

Abstract
Poly(ADP-ribose) polymerase-1 (PARP-1) is activated in response to DNA injury in eukaryotic cells and has been implicated in cell dysfunction in reperfusion injury. In this study we investigated the role of PARP-1 on apoptosis in early myocardial reperfusion injury. Mice genetically deficient of PARP-1 (PARP-1−/−) and wild-type littermates were subjected to myocardial ischemia and reperfusion. Myocardial injury was assessed by measuring the serum levels of creatine phosphokinase and oligonucleosomal DNA fragments in the infarcted area. Expression of the anti-apoptotic protein, Bcl-2, and the pro-apoptotic protein, Bax, was analyzed by Western blot. Activation of caspases, important executioners of apoptosis, and activation of the nuclear factor κB (NF-κB) pathway were evaluated. Gene expression profiles for apoptotic regulators between PARP-1−/− and wild-type mice also were compared. Myocardial damage in PARP-1−/− mice was reduced significantly, as indicated by lower serum creatine phosphokinase levels and reduction of apoptosis, as compared with wild-type mice. Western blot analyses showed increased expression of Bcl-2, which was associated with reduction of caspase-1 and caspase-3 activation. This cardioprotection was associated with significant reduction of the activation of IκB kinase complex and NF-κB DNA binding. Microarray analysis demonstrated that the expression of 29 known genes of apoptotic regulators was significantly altered in PARP-1−/− mice compared with wild-type mice, whereas 6 known genes were similarly expressed in both genotypes. The data indicate that during reperfusion absence of PARP-1 leads to reduction of myocardial apoptosis, which is associated with reduced NF-κB activation and altered gene expression profiles.
INTRODUCTION
Coronary reperfusion after ischemia may exacerbate myocardial injury by the release of potent reactive oxygen and nitrogen species, thus influencing the degree of recovery and, eventually, cell death by necrosis and apoptosis (1–3). Current evidence suggests that these reactive species activate signal transduction pathways, which lead to a rapid activation of the nuclear factor-κB (NF-κB), through interaction with IκB kinase (IKK). Activation of this transcription factor induces a coordinate expression of genes of several inflammatory mediators, including cytokines and adhesion molecules (4–6).
Previously, our laboratory and other investigators have shown that oxidative stress may cause metabolic disarrangement through a pathway mediated by poly (ADP-ribose) polymerase (PARP) (7–11). PARP is a chromatin-associated nuclear enzyme, which is activated by stranded DNA nicks and breaks in damaged cells. It modifies nuclear proteins through attachment of poly (ADP-ribose) units (8,9). Active PARP causes ADP-ribosylation of a wide variety of proteins including histones, topoisomerases I and II, and molecule Ap4A, which associates with DNA polymerase, DNA polymerases α and β, DNA ligase, and PARP itself (10,12–15). However, the role of PARP in functions of nuclear proteins and transcription factors is not completely understood.
Recently, it has been reported that PARP is a family of enzymes comprised of several members such as PARP-1, short PARP, PARP-2, PARP-3, vault PARP, and tankyrase (14). Evidence has accumulated that activation of PARP-1 is a major cytotoxic pathway for tissue injury in different pathologies associated with inflammation (16). In previous in vivo studies, we have demonstrated that pharmacological inhibition of PARP or genetic ablation of PARP-1 ameliorates the pathophysiologic changes of myocardial reperfusion injury (17–19). Abolition of poly(ADP-ribosyl)ation, with the use of pharmacological inhibitors or genetic deletion of PARP-1, also has been shown to attenuate tissue injury after endotoxic and hemorrhagic shock, cerebral ischemia, streptozocin-induced diabetes, arthritis, and localized colon inflammation (7,20–24).
In this study, we provide insight into the cardioprotective mechanism of PARP-1 inhibition. We demonstrated that the extent of myocardial cell apoptosis is reduced substantially in PARP-1−/− mice. Furthermore, our results indicate that PARP-1–induced changes of NF-κB activation and gene expression of apoptotic regulators may contribute to myocardial reperfusion injury.
MATERIALS AND METHODS
Myocardial Ischemia and Reperfusion
The investigation conformed to the Guide for the Care and Use of Laboratory Animals published by US National Institutes of Health (NIH Publication No. 85–23, revised 1996) and commenced with the approval of the Institutional Animal Care and Use Committee. Mice genetically deficient of PARP-1 (PARP-1−/−) and their wild-type littermates PARP-1+/+ (129/SV × C57BL/6, 20 to 22 g) were anesthetized with thiopentone sodium (40 μg/g). The trachea was cannulated with a PE-90 catheter and artificial respiration was provided by a respirator with FiO2 of 0.80, a frequency of 100 strokes/min and a tidal volume of 0.8 to 1.2 mL to maintain normal PO2, PCO2, and pH. Rectal temperature was monitored with a rectal probe and was maintained within 36.5 and 37 °C. Coronary occlusion and reperfusion was performed as previously described (19). The chest was opened by a cut along the left side of the sternum through the ribs. The animal was rotated to expose the left ventricle. The left anterior descending branch (LAD) of the left coronary artery was occluded by ligation with a 7–0 silk suture over a 1-mm section of PE-10 tubing, which was placed on top of the vessel. After 30 min occlusion, reperfusion occurred by cutting the suture. In the heart, the cardiac venous network was clearly visible with a dissection microscope, and no veins were occluded with this maneuver. Different groups of mice were sacrificed at the end of the ischemia (30 min) or at various time points after reperfusion (5, 15, 30, 45, 60 min, and 24 h). Blood samples were collected. Hearts were rapidly harvested, and the left ventricles were used for histological and biochemical studies. A group of mice underwent the above surgical procedure with the exception of LAD occlusion and reperfusion and served as sham control group.
Measurement of Serum Creatine Phosphokinase Activity
Serum levels of creatine phosphokinase were evaluated as an index of cardiac cellular damage by using a commercial kit (Sigma Chemical Co., St. Louis, MO, USA).
Determination of Apoptosis
Cell death by apoptosis was evaluated by measuring oligonucleosomal DNA fragments using a histochemical terminal deoxynucleotidyl transferase (TdT) “Tunel”-like staining (TdT-FragEL kit, Oncogene Research Products, Cambridge, MA, USA) and by a DNA laddering assay as previously described (25). For the in situ Tunel staining, frozen cardiac sections were permeabilized with protease K (2 mg/mL) in 10 mM Tris (pH 8) at room temperature for 20 min. Endogenous peroxidase was quenched with 3% H2O2 in methanol for 5 min. Sections were incubated with a reaction buffer composed by biotin-dCTP and unlabeled dCTP and TdT enzyme in a humidified chamber at 37 °C. In this assay, TdT binds to exposed 3′OH ends of DNA fragments and catalyzes the addition of biotin-labeled and unlabeled deoxynucleotides. Then byotinilated nucleotides were detected using a streptavidin-horseradish peroxidase conjugate and diaminobenzidine. To quantitate the degree of apoptosis, the numbers of apoptotic cells were counted by 3 independent observers blinded to experimental protocol. The apoptotic index (number of stained myocardial nuclei/number of total myocardial nuclei) was calculated. For the DNA laddering assay, hearts were homogenized in a buffer containing 137 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 1.0 mM MgCl2, 0.44 mM KH2PO4, 0.34 mM Na2HPO4, 5.6 mM glucose, and 20 mM HEPES (pH 7.5). After centrifugation (1000 × g, 10 min), the pellets were solubilized at 4 °C with lysis buffer (50 mM Tris, 10 mM EDTA, 0.5% Sarkosyl, pH 8.0) and digested in the presence of protease K (1 mg/mL) for 2 h at 50 °C. Samples were then treated with 20 μg RNase at 37 °C for 1 h. Equal quantities of sample along with 1-kb DNA ladder (Gibco Technologies, Grand Island, NY, USA) were subjected to electrophoresis on a 1.8% agarose gel containing ethidium bromide. DNA staining was visualized using the Bio-Rad gel documentation system (Bio-Rad Laboratories, Hercules, CA, USA).
Subcellular Fractionation and Nuclear Protein Extraction
Hearts were homogenized in a buffer containing 0.32 M sucrose, 10 mM Tris HCl, pH 7.4, 1 mM EGTA, 2 mM EDTA, 5 mM NaN3, 10 mM β-mercaptoethanol, 20 μM leupeptin, 0.15 μM pepstatin A, 0.2 mM phenylmethylsuphonyl fluoride, 50 mM NaF, 1 mM sodium orthovanadate, and 0.4 nM microcystin. The homogenates were centrifuged (1000 × g, 10 min) and the supernatant (cytosol + membrane extract) was collected to evaluate Bax, Bcl-2, and IκBα content and activity of IKK and caspase-1 and caspase-3 as described below. The pellets were solubilized in Triton buffer (1% Triton X-100, 150 mM NaCl, 10 mM Tris HCl, pH 7.4, 1 mM EGTA, 1 mM EDTA, 0.2 mM sodium orthovanadate, 20 μM leupeptin A, 0.2 mM phenylmethylsuphonyl fluoride. The lysates were centrifuged (15000 × g, 30 min, 4 °C), and the supernatant (nuclear extract) was collected to evaluate the activity of NF-κB.
Measurement of Activity of Caspase-1 and Caspase-3
Activity of caspase-1 and caspase-3 was measured by the cleavage of the fluorogenic tetrapeptide-amino-4-methylcoumarine conjugates, YVAD-amino-fluoro-coumarine (AFC) for caspase-1 and DEVD-AFC for caspase-3, as described (26). Cytosol extracts and substrates (50 μM) were combined in the caspase reaction buffer (100 mM HEPES, 10% sucrose, 5 mM dithiothreitol, 0.1% CHAPS, pH 7.25). AFC liberation was monitored for more than 30 sec with a Perkin-Elmer fluorimeter using 400-nm excitation and 505-nm emission wavelength. Fluorescence units were converted to pmols of AFC using a calibration curve generated with free AFC. Data are given as YVAD-fluoro-methyl-ketone or DEVD-fluoro-methyl-ketone inhibitable AFC generation.
Western Blot Analyses
Cytosol content of Bax and Bcl-2 and cytosol degradation of IκBα were determined by immunoblot analyses. Cytosol extracts were boiled in loading buffer (125 mM Tris HCl, pH 6.8, 4% SDS, 20% glycerol, and 10% 2-mercaptoethanol) and 50 μg of protein were loaded per lane onto an 8% to 16% Tris-glycine gradient gel. Proteins were separated electrophoretically and transferred to nitro-cellulose membranes. For immunoblotting, membranes were blocked with 5% nonfat dried milk in Tris buffered saline for 1 h and then incubated with primary antibodies against Bax, Bcl-2, or IκBα for 1 h. The membranes were washed in Tris-buffered saline with 0.1% Tween 20 and incubated with secondary peroxidase-conjugated antibody. Immunoreaction was visualized by chemiluminescence. Densitometric analysis of blots was performed using ImageQuant (Molecular Dynamics, Sunnyvale, CA, USA).
Assay of IKK Activity
Activity of IKK was determined by immune complex kinase assay and was estimated as the ability to phosphorylate glutathion-S-transferase-IκBα (25). After immunoprecipitation of lysate with specific antibody directed to IKKγ, the immunoprecipitate was incubated for 30 min at 30 °C in 40 μL of reaction buffer (25 mM HEPES, pH 7.6, 20 mM MgCl2, 20 mM glycerol phosphate, 0.1 mM sodium orthovanadate, 2 mM dithiothreitol, 25 μM ATP, and 5 μCi of [μ-32P]ATP. Glutathion-S-transferase-IκBα (1–54) (4 μg) was used as substrates for IKK complex. Reaction products were separated by SDS-polyacrylamide gel electrophoresis and visualized by autoradiography. Densitometric analysis was performed using Image-Quant (Molecular Dynamics).
Electrophoretic Mobility Shift Assay
Electrophoretic mobility shift assay was performed as previously described (25). Oligonucleotide probes corresponding to NF-κB consensus sequence (5′-AGT TGA GGG GAC TTT CCC AGG C-3′) were labeled with γ-[32P]ATP using T4 polynucleotide kinase and purified in Bio-Spin chromatography columns (Bio-Rad). Ten micrograms of nuclear protein were incubated with electrophoretic mobility shift assay buffer (12 mM HEPES pH 7.9, 4 mM Tris HCl pH 7.9, 25 mM KCl, 5 mM MgCl2, 1 mM EDTA, 1 mM dithiothreitol, 50 ng/mL poly [d(I-C)], 12% glycerol vol/vol, and 0.2 mM phenylmethylsuphonyl fluoride) and radiolabeled oligonucleotide. Excess of unlabeled oligonucleotide was added in some samples for competition to verify the specificity of NF-κB binding (data not shown). Protein-nucleic acid complexes were resolved using a non-denaturing polyacrylamide gel consisting of 5% acrylamide (29:1 ratio of acrylamide:bisacrylamide) and run in 0.5X TBE (45 mM Tris HCl, 45 mM boric acid, 1 mM EDTA) for 1 h at constant current (30 mA). Gels were transferred to Whatman 3M paper, dried under a vacuum at 80 °C for 1 h, and exposed to photographic film at −70 °C with an intensifying screen. Densitometric analysis was performed using ImageQuant (Molecular Dynamics).
DNA Microarray Analysis
Total RNAs were isolated from hearts, 10 μg of total RNA were quantitatively amplified, and biotin was labeled as previously described (27,28). Briefly, RNA was converted to double-stranded cDNA with an oligo(dT) primer that had a T7 RNA polymerase site at the 5′ end. The cDNA was used directly in an in vitro transcription reaction in the presence of biotinylated nucleotides to produce antisense RNA, which was hybridized overnight to Genechips (Affymetrix, San Jose, CA, USA) displaying probes for 11000 genes and expressed sequence tags. Different groups of mice (n = 2 animals for each genotype) were sacrificed before ischemia (control group at time 0) or at the end of reperfusion (60 min). One chip for each animal was used. Chips were washed, stained with phycoerythrin-streptavidin, and read with Affymetrix Genechip scanner and accompanying gene expression software. Labeled bacterial RNAs of known concentration were spiked into each chip hybridization mix to generate an internal standard curve, allowing normalization between chips and conversion of raw hybridization intensity values to mRNA frequency (mRNA molecules per million). DNA microarray data were filtered for genes with RNA expression changes of 2-fold or greater at 1 h after reperfusion in PARP-1−/− mice compared with wild-type mice. This screen resulted in 402 genes. With the K-means algorithm in the GeneSpring software (Silicon Genetics, Redwood City, CA, USA), clustering approaches with hierarchical tree analysis applied to the expression ratio measurements resulted in the selection of 35 genes with known functions as cell death regulators.
Materials
The primary antibodies directed at Bcl-2, Bax, IκBα and IKKγ and the oligonucleotide for NF-κB were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). All other chemicals were from Sigma/Aldrich.
Data Analysis
All values in the figures and text are expressed as mean ±SEM of n observations (n = 3 to 12 animals for each group). The results were examined by analysis of variance with individual comparisons performed by t-test. For microarray analysis (n = 2), P values were derived from t-test analysis using the default statistical comparison provided by GeneSpring (Silicon Genetics). A P value < 0.05 was considered significant.
RESULTS
Absence of PARP-1 Reduces Myocardial Damage and Apoptosis
In wild-type PARP-1+/+ mice, occlusion (30 min) of the LAD followed by reperfusion (60 min) resulted in a marked elevation of serum levels of creatine phosphokinase activity (2774 ± 513 U/mL). Absence of a functional PARP-1 gene in PARP-1−/− mice resulted in a significant reduction of serum levels of creatine phosphokinase activity (687 ±301 U/mL; P < 0.05), suggesting a reduction of myocardial damage. Basal levels of creatine phosphokinase activity were similar in PARP-1+/+ and PARP-1−/− mice (226 ± 39 and 385 ± 165 U/mL, respectively). These current data are in agreement with our previous studies demonstrating that inhibition of PARP-1 results in significant cardioprotection as evaluated by histology, creatine phosphokinase activity, and infarct size in rodents subjected to early (60 min) or late reperfusion injury (24 h) (17–19).
To test whether myocardial injury was associated with cell death by apoptosis, we measured oligonucleosomal DNA fragmentation. In-gel electrophoresis of DNA revealed significant fragmentation in hearts of PARP-1+/+ mice at 60 min after reperfusion (Figure 1A). This finding was confirmed by in situ Tunel staining, which showed the marked appearance of dark brown apoptotic cells scattered throughout the infarcted area of PARP-1+/+ mice (Figure 1B). On the contrary, DNA fragmentation was abrogated and only a small number of cells were stained dark brown in the left ventricle of PARP-1−/− mice (Figure 1A and 1C), indicating a significant decrease of apoptotic cell death. Apoptotic indices of hearts from PARP-1+/+ mice compared with PARP-1−/−mice were 0.70 ± 0.09% compared with 0.46 ± 0.04%, respectively (P < 0.05). Few or no apoptotic cells were observed in sections from sham mice of both genotypes.
Figure 1.
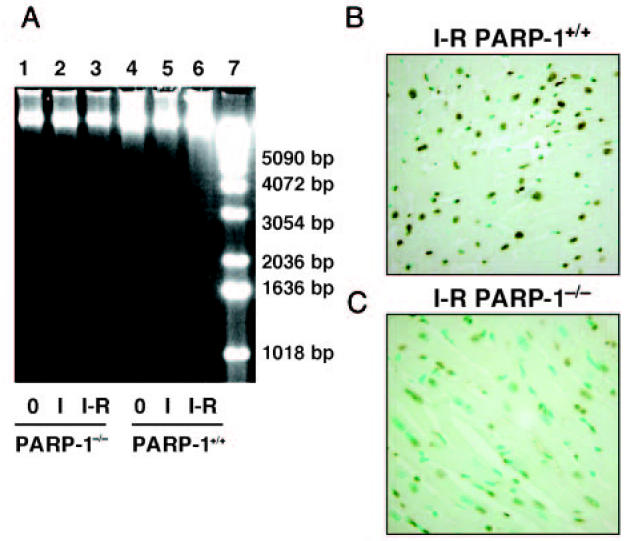
Apoptosis in cardiac tissue after in vivo ischemia and reperfusion. A: Representative in gel electrophoresis of DNA fragmentation. Lanes 1 and 4 represent DNA from sham operated PARP-1−/− (lane 1) or PARP-1+/+ (lane 4) mice; lanes 2 and 5 represent DNA from PARP-1−/− (lane 2) or PARP-1+/+ (lane 5) mice subjected to 30-min ischemia; lanes 3 and 6 represent DNA from PARP-1−/− (lane 3) or PARP-1+/+ (lane 6) mice subjected to 30-min ischemia and 60-min reperfusion; lane 7 represents DNA size markers. B and C: Representative photomicrographs of in situ Tunel method of cardiac sections. Following 30 min occlusion and 60 min reperfusion, a massive staining for apoptosis was observed throughout the infarcted area in cardiac sections from PARP-1+/+ mice (B). In PARP-1−/− mice, a dark staining was observed in a few apoptotic nuclei (C). Magnification ×1000; 1 cm = 7.9 μm. A similar pattern was seen in n = 5 to 6 different tissue sections in each experimental group.
Levels of Bcl-2 Are Increased in PARP-1−/− Mice
The change in expression of apoptosis-related proteins was analyzed during the course of reperfusion by Western blotting. The anti-apoptotic protein Bcl-2 diminished after reperfusion in hearts of wild-type mice, whereas it increased in PARP-1−/− mice. Expression of the pro-apoptotic Bax remained unchanged after ischemia and reperfusion in mice of both genotypes (Figure 2).
Figure 2.

Western blot analysis for Bcl-2 and Bax. Representative Western blot analysis for Bcl-2 (A) and Bax (C) in PARP-1+/+ mice (upper panel) and in PARP-1−/− mice (lower panel). B and D: Image analysis of Bcl-2 and Bax determined by densitometry. Fold increase was calculated compared with respective sham value (time 0) set to 1.0. Results are representative of 3 separate time-course experiments. *P < 0.05 compared with PARP-1+/+ mice.
Activity of Caspase-1 and Caspase-3 Is Reduced in PARP-1−/− Mice
Downstream effectors of apoptosis are cysteine proteases or caspases (29). Therefore, we determined the activity of caspase-1 and caspase-3. The proteolytic activity of both caspases increased at the end of ischemia, but returned to basal activity at 1 h after reperfusion in wild-type mice. On the contrary, we observed a significant reduction of activity of both caspases after ischemia in PARP-1−/− mice (Figure 3). At later stages of reperfusion (24 h), caspase-3 activity was below basal levels and ranged between 2.38 ± 0.03 and 1.47 ± 0.84 μmol/100 mg tissue in PARP-1+/+ and PARP-1−/− mice, respectively, whereas caspase-1 activity was undetectable in both genotypes.
Figure 3.

Catalytic activity of caspase-1 (A) and caspase-3 (B) in the heart after ischemia (30 min) and reperfusion (60 min). Each data point is the mean ± SEM of 3 to 6 animals for each group. *P < 0.05 compared with time 0 of the same genotype; #P < 0.05 compared with PARP-1+/+ mice at the same time point.
Activation of NF-κB, Degradation of IκBα, and Activity of IKK Are Depressed in the Absence of PARP-1
To investigate the molecular mechanisms by which absence of PARP-1 may attenuate reperfusion injury, we evaluated the nuclear activation of NF-κB. In PARP-1+/+ mice, DNA binding of NF-κB increased after reperfusion in a biphasic fashion, increasing early at 5 min after reperfusion, then decreasing at 15 min, and increasing again at later reperfusion. In PARP-1−/− mice, activity of NF-κB exhibited similar biphasic kinetics. However, the degree of activation was significantly reduced when compared with wild-type mice (Figure 4). At later stages of reperfusion (24 h), NF-κB activation was similar in both experimental groups. The fold increase of NF-κB activity was 1.10 ± 0.06 and 1.41 ± 0.19 compared with basal levels at time 0 (set to 1), respectively, in PARP-1+/+ and PARP-1−/− mice.
Figure 4.
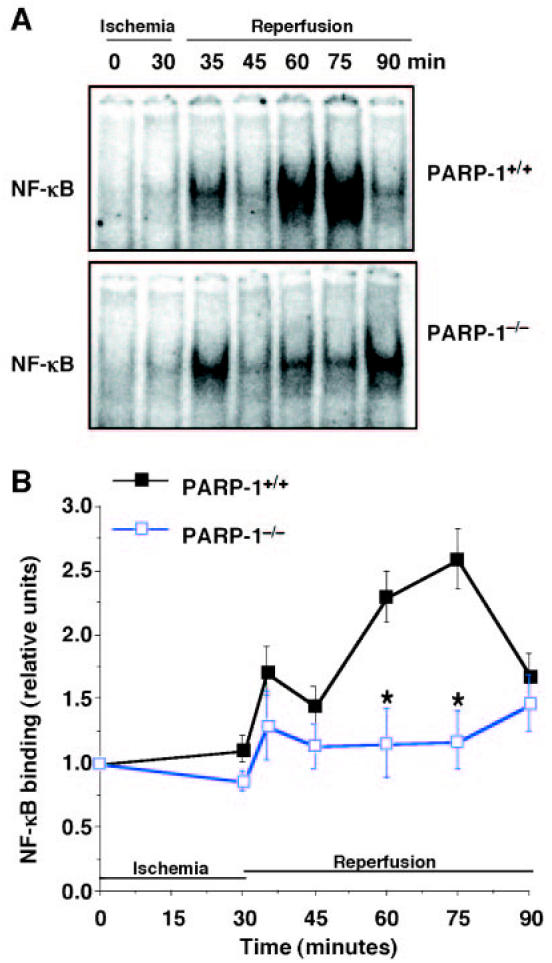
DNA binding of NF-κB. Representative autoradiographs of electrophoretic mobility shift assay for NF-κB (A) in PARP-1+/+ mice (upper panel) and PARP-1−/− mice (lower panel). Image analysis of activation of NF-κB (B) determined by densitometry. Fold increase was calculated compared with respective sham value (time 0) set to 1.0. Results are representative of 5 separate time-course experiments. *P < 0.05 compared with PARP-1+/+ mice.
Because degradation of IκBα by a phosphorylation- and ubiquitination-dependent pathway, represents an important event for NF-κB nuclear translocation and the initiation of transcription (4–6), we further determined the reperfusion-induced degradation of IκBα and activity of IKK. We found that in PARP-1+/+ mice IκBα content was degraded at 5 and 15 min after reperfusion. It was then replenished at 30 min and degraded again at later time points after reperfusion (Figure 5). The events were preceded by an increase of the phosphorylative activity of IKK. The enzyme activity rose at 30 min after ischemia, reached a peak at 5 min after reperfusion, partially declined at 30 min, and rose again at 45 and 60 min after reperfusion (Figure 6). In contrast, in PARP-1−/− mice IκBα degradation was inhibited after reperfusion. After an initial increase, activity of IKK declined rapidly, reaching basal levels at the end of reperfusion (Figures 5 and 6). At later stages of reperfusion (24 h), IKK was similarly activated in both experimental groups. The fold increase of IKK activity was 1.35 ± 0.04 and 1.30 ± 0.11 compared with basal levels at time 0 (set to 1), respectively, in PARP-1+/+ and PARP-1−/− mice.
Figure 5.
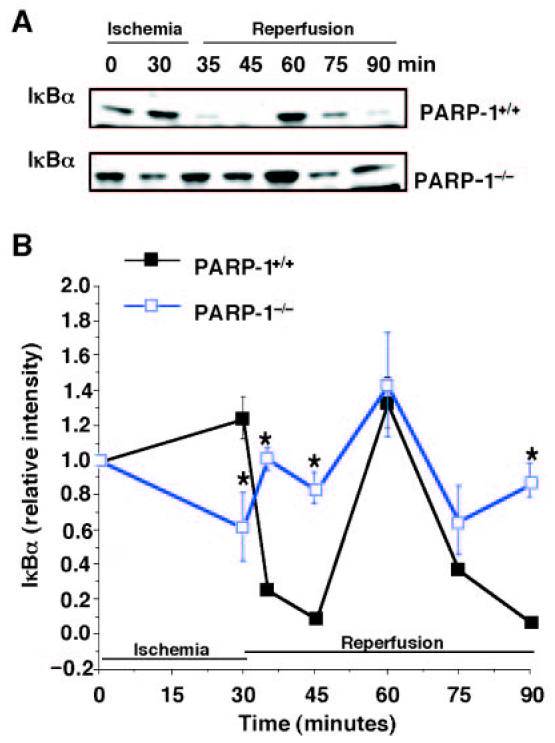
Degradation of IκBα. Representative Western blot analysis for cytosolic content of IκBα (A) in PARP-1+/+ mice (upper panel) and PARP-1−/− mice (lower panel). Image analysis of activation of IκBα content (B) determined by densitometry. Fold increase was calculated compared with respective sham value (time 0) set to 1.0. Results are representative of 5 separate time-course experiments. *P < 0.05 compared with PARP-1+/+ mice.
Figure 6.
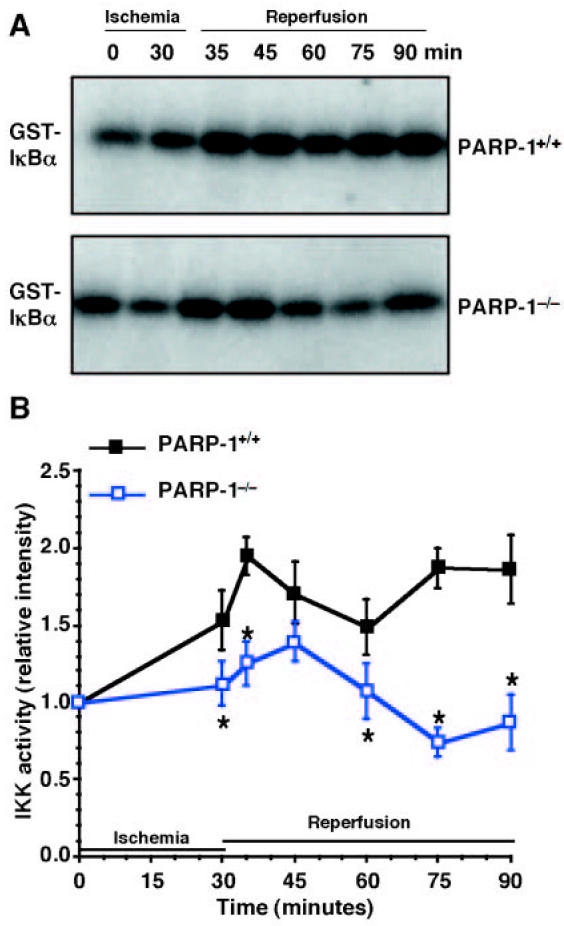
Activity of IKK. Representative autoradiographs of analysis for IKK activity (A) in PARP-1+/+ mice (upper panel) and PARP-1−/− mice (lower panel). Image analysis of IKK activity (B) determined by densitometry. Fold increase was calculated compared with respective sham value (time 0) set to 1.0. Results are representative of 5 separate time-course experiments. *P < 0.05 compared with PARP-1+/+ mice.
Gene Expression of Apoptotic Regulators Is Altered in PARP-1−/− Mice Subjected to Myocardial Ischemia and Reperfusion
To further examine the role of PARP-1 on gene expression during reperfusion, microarray analysis was performed with heart mRNA from PARP-1−/− and wild-type mice subjected to myocardial ischemia (30 min) and reperfusion (60 min). The total number of genes differing between PARP-1−/− and wild-type mice was 402. Genes with known function as cell death regulators were selected for analysis. The results for these known genes and annotated expressed sequence tags are shown in Table 1 and Figure 7. A similar increase of gene expression of Bcl-2-related proteins, growth arrest, and DNA-damage-inducible proteins, CCAAT-enhancer-binding protein δ, cell death-inducing DNA fragmentation factor α, mitogen-activated protein kinase kinase kinase 6, and TNF receptor superfamily member 6 was observed in both genotypes (Figure 7B and Table 1). A large increase of the TNFα-induced protein 3, the mitogen-activated protein kinase kinase kinase 8, serine proteinase inhibitor clade E member 1, B-cell leukemia/lymphoma 3, quiescin 6, and CCAAT-enhancer-binding protein β was observed in wild-type mice at 1 h after reperfusion. In contrast, PARP-1−/− mice exhibited a significant lower expression of these genes. Several cDNAs for other known genes for apoptosis regulators, such as serine proteinase inhibitor clade B member 2, adipsin (BAD protein), calpain 3, caspase 12, growth arrest specific 5, growth differentiation factor 5, histocompatibility 2 Q region locus 7, insulin-like growth factor 2 receptor, peroxiredoxin 2 transformation related protein 53 and clusterin, were significantly upregulated in wild-type mice, but they were down-regulated below constitutive levels in PARP-1−/− mice. The genes for caspase 1, caspase 8–like apoptosis regulator and death-associated kinase 3 were downregulated after 1 h reperfusion in both genotypes, however the downregulation was significantly more pronounced in PARP-1−/− mice (Figure 7A and Table 1). On the contrary, absence of PARP-1 resulted in increased expression of several genes of cell growth and death regulators, such as amphiregulin, fibroblast growth factor receptor 3, early growth response 2, mitogen-activated protein kinase kinase 7, and T-cell death associated gene, when compared with genes of wild-type mice (Figure 7C and Table 1).
Table 1.
Differential gene expression of apoptosis regulators
| GenBank Accession | Gene | Symbol | Ratioa |
|---|---|---|---|
| Genes significantly increased in wild-type mice | |||
| X04673 | Adipsin (Bad protein) | Adn | 0.24 |
| NM_033601 | B-cell leukemia/lymphoma 3 | Bcl3 | 0.24 |
| NM_007601 | Calpain 3 | Capn3 | 0.07 |
| NM_009807 | Caspase 1 | Casp1 | 0.57 |
| NM_009808 | Caspase 12 | Casp12 | 0.17 |
| NM_009883 | CCAAT/enhancer binding protein (C/EBP), β | Cebpb | 0.81 |
| BC029223 | Caspase 8 and Fas associated death domain–like apoptosis regulator | Cflar | 0.58 |
| BM118501 | Clusterin | Clu | 0.56 |
| NM_007828 | Death-associated kinase 3 | Dapk3 | 0.41 |
| NM_013525 | Growth arrest specific 5 | Gas5 | 0.09 |
| NM_008109 | Growth differentiation factor 5 | Gdf5 | 0.12 |
| X16202 | Histocompatibility 2, Q region locus 7 | H2-Q7 | 0.53 |
| NM_010515 | Insulin-like growth factor 2 receptor | Igfr2 | 0.56 |
| NM_007746 | Mitogen activated protein kinase kinase kinase 8 | Map3k8 | 0.14 |
| NM_011563 | Peroxiredoxin 2 | Prdx2 | 0.53 |
| NM_023268 | Quiescin 6 | Qscn6 | 0.54 |
| NM_008871 | Serine (or cysteine) proteinase inhibitor, clade E, member 1 | Serpine1 | 0.38 |
| NM_011111 | Serine (or cysteine) proteinase inhibitor, clade B, member 2 | Serpinb2 | 0.04 |
| NM_009397 | Tumor necrosis factor α–induced protein 3 | Tnfaip3 | 0.45 |
| NM_011640 | Transformation related protein 53 | Trp53 | 0.67 |
| Genes significantly increased in mice of both genotypes | |||
| U23778 | B-cell leukemia/lymphoma 2 related protein A1b | Bcl2a1b | 1.14 |
| U23781 | B-cell leukemia/lymphoma 2 related protein A1d | Bcl2a1d | 0.86 |
| NM_007679 | CCAAT/enhancer binding protein (C/EBP), delta | Cebpd | 0.74 |
| NM_007702 | Cell death-inducing DNA fragmentation factor, alpha | Cidea | 0.57 |
| U00937 | Growth arrest and DNA-damage-inducible 45 alpha | Gadd45a | 0.77 |
| NM_008655 | Growth arrest and DNA-damage-inducible 45 beta | Gadd45b | 0.99 |
| NM_011817 | Growth arrest and DNA-damage-inducible 45 gamma | Gadd45g | 1.5 |
| NM_016693 | Mitogen-activated protein kinase kinase kinase 6 | Map3k6 | 0.85 |
| NM_007987 | Tumor necrosis factor receptor superfamily, member 6 | Tnfrsf6 | 0.9 |
| Genes significantly increased in PARP-1−/− mice | |||
| L41352 | Amphiregulin | Areg | 2.76 |
| M24377 | Early growth response 2 | Egr2 | 3.7 |
| M81342 | Fibroblast growth factor receptor 3 | Fgfr3 | 6.57 |
| NM_011944 | Mitogen activated protein kinase kinase 7 | Map2k7 | 3.42 |
| NM_009344 | T-cell death associated gene | Tdag | 2.35 |
Ratios indicate the differential expression of genes in PARP-1−/− mice compared with those in wild-type animals subjected to myocardial ischemia (30 min) and reperfusion (60 min).
Figure 7.
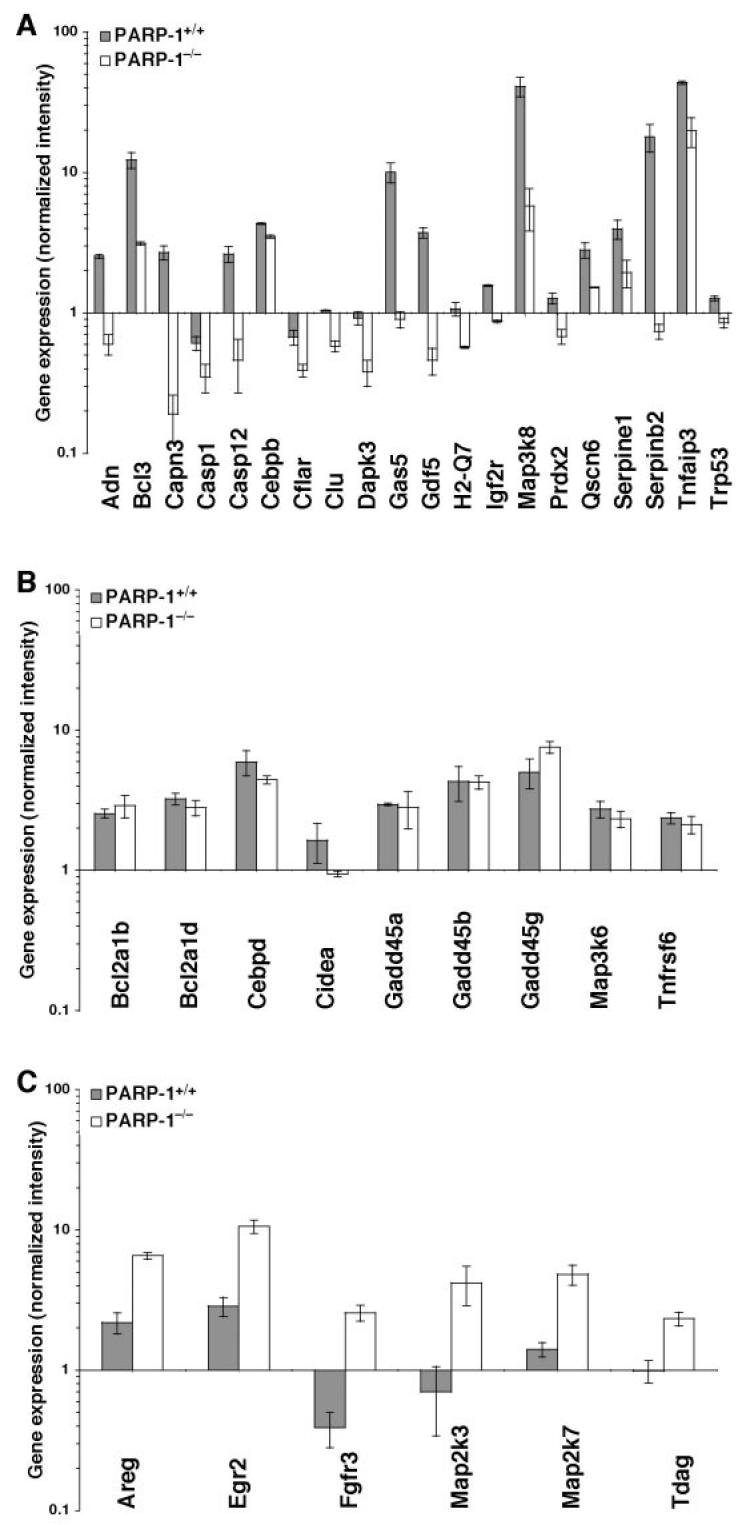
Gene expression of cell death regulators. Genes of cell death regulators increased in wild-type mice (A), in PARP-1−/− mice (B), or similarly expressed in both genotypes (C). Gene expression levels are shown as normalized intensity relative to the mRNA expression of the gene seen in the control mice (set to 1 at time 0) of the respective genotype. Values of PARP-1−/− mice shown in A and C are significantly different compared with PARP-1+/+ mice (P < 0.05). GenBank accession numbers and names of the 35 genes are shown in Table 1.
DISCUSSION
We previously reported that genetic or pharmacological inhibition of PARP-1 confers resistance to myocardial ischemia and reperfusion injury in rodents (17–19). The present study further demonstrates that inhibition of PARP-1 provides cardioprotection and that amelioration of damage is associated with reduction of apoptotic cell death. Furthermore, our data demonstrate for the 1st time that in the absence of PARP-1, cardioprotection is associated with downregulation of the signal transduction pathway of NF-κB and modification of gene expression of apoptotic regulators.
Genetic Inhibition of PARP-1 Preserves Cardiomyocytes from Apoptotic Death
Following ischemia and reperfusion, cardiomyocytes die via necrosis and apoptosis (3). Caspases, the cysteine aspartyl pro-teases, are a class of programmed cell death genes that have been characterized as a major executioner of apoptosis (29). During oxidative stress reactive oxygen species trigger the release of cytochrome c from mitochondria and, subsequently, caspase activation. Active caspases promote cellular demolition by activating other destructive enzymes, such as DNAses, and by directly targeting key structural proteins, such as lamin and actin, and regulatory proteins, thus leading to chromatin margination, DNA fragmentation, and nuclear collapse (29). In our experiments, we found that caspase-1 and caspase-3 were already activated at the end of the ischemia, thus suggesting that the mitochondrial pathway of apoptosis is a very early event in myocardial injury. Caspase-3, but not caspase-1, has been shown to cleave the 112 kDa nuclear protein PARP into an 85 kDa apoptotic fragment (30,31), and this cleavage by caspase-3 has been shown to be necessary for apoptosis (32). In this regard, the nuclear presence of proteolytic fragments of PARP has been considered a hallmark of an apoptotic cell. However, the role of PARP-1 in apoptosis remains to be determined because conflicting data have been reported. Some investigators have shown that neurons or hepatocytes from PARP-deficient mice do not exhibit any altered sensitivity to apoptotic stimuli, whereas others have demonstrated that pharmacological or genetic inhibition may increase apoptosis in cells subjected to alkylating agents (33,34). The family of Bcl-2-related proteins constitutes the most relevant class of apoptotic regulators and, more specifically, the ratio of anti- or pro-apoptotic proteins determines whether the cell will survive or die (35,36). Members of the Bcl-2 family physically interact with other proteins, including other family members and these interactions appear to modulate their function. For example, active (unphosphorylated) Bad protein induces apoptosis by inhibiting anti-apoptotic Bcl-2 family members, such as Bcl-xl, thereby allowing 2 other proapoptotic members, Bak and Bax, to aggregate, inducing the release of cytochrome c, activation of caspases, and apoptosis (37,38). On the other hand, expression of Bcl-2 protein prevents the induction of apoptosis caused by a variety of oxidative stresses, and it can influence the level of caspase activation (35). In our study, we found that genetic inhibition of PARP-1 reduced apoptosis. This event was associated with reduced activity of both caspase-1 and caspase-3. Furthermore, we found that the Bcl-2/Bax ratio was remarkably increased in PARP-1−/− mice, shifting the balance toward cell survival and well paralleling with the reduction of DNA fragmentation. To further understand the role of PARP-1 in the regulation of apoptosis, we studied gene expression patterns of known cell death regulators. In agreement with our biochemical and histochemical data, DNA microarray analysis showed that loss of PARP-1 gene is associated with markedly reduced expression of apoptotic regulators. Interestingly, we found that other growth factors and cell death regulators that participate in tissue regeneration and wound repair (such as amphiregulin, fibroblast growth factor receptor 3, and early growth response 2) were increased in PARP-1−/− mice when compared with wild-type animals. Taken together, our data demonstrated that during reperfusion injury apoptosis is due to imbalance between anti-apoptotic and pro-apoptotic factors. In this regard, inhibition of PARP-1 may reduce ideal triggers for the apoptotic process by shifting the ratio of apoptotic regulators at the genetic level. However, the modest, though significant, changes of apoptotic index in PARP-1−/− mice (which exhibited approximately 40% reduction when compared with wild-type mice) cannot fully explain the remarkable cardioprotective effects observed. This finding may suggest that reduction of apoptosis may play only a moderate role in the prevention of myocardial damage provided by PARP-1 inhibition. In our study we also found that serum levels of creatine phosphokinase activity were elevated at the end of reperfusion period. It is noteworthy that serum creatine phosphokinase should be considered a standard biomarker for cell necrosis as the enzyme is released from the cytosol after the destruction of the plasma membrane of dead myocytes (39). On the other hand, in contrast to the swelling and membrane rupture associated with necrosis, apoptotic cells shrink and maintain their membrane integrity (3). It is conceivable that elevated levels of creatine phosphokinase do not reflect the apoptotic cell death, as apoptotic cells, which maintain an intact membrane, may still retain the enzyme. Therefore, these data suggest that during early reperfusion cell death may occur via necrotic and apoptotic processes. Because genetic ablation of PARP-1 is associated with very mild architectural derangement (17–19) and reduced serum creatine phosphokinase activity, it is possible that the cardioprotective effects of PARP-1 inhibition may be due to prevention of both necrosis and apoptosis.
PARP-1 Participates in the Regulation of Signal Transduction Mechanisms
The overwhelming inflammatory response during reperfusion is regulated at the transcription level. During reperfusion oxidative stress is an important signal for the activation of a complex cascade of protein kinases. At the downstream of the cascade, phosphorylation of IκBα by IKK complex is a prerequisite for the degradation of IκBα and activation of NF-κB (4,6). Activation of NF-κB leads to upregulation of adhesion molecules along with a sustained production of pro-inflammatory cytokines, thus creating a pathological loop that may eventually progress to necrotic and apoptotic tissue death (4–6). Numerous experimental studies have proven that activation of NF-κB is implicated in myocardial reperfusion injury. Enhancement of NF-κB DNA binding activity has been found in areas of infarction in rodents subjected to myocardial ischemia and reperfusion (25,40). With particular clinical relevance, nuclear translocation of NF-κB has been found in cardiac biopsies of patients with unstable angina (41). In our study, we found that in the absence of PARP-1, the nuclear activation of the transcription factor is depressed during reperfusion in the ischemic myocardial tissue. The inhibition of NF-κB DNA binding may explain our previous data demonstrating that genetic or pharmacological inhibition of PARP-1 reduced the expression of NF-κB-dependent inflammatory mediators. Specifically, inhibition of PARP-1 significantly reduced expression of intercellular adhesion molecule-1 and P-selectin and reduced the release of TNFα and interleukin-10 in in vivo myocardial reperfusion injury (18–19). Under the experimental conditions used in our laboratory, the activation of the NF-κB pathway appears to be a very early event within 1 h, as at late reperfusion (24 h), NF-κB was not markedly increased when compared with basal levels. Although it is difficult to translate our findings on time course to the human kinetics of NF-κB activation, clinical observations suggest that reperfusion leads to irreversible injury in a relative short period (42). Gene expression of cytokines, adhesion molecules, and vasoactive substances, along with increased activity of NF-κB, has been demonstrated in left ventricular biopsies taken after early reperfusion (approximately 40 min) from patients with stable angina pectoris or unstable angina (41). Thus, these data suggest that therapeutic approaches targeting PARP-1 in the very early stage of reperfusion may exert beneficial effect in the salvage of the ischemic myocardium.
Our findings are in line with other reports demonstrating a role of PARP-1 and/or poly(ADP-ribosyl)ation in signal transduction. It has been demonstrated that PARP-1-deficient cells are defective in NF-κB–dependent transcriptional activation and show a down-regulation of inducible nitric oxide synthase (iNOS) after genotoxic stress (43,44). In our study, the role of PARP-1 in the regulation of transcription was further supported by the microarray analysis, as we identified several genes with known functions, whose expression patterns were markedly altered in PARP-1−/−mice compared with wild-type animals. Whether the expression of these genes is secondarily affected by NF-κB activation or other transcription factors remains to be elucidated. It is possible that PARP-1 may regulate several transcription factors other than NF-κB. For example, we have recently demonstrated that activation of activator protein-1 (AP-1) is significantly reduced in immunostimulated PARP-1−/− fibroblasts (45) and in PARP-1−/− mice subjected to myocardial reperfusion injury (46). In a similar report, Ha and colleagues have shown that activation of the transcription factors AP-1, specificity protein-1 (SP-1), octamer transcription factor-1 (Oct-1), Yin-Yang-1 (YY-1), and Stat-1 is downregulated in PARP-1-deficient glial cells (47). Interestingly, in our study, when examined at a more molecular level, the reduction of NF-κB activity observed in PARP-1−/− mice was associated with inhibition of IκBα degradation and reduction of IKK activity. As activation of IKK and phosphorylation and ubiquination of IκBα take place in the cytoplasm, it is very unlikely that the nuclear PARP-1 may affect directly IKK. It is possible that reduction of NF-κB activation and/or other transcription factors reduces the expression of pro-inflammatory molecules necessary for further activation of the cascade of cytosolic kinases.
However, our study did not address the precise mechanism by which PARP-1 may modulate transcription. Several scientific reports suggest the hypothesis that PARP-1 may affect transcription through a poly (ADP-ribosyl)ation-dependent and a poly (ADP-ribosyl)ation-independent mechanism. For example, poly (ADP-ribosyl)ation of nuclear extracts in vitro suppressed the ability of NF-κB to form a complex with its specific DNA probe. Recombinant p50 and p65 were poly (ADP-ribosyl)ated, whereas IκBα was not (48). Other studies have also proven that PARP-1 modulates transcription by direct interaction with activating protein-2 (49), Oct-1 (50), YY-1 (51), and transcription enhancer factor-1 (52). PARP-1 may function as a specific transcriptional coactivator through direct interaction with NF-κB (53). This dual regulatory role of PARP-1, that is, dependent or independent of the polymerase activity, is further supported by recent findings demonstrating that genetic deletion of PARP-1, but not pharmacological inhibition of its polymerase activity, reduced NF-κB binding in glial cells. However, pharmacological inhibition of the catalytic activity of PARP-1 was able to reduce gene expression of iNOS (47). Similarly to these findings, we have demonstrated that genetic deletion of PARP-1, but not pharmacological inhibition, inhibited DNA binding of the heat shock factor-1 in fibroblasts. On the contrary, pharmacological PARP-1 inhibition abolished DNA binding of AP-1 (45).
In conclusion, we have shown that inhibition of PARP-1 reduces NF-κB pathway and is associated with a reduction in apoptosis. This suggests an important regulatory role of PARP-1 in signal transduction and highlights the value of inhibition of PARP-1 as a promising strategy for myocardial preservation after ischemia and reperfusion.
Acknowledgments
Funding for this study was provided by the Natl. Institutes of Health (grant R01 HL-60730) to Dr. Basilia Zingarelli.
REFERENCES
- 1.Ferrari R, et al. Oxidative stress during myocardial ischaemia and heart failure. Eur Heart J. 1998;19:B2–11. [PubMed] [Google Scholar]
- 2.Grisham MB, Granger DN, Lefer DJ. Modulation of leukocyte-endothelial interactions by reactive metabolites of oxygen and nitrogen: relevance to ischemic heart disease. Free Radic Biol Med. 1998;25:404–33. doi: 10.1016/s0891-5849(98)00094-x. [DOI] [PubMed] [Google Scholar]
- 3.Colucci WS. Apoptosis in the heart. N Engl J Med. 1996;335:1224–6. doi: 10.1056/NEJM199610173351610. [DOI] [PubMed] [Google Scholar]
- 4.Baeuerle PA. IκB-NF-κB structures: at the interface of inflammation control. Cell. 1998;11:729–31. doi: 10.1016/s0092-8674(00)81694-3. [DOI] [PubMed] [Google Scholar]
- 5.Maulik N, Goswami S, Galang N, Das DK. Differential regulation of Bcl-2, AP-1 and NF-κB on cardiomyocyte apoptosis during myocardial ischemic stress adaptation. FEBS Lett. 1999;443:331–6. doi: 10.1016/s0014-5793(98)01719-0. [DOI] [PubMed] [Google Scholar]
- 6.Karin M, Delhase M. The IκB kinase (IΚK) and NF-κB: key elements of proinflammatory signaling. Semin Immunol. 2000;12:85–98. doi: 10.1006/smim.2000.0210. [DOI] [PubMed] [Google Scholar]
- 7.Zingarelli B, O’Connor M, Wong H, Salzman AL, Szabó C. Peroxynitrite-mediated DNA strand breakage activates poly-adenosine diphosphate ribosyl synthetase and causes cellular energy depletion in macrophages stimulated with bacterial lipopolysaccharide. J Immunol. 1996;156:350–8. [PubMed] [Google Scholar]
- 8.Althaus FR, Richter C. ADP-ribosylation of proteins. Enzymology and biological significance. Mol Biol Biochem Biophys. 1987;37:1–237. [PubMed] [Google Scholar]
- 9.Lautier D, Lagueux J, Thibodeau J, Ménard L, Poirier GG. Molecular and biochemical features of poly (ADP-ribose) metabolism. Mol Cell Biochem. 1993;122:171–93. doi: 10.1007/BF01076101. [DOI] [PubMed] [Google Scholar]
- 10.Cochrane CG. Mechanisms of oxidant injury of cells. Molec Aspects Med. 1991;12:137–47. doi: 10.1016/0098-2997(91)90009-b. [DOI] [PubMed] [Google Scholar]
- 11.Zhang J, Dawson VL, Dawson TM, Snyder SH. Nitric oxide activation of poly (ADP-ribose) synthetase in neurotoxicity. Science. 1994;263:687–9. doi: 10.1126/science.8080500. [DOI] [PubMed] [Google Scholar]
- 12.Kasid UN, Halligan B, Liu LF, Dritschilo A, Smulson ME. Poly (ADP-ribose) mediated post-translational modification of chromatin-associated human topoisomerase I. Inhibitory effects on catalytic activity. J Biol Chem. 1989;264:18687–92. [PubMed] [Google Scholar]
- 13.Desmarais Y, Ménard L, Lagueux J, Poirier GG. Enzymological properties of poly (ADP-ribose) polymerase: Characterization of automodification sites and of its NADase activity. Biochem Biophys Acta. 1991;1078:179–86. doi: 10.1016/0167-4838(91)99007-f. [DOI] [PubMed] [Google Scholar]
- 14.D’Amours D, Desnoyers S, D’Silva I, Poirier GG. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J. 1999;342:249–68. [PMC free article] [PubMed] [Google Scholar]
- 15.D’Silva I, et al. Relative affinities of poly (ADP-ribose) polymerase and DNA-dependent protein kinase for DNA strand interruptions. Biochim Biophys Acta. 1999;1430:119–26. doi: 10.1016/s0167-4838(98)00278-7. [DOI] [PubMed] [Google Scholar]
- 16.Szabó C, Dawson VL. Role of poly(ADP-ribose) synthetase in inflammation and ischaemia-reperfusion. Trends Pharmacol Sci. 1998;19:287–98. doi: 10.1016/s0165-6147(98)01193-6. [DOI] [PubMed] [Google Scholar]
- 17.ZingarelliB, Cuzzocrea S, Zsengeller Z, Salzman AL, Szabó C. Protection against myocardial ischemia and reperfusion injury by 3-aminobenzamide, an inhibitor of poly (ADP-ribose) synthetase. Cardiovasc Res. 1997;36:205–12. doi: 10.1016/s0008-6363(97)00137-5. [DOI] [PubMed] [Google Scholar]
- 18.Zingarelli B, Salzman AL, Szabó C. Genetic disruption of poly (ADP-ribose) synthetase inhibits the expression of P-selectin and intercellular adhesion molecule-1 in myocardial ischemia/reperfusion injury. Circ Res. 1998;83:85–94. doi: 10.1161/01.res.83.1.85. [DOI] [PubMed] [Google Scholar]
- 19.Yang Z, Zingarelli B, Szabó C. Effect of genetic disruption of poly (ADP-ribose) synthetase on delayed production of inflammatory mediators and delayed necrosis during myocardial ischemia-reperfusion injury. Shock. 2000;13:60–6. doi: 10.1097/00024382-200013010-00011. [DOI] [PubMed] [Google Scholar]
- 20.Liaudet L, Soriano FG, Szabó E, Virag L, Mabley JG, Salzman AL, Szabó C. Protection against hemorrhagic shock in mice genetically deficient in poly (ADP-ribose)polymerase. Proc Natl Acad Sci USA. 2000;97:10203–8. doi: 10.1073/pnas.170226797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eliasson M, et al. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med. 1997;3:1089–95. doi: 10.1038/nm1097-1089. [DOI] [PubMed] [Google Scholar]
- 22.Pieper AA, et al. Poly (ADP-ribose) polymerase deficient mice are protected from streptozotocin-induced diabetes. Proc Natl Acad Sci USA. 1999;96:3059–64. doi: 10.1073/pnas.96.6.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Szabó C, et al. Protection against peroxynitrite-induced fibroblast injury and arthritis development by inhibition of poly (ADP-ribose) synthase. Proc Natl Acad Sci USA. 1998;95:3867–72. doi: 10.1073/pnas.95.7.3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zingarelli B, Szabó C, Salzman AL. Blockade of poly (ADP-ribose) synthetase inhibits neutrophil recruitment, oxidant generation, and mucosal injury in murine colitis. Gastroenterology. 1999;116:335–45. doi: 10.1016/s0016-5085(99)70130-7. [DOI] [PubMed] [Google Scholar]
- 25.Zingarelli B, Hake PW, Yang Z, O’Connor M, Denenberg A, Wong HR. Absence of inducible nitric oxide synthase modulates early reperfusion-induced NF-κB and AP-1 activation and enhances myocardial damage. FASEB J. 2002;16:327–42. doi: 10.1096/fj.01-0533com. [DOI] [PubMed] [Google Scholar]
- 26.Vanags DM, Orrenius S, Aguilar-Santelises M. Alterations in Bcl-2/Bax protein levels in platelets form part of an ionomycin-induced process that resembles apoptosis. Br J Haematol. 1997;99:824–31. doi: 10.1046/j.1365-2141.1997.4813284.x. [DOI] [PubMed] [Google Scholar]
- 27.Byrne MC, Whitley MZ, Follettie MT. (2000) Preparation of mRNA for expression monitoring. In: Current protocols in molecular biology 4th ed. Ausubel FM (ed.) John Wiley and Sons, Inc., New York, N.Y. p. 22.2.1–22.2.13. [DOI] [PubMed]
- 28.Zhang J, Xu M, Aronow B. Expression profiles of 109 apoptosis pathway-related genes in 82 mouse tissues and experimental conditions. Biochem Biophys Res Commun. 2002;297:537–44. doi: 10.1016/s0006-291x(02)02240-4. [DOI] [PubMed] [Google Scholar]
- 29.Villa P, Kaufmann SH, Earnshaw WC. Caspases and caspase inhibitors. Trends Biochem Sci. 1997;22:388–93. doi: 10.1016/s0968-0004(97)01107-9. [DOI] [PubMed] [Google Scholar]
- 30.Duan H, Orth K, Chinnaiyan AM, Poirier GG, Froelich CJ, He WW, Dixit VM. ICE-LAP6, a novel member of the ICE/Ced-3 gene family, is activated by the cytotoxic T cell protease granzyme B. J Biol Chem. 1996;271:16720–4. doi: 10.1074/jbc.271.28.16720. [DOI] [PubMed] [Google Scholar]
- 31.Casciola-Rosen L, Nicholson DW, Chong T, Rowan KR, Thornberry NA, Miller DK, Rosen A. Apopain/CPP32 cleaves proteins that are essential for cellular repair: a fundamental principle of apoptotic death. J Exp Med. 1996;183:1957–64. doi: 10.1084/jem.183.5.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tewari M, et al. Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81:801–9. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- 33.Pieper AA, Verma A, Zhang J, Snyder SH. Poly (ADP-ribose) polymerase, nitric oxide and cell death. Trends Pharmacol Sci. 1999;20:171–81. doi: 10.1016/s0165-6147(99)01292-4. [DOI] [PubMed] [Google Scholar]
- 34.Oliver FJ, de la Rubia G, Rolli V, Ruiz-Ruiz MC, de Murcia G, Murcia JM. Importance of poly(ADP-ribose) polymerase and its cleavage in apoptosis. Lesson from an uncleavable mutant. J Biol Chem. 1998;273:33533–9. doi: 10.1074/jbc.273.50.33533. [DOI] [PubMed] [Google Scholar]
- 35.Plas DR, Thompson CB. Cell metabolism in the regulation of programmed cell death. Trends Endocrinol Metab. 2002;13:75–8. doi: 10.1016/s1043-2760(01)00528-8. [DOI] [PubMed] [Google Scholar]
- 36.Kroemer G. The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat Med. 1997;3:614–20. doi: 10.1038/nm0697-614. [DOI] [PubMed] [Google Scholar]
- 37.Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, Korsmeyer SJ. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell. 2001;8:705–11. doi: 10.1016/s1097-2765(01)00320-3. [DOI] [PubMed] [Google Scholar]
- 38.Wei MC, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–30. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bais R, Edwards JB. Creatine kinase. Crit Rev Clin Lab Sci. 1982;16:291–335. doi: 10.3109/10408368209107030. [DOI] [PubMed] [Google Scholar]
- 40.Chandrasekar B, Freeman GL. Induction of nuclear factor-κB and activation protein 1 in postischemic myocardium. FEBS Lett. 1997;401:30–4. doi: 10.1016/s0014-5793(96)01426-3. [DOI] [PubMed] [Google Scholar]
- 41.Valen G, Hansson GK, Dumitrescu A, Vaage J. Unstable angina activates myocardial heat shock protein 72, endothelial nitric oxide synthase, and transcription factors NF-κB and AP-1. Cardiovasc Res. 2000;47:49–56. doi: 10.1016/s0008-6363(00)00071-7. [DOI] [PubMed] [Google Scholar]
- 42.Schwartz GG. Exploring new strategies for the management of acute coronary syndromes. Am J Cardiol. 2000;86:44J–9J. doi: 10.1016/s0002-9149(00)01227-3. [DOI] [PubMed] [Google Scholar]
- 43.Hassa PO, Hottiger MO. A role of poly (ADP-ribose) polymerase in NF-κB transcriptional activation. Biol Chem. 1999;380:953–9. doi: 10.1515/BC.1999.118. [DOI] [PubMed] [Google Scholar]
- 44.Oliver FJ, et al. Resistance to endotoxic shock as a consequence of defective NF-κB activation in poly (ADP-ribose) polymerase-1 deficient mice. EMBO J. 1999;18:4446–54. doi: 10.1093/emboj/18.16.4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Andreone TL, Denenberg A, O’Connor M, Hake PW, Zingarelli B. Poly (ADP-ribose) polymerase-1 regulates activation of activator protein-1 in murine fibroblasts. J Immunol. 2003;170:2113–20. doi: 10.4049/jimmunol.170.4.2113. [DOI] [PubMed] [Google Scholar]
- 46.Zingarelli B, Hake PW, O’Connor M, Denenberg A. (2002) Absence of poly (ADP-ribose) polymerase-1 reduces apoptosis by modulation of activator protein-1 and heat shock factor-1 pathways during myocardial ischemia and reperfusion in mice. Circulation 106(Suppl.):II-91 (abs. 461), 2002.
- 47.Ha HC, Hester LD, Snyder SH. Poly(ADP-ribose) polymerase-1 dependence of stress-induced transcription factors and associated gene expression in glia. Proc Natl Acad Sci USA. 2002;99:3270–5. doi: 10.1073/pnas.052712399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kameoka M, Ota K, Tetsuka T, Tanaka Y, Itaya A, Okamoto T, Yoshihara K. Evidence for regulation of NF-κB by poly(ADP-ribose) polymerase. Biochem J. 2000;346:641–9. [PMC free article] [PubMed] [Google Scholar]
- 49.Kannan P, Yu Y, Wankhade S, Tainsky MA. PolyADP-ribose polymerase is a coactivator for AP-2-mediated transcriptional activation. Nucleic Acids Res. 1999;27:866–74. doi: 10.1093/nar/27.3.866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nie J, Sakamoto S, Song D, Qu Z, Ota K, Taniguchi T. Interaction of Oct-1 and automodification domain of poly(ADP-ribose) synthetase. FEBS Lett. 1998;424:27–32. doi: 10.1016/s0014-5793(98)00131-8. [DOI] [PubMed] [Google Scholar]
- 51.Oei SL, Griesenbeck J, Schweiger M, Babich V, Kropotov A, Tomilin N. Interaction of the transcription factor YY1 with human poly(ADP-ribosyl) transferase. Biochem Biophys Res Commun. 1997;240:108–11. doi: 10.1006/bbrc.1997.7621. [DOI] [PubMed] [Google Scholar]
- 52.Butler AJ, Ordahl CP. Poly(ADP-ribose) polymerase binds with transcription enhancer factor 1 to MCAT1 elements to regulate muscle-specific transcription. Mol Cell Biol. 1999;19:296–306. doi: 10.1128/mcb.19.1.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hassa PO, Covic M, Hasan S, Imhof R, Hottiger MO. The enzymatic and DNA binding activity of PARP-1 are not required for NF-κB coactivator function. J Biol Chem. 2001;276:45588–97. doi: 10.1074/jbc.M106528200. [DOI] [PubMed] [Google Scholar]