds and provide additional metal ion binding ligands. Two more are in P14 and form an extra base pair (C201–G212). This base pair may allow better juxtaposition of the intertwined J11/12–J12/13 loop structure with the L14-P8 peripheral interactions.
To examine how these 12 sequence changes influence stability and folding cooperativity, four mutants were constructed starting from the E. coli S-domain sequence (Table 1 ▶). Three mutants substitute or insert a subset of these 12 nucleotide sequences from T. thermophilus S-domain into the E. coli S-domain as dictated by their common locations. Mutant “P12” contains two substitutions (E. coli numbering), A148 → G and U172 → C. Mutant “P9 + P11” contains two substitutions, A129 → C and U231 → C, and two insertions, C107′–G117′. Mutant “J11/12 + P14” contains five substitutions—C138 → G, G183 → C, G205 → C, U223 → G, and U219 → G—and one insertion, C208′, which pairs with G219. The fourth mutant, “All 12,” contains all 12 sequence changes in order to examine possible cumulative effects.
The folding behavior of the “All 12” mutant confirms our primary prediction from phylogenetic comparisons and the crystal structure of the thermophilic S-domain. Introduction of the 12 mutations into the E. coli S-domain converted it into a thermophilic S-domain with essentially the same stability and folding cooperativity as the T. thermophilus S-domain (Table 1 ▶; Fig. 7 ▶).
FIGURE 7.

Correlating changes in stability (KMg) to folding cooperativity (Hill constant, •) and the number of new and altered interactions for E. coli S-domain mutants (□). Mutations in a local structure can increase stability (P9 + P11 and J11/12 + P14) but are insufficient to change folding cooperativity. Simultaneous mutations in more than one local structure can increase folding cooperativity but do not gain additional stability as defined by the KMg value.
A subset of the 12 mutations affected folding differently (Table 1 ▶; Fig. 7 ▶). The “P12” mutant folded like the wild-type E. coli S-domain. Both “P9 + P11” and “J11/12 + P14” mutants achieved greater stability (similar KMg as the T. thermophilus S-domain) but did not alter folding cooperativity (similar Hill constant as the E. coli S-domain).
Mutations of these 12 positions clearly act in two distinct ways in altering S-domain folding. For this RNA structure, the introduction of four or five new or altered interactions into a mesophilic S-domain are sufficient to lower the Mg2+ requirement for folding to the same level as the thermophilic S-domain (Fig. 7 ▶). This result confirms once again that increasing the stability of a tertiary RNA does not require a large number of sequence changes, as was demonstrated previously for a group I ribozyme (Guo and Cech 2002) and a catalytic domain of RNase P RNA (Fang et al. 2003). However, increasing the folding cooperativity to a significant extent requires mutations in more than one local structure (Fig. 7 ▶). This result suggests that folding cooperativity is a property of structural networks involving multiple structural units. Stability, on the other hand, can be incrementally improved by separately tuning each structural unit.
Conclusions
The availability of the high-resolution structure and extensive phylogenetic information coupled with folding studies allowed for detailed evaluation of the structural basis of altering RNA stability. In the case of the thermophilic S-domain, numerous altered or additional interactions lead to more intricate and stronger hydrogen bonding, stacking, and metal ion-binding networks in pertinent regions throughout the molecule. These interactive networks, particularly those in the P14/P8 peripheral structure and the core, allow better packing, decrease surface accessibility, and likely reduce the fluctuations of the native structure.
MATERIALS AND METHODS
Sample preparation
All RNAs were synthesized by in vitro transcription using T7 RNA polymerase (Milligan and Uhlenbeck 1989), purified on denaturing polyacrylamide gels, and stored in water at −20°C. Transcription templates for the mutant RNAs were made by PCR amplification using synthetic DNA oligonucleotides incorporating the desired mutations. Prior to all experiments, RNA was heated in 20 mM TrisHCl (pH 8.1) at 85°C for 2 min, followed by 5 min at room temperature. The RNA at this stage was designated as the unfolded, or U, state.
Mg2+-titration monitored by CD spectroscopy
These are performed as described previously (Sosnick et al. 2000). Briefly, folding rates were obtained for both S-domains over a wide range of Mg2+ concentrations in order to determine waiting times for equilibrium titrations (data not shown). In a thermostated cuvette holder, 0.3 μM RNA in the U state was pre-equilibrated at 37°C. Aliquots of MgCl2 stock solutions were dispensed using a Hamilton titrator connected to the Jasco J715 spectropolarimeter. Based on the folding rates, waiting times were programmed after the injection of each aliquot prior to the collection of CD and absorbance signals for 30 sec. The waiting times varied from 3 to 70 min for T. thermophilus and from 3 to 9 min for E. coli S-domains, depending on the [Mg2+] in the cuvette. Waiting times for the S-domain mutants were similar to T. thermophilus S-domain to ensure complete equilibration.
Depending on whether the buffer contained urea, various transitions (U-to-I, I-to-N) were observed by CD and UV absorbance at 260, 278, and 287 nm. The two Mg2+-dependent transitions were described according to a semi-empirical cooperative binding model
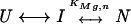 |
where n and KMg are the Hill coefficient and Mg2+-midpoint of the transitions. The CD traces at 287 nm contained the I-to-N transition and were fit to
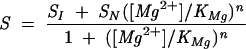 |
(1) |
where SI and SN are the observed signals for the I and the N states, respectively.
Mg2+-titration monitored by partial nuclease T1 digestion
A total of 0.3 μM 5′ 32P-labeled S-domain RNA in the U state was mixed with MgCl2 in individual tubes, each with a different [Mg2+]. The mixture was incubated at 37°C for up to 70 min to allow approach to equilibrium. The nuclease reaction was performed at 37°C for 5 min at final concentrations of 0.1 U/μL T1 nuclease. The solutions were mixed with an equal volume of 9 M urea and 100 mM EDTA and loaded immediately onto denaturing polyacrylamide gels. The amount of cleavage product was quantified using a Fuji PhosphorImager.
Phylogenetic comparison
BioEdit (Hall 1999) was used to investigate the sequence differences between mesophilic and thermophilic S-domains. Aligned sequences of A-type RNAs were obtained from the RNase P database (Brown 1999).
Acknowledgments
This work was supported by a grant from the NIH (GM57880). Three-dimensional figures are generated using DRAWNA, kindly made available by Dr. Eric Westhof. We thank the reviewers for insightful comments.
Abbreviations
CD, circular dichroism
KMg, Mg2+ concentration at the midpoint of an RNA folding transition
m, the free energy dependence on the urea concentration
n, the Hill coefficient
U, unfolded
I, equilibrium intermediate
N, native
Article and publication are at http://www.rnajournal.org/cgi/doi/10.1261/rna.2186506.
REFERENCES
- Altman, S. and Kirsebom, L. 1999. Ribonuclease P. In The RNA world, 2d ed. (eds. R.F. Gesteland et al.), pp. 351–380. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
- Brion, P. and Westhof, E. 1997. Hierarchy and dynamics of RNA folding. Annu. Rev. Biophys. Biomol. Struct. 26: 113–137. [DOI] [PubMed] [Google Scholar]
- Brown, J.W. 1999. The ribonuclease P database. Nucleic Acids Res. 27: 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, J.W., Haas, E.S., and Pace, N.R. 1993. Characterization of ribonuclease P RNAs from thermophilic bacteria. Nucleic Acids Res. 21: 671–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cate, J.H., Gooding, A.R., Podell, E., Zhou, K., Golden, B.L., Kundrot, C.E., Cech, T.R., and Doudna, J.A. 1996. Crystal structure of a group I ribozyme domain: Principles of RNA packing. Science 273: 1678–1685. [DOI] [PubMed] [Google Scholar]
- Fang, X.W., Golden, B.L., Littrell, K., Shelton, V., Thiyagarajan, P., Pan, T., and Sosnick, T.R. 2001. The thermodynamic origin of the stability of a thermophilic ribozyme. Proc. Natl. Acad. Sci. 98: 4355–4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, X.W., Srividya, N., Golden, B.L., Sosnick, T.R., and Pan, T. 2003. Stepwise conversion of a mesophilic to a thermophilic ribozyme. J. Mol. Biol. 330: 177–183. [DOI] [PubMed] [Google Scholar]
- Frank, D.N. and Pace, N.R. 1998. Ribonuclease P: Unity and diversity in a tRNA processing ribozyme. Annu. Rev. Biochem. 67: 153–180. [DOI] [PubMed] [Google Scholar]
- Guo, F. and Cech, T.R. 2002. Evolution of Tetrahymena ribozyme mutants with increased structural stability. Nat. Struct. Biol. 9: 855–861. [DOI] [PubMed] [Google Scholar]
- Guo, F., Gooding, A.R., and Cech, T.R. 2004. Structure of the Tetrahymena ribozyme: Base triple sandwich and metal ion at the active site. Mol. Cell 16: 351–362. [DOI] [PubMed] [Google Scholar]
- Hall, T.A. 1999. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41: 95–98. [Google Scholar]
- Hartmann, R.K. and Erdmann, V.A. 1991. Analysis of the gene encoding the RNA subunit of ribonuclease P from T. thermophilus HB8. Nucleic Acids Res. 19: 5957–5964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazantsev, A.V., Krivenko, A.A., Harrington, D.J., Carter, R.J., Holbrook, S.R., Adams, P.D., and Pace, N.R. 2003. High-resolution structure of RNase P protein from Thermotoga maritima. Proc. Natl. Acad. Sci. 100: 7497–7502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasilnikov, A.S., Xiao, Y., Pan, T., and Mondragón, A. 2004. Basis for structural diversity in homologous RNAs. Science 306: 104–107. [DOI] [PubMed] [Google Scholar]
- Leontis, N.B. and Westhof, E. 2003. Analysis of RNA motifs. Curr. Opin. Struct. Biol. 13: 300–308. [DOI] [PubMed] [Google Scholar]
- Loria, A. and Pan, T. 1996. Domain structure of the ribozyme from eubacterial ribonuclease P. RNA 2: 551–563. [PMC free article] [PubMed] [Google Scholar]
- Massire, C., Jaeger, L., and Westhof, E. 1998. Derivation of the three-dimensional architecture of bacterial ribonuclease P RNAs from comparative sequence analysis. J. Mol. Biol. 279: 773–793. [DOI] [PubMed] [Google Scholar]
- Milligan, J.F. and Uhlenbeck, O.C. 1989. Synthesis of small RNAs using T7 RNA polymerase. Methods Enzymol. 180: 51–62. [DOI] [PubMed] [Google Scholar]
- Paul, R., Lazarev, D., and Altman, S. 2001. Characterization of RNase P from Thermotoga maritima. Nucleic Acids Res. 29: 880–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, H., Sosnick, T.R., and Pan, T. 2001. Modular construction of a tertiary RNA structure: The specificity domain of the Bacillus subtilis RNase P RNA. Biochemistry 40: 11202–11210. [DOI] [PubMed] [Google Scholar]
- Shelton, V.M., Sosnick, T.R., and Pan, T. 1999. Applicability of urea in the thermodynamic analysis of secondary and tertiary RNA folding. Biochemistry 38: 16831–16839. [DOI] [PubMed] [Google Scholar]
- Sosnick, T.R. and Pan, T. 2003. RNA folding: Models and perspectives. Curr. Opin. Struct. Biol. 13: 309–316. [DOI] [PubMed] [Google Scholar]
- Sosnick, T.R., Fang, X., and Shelton, V.M. 2000. Application of circular dichroism to study RNA folding transitions. Methods Enzymol. 317: 393–409. [DOI] [PubMed] [Google Scholar]