

Abstract
The structural difference in proteins between unbound and bound forms directly suggests the importance of the conformational plasticity of proteins. However, pathways that connect two-end structures and how they are coupled to the binding reaction are not well understood at atomic resolution. Here, we analyzed the free-energy landscape, explicitly taking into account coupling between binding and conformational change by performing atomistic molecular dynamics simulations for Ca2+ binding to a calmodulin loop. Using the AMBER force field with explicit water solvent, we conducted umbrella sampling for the free-energy surface and steered molecular dynamics for the pathway search. We found that, at an early stage of binding, some key residue side chains extend their “arms” to catch Ca2+ and, after catching, they carry the Ca2+ to the center of the binding pocket. This grabbing motion resulted in smooth and stepwise exchange in coordination partners of Ca2+ from water oxygen to atoms in the calmodulin loop. The key residue that first caught the ion was one of the two acidic residues, which are highly conserved. In the pathway simulations, different pathways were observed between binding and dissociation reactions: The former was more diverse than the latter.
INTRODUCTION
It is broadly believed that conformational plasticity is obligatory for proteins to function (1). First, many proteins are known to be natively unfolded before binding to their partners, which may be beneficial for fast binding (2), apparently implying importance of plasticity (3). Second, in structural studies, significant differences between unbound conformations and those in complex are often seen, indicating that most proteins are plastic (4). Cocrystal structures of some enzymes with bound substrates themselves suggest that substrates can find their pathways to the binding sites only by opening enzyme conformations. In contrast to the wealth of structural information about two-end structures, i.e., unbound and bound forms, it is rather surprising that how binding reactions proceed by coupling to structural change has not been well addressed at atomic resolution. Some physiochemical studies elucidated critical roles of fluctuation (5–7); an example of a well-characterized reaction is CO binding to myoglobin (5). However, direct information at atomic resolution is rarely available (6,7). Even for very simple reactions of proteins, such as ion binding reactions, there are many interesting and fundamental questions that need to be addressed: How do proteins change their conformation upon binding? Ligands need to expel water molecules before binding to proteins, which costs much, especially in the case of ions. How does this occur? How specific are binding pathways at atomic resolution? Here, we address these questions based on molecular dynamics simulations of an ion-binding process.
For addressing these fundamental issues of biological reactions, we here chose the Ca2+ binding reaction to a calmodulin (CaM) loop, as a model system. Calmodulin is well-known as a signal transduction protein and exists broadly in eukaryotes. The function is activated by binding four Ca2+ to their binding sites. Calmodulin is dumbbell-shaped with two domains, each of which has two Ca2+ binding sites (EF-hand loop) (Fig. 1). The binding reaction and conformational change upon binding have been relatively well-studied experimentally (8–19) as well as theoretically (20–23). Especially, Bierzynski and co-workers (15,16) showed that short peptides of some 12 residues, which correspond to Ca2+ binding loop of CaM, can bind to lanthanum (La3+), forming the structure that is analogous to the structure of the intact sequence bound to Ca2+. They also indicated that this peptide weakly binds to Ca2+, although the Ca2+-saturated loop still preserves some conformational flexibility. Ye et al. (18,19) developed an approach for investigating the site-specific Ca2+ binding affinity of each binding site by grafting the binding loop of CaM. The success of the grafting approach indicates that the loop itself, once structurally buttressed by the scaffold, is the functional motif independent of other parts. These two observations together make a CaM loop one of the smallest functional motifs that were experimentally shown to work “as is”, and thus the CaM loop is an ideal system for detailed simulation study of an ion-binding reaction.
FIGURE 1.

Structure of CaM (right) and its loop III (inside left box). In the latter, the five colored residues coordinate to Ca2+, Asp-93 (green), Asn-95 (blue), Asn-97 (magenta), Tyr-99 (yellow), and Glu-104 (cyan). The figure is prepared with MOLSCRIPT (42), Raster3D (43), and PyMOL (44).
In this article, we investigate the free-energy landscape and pathways of Ca2+ binding reaction to a CaM loop, using molecular dynamics (MD) simulation of an all-atom model and explicit solvent molecules. First, a two-dimensional free-energy surface that explicitly deals with coupling between loop conformation and ion-binding was calculated by the umbrella sampling method. Second, binding/dissociation pathways were investigated by steered MD simulations. Both in the free-energy analysis and in the pathway search, we found that, at an early stage of binding, some side chains in the binding site extend their “arms” for catching Ca2+, at which conformations are more extended than apo as well as holo conformations of the side chain. We also saw that dehydration and binding are tightly coupled and stepwise, which results in a relatively smooth free-energy landscape in the course of the binding reaction. At atomic resolution, binding pathways are intrinsically stochastic but are reasonably well biased, making the anchor residues limited to only two acidic residues.
MATERIALS AND METHODS
CaM has four binding sites of Ca2+, each of which forms a loop with 12 amino acids and often called I–IV (Fig. 1) from N- to C-termini. Here, as a model system, we picked up loop III, which is in the C-terminal domain and is known to have the highest affinity for Ca2+ (8,9). The loop contains 12 amino acids (DKDGNGYISAAE), three of which are acidic (Asp-93, Asp-95, and Glu-104,) and one is basic (Lys-94), with its net charge equal to −2. As the reference loop structure of the holo form (bound Ca2+), we used the NMR structure of Xenopus laevis CaM, 1mux entry of the Protein Data Bank. For comparison, we referred the unbound form, which we call the apo form, of the same loop, taken from the NMR structure of X. laevis CaM without bound Ca2+, 1cfd entry. N- and C-termini were capped by acetyl and methylamine, respectively, when MD was performed.
The MD simulations were performed for this loop III of CaM (Fig. 1) and a Ca2+ atom, with explicit water solvents using the AMBER 7 program (24), in which the parm99 force field for protein atoms (25), the parameters of Bradbrook et. al. (26) for Ca2+, and the TIP3P potential for water molecules (27) were used. The total number of atoms contained was 5618, of which 5445 atoms were from water. We performed equilibration simulation with NPT and NVT conditions before sampling. We used the periodic boundary condition where the size of unit box is ∼38 × 38 × 38 Å3, which is obtained by the equilibration calculation. Electrostatic interactions were calculated by the particle-mesh Ewald algorithm. To retain the loop backbone shape, the harmonic constraint was applied to all main-chain heavy atoms. The force constant kbb for this constraint was 0.1 kcal/mol/Å2 for the body of simulations, although we employed stronger (1.0 kcal/mol/Å2 and 10 kcal/mol/Å2) constants to better check how backbone flexibility would affect the binding reaction. Comparing with flexibility of the whole C-domain of CaM, we consider that kbb = 0.1 kcal/mol/Å2 is the most appropriate among the three constraints studied. With kbb = 1.0 kcal/mol/Å2, the backbone apo structure of the loop has ∼100 kcal/mol higher energy than the reference holo structure, and thus the CaM loop cannot reach the near-apo structure. With kbb = 0.1 kcal/mol/Å2, however, as we will see, the CaM loop structure becomes closer to the apo form rather than to the holo form after Ca2+ dissociation.
The coordination to Ca2+ is quite subtle. In the NMR structure of the holo form, Ca2+ is coordinated by six atoms of the CaM loop (Oδ of Asp-93, Oδ of Asp-95, Oδ of Asn-97, backbone O of Tyr-99, and bidentate coordination by Glu-104 at two Oɛ) and an oxygen atom of water (a total of seven atoms are coordinated). As a preparatory simulation, using the current force field, we performed a short equilibrium simulation of the whole C-terminal domain of CaM from the NMR holo structure finding that the coordination number to Ca2+ fluctuates between seven and eight atoms. More specifically, coordination by Asp-95 frequently flips between bidentate and monodentate, then the coordination number from the CaM loop is 6 or 7 and that from water is 1. We concluded that, with the current van der Waals parameter of Ca2+, coordination numbers of 7 and 8, are quasi-equilibrium. Interestingly, when we simulated the cleaved loop III of 12 residues, the equilibrium was shifted toward the coordination number of 8 although that of 7 was metastable as described below. A similar bidentate was observed in an earlier report (28). This subtle difference in the coordination of the holo structure, which is highly sensitive to the parameter of simulations, is not the focus of this work. In the analysis below, coordination by eight atoms (seven from the CaM loop and one from water) is used as a reference holo state of the cleaved loop. The coordinated atoms in the CaM loop are Oδ of Asp-93, two Oδ of Asp-95, Oδ of Asn-97, backbone O of Tyr-99, and two Oɛ of Glu-104.
For calculating the free-energy surface via umbrella sampling (29), we define the reaction coordinate, R, as the sum of seven distances between Ca2+ and the coordinated atoms in the protein (i.e., any oxygen atoms of water are not included here):
 |
A harmonic restraining potential, 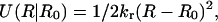 was added to the potential energy for constraining proteins to a certain range of R, where kr = 10 kcal/mol/Å2 was used throughout this work. In the umbrella sampling simulation, the R0 value specifies the window. The free-energy surface, or the potential of mean force, was calculated from umbrella sampling simulations where a series of simulations with a broad range of R0 (windows) were performed. At first, the system was equilibrated for 0.17 ns in the holo state, and one of structures in this ensemble was used as an initial configuration. Starting from R0 = 16.8 Å, which corresponds to the holo structure, we shifted windows R0 by
was added to the potential energy for constraining proteins to a certain range of R, where kr = 10 kcal/mol/Å2 was used throughout this work. In the umbrella sampling simulation, the R0 value specifies the window. The free-energy surface, or the potential of mean force, was calculated from umbrella sampling simulations where a series of simulations with a broad range of R0 (windows) were performed. At first, the system was equilibrated for 0.17 ns in the holo state, and one of structures in this ensemble was used as an initial configuration. Starting from R0 = 16.8 Å, which corresponds to the holo structure, we shifted windows R0 by 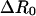 toward both smaller and larger R0 values. In each window, R0, the simulation was started with one of conformations in the previous window, equilibration was performed for 50 ps, and then configurations were collected for 0.2 ns. The simulations were done at 300 K. We used
toward both smaller and larger R0 values. In each window, R0, the simulation was started with one of conformations in the previous window, equilibration was performed for 50 ps, and then configurations were collected for 0.2 ns. The simulations were done at 300 K. We used 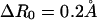 intervals for
intervals for 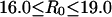 windows, and 0.25 Å intervals for
windows, and 0.25 Å intervals for 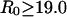 windows. In the dissociation direction, simulations were completed when
windows. In the dissociation direction, simulations were completed when 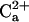 dissociated from the CaM loop. (In total, we used 289 windows for kbb = 0.1 kcal/mol/Å2, 190 windows for kbb = 1.0 kcal/mol/Å2, and 224 windows for kbb = 10 kcal/mol/Å2, and thus one series contains ∼60 ns cumulative simulation time.) The series of simulations were combined by the weighted histogram analysis method (30) for calculating the free-energy surfaces. We repeated identical series of MD simulations twice from different initial structures for each of three loop constraints kbb, thus resulting in six series in total (∼300 ns cumulative simulation time). With the same constraint kbb, the overall free-energy surface looked identical between two series, although atomistic details were slightly different. Mostly, we will show results from one of two series, and arguments will be given when differences between two series were found.
dissociated from the CaM loop. (In total, we used 289 windows for kbb = 0.1 kcal/mol/Å2, 190 windows for kbb = 1.0 kcal/mol/Å2, and 224 windows for kbb = 10 kcal/mol/Å2, and thus one series contains ∼60 ns cumulative simulation time.) The series of simulations were combined by the weighted histogram analysis method (30) for calculating the free-energy surfaces. We repeated identical series of MD simulations twice from different initial structures for each of three loop constraints kbb, thus resulting in six series in total (∼300 ns cumulative simulation time). With the same constraint kbb, the overall free-energy surface looked identical between two series, although atomistic details were slightly different. Mostly, we will show results from one of two series, and arguments will be given when differences between two series were found.
We also investigated binding/dissociation pathways by performing steered MD simulations (31,32), where we applied a time-dependent external force to accelerate the binding/dissociation reactions. The external force applied is between Ca2+, and each coordinated atom in the CaM loop that we described earlier. The oxygen atoms of water that coordinate to Ca2+ are also not included. The external potential applied is defined as
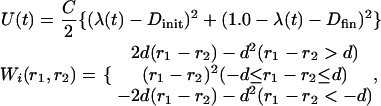 |
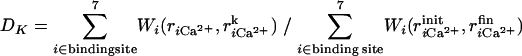 |
where init and fin correspond to initial and final conformations of the steered simulations. 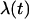 varies, linearly in time, from 0 to 1 through the 1 ns simulation.
varies, linearly in time, from 0 to 1 through the 1 ns simulation. 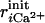 is the distance between the ith coordinated atom of CaM and Ca2+ at the initial structure, and
is the distance between the ith coordinated atom of CaM and Ca2+ at the initial structure, and 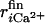 is one at the final structure. For a binding (apo to holo) simulation, the initial structure was picked up from snapshots obtained from the apo state simulations (see below), whereas the final structure was the holo (bound) state. The correspondence is opposite to it in a dissociation simulation. The effective force constant varies between 6 and 7 kcal/mol/Å2. We repeated six simulations with different apo structures for both binding and dissociation, thus resulting in 12 simulations.
is one at the final structure. For a binding (apo to holo) simulation, the initial structure was picked up from snapshots obtained from the apo state simulations (see below), whereas the final structure was the holo (bound) state. The correspondence is opposite to it in a dissociation simulation. The effective force constant varies between 6 and 7 kcal/mol/Å2. We repeated six simulations with different apo structures for both binding and dissociation, thus resulting in 12 simulations.
Binding pathways are quantified as the probability that the ith residue coordinates to Ca2+ when the total coordination number is ntot . The probability is defined by the following;
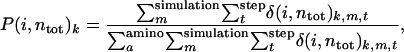 |
where k is either binding or dissociation and 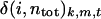 is unity if the ith residue coordinates to Ca2+ in the snapshot of the mth simulation at time = t and 0 otherwise.
is unity if the ith residue coordinates to Ca2+ in the snapshot of the mth simulation at time = t and 0 otherwise.
In this study, an oxygen atom in the CaM loop is considered as coordinated to Ca2+ when a distance between oxygen and Ca2+ is <2.9 Å.
RESULTS
Overview of the free-energy surface
Characteristics of how Ca2+ binding is coupled to conformational change of the CaM loop are best summarized in the two-dimensional free-energy surface shown in Fig. 2, where Ca2+ binding and conformational change are monitored in the y and x axes, respectively. Namely, the x axis is the root mean-square deviation (RMSD) of side chain heavy atoms in the CaM loop measured from NMR structure in the holo, and the y axis is the direct interaction energy Ebind between Ca2+ and the CaM loop. The global free-energy minimum is at (RMSD = 1.9, Ebind = −695.0), which corresponds to R = 16.4 Å (see the open labels in Fig. 2) and thus is essentially the complete bound state. During the dissociation reaction, first the CaM loop deforms substantially, reaching (RMSD = 5.5, Ebind = −600) at R ∼ 50 Å, where RMSD is larger than that in the apo structure. As the dissociation further goes on, RMSD was reduced to ∼4 Å at R ∼ 80 Å, and thus the CaM loop is less extended. An estimate of the RMSD value after complete dissociation is further reduced to ∼3Å (see the solid arrow at the top of the figure). In the direction of Ca2+ binding, the free energy is mostly downhill. Most interestingly, in the middle of binding, the CaM loop side chains are substantially more extended than either apo or holo structures.
FIGURE 2.

Two-dimensional free-energy surface (potential of mean force) of Ca2+ binding to the CaM loop, where the x axis monitors the CaM loop deformation, whereas the y axis represents the strength of Ca2+ binding. More precisely, the x axis is the RMSD of heavy atoms in the side chain of CaM measured from the holo form, and the y axis is the direct interaction energy between Ca2+ and the CaM loop. The chart on the right side indicates mapping to the free-energy value (in kcal/mol). The arrow at the top indicates RMSD of the apo form. Characters on the map indicate corresponding values of the reaction coordinate R in the umbrella sampling simulations.
The coordination numbers to Ca2+ are plotted in Fig. 3 A as a function of the reaction coordinate, R. Here, we found that, in the dissociation direction, a decrease in the number of coordination by CaM loop atoms and an increase in that by water oxygen atoms are quite well anticorrelated; Namely, coordination to Ca2+ switches from CaM atoms to water atoms, resulting in the sum being almost constant between 7 and 8. In the early stage of dissociation, R < 22 Å, this switch in coordination numbers is quite rapid, whereas in the middle range, 22 Å < R < 48 Å, coordination number does not change significantly. (Fluctuation here is primarily from statistical error. Since the coordination number is inherently rounded to an integer, it is thus is unavoidably sensitive to the cutoff value used.) Interestingly, this stage corresponds to where the CaM loop RMSD grows very rapidly in Fig. 2. Thus, 3–4 atoms of acidic residues in the CaM loop carry Ca2+ from the center of the binding pocket to the rim of the loop. The number of acidic residues (Asp-93, Asp-95, and Glu104) in the loop that bind to Ca2+ is shown in Fig. 3 B, which is robust in statistics. Here, we found that two (one) of the three residues coordinate to Ca2+ for 48 Å < R < 63 Å (R > 63 Å). We note that in the initial stage of dissociation, three atoms separate from the ion in a narrow range of R, whereas in the later stage, dissociation proceeds slowly with R. This is because once some side chains dissociated from Ca2+, they can easily change their conformation (Fig. 2), which contributes to a large increase in R.
FIGURE 3.

Coordination numbers to Ca2+ along the reaction coordinate, R. (A) Coordination numbers to Ca2+ by water (solid line) and by the CaM loop (dashed line) as a function of the reaction coordinate, R. (B) Number of the acidic residues (Asp-93, Asp-95, and Glu-104) that coordinate to Ca2+ versus the reaction coordinates, R.
Based on these observations, for convenience, we divided the binding/dissociation process into four states: 1), R = 16.4 Å, the holo state; 2), R = 18.2 Å, one of the atoms takes off and is in a free-energy local minima near the holo state; 3), R = 50.0 Å, near the transition region where the CaM loop side-chain RMSD reaches the maximum (Fig. 2); and 4), R = 80.0 Å, the side-chain RMSD is ∼4–5 Å and only one acidic residue remains coordinated.
The holo state with R = 16.4 Å
In the stage R = 16.4 Å, the free-energy surface reaches a minimum. Fluctuation of Ca2+ and each atom that coordinates to Ca2+ are shown in Fig. 4. We saw that the coordination here is identical to that in the initial structure: the seven CaM atoms coordinate to Ca2+ (Fig. 4). Atomic fluctuation is relatively small, as expected.
FIGURE 4.

Structures, fluctuation of Ca2+, and fluctuation of the CaM side chains at several stages of the dissociation/binding reaction. The first, third, fifth, and seventh figures represent fluctuation of Ca2+ at the reaction coordinate R = 16.4, 18.0, 50.0, and 80.0, respectively, in which the colored balls indicate binding site atoms: Asp-93 (green), Asp-95 (blue), Asn-97 (magenta), Tyr-99 (yellow), and Glu-104 (cyan). The second, fourth, sixth, and eighth figures represent fluctuations of binding site atoms at the same four stages as above. The color scheme is the same as before. The last snapshot is a representative structure of the CaM loop, at which Ca2+ is far from the binding sites (see text). The figures are prepared with MOLSCRIPT (42) and Raster3D (43).
A free-energy local minimum with R = 18.2 Å
From the holo state to a local minimum at R = 18.2 Å, at first one of the oxygen atoms in the bidentate coordination (Asp-95 or Glu-104) dissociates. Of two residues, Asp-95 is more likely to lose one coordinate bond, but the choice is to some extent stochastic.
At the stage R = 18.2 Å, the six CaM atoms are coordinated and the free-energy surface reaches a local minimum. A representative snapshot in Fig. 4 is the case where Asp-95 loses one coordinate bond to Ca2+. This structure is in a similar coordination mode to that of the NMR structure of the whole CaM. The fluctuation of the dissociated atoms (Oδ of Asp-95) becomes larger. However, the rest is relatively weakly fluctuated (Fig. 4) and the side-chain RMSD is ∼2 Å.
Near the transition region with R = 50.0 Å
On the way from the local minimum at R = 18.2 Å to near the transition region R = 50.0 Å, the CaM side-chain RMSD rapidly increases whereas the interaction energy between Ca2+ and CaM is virtually unchanged (Fig. 2). As is shown in Fig. 4, Ca2+ moves from the central binding pocket to the rim (left side in Fig. 4). Throughout this loop deformation, Ca2+ is coordinated by ∼3–4 CaM loop atoms from two acidic residues, as in Fig. 3 A. In the particular snapshot in Fig. 4, Ca2+ is carried by Asp-93 and Glu-104. Which of the two acidic residues carry Ca2+ away is stochastic, but the majority is the pair of Asp-93 and Glu-104. Near the transition region R = 50.0 Å, the CaM side-chain RMSD reaches 6–7 Å, substantially larger than the RMSD value at the apo structure (=3.0 Å). Interestingly, the number of acidic residues that bind to Ca2+ drops from three to two at R ∼ 48 Å very sharply (Fig. 3). Calmodulin atoms that lose contacts with Ca2+ are Oδ of Asn-97 and backbone O of Tyr-99, as well as one in acidic residues (in the snapshot, Asp-93). The Ca2+ tries to keep coordinated by a few acidic residues while the loop partially deforms.
In the stage R = 50.0 Å, two acidic residues coordinate to Ca2+; however, the member of atoms involved in the coordination changes frequently. By losing the contact with Ca2+, the backbone O of Tyr-99 rotates, which leads to a complete flip of the Tyr-99 side chain as well. Moreover, the side chain of Lys-94 is flipped over, too (in Fig. 4, from down to up conformations). These together contribute to large RMSD of the CaM loop side chain at this state. Fluctuation of Ca2+ is significantly magnified. Asp-95 and Asn-97 lose their contacts with Ca2+, resulting in larger fluctuation.
The R = 80 Å stage
From R = 50.0 Å to R = 80.0 Å, all but one atom that coordinates to Ca2+ goes away and thus the interaction energy between CaM and Ca2+ is substantially weakened from ∼−600 kcal/mol to −300 kcal/mol. During this process, the CaM side-chain RMSD decreases to ∼4 Å. This RMSD is close to the value of the side-chain RMSD between the apo and holo states, which is indicated by the black arrow on the top of Fig. 2.
In a snapshot at R = 80.0 Å (Fig. 4), only one oxygen of Glu-104 coordinates to Ca2+ (at the bottom left of Fig. 4, R = 80.0 loop), whereas other atoms that coordinate to Ca2+ in the holo state are dissociated. In five out of six simulations, Glu-104 was the last residue that coordinates to Ca2+, whereas in only one case it was Asp-95. Interestingly, side-chain orientations of Asp-93, Lys-94, and Tyr-99 roughly go back to those at the holo state. Near the transition region R = 50.0 Å, to move Ca2+ away to the rim of the binding pocket, two acidic residues have to move away carrying Ca2+, which perhaps induces side-chain deformation larger than that in R = 80.0 Å.
The apo state
We investigated CaM loop conformations after complete dissociation of Ca2+ by performing MD simulations of the loop with Ca2+ placed sufficiently far from the loop. We picked up a snapshot from the ensemble of the R0 = 70 Å window, and Ca2+ located near Glu-104 was exchanged with one water molecule sufficiently far from the CaM loop (the resulting R value was 187.5 Å). As in former simulations, we applied the loop constraint (kbb = 0.1 kcal/mol/Å2) and performed 0.5 ns simulations. The side-chain RMSD from the holo structure fluctuates around 4–5.5 Å (the red curve in Fig. 7 in the Supplementary Material), which is about the same as that in the R = 80 Å window above, and thus has significantly smaller RMSD than that near the transition region R = 50 Å. As expected, the orientation of CaM side chains is closer to the apo form (the RMSD between the simulated and the apo structures is ∼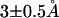 as shown in the blue curve in Fig. 7) rather than to the holo form (the corresponding RMSD
as shown in the blue curve in Fig. 7) rather than to the holo form (the corresponding RMSD 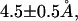 the green curve in Fig. 7). This is primarily because coordinated side-chain atoms that have either negatively charged or polar groups prefer pointing toward solvent waters.
the green curve in Fig. 7). This is primarily because coordinated side-chain atoms that have either negatively charged or polar groups prefer pointing toward solvent waters.
Flexibility of loop
We also calculated the free-energy surfaces with stronger (kbb = 1.0 kcal/mol/Å2 and 10 kcal/mol/Å2) constraints with the same umbrella sampling method and compared them with the free-energy surface of the weakest constraint shown in Fig. 2 (Fig. 8, A and B, in the Supplementary Material). Regardless of the strength of constraint, we found that, in the Ca2+ binding reaction, the CaM loop side-chain RMSD from the holo structure first increases and then decreases up to the holo structure, and thus initiation of binding occurs via extended side-chain conformation. However, the extension of the loop side chains is less prominent with stronger constraints, as expected: The RMSD value at R = 50.0 Å decreases as the constraint becomes stronger. Thus, flexibility of the loop influences the binding reaction.
Looking into coordination pathways, we found that, regardless of the strength of the constraint, one of the acidic residues catches Ca2+ at the very beginning of the binding reactions. However, in a more detailed comparison of which residue binds to Ca2+ in what order, we saw some differences. (i.e., the value of R, at which acidic residues dissociates are dependent on the strength of the constraint. With stronger constraints, the dissociation occurs at small value of R without changing side-chain orientation of nonacidic residues.) With the kbb = 0.1 kcal/mol/ Å2 or 1.0 kcal/mol/ Å2 constraint, all three acidic residues bind Ca2+, which are followed by nonacidic residues binding. However, in the strongest constraint (kbb = 10 kcal/mol/ Å2), Tyr-99 binds right after the first coordination by Glu-104, which is then followed by the other two acidic residues, Asp-93 and Asp-95. The sequential order of coordination is inherently stochastic and thus seems to be sensitive to change in constraint.
Binding/dissociation pathways by steered MD simulations
We searched binding and dissociation pathways by steered MD. As in Methods, external forces that drive the system between apo and holo structures were introduced on seven distances 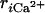 between Ca2+ and coordinated atoms in the CaM loop. We note that
between Ca2+ and coordinated atoms in the CaM loop. We note that 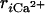 at the apo form depends on the dissociated position of Ca2+, which is somewhat arbitrary. Therefore, we repeated both binding and dissociation simulations six times, starting from different Ca2+ positions in the apo state.
at the apo form depends on the dissociated position of Ca2+, which is somewhat arbitrary. Therefore, we repeated both binding and dissociation simulations six times, starting from different Ca2+ positions in the apo state.
Both in binding and dissociation simulations, coordination numbers to Ca2+ by the CaM loop and that by water molecules are anti-correlated and change stepwise as we found in the free-energy analysis. Thus, the sum of the two coordination numbers is nearly constant between 7 and 8 (data not shown).
Fig. 5 plots some of simulated binding and dissociation pathways on the two-dimension, which is the same two-dimension as in Fig. 2; the x axis is the RMSD of side-chain heavy atoms in the CaM loop measured from the holo form and the y axis is the direct interaction energy Ebind between Ca2+ and the CaM loop. The green curve depicts one of the dissociation trajectories. Consistent with the free-energy surface in Fig. 2, upon dissociation the loop RMSD increases rapidly up to values larger than that at the apo structure, indicating that the loop is more extended in the near-transition region than that in the apo form. As dissociation goes on further, the loop RMSD gets back to reduced values approaching the RMSD in the apo form. Among six trajectories of dissociation, the pathways are quite similar to each other. In contrast, the binding pathways (two examples depicted in red and blue) look somewhat different from dissociation pathways and are more diverse. Some binding trajectories reach the structure where all five amino acids coordinate to Ca2+ (the red curve is an example), whereas other trajectories end up with structures where only four amino acids coordinate to Ca2+ (the blue curve is an example). Interestingly, for the trajectories that the side-chain RMSD significantly increase at an early stage of binding (an example is the red curve), the trajectory can reach coordination by five amino acids. For the trajectory that Ca2+ ends up with poorer coordination, we found that the side-chain RMSD does not increase very significantly at an early stage.
FIGURE 5.

Some trajectories of binding (red and blue curves) and dissociation (green curve) by the steered MD simulations. The x axis means the RMSD of heavy atoms on the loop's side chain from the holo form and the y axis is the direct interaction energy between Ca2+ and the CaM loop.
We then investigated which side chains coordinate to Ca2+ in what order in the steered MD trajectories. Fig. 6 plots the probability 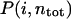 that the ith residue coordinates to Ca2+ when a total of ntot residues coordinate. Here, ntot plays the role of net reaction coordinate (see Methods for the complete definition). From Fig. 6 A, which is for binding reaction, two acidic residues (Asp-95 and Glu-104) prefer to coordinate to Ca2+ at an early stage (1 ∼ 3); however, the precise order of coordination looks somewhat stochastic. Pathways for dissociation are depicted in Fig. 6 B, which is less diverse. The nonacidic residues dissociate from Ca2+ first and then the three acidic residues dissociate stepwise; this character is similar to that found in the free-energy analysis above. Especially, Glu-104 keeps coordinated up to the latest stage. It is partly because the side chain of Glu is longer than that of Asp. We found that the process looks slightly different between binding and dissociation—the order of coordination looks more diverse in binding than that in dissociation. We guess that the coordination order for the binding process depends more sensitively on the initial apo position of the Ca2+.
that the ith residue coordinates to Ca2+ when a total of ntot residues coordinate. Here, ntot plays the role of net reaction coordinate (see Methods for the complete definition). From Fig. 6 A, which is for binding reaction, two acidic residues (Asp-95 and Glu-104) prefer to coordinate to Ca2+ at an early stage (1 ∼ 3); however, the precise order of coordination looks somewhat stochastic. Pathways for dissociation are depicted in Fig. 6 B, which is less diverse. The nonacidic residues dissociate from Ca2+ first and then the three acidic residues dissociate stepwise; this character is similar to that found in the free-energy analysis above. Especially, Glu-104 keeps coordinated up to the latest stage. It is partly because the side chain of Glu is longer than that of Asp. We found that the process looks slightly different between binding and dissociation—the order of coordination looks more diverse in binding than that in dissociation. We guess that the coordination order for the binding process depends more sensitively on the initial apo position of the Ca2+.
FIGURE 6.

The ensemble of coordination pathways. The probability P(i, ntot) that the ith residue joins to coordination to Ca2+ for a given total number of amino acids ntot that coordinate to Ca2+ is represented by darkness in each block. We note that ntot plays the role of gross reaction coordinate here. (A) Binding reaction. (B) Dissociation reaction.
In contrast to Asp-95 and Glu-104, the role of Asp-93 is subtle. In the dissociation reaction, it keeps coordinated until the latest stage. (The sequential order is second last.) In binding trajectories, however, when Ca2+ is first coordinated by Asp-93, the trajectory fails to make further coordination by other residues, thus leading to a poorer final structure (data not shown). This is one of the examples that the initial apo position of the Ca2+ affects binding process. Asp-93 in the holo structure of the whole CaM domain is relatively buried and thus Ca2+ cannot access Asp-93 directly from the surface (Fig. 1). Thus, when Ca2+ is coordinated first by Asp-93 in the CaM loop simulation, the access path is from the backside of the loop where it is occupied by other residues in the intact CaM. From this direction, further coordination by other atoms is impossible.
Finally, we note that the order of coordination can, to some extent, be altered by the condition of how the exerting force is added.
DISCUSSIONS AND CONCLUSIONS
Camacho and co-workers (33) suggested that there are a few anchor residues in interacting proteins that first recognize partners, and conformations of these anchor residues in the unbound form are similar to those in the bound complex. In the loop that we studied here, indeed, rotamers of Asp-93 and Glu-104 are similar in holo and in apo states, and thus conformations of them in the apo state are ready for recognizing the ligand, whereas the rotamer of Asp-95 is different in the holo and apo states. Even though these key resides in the apo state have the preset side-chain conformations ready for binding, our work suggests that these key side chains extend their conformation once, for recruiting the ligand.
In evolutionary aspects, not surprisingly, the acidic residues on the CaM loop; Asp-93, Asp-95, and Glu-104, are highly conserved among virtually all EF-hand Ca2+ binding motifs (34). Point mutations in these residues of the EF-hand motif basically eliminate the Ca2+ binding ability unless extremely high Ca2+ concentrations are explored (35–39).
Even though the simulated system here is relatively small in the standard of today's biomolecular simulations, the accurate estimation of a free-energy surface is still extremely difficult. Especially, Ca2+ has two positive charges that make coordination so strong both in the bound and the unbound states. Exchange of coordination partners through ion-binding reaction involves exchange in strong interactions, which makes unbiased conformational sampling very difficult. Moreover, the ion induces relatively large polarization in surrounding molecules, which may require a higher order description in energy function, such as the polarizable force field and quantum chemical approach, as has been suggested (40,41). Unavoidably, thus, estimates of the binding energy, the free-energy difference between holo and apo forms, may not be accurate, quantitatively speaking. More accurate and rigorous simulations need to be done in the future. Work is now in progress to develop more efficient sampling techniques. However, despite some uncertainty in free-energy values, we insist that the grabbing mechanism, which is a qualitative perspective, holds true in many relevant ion-binding reactions.
In summary, we have addressed how conformational change in protein side chains is coupled to ion binding by simulating Ca2+ binding to a CaM loop. We found that side chains of some key residues extend their “arms” to catch the ion, which we would like to call the grabbing mechanism. Through this mechanism, stepwise dehydration of water molecules coordinated to Ca2+ and concerted stepwise binding of protein side chains occur, resulting in a smooth free-energy surface. We postulate that the same sort of mechanisms would be found widely in other bindings, especially in the case of binding of charged ligands.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
The authors thank to Prof. David Leitner for reading the article and making a number of helpful suggestions.
C.K. is supported by the Research Fellowship of the Japan Society for the Promotion of Science for Young Scientists. This work was partly supported by the “Water and Biomolecules” program from the Japan Ministry of Education, Culture, Sports, Science and Technology.
Chigusa Kobayashi's present address is Dept. of Computational Molecular Science, Institute for Molecular Science, Okazaki, Aichi, 444–8585, Japan.
References
- 1.Frauenfelder, H., S. G. Sligar, and P. G. Wolynes. 1991. The energy landscapes and motions of proteins. Science. 254:1598–1603. [DOI] [PubMed] [Google Scholar]
- 2.Shoemaker, B. A., J. J. Portman, and P. G. Wolynes. 2000. Speeding molecular recognition by using the folding funnel: the fly-casting mechanism. Proc. Natl. Acad. Sci. USA. 97:8868–8873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bracken, C., L. M. Iakoucheva, P. R. Romero, and A. K. Dunker. 2004. Combining prediction, computation and experiment for the characterization of protein disorder. Curr. Opin. Struct. Biol. 14:570–576. [DOI] [PubMed] [Google Scholar]
- 4.Gerstein, M., and N. Echols. 2004. Exploring the range of protein flexibility, from a structural proteomics perspective. Curr. Opin. Chem. Biol. 8:14–19. [DOI] [PubMed] [Google Scholar]
- 5.Austin, R. H., K. W. Beeson, L. Eisenstein, H. Frauenfelder, and I. C. Gunsalus. 1975. Dynamics of ligand binding to myoglobin. Biochemistry. 14:5355–5373. [DOI] [PubMed] [Google Scholar]
- 6.Schmidt, M., K. Nienhaus, R. Pahl, A. Krasselt, S. Anderson, F. Parak, G. U. Nienhaus, and V. Srajer. 2005. Ligand migration pathway and protein dynamics in myoglobin: a time-resolved crystallographic study on L29W MbCO. Proc. Natl. Acad. Sci. USA. 102:11704–11709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thorpe, I. F., and C. L. Brooks 3rd. 2004. The coupling of structural fluctuations to hydride transfer in dihydrofolate reductase. Proteins. 57:444–457. [DOI] [PubMed] [Google Scholar]
- 8.Pedigo, S., and M. A. Shea. 1995. Quantitative endoproteinase GluC footprinting of cooperative Ca2+ binding to calmodulin: proteolytic susceptibility of E31 and E87 indicates interdomain interactions. Biochemistry. 34:1179–1196. [DOI] [PubMed] [Google Scholar]
- 9.Sorensen, B. R., and M. A. Shea. 1998. Interactions between domains of apo calmodulin alter calcium binding and stability. Biochemistry. 37:4244–4253. [DOI] [PubMed] [Google Scholar]
- 10.Pedigo, S., and M. A. Shea. 1995. Discontinuous equilibrium titrations of cooperative calcium binding to calmodulin monitored by 1-D 1H-nuclear magnetic resonance spectroscopy. Biochemistry. 34:10676–10689. [DOI] [PubMed] [Google Scholar]
- 11.Jaren, O. R., S. Harmon, A. F. Chen, and M. A. Shea. 2000. Paramecium calmodulin mutants defective in ion channel regulation can bind calcium and undergo calcium-induced conformational switching. Biochemistry. 39:6881–6890. [DOI] [PubMed] [Google Scholar]
- 12.Faga, L. A., B. R. Sorensen, W. S. VanScyoc, and M. A. Shea. 2003. Basic interdomain boundary residues in calmodulin decrease calcium affinity of sites I and II by stabilizing helix-helix interactions. Proteins. 50:381–391. [DOI] [PubMed] [Google Scholar]
- 13.Ouyang, H., and H. J. Vogel. 1998. Metal ion binding to calmodulin: NMR and fluorescence studies. Biometals. 11:213–222. [DOI] [PubMed] [Google Scholar]
- 14.Snyder, E. E., B. W. Buoscio, and J. J. Falke. 1990. Calcium(II) site specificity: effect of size and charge on metal ion binding to an EF-hand-like site. Biochemistry. 29:3937–3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wojcik, J., J. Goral, K. Pawlowski, and A. Bierzynski. 1997. Isolated calcium-binding loops of EF-hand proteins can dimerize to form a native-like structure. Biochemistry. 36:680–687. [DOI] [PubMed] [Google Scholar]
- 16.Siedlecka, M., G. Goch, A. Ejchart, H. Sticht, and A. Bierzyski. 1999. Alpha-helix nucleation by a calcium-binding peptide loop. Proc. Natl. Acad. Sci. USA. 96:903–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lopez, M. M., D. H. Chin, R. L. Baldwin, and G. I. Makhatadze. 2002. The enthalpy of the alanine peptide helix measured by isothermal titration calorimetry using metal-binding to induce helix formation. Proc. Natl. Acad. Sci. USA. 99:1298–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ye, Y., S. Shealy, H. W. Lee, I. Torshin, R. Harrison, and J. J. Yang. 2003. A grafting approach to obtain site-specific metal-binding properties of EF-hand proteins. Protein Eng. 16:429–434. [DOI] [PubMed] [Google Scholar]
- 19.Ye, Y., H. W. Lee, W. Yang, S. Shealy, and J. J. Yang. 2005. Probing site-specific calmodulin calcium and lanthanide affinity by grafting. J. Am. Chem. Soc. 127:3743–3750. [DOI] [PubMed] [Google Scholar]
- 20.Wriggers, W., E. Mehler, F. Pitici, H. Weinstein, and K. Schulten. 1998. Structure and dynamics of calmodulin in solution. Biophys. J. 74:1622–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vigil, D., S. C. Gallagher, J. Trewhella, and A. E. Garcia. 2001. Functional dynamics of the hydrophobic cleft in the N-domain of calmodulin. Biophys. J. 80:2082–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Komeiji, Y., Y. Ueno, and M. Uebayasi. 2002. Molecular dynamics simulations revealed Ca(2+)-dependent conformational change of calmodulin. FEBS Lett. 521:133–139. [DOI] [PubMed] [Google Scholar]
- 23.Shepherd, C. M., and H. J. Vogel. 2004. A molecular dynamics study of Ca(2+)-calmodulin: evidence of interdomain coupling and structural collapse on the nanosecond timescale. Biophys. J. 87:780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Case, D. A., D. A. Pearlman, J. W. Caldwell, T. E. Cheatham III, J. Wang, W. S. Ross, C. L. Simmerling, T. A. Darden, K. M. Merz, R. V. Stanton, A. L. Cheng, J. J. Vincent, M. Crowley, V. Tsui, H. Gohlke, R. J. Radmer, Y. Duan, J. Pitera, I. Massova, G. L. Seibel, U. C. Singh, P. K. Weiner, and P. A. Kollman. 2002. AMBER. Version 7. University of California, San Francisco.
- 25.Wang, J. M., P. Cieplak, and P. A. Kollman. 2000. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 21:1049–1074. [Google Scholar]
- 26.Bradbrook, G. M., T. Gleichmann, S. J. Harrop, J. Habash, J. Raftery, J. Kalb, J. Yariv, I. H. Hillier, and J. R. Helliwell. 1998. X-ray and molecular dynamics studies of concanavalin-A glucoside and mannoside complexes: relating structure to thermodynamics of binding. J. Chem. Soc. Faraday T. 94:1603–1611. [Google Scholar]
- 27.Jorgensen, W. L., J. Chandrasekhar, J. Madura, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- 28.Marchand, S., and B. Roux. 1998. Molecular dynamics study of calbindin D9k in the apo and singly and doubly calcium-loaded states. Proteins. 33:265–284. [PubMed] [Google Scholar]
- 29.Torrie, G. M., and J. P. Valleau. 1977. Nonphysical sampling distributions in Monte Carlo free-energy estimation: umbrella sampling. J. Comput. Phys. 23:187–199. [Google Scholar]
- 30.Kumar, S., D. Bouzida, R. H. Swendsen, P. A. Kollman, and J. M. Rosenberg. 1992. THE weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 13:1011–1021. [Google Scholar]
- 31.Grübmuller, H., B. Heymann, and P. Tavan. 1996. Ligand binding: molecular mechanics calculation of the streptavidin-biotin rupture force. Science. 271:997–999. [DOI] [PubMed] [Google Scholar]
- 32.Izrailev, S., S. Stepaniants, M. Balsera, Y. Oono, and K. Schulten. 1997. Molecular dynamics study of unbinding of the avidin-biotin complex. Biophys. J. 72:1568–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rajamani, D., S. Thiel, S. Vajda, and C. J. Camacho. 2004. Anchor residues in protein-protein interactions. Proc. Natl. Acad. Sci. USA. 101:11287–11292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Falke, J. J., S. K. Drake, A. L. Hazard, and O. B. Peersen. 1994. Molecular tuning of ion binding to calcium signaling proteins. Q. Rev. Biophys. 27:219–290. [DOI] [PubMed] [Google Scholar]
- 35.Maune, J. F., C. B. Klee, and K. Beckingham. 1992. Ca2+ binding and conformational change in two series of point mutations to the individual Ca(2+)-binding sites of calmodulin. J. Biol. Chem. 267:5286–5295. [PubMed] [Google Scholar]
- 36.Babu, A., H. Su, Y. Ryu, and J. Gulati. 1992. Determination of residue specificity in the EF-hand of troponin C for Ca2+ coordination, by genetic engineering. J. Biol. Chem. 267:15469–15474. [PubMed] [Google Scholar]
- 37.Matsuura, I., E. Kimura, K. Tai, and M. Yazawa. 1993. Mutagenesis of the fourth calcium-binding domain of yeast calmodulin. J. Biol. Chem. 268:13267–13273. [PubMed] [Google Scholar]
- 38.Drake, S. K., M. A. Zimmer, C. Kundrot, and J. J. Falke. 1997. Molecular tuning of an EF-hand-like calcium binding loop contributions of the coordinating side chain at loop position 3. J. Gen. Physiol. 110:173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hobson, K. F., N. A. Housley, and S. Pedigo. 2005. Ligand-linked stability of mutants of the C-domain of calmodulin. Biophys. Chem. 114:43–52. [DOI] [PubMed] [Google Scholar]
- 40.Bliznyuk, A. A., and A. A. Rendell. 2004. Electronic effects in biomolecular simulations; investigation of KcsA potassium ion channel. J. Phys. Chem. B. 108:13866–13873. [Google Scholar]
- 41.Allen, T. W., O. S. Andersen, and B. Roux. 2004. Energetics of ion conduction through the gramicidin channel. Proc. Natl. Acad. Sci. USA. 101:117–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kraulis, P. J. 1991. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Cryst. 24:946–950. [Google Scholar]
- 43.Merritt, E. A., and D. J. Bacon. 1997. Raster3D photorealistic molecular graphics. Methods Enzymol. 277:505–524. [DOI] [PubMed] [Google Scholar]
- 44.DeLano, W. L. 2002. The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, CA.