

Abstract
Most xenobiotics that enter the body are subjected to metabolism that functions primarily to facilitate their elimination. Metabolism of certain xenobiotics can also result in the production of electrophilic derivatives that can cause cell toxicity and transformation. Many xenobiotics can also activate receptors that in turn induce the expression of genes encoding xenobiotic-metabolizing enzymes and xenobiotic transporters. However, there are marked species differences in the way mammals respond to xenobiotics, which are due in large part to molecular differences in receptors and xenobiotic-metabolizing enzymes. This presents a problem in extrapolating data obtained with rodent model systems to humans. There are also polymorphisms in xenobiotic-metabolizing enzymes that can impact drug therapy and cancer susceptibility. In an effort to generate more reliable in vivo systems to study and predict human response to xenobiotics, humanized mice are under development.
Keywords: CYP, nuclear receptor, transgenic mouse model, drug metabolism, pharmacokinetics, pharmacogenetics, cancer, toxicity, ligands, gene activation
INTRODUCTION
Upon entering the body, a foreign compound is subjected to metabolism by a large group of enzymes, collectively referred as xenobiotic-metabolizing enzymes, that includes the phase 1 oxidative enzymes and the phase 2 conjugating enzymes. The cytochrome P450 (CYP) enzymes are among the most important phase 1 enzymes (1–3); they metabolize most clinically used drugs and are required for metabolic activation of chemical carcinogens and toxins. The phase 2 enzymes facilitate the elimination of drugs and the inactivation of carcinogenic metabolites produced by CYPs. The balance between the phase 1 and phase 2 enzymes will determine the metabolic fate of a particular chemical. The expression of theses enzymes can markedly vary between individuals owing to differences in the extent of induction and by polymorphisms.
Most CYPs are inducible except the well-known polymorphic CYP2D6. Transcriptional regulation of genes in the CYP1A, CYP1B, CYP2B, CYP2C, CYP3A, and CYP4A subfamilies are primarily controlled by aryl hydrocarbon receptor (AHR), constitutive androstane receptor (CAR, NR1I4), pregnant X receptor (PXR, NR1I2) and peroxisome proliferator-activated receptor α (PPARα, NR1C1) (4–10). These receptors consist of a core DNA-binding domain and a ligand-binding domain. After binding with a ligand, the receptors undergo certain conformational changes that coordinately release corepressors and recruit coactivators to enable transcriptional activation of their target genes, including those encoding the xenobiotic-metabolizing CYPs.
There are marked species differences in the response to xenobiotics (11–14). This is especially notable between humans and rodents such as rats and mice, the most commonly used experimental models for studies in pharmacology and toxicology. For instance, the CYP2D subfamily in humans has a single active member CYP2D6 that is highly polymorphic; rats and mice have at least five genes, none of which encodes a protein having the same enzymatic activities as CYP2D6 (11, 14). Among the nuclear receptors, the human and mouse PPARα have different ligand-binding affinities (15, 16) and expression levels in liver (17). Consequently, striking species differences have been observed in the response to xenobiotics, particularly between mice and humans.
One approach to overcome the gap of species difference is to generate humanized transgenic mice by introducing a human gene into the mouse genome, thus offering a better animal system to predict the human response to foreign chemicals and understand the underlying mechanisms (18–21). There are a number of approaches that can be used to generate a humanized mouse. The most common is to fuse the human cDNA to a promoter that drives expression of the cDNA in the mouse. For example, the serum albumin promoter can be used to deliver expression of a protein specifically in the liver. Another approach is to use the complete human genomic clone as a transgene. Use of a bacterial artificial chromosome (BAC) is ideal because one can obtain clones that contain the complete gene and all of the regulatory elements that drive expression of the gene. The human transgene can then be bred onto a mouse line in which the endogenous mouse gene has been disrupted. The third approach is to “knock-in” the human gene or cDNA into the endogenous mouse gene. This would result in disruption of the mouse gene and introduction of the human gene. In this case, a cDNA is commonly used because production of a recombination vector containing a complete human gene and sufficient mouse flanking sequence to promote recombination with the native mouse gene would be technically difficult. All of these approaches have advantages and disadvantages, and the use of one approach above another would depend on the questions that need to be addressed in the study.
BIOLOGICAL MEDIATORS IN RESPONSE TO XENOBIOTICS
Cytochrome P450s are Responsible for the Metabolism of Xenobiotics
The phase 1 enzymes largely consist of the flavin-containing monooxygenase (FMO) superfamily and the CYP superfamily. The CYP superfamily is the most important contributor to the metabolism of drugs and the metabolic activation of toxicants and chemical carcinogens (1, 22). Although there are a number of CYP families that are involved in critical pathways of sterol and bile acid synthesis, four families primarily function to metabolize foreign compounds (2, 3). These include families CYP1 through CYP4. The CYP1 family is most notable for carcinogen and toxicant metabolism, whereas the CYP2 and CYP3 families metabolize drugs and other compounds, ultimately resulting, after phase 2 metabolism, in more stable and hydrophilic derivatives, although there are exceptions.
CYP-mediated oxidation is the principal means of eliminating clinically administered drugs, and thus the extent of metabolism governs the plasma half-lives of drugs. Most drugs are given in chemical forms that have biological activity, and metabolism serves to inactivate this activity by converting the drug to a derivative that can no longer bind to its cellular target and can be easily excreted from human body. However, a few drugs are actually prodrugs that require metabolism to convert them to active forms. The xenobiotic-metabolizing enzymes are expressed at high levels in liver, and thus orally administered drugs are subjected to what is commonly referred to as a “first-pass metabolism,” which can reduce drug bioavailability. As the drug continues to circulate through the liver, its plasma concentration becomes lower and the extent of metabolism decreases. For drug therapy, phase I clinical trials serve to determine the optimum dosing for a drug that leads to a favorable therapeutic outcome and no side effects. For most orally administered drugs, this experimentally determined dose is the average, and depending on the drugs safety index, it can be used to treat most adult patients with adjustments for juveniles and infants. Differences in the extent of metabolism can be tolerated with drugs exhibiting wide safety margins or therapeutic indexes. However, drugs with narrow therapeutic indexes must be used cautiously, and variation in metabolism can have serious consequences. Because metabolism can markedly influence the efficacy and safety of a drug, pharmaceutical companies determine the route of metabolism of drug candidates very early in the drug development or drug discovery process.
Adverse drug reactions can occur when a drug is not metabolized at a rate that leads to favorable pharmacokinetics. Differences in rate of metabolism of a drug can be due to drug interactions with another coadministered drug that is metabolized by the same enzyme. Thus, it becomes important to determine the identity of the CYP that metabolizes a particular drug and to avoid coadministering drugs that are metabolized by the same enzyme. Concomitant use of a CYP substrate and inhibitor can also result in harmful events as the inhibitor diminishes the metabolism of the substrate drug. For example, some selective serotonin reuptake inhibitors (SSRIs), such as paroxetine and fluoxetine, are potent inhibitors of CYP2D6. These SSRIs have been known to cause unwanted drug-drug interactions with other psychotropic drugs metabolized by CYP2D6 (23). Therefore, it is advised not to use these drug substrates and inhibitors concomitantly, and it is important to determine the potential inhibition of new drug candidates in preclinical studies. However, in certain instances interactions can be directed as an effective approach to increase the bioavailability of the substrate drug and has become a potential therapeutic strategy. Moreover, some drugs are CYP inducers and can not only induce their own metabolism but induce metabolism of other coadministered drugs. For example the gastritis drug omeprazole is a ligand for the AHR and can induce CYP1A1 and CYP1A2 (24, 25). Finally, drug metabolism can also be influenced by diet. CYP inhibitors and inducers are commonly found in diets and in some cases these can influence the toxicity and efficacy of a drug. Components found in grapefruit juice are potent inhibitors of CYP3A4 (26, 27). Steroid hormones and components of herbal products such as St. John’s wort can induce hepatic levels of CYP3A4 (28). In fact, much information has been acquired about drug substrates and inhibitors and inducers of various CYPs (Table 1), and this information should be used as a resource to avoid harmful drug-drug interactions.
TABLE 1.
Listed are some drug substrates, inhibitors, and inducers of human CYP2D6, CYP2E1, and CYP3A4 isozymes
| CYP2D6 | Substrate | Psychotropic drugs:
Aimtriptyline, citalopram, clomipramine, clozapine, desipramine, fluoxetine, imipramine, mianserine, mirtazapine, nortriptyline, paroxetine, venlafaxine Cardiovascular drugs: Alprenolol, bufuralol, encainide, flecainide, metoprolol, propafenone, propranolol, timolol Miscellaneous drugs: Codeine, debrisoquine, dextromethorphan, phenformin |
| Inhibitor | Fluoxetine, fluvoxamine, methadone, norfluoxetine, paroxetine, quinidine, sertraline | |
| Inducer | None | |
| CYP2E1 | Substrate | Acetaminophen, aniline, benzene, chlorzoxazone, dapsone, enflurane, ethanol, N,N-dimethyl formamide, halothane, isoflurane, methoxyflurane, sevoflurane, theophylline |
| Inhibitor | Diethyldiethiocarbamate, disulfiram | |
| Inducer | Ethanol, isoniazid | |
| CYP3A4 | Substrate | Benzodiazepines:
Alprazolam, clonazepam, clorazepate, diazepam, flurazepam, halazepam, midazolam, prazepam, triazolam Calcium channel blockers: Amlodipine, diltiazem, felodipine, lercanidipine, nifedipine, nisoldipine, nitrendipine, verapamil HIV antivirals: Indinavir, nelfinavir, ritonavir, saquinavir Immune modulators: Cyclosporine, tacrolimus (FK506) Macrolide antibiotics: Clarithromycin, erythromycin Steroids: Estradiol, hydrocortisone, progesterone, testosterone Miscellaneous drugs: Codeine, dextromethorphan, methadone, taxol |
| Inhibitor | Azole antifungals:
Clotrimazole, fluconazole, itraconazole, ketoconazole, miconazole HIV antivirals: Amprenavir, atazanavir, indinavir, nelfinavir, ritonavir, saquinavir Miscellaneous drugs: Gestodene, troleandomycin |
|
| Inducer | Carbamazepine, dexamethasone, phenobarbital, rifampicin |
As noted earlier, interindividual differences in drug metabolism can be influenced by polymorphisms in CYPs. The CYP2D6 polymorphism (29–31) led to the withdrawal of a few clinically used drugs and the cautious use of others that are known CYP2D6 substrates. The study of this and other polymorphisms in drug metabolism also led to the concept of individualized medicine by adjusting dosing based on the extent of metabolism as determined by therapeutic drug monitoring (32). Polymorphisms also exist in the CYP2A6, CYP2C9, and CYP2C19 genes (29, 33). A rare genetic deficiency exists in the CYP1B1 gene that leads to congenital glaucoma (34) and the role of this CYP in ocular development was confirmed by examining the Cyp1b1-null mice (35).
Xenobiotic Receptors Control the Regulation of Metabolism
There are a number of signal transduction pathways that are responsive to drugs and other foreign compounds (6, 36–39). As a general paradigm, a compound can induce its own metabolism by activation of a receptor leading to induction of the expression of CYPs and other phase 2 genes that result in enhanced metabolism of the compound. Although there are a few examples of stimulation of metabolism by various chemicals, the numbers of foreign chemical inducers of metabolism are more limited than the number of CYP substrates. There are several intracellular receptors that appear to be activated by foreign chemicals. These include the AHR (40, 41), CAR (42, 43), PXR (44–46), and PPARα (15,47).
The AHR, a member of the Per-Arnt-Sim (PAS), beta helix-loop-helix superfamily, activates the expression of CYP1A1, CYP1A2, and CYP1B1 genes (48). The AHR resides in the nucleus bound to heat shock protein 90 (HSP90), hepatitis B virus X-associated protein 2 (XAP2), and perhaps other chaperones that upon ligand binding to the AHR, are released coincident of nuclear translocation and heterodimerization with the AHR nuclear translocator (ARNT), culminating in target gene activation by binding of the AHR-ARNT to the xenobiotic response element (XRE) site. AHR ligands include drugs such as omeprazole; natural products found in plants including β-napthoflavone and β-carboline; and carcinogens such as benzo[a]pyrene, 3-methylcholanthrese, 7,12-dimethylbenz[a]anthracene, and 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). There are polymorphisms in the AHR, particularly the well-studied aryl hydrocarbon (AH) locus in mice that is due to allelic variants that results in altered ligand binding (48). There are also a number of AHR allelic variants in humans but their functional significance and role in susceptibility to environmentally based disease have not been established (49).
PXR, CAR, and PPARα are members of the Type 2 nuclear receptors, historically referred to as orphan receptors until their ligands were identified. Unlike steroid and other nuclear receptors, these proteins are activated by a large number of xenobiotics, including drugs (50). These receptors all use the retinoid X receptor (RXR) as a partner for heterodimerization and bind to various cis-acting elements, usually direct repeats upstream of target genes.
PXR, also called the steroid X receptor (SXR), activates the expression of CYPs involved in the metabolism of a large number of xenobiotics, including numerous clinically used drugs. In particular, PXR induces expression of the CYP3A4 gene, and the phase 2 sulfotransferase (SULT1A) (51) and UDP-glucuronosyltransferase 1A (UGT1A) genes (52). This receptor exhibits species differences in ligand-binding specificities between humans and mice (12). For example, the human PXR is activated by the drug rifampicin, whereas the mouse receptor does not bind this ligand. Conversely, the mouse Pxr is activated by pregnenolone 16α-carbonitrile (PCN), whereas this compound does not activate the human receptor.
CAR is a constitutively activated receptor that increases transcription of the CYP2B and CYP3A genes (53–56) and the phase 2 glutathione S-transferase (GST) and UGT1A genes (52, 57, 58). The inverse agonist androstanol, an endogenous steroid, binds to CAR and inhibits its activity. There is also evidence that CAR regulates the degradation of bilirubin, the major product of heme degradation (59). Among the activators of CAR are the drug phenobarbital (56) and the insecticide contaminant 1,4-bis[2(3,5-dichloropyridyloxy)]benzene (TCPOBOP) (60). Although TCPOBOP is a direct CAR ligand, phenobarbital and other CAR activators do not directly bind CA but trigger a signal transduction pathway resulting in translocation of the receptor from the cytoplasm to the nucleus (61). There is also species difference in the ligand specificity of CAR. TCPOBOP is a potent murine Car ligand but does not activate rat or human CAR (62). The compound 6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime (CITCO) was identified as a specific human CAR agonist (63). Meclizine, an antiemetic drug, is a potent agonist of the murine receptor, but it is an inverse agonist of human CAR, similar to androstanol (64). On the other hand 6,7-dimethylesculetin, the putative active component of the Chinese herbal medicine Yin Zhi Huang used in the treatment of neonatal jaundice, activates both the mouse and human CAR (65).
PPARα is responsible for the phenomenon known as peroxisome proliferation that has been described in rats and mice (66). PPARα also exhibits a major species difference that accounts for the response to peroxisome proliferators (13). Rats and mice exhibit marked peroxisome proliferation and hepatomegaly that is a result of increased hepatocyte size and increased cell proliferation. Most of the marked physiological consequences of PPARα activation are due to increases in enzymes involved in peroxisomal and mitochondrial fatty acid β-oxidation. After long-term administration, peroxisome proliferator chemicals cause liver cancer in susceptible species. Fibrate drugs, lowering serum triglycerides and cholesterol, have been in clinical use for more than 50 years. In stark contrast to rats and mice, these agents do not cause peroxisome proliferation and hepatomegaly in humans or monkeys and epidemiology studies have revealed no increase in liver cancers (8, 67).
SPECIES DIFFERENCES IN DRUG METABOLISM
Rats and mice can metabolize a particular compound differently from humans (68), which is due in large part to marked species differences in the expression and catalytic activities of CYPs. It is particularly notable between human CYP2D6 and rodent Cyp2d, and CYP3A4 and rodent Cyp3a, respectively (11, 69). This is of particular importance because CYP2D6 and CYP3A4 collectively metabolize more than 70% of the drugs on the market. There are striking differences in the complexity of the CYP genes between humans and rats or mice, and in the number of CYP genes expressed and their catalytic activities. There are also species differences in the CYP3A genes, in particular, in their expression distribution in liver and extrahepatic tissues. As discussed above, there are species differences in the nuclear receptors that regulate the CYP genes that also account for species differences in the response to xenobiotics. These differences reside in the ligand specificities between human and rodent receptors. The practical result of these differences is that the metabolism and response of a particular compound in rodents does not necessarily reflect its veracity in humans. Thus, animal models may not be of value in predicting human responses to xenobiotics such as drugs. This problem can potentially be circumvented through the production of humanized mice.
CYTOCHROME P450 AND XENOBIOTIC RECEPTOR HUMANIZED MICE
CYP2D6-Humanized Mice
CYP2D6 is involved in the metabolism of a large number of clinically used drugs (30, 70) (Table 1); it does not produce any electrophilic metabolites that would cause toxicity or cancer. Two pseudogenes, designated CYP2D7P and CYP2D8P, are also found in humans. In contrast, rats and mice contain nine Cyp2d genes, several of which are expressed (http://www.icgeb.org/~p450srv/new/p450.html). Most importantly, none of the mouse genes appear to encode a Cyp2d with a catalytic activity similar to CYP2D6 (14). This is one of the clearest and best defined examples of a species difference in a CYP between mice and humans (11, 68). In the absence of this activity, humanized mice can be developed expressing the CYP2D6 gene in a wild-type mouse containing the Cyp2d genes. A CYP2D6-humanized mouse was produced using a lambda phage genomic clone containing the wild-type CYP2D6 gene. Out of several founder lines, one expressed CYP2D6 in the liver, kidney, and small intestine, known sites of expression of this CYP in humans (71). This line was used to determine the activity of CYP2D6 toward debrisoquine, an antihypertensive β-adrenoceptor blocking drug that is metabolized by CYP2D6 primarily through 4-hydroxylation. Debrisoquine was originally used in the discovery of the debrisoquine polymorphism in humans and the urinary ratio of debrisoquine/4-hydroxydebrisoquine has been used for many years as a measure of CYP2D6 activity in humans. The CYP2D6-humanized mice were able to efficiently metabolize debrisoquine (Figure 1), with pharmacokinetic parameters and urinary metabolite levels reflective of human extensive metabolizers (see below) of debrisoquine. To analyze the metabolism of debrisoquine in vivo, pharmacokinetic (PK) analysis revealed that mice having five copies of the transgene had a low area under the curve (AUC) for debrisoquine and a high AUC for the 4-hydroxydebrisoquine, whereas wild-type mice produced little of the metabolite and had a high AUC for the parent compound. Mice hemizygous for the transgene cluster were intermediate between wild-type and homozygote.
Figure 1.
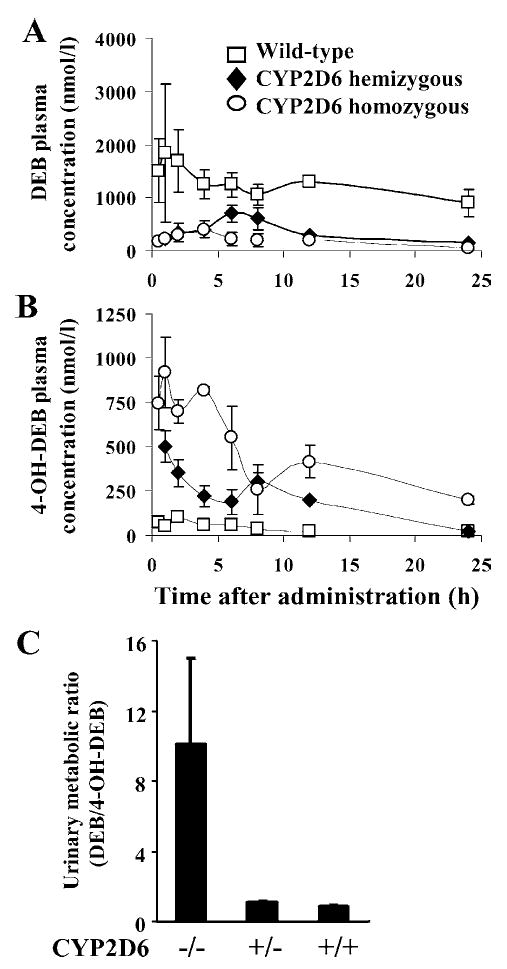
Serum concentrations of debrisoquine (DEB) (A) and 4-hydroxydebrisoquine (4-OH-DEB) (B) versus time curves for the wild-type, CYP2D6-humanized heterozygous, and homozygous mice after single oral administration of DEB (2.5 mg/kg). (C) DEB urinary metabolic ratio (MR) in wild-type and CYP2D6-humanized mice.
The debrisoquine/4-hydroxydebrisoquine excreted in the urine of subjects given a dose of debrisoquine has been used to estimate the extent of debrisoquine metabolism in humans. This ratio, called the metabolic ratio (MR), is the standard method for analyzing debrisoquine metabolism in humans because the parent compound and its principle metabolite are excreted in the urine. This analysis was performed on wild-type and CYP2D6-humanized mice and revealed that the MR in wild-type mice was approximately 10 and that of CYP2D6-humanized mice, both homozygous and hemizygous, was approximately 1 (Figure 1); the latter value is similar to that found in human extensive metabolizers. These studies confirm that wild-type mice are similar to human poor metabolizers of debrisoquine, and that CYP2D6-humanized mice are similar to human extensive metabolizers. The CYP2D6-humanized mice establish the feasibility of using human genomic clones for the production of CYP-humanized mice that exhibit metabolic activity in vivo that is similar to humans.
The CYP2D6-humanized mice can also be used to determine the mechanism of regulation of the human gene (71) by introducing the transgene into a liver-specific null mouse for hepatocyte nuclear factor 4α (HNF4α) (72). Mice lacking expression of HNF4α in the liver have decreased expression of the CYP2D6 transgene, indicating a role for this factor in the control of the liver-specific expression of CYP2D6. Expression is not lost in the absence of HNF4α, however, indicating that other liver-enriched factors may have a role in its expression.
This CYP2D6-humanized mouse model has been further applied to the search for potential endogenous substrates for CYP2D6. Polymorphic CYP2D6 is expressed in several types of neurons in the central nervous system (73–78), but its function in the human brain remains elusive even though personality differences have been reported between EMs and PMs, suggesting the potential existence of endogenous substrates for CYP2D6 (79). It was revealed that CYP2D6 in this model catalyzed the O-demethylation of a number of psychotropic methoxyin-dolethylamines derived from serotonin (5-hydroxytryptamine, 5-HT) (70, 80, 81). Serum 5-HT levels were markedly higher in CYP2D6-humanized mice than wild-type mice dosed with 5-methoxytryptamine (5-MT). Some were directly produced from exogenously administrated 5-MT by CYP2D6 catalysis, which was further confirmed by the finding that deuterated 5-HT was produced from deuterated 5-MT in CYP2D6-humanized mice but not from wild-type mice. Upon pretreatment with quinidine, a potent CYP2D6 inhibitor, the CYP2D6-humanized mice did not produce any deuterated 5-HT from deuterated 5-MT. The regeneration of 5-HT from 5-MT catalyzed by CYP2D6 was suggested as a missing link in a serotonin-melatonin cycle (81). Taken together, this CYP2D6-humanized mouse model provides the possibility to investigate the functional significance of CYP2D6 under controlled conditions and at systemic levels, and it would be a unique addition for studying CYP2D6 pharmacogenetics.
CYP3A4-Humanized Mice
CYP3A4 is the most abundantly expressed CYP in human liver, although there is a wide degree of interindividual variability in expression that is likely due to the differences in extent of regulation of the CYP3A4 gene (82). CYP3A4 is known to metabolize more than 60% of all therapeutic drugs used in the treatment of many disorders, including hypercholesterolemia (statin drugs), bacterial infections (erythromycin), cancer (tamoxifen), and immune suppression (cyclosporine) (Table 1). Because many drugs that are metabolized by CYP3A4 are coadministered, there is a potential for adverse drug reactions owing to drug interactions. This is of great concern for the development of new drugs, particularly those that are used in combination therapies with other drugs. Four CYP3A forms are expressed in humans: CYP3A4, CYP3A5, CYP3A7, and CYP3A43. Interestingly, mice have eight CYP3A genes (http://www.icgeb.org/~p450srv/new/p450.html), which again illustrates the marked species differences in complexity of CYP gene families. The most abundantly expressed CYP3A gene in humans is CYP3A4. This CYP enzyme is especially of interest because it is not only expressed in liver but also the most abundantly expressed in the gut (83–86), where it can metabolize a large number of orally administered drugs (Table 1).
To generate mice that express human CYP3A4, a BAC clone was used as a transgene (87). These mice expressed high levels of CYP3A4 protein in the small intestine. Surprisingly, little expression was found in the liver, a major site of CYP3A4 expression in humans. However, recent studies have revealed low constitutive and inducible expression in the livers of mature female mice; no expression was found in adult males (88). Expression was observed in livers of immature males and female mice. These studies revealed that CYP3A4 transgene expression is not only dependent on sex but also on age in the humanized mice. Sex-dimorphism in the expression of CYP3A4 was also observed in human livers (89). The CYP3A4 catalytic activity in humanized mouse was demonstrated using the midazolam, which is commonly used as a standard probe for CYP3A4 activity in humans. Midazolam is oxidized at the 4- and 1′- position by CYP3A4. The pharmacokinetics of midazolam metabolism was determined upon oral administration and differences in AUC for the parental compound and the primary 1′-hydroxymidazolam were detected between the CYP3A4-humanized and wild-type mice, indicating that the transgenics have a higher rate of midazolam metabolism and clearance. No differences were observed between the transgenic mice and wild-type mice when the drug was administered intravenously (87). Interestingly, these humanized mice exhibited an impaired lactation phenotype, which was found to be associated with underdeveloped mammary alveoli, deficient milk protein gene mRNA expression, and lower serum estradiol levels (Figure 2) (88). One interpretation is that this phenotype is due to low estrogen as a result of enhanced metabolism of estradiol and its precursor, testosterone (Figure 2). It is also an intriguing possibility that this increased metabolism occurs in the gut during the course of enterohepatic circulation of estradiol (Figure 2) because in untreated CYP3A4-humanized mice the enzyme is expressed at highest levels in the intestine (87). Indeed, enterohepatic recycling of estrogen has been demonstrated in humans (90). These observations from the CYP3A4-humanized mice suggest that CYP3A4 may play an important role in the homeostasis of sex steroids. These mice will aid in the in vivo analysis of orally administered drugs that are substrates for CYP3A4 and potentially be of great value in the prediction of drug interactions. They will also be of value in determining the mechanism of tissue-specific and inducible regulation of the CYP3A4 gene.
Figure 2.
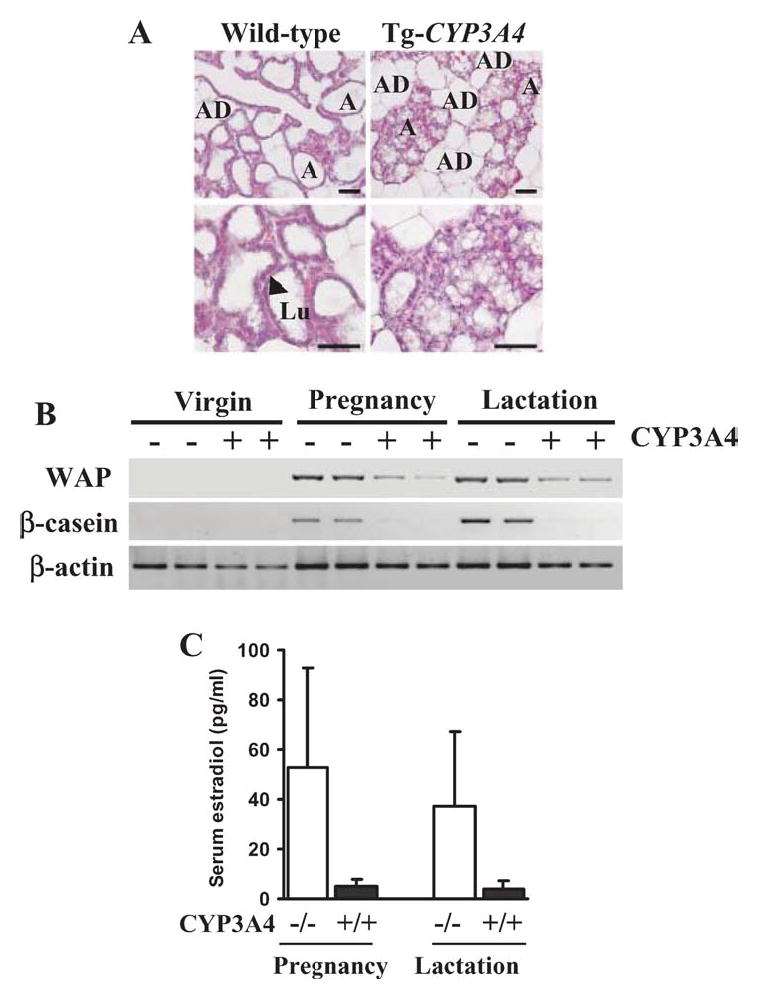
Impaired lactogenesis of CYP3A4-humanized mice associated with low estradiol levels. (A) Histological examination of mammary glands. In transgenic nursing mothers, it was sparsely filled with underdeveloped alveoli. In wild-type mice, the alveoli were fully distended by the accumulation of milk and minimal volume of adipose tissue (AD) was present. At higher magnification, the lumen (Lu) and epithelial cells (black arrow) of the alveoli are indicated. Scale bar: 50 μm. (B) Expression of milk protein genes in mammary glands as examined by RT-PCR. In both wild-type and CYP3A4-humanized virgin mouse mammary glands, whey acid protein (WAP) and β-casein were not detectable. During pregnancy and lactation, WAP and β-casein were abundant in wild-type mice and reduced or undetectable in transgenic mice. (C) Serum estradiol levels were significantly (*P < 0.05, n = 5 in each group) decreased in pregnant and lactating humanized mice. (D) Catabolism of testosterone and estradiol by CYP3A4. (E) Estradiol under enterohepatic circulation hydroxylated by intestinal CYP3A4.
CYP2E1-Humanized Mice
CYP2E1 is an ethanol-inducible CYP abundantly expressed in human liver (91, 92). It is also expressed in extrahepatic tissues such as kidney, lung, and brain. CYP2E1 catalyzes the oxidation of many toxicologically important low-molecular-weight chemicals to more toxic electrophilic metabolites (1, 22, 93–95). CYP2E1 substrates include organic solvents (ethanol, chloromethane, and benzene), nitrosamines (N-nitrosodimethylamine), and therapeutic drugs [acetaminophen (APAP)] (Table 1). CYP2E1 is also involved in the oxidation of physiologically important fatty acid metabolites (96–99). There are no reports of impaired CYP2E1 expression or activity or polymorphisms in humans other than synonymous single nucleotide polymorphisms and differ among ethnic populations (100–102). CYP2E1 is believed to play an important role in alcohol-induced liver injury associated with oxidative stress, mitochondrial damage and glutathione depletion (22, 103), and alcohol-induced liver disease (104, 105).
A BAC clone containing the human CYP2E1 gene was used to make a transgenic mouse line that was bred to the Cyp2e1-null mice (106) to generate CYP2E1-humanized mice (107). Human CYP2E1 protein and mRNA was largely expressed in the livers of the humanized mice, and the mRNA was found in kidney, lung, and small intestine, indicating a similar tissue distribution pattern as that observed in human. The human CYP2E1 enzymatic activity in mouse livers was demonstrated by chlozoxazone and p-nitrophenol hydroxylations, which were blocked by a CYP2E1 monoclonal antibody. There is no significant difference in chlozoxa-zone 6-hydroxylase activity between human CYP2E1 and mouse Cyp2e1 (11). By contrast, o-hydroxylation of p-nitrophenol revealed a difference between CYP2E1-humanized and wild-type control mice (107). Additionally, the mRNA and protein were inducible by treating the CYP2E1-humanized mice with acetone.
Therapeutic doses of the over-the-counter analgesic APAP are generally safe for patients; however, an overdose can result in severe and sometimes fatal hepatotoxicity. CYP2E1 metabolizes APAP to a highly reactive metabolite, N-acetyl-p-benzoquinone imine (NAPQI), that can covalently bind to cellular nucleophiles such as DNA, RNA, and proteins. NAPQI is usually inactivated by GST; depletion of hepatic reduced glutathione (GSH) the cosubstrate for GST, increases APAP toxicity. A critical role of CYP2E1 in APAP toxicity was demonstrated by using Cyp2e1-null mice, which were found to be resistant to APAP-induced hepatotoxicity (106). Studies using the CYP2E1-humanized mouse model revealed a potential functional difference between human and mouse CYP2E1 in APAP toxicity (107). After administration of 200 to 400 mg/kg APAP, most of the wild-type mice showed mild-to-moderate degrees of centrilobular hepatocyte necrosis, whereas none of the Cyp2e1-null or CYP2E1-humanized mice exhibited hepatic necrosis lesions (Table 2). These results were in agreement with measurements of serum alanine aminotransferase (ALT) activity (107), a surrogate biomarker for hepatotoxicity. These results demonstrated that the CYP2E1-humanized mice allow the direct assessment of human CYP2E1 function in a whole animal model, excluding any potential species difference caused by murine Cyp2e1, and thus they may be of value in pharmacological and toxicological studies aimed to predict human risk assessment.
TABLE 2.
Degrees of centrilobular hepatic necrosis in mice treated with saline or APAP. The livers were examined histologically and classed according to the degree of centrilobular hepatocyte necrosis, marked as none, mild, moderate, or severe. Mice found dead 24 h after the APAP treatment were also noted. The numbers indicate how many mice were classed with which degree of hepatocyte necrosis per total number of mice examined in each group (total numbers of mice in each group, n = 5–10)
| Genotype | Saline | APAP (200 mg/kg) | APAP (400 mg/kg) |
|---|---|---|---|
| Wild-type | None 5/5 | None 3/5 | Moderate 5/7 |
| Mild 1/5 | Found dead 2/7 | ||
| Moderate 1/5 | |||
| Cyp2e1-null | None 5/5 | None 6/6 | None 5/5 |
| CYP2E1-humanized | None 8/8 | None 9/9 | Mild 3/10 |
| Moderate 2/10 | |||
| Severe 4/10 | |||
| Found dead 1/10 |
AHR-Humanized Mice
One of the most well-studied genetic differences in response to xenobiotics is the AH locus in mice, where there exists approximately a tenfold difference between strains of mice in response to certain AHR ligands owing to a polymorphism that is a result of amino acid differences in the C-terminal and ligand domain of the receptor (48). Humans appear to have a receptor type that is more similar to the resistant mouse phenotype, yet there are distinct differences (49). An AHR-humanized mouse was generated by knocking the human AHR cDNA into the mouse Ahr gene promoter to develop a more predictive model to study dioxin toxicity and susceptibility to birth defects and carcinogens as a result of exposure to AHR ligands (108). Although the AHR-humanized mice exhibited similar target gene induction potential to the classic resistant strain of mice in response to the “low-affinity” ligand 3-methylcholanthrene, as expected from in vitro transfection studies, the humanized mice were more resistant to target gene induction by TCDD. Neonatal exposure to TCDD resulted in cleft palate in the wild-type mice of both the sensitive and resistant strains; the AHR-humanized mouse fetuses did not display this teratogenic end-point, although they did develop hydronephrosis (108). The AHR-humanized mouse could be a novel tool for use in human risk assessment for dioxin toxicity and carcinogenicity.
PXR-Humanized Mice
PXR regulates the expression of the CYP3A and several phase 2 enzymes and thus has a major impact on drug metabolism. There are major species differences in ligand specificities of PXR (12, 109). PCN activates the mouse and rat receptor but does not activate human PXR. In contrast, the human receptor is activated by rifampicin and clotrimazole, whereas the mouse and rat Pxr are not activated by these drugs. A PXR-humanized mouse was produced with a transgene in which the human PXR cDNA was fused to the albumin promoter (109). The transgene was bred onto the murine Pxr-null background. Target gene induction with various ligands revealed induction of the endogenous mouse Cyp3a11 gene by rifampicin and clotrimazole; minimal induction was observed with the mouse Pxr ligand PCN in humanized mice. Another transgenic mouse line was also produced in which the human PXR cDNA was fused with the VP16 coactivator to yield a constitutively activated receptor. As expected, mice expressing the fusion protein are resistant to tribromoethanol and zoxazolamine (109). These mice represent an in vivo model to test for potential PXR ligands and possible endogenous functions of this receptor, such as in controlling bilirubin metabolism (110) and protecting against bile acid toxicity (111).
CAR-Humanized Mice
Similar to PXR, there are species differences in the ligand specificity of CAR. Therefore a CAR-humanized mouse was produced with a transgene composed of the albumin promoter fused to the human CAR cDNA (58). Treating wild-type mice with the drug meclizine activated the Cyp2b10 target gene; there was no induction in Car-null mice, in agreement with the in vitro findings that this drug is a murine Car agonist (64). However, meclizine did not induce murine Cyp2b10 but suppressed its induction by phenobarbital in humanized mouse hepatocytes. Indeed, meclizine, an antimetics that is used to treat the nausea associated with acute APAP toxicity, was able to protect CAR-humanized mice against APAP-induced hepatotoxicity. The CAR-humanized mice could be used to evaluate drugs under development that target CAR for the control of bilirubin metabolism and to treat APAP-induced hepatotoxicity in cases of APAP overdose.
PPARα-Humanized Mice
Peroxisome proliferation is a rodent-specific response when PPARα is activated by a diverse group of chemicals termed peroxisome proliferators; a critical role for PPARα in this process was demonstrated by using the Pparα-null mouse model (112, 113). Peroxisome proliferators include organic solvents (trichloroacetic acid, trichloroethylene), herbicides (haloxyfab, lactyofen), and hypolipidemic fibrate drugs (clofibrate, fenofibrate, gemfibrozile). Coincident with an increase in numbers of peroxisome is an elevation in the levels of a number of enzymes involved in fatty acid β-oxidation, including peroxisomal acyl-CoA oxidase, thiolase and bifunctional (hydratase + 3-hydroxyacyl-CoA dehydrogenase), numerous mitochondrial fatty acid oxidizing enzymes, microsomal CYP4A (ω-oxidation), fatty acid binding protein, and fatty acid transporters (114). Induction of acyl-CoA oxidase leads to generation of hydrogen peroxide (H2O2) as a byproduct, causing oxidative stress. As a result, DNA can be damaged or mutated, potentially resulting in cell transformation and hepatocarcinogenesis (115, 116). Chronic exposure to peroxisome proliferators leads to hepatocellular carcinomas (6, 8, 117). However, there is no evidence of peroxisome proliferation in livers from humans and monkeys given fibrate drugs (118–122). This is also supported by the studies using human hepatocyte cultures. In contrast to rats and mice, human hepatocytes are unresponsive to peroxisome proliferators (123). Humans do have a functional PPARα (124–126), but there exist striking species differences in levels of expression PPARα (17) and ligands affinities between human and mouse PPARα forms (15, 16).
To better understand the underlying molecular mechanism of this species difference, a PPARα-humanized mouse line was generated (127). Under the control of the tetracycline responsive regulatory system, the human PPARα was expressed in the livers of murine Pparα-null mice (112). The PPARα-humanized mice functionally responded to the PPARα ligands Wy-14643 and fenofibrate in a manner similar to wild-type mice, as demonstrated by the induction of genes encoding fatty acid synthesis, oxidation, and transport proteins and the decrease of serum triglycerides (127). However, the PPARα-humanized mice did not exhibit hepatocellular proliferation that was observed in wild-type mice. This was associated with the lack of high-level increases of catalase-enriched peroxisomes, incorporation of 5-bromo-2′-deoxyuridine into hepatocyte nuclei, and induction of cell cycle control genes (Figure 3). Because the expression levels of human PPARα in the humanized mouse hepatocytes were comparable with murine PPARα in wild-type mice (127), the structural and functional difference between human and mouse PPARα should be the determinant for the differential susceptibility to hepatocarcinomas between species (15). To eliminate the potential species difference, the PPARα-humanized mice would serve as an invaluable model to assess human risk to these peroxisome proliferators, including the lipid and cholesterol-lowering fibrate drugs.
Figure 3.
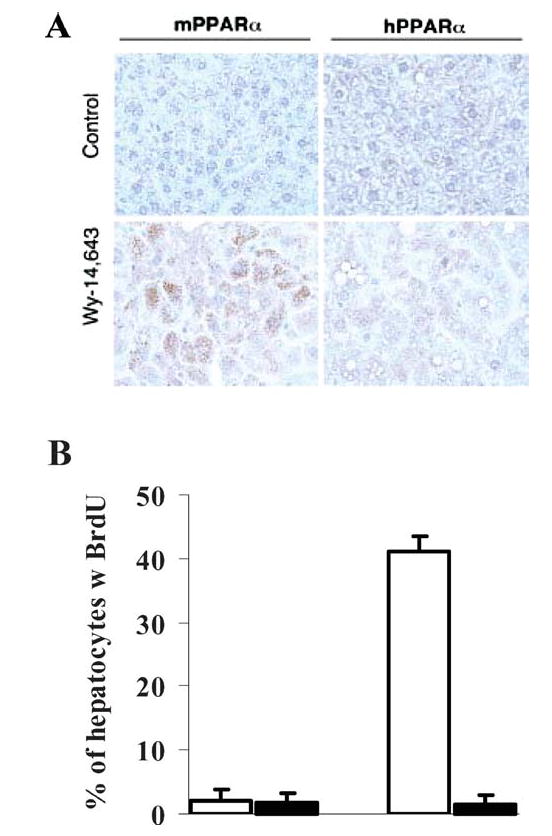
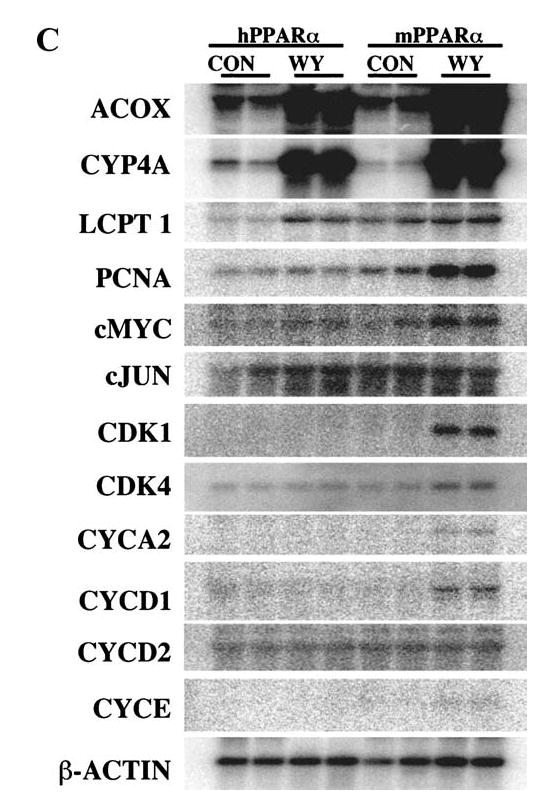
Differential peroxisome proliferation, replicative DNA synthesis, and regulation of genes involved in cell cycle control in livers of PPARα-humanized and wild-type mice treated with Wy-14643. (A) Immunohistochemical staining with anti-catalase antibody indicated that peroxisomes (brown granular structures) were obviously increased in the wild-type mice treated with Wy-14643. Magnification: 400×. (B) Labeling index of Wy-14643-induced incorporation of 5-bromo-2′-deoxyuridine (BrdU) into hepatocyte nuclei as examined by immunohistochemical analysis with anti-BrdU antibody. (C) Regulation of genes involved in cell cycle control as revealed by Northern hybridization. ACOX, peroxisome acyl-CoA oxidase; LCPT, liver carnitine palmitoyltransferase; PCNA, proliferating cellular nuclear antigen; CDK, cyclin-dependent kinase.
Other Mice Bearing Human Cytochrome P450 or Xenobiotic Receptor Transgene
Although beyond the scope of this review, other transgenic mice have been reported that express human CYP1A1 (128), CYP1A2 (128, 129), CYP1B1 (130), CYP2E1 (131), CYP3A7 (132), CYP4B1 (133), CYP7A1 (134–136), CYP19 (137), and CYP27 (138). Mice bearing the regulatory sequence of human CYP1A2 (139) and CYP3A4 (140, 141) have also been developed to investigate the regulation of human CYPs in mice. These unique models have been widely used and will continue to be used for studying their functional roles in physiology, pharmacology, pathology, and toxicology that contribute to better understanding, prediction, and management of the situations in humans.
CONCLUSIONS AND PERSPECTIVES
Recent development of CYP and nuclear receptor humanized mouse models overcome the species differences caused by the intrinsic genes. These humanized mouse models have proven a valuable addition to the existing in vitro and in vivo assays for studying the function of CYPs and xenobiotic receptors in a whole-animal system, directly assessing the consequent functional significance under controlled conditions, and delineating the regulatory networks and mechanistic basis for the responses to xenobiotics. These models should have broad application in the understanding of hormone homeostasis and human disease and the evaluation and prediction of the metabolism, pharmacokinetics, pharmacodynamics, and toxicity of drug candidates in preclinical studies. To achieve quantitative assessments, a precise determination of the functional proteins expressed in the humanized mice is essential. Because of the physiological differences between mice and humans, particularly in body weight and blood flow rate, an allometric scaling approach can be very helpful. Ultimately, a combined CYP and nuclear receptor humanized mouse will allow us to investigate both the function and regulation of CYP drug-metabolizing enzyme in an integrated biological system.
Footnotes
The U.S. Government has the right to retain a nonexclusive, royalty-free license in and to any copyright covering this paper.
References
- 1.Gonzalez FJ, Gelboin HV. Role of human cytochromes P450 in the metabolic activation of chemical carcinogens and toxins. Drug Metab Rev. 1994;26:165–83. doi: 10.3109/03602539409029789. [DOI] [PubMed] [Google Scholar]
- 2.Guengerich FP. Cytochromes P450, drugs, and diseases. Mol Interv. 2003;3:194–204. doi: 10.1124/mi.3.4.194. [DOI] [PubMed] [Google Scholar]
- 3.Nebert DW, Russell DW. Clinical importance of the cytochromes P450. Lancet. 2002;360:1155–62. doi: 10.1016/S0140-6736(02)11203-7. [DOI] [PubMed] [Google Scholar]
- 4.Willson TM, Kliewer SA. PXR, CAR and drug metabolism. Nat Rev Drug Discov. 2002;1:259–66. doi: 10.1038/nrd753. [DOI] [PubMed] [Google Scholar]
- 5.Gonzalez FJ, Liu SY, Yano M. Regulation of cytochrome P450 genes: molecular mechanisms. Pharmacogenetics. 1993;3:51–57. doi: 10.1097/00008571-199302000-00006. [DOI] [PubMed] [Google Scholar]
- 6.Gonzalez FJ. The peroxisome proliferator-activated receptor alpha (PPARalpha): role in hepatocarcinogenesis. Mol Cell Endocrinol. 2002;193:71–79. doi: 10.1016/s0303-7207(02)00098-9. [DOI] [PubMed] [Google Scholar]
- 7.Akiyama TE, Gonzalez FJ. Regulation of P450 genes by liver-enriched transcription factors and nuclear receptors. Biochim Biophys Acta. 2003;1619:223–34. doi: 10.1016/s0304-4165(02)00480-4. [DOI] [PubMed] [Google Scholar]
- 8.Gonzalez FJ, Peters JM, Cattley RC. Mechanism of action of the nongenotoxic peroxisome proliferators: role of the peroxisome proliferator-activator receptor alpha. J Natl Cancer Inst. 1998;90:1702–9. doi: 10.1093/jnci/90.22.1702. [DOI] [PubMed] [Google Scholar]
- 9.Waxman DJ. P450 gene induction by structurally diverse xenochemicals: central role of nuclear receptors CAR, PXR, and PPAR. Arch Biochem Biophys. 1999;369:11–23. doi: 10.1006/abbi.1999.1351. [DOI] [PubMed] [Google Scholar]
- 10.Denison MS, Nagy SR. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol. 2003;43:309–34. doi: 10.1146/annurev.pharmtox.43.100901.135828. [DOI] [PubMed] [Google Scholar]
- 11.Bogaards JJ, Bertrand M, Jackson P, Oudshoorn MJ, Weaver RJ, et al. Determining the best animal model for human cytochrome P450 activities: a comparison of mouse, rat, rabbit, dog, micropig, monkey and man. Xenobiotica. 2000;30:1131–52. doi: 10.1080/00498250010021684. [DOI] [PubMed] [Google Scholar]
- 12.Jones SA, Moore LB, Shenk JL, Wisely GB, Hamilton GA, et al. The pregnane X receptor: a promiscuous xenobiotic receptor that has diverged during evolution. Mol Endocrinol. 2000;14:27–39. doi: 10.1210/mend.14.1.0409. [DOI] [PubMed] [Google Scholar]
- 13.Choudhury AI, Chahal S, Bell AR, Tomlinson SR, Roberts RA, et al. Species differences in peroxisome proliferation; mechanisms and relevance. Mutat Res. 2000;448:201–12. doi: 10.1016/s0027-5107(99)00237-7. [DOI] [PubMed] [Google Scholar]
- 14.Masubuchi Y, Iwasa T, Hosokawa S, Suzuki T, Horie T, et al. Selective deficiency of debrisoquine 4-hydroxylase activity in mouse liver microsomes. J Pharmacol Exp Ther. 1997;282:1435–41. [PubMed] [Google Scholar]
- 15.Sher T, Yi HF, McBride OW, Gonzalez FJ. cDNA cloning, chromosomal mapping, and functional characterization of the human peroxisome proliferator activated receptor. Biochemistry. 1993;32:5598–604. doi: 10.1021/bi00072a015. [DOI] [PubMed] [Google Scholar]
- 16.Ljung B, Bamberg K, Dahllof B, Kjellstedt A, Oakes ND, et al. AZ 242, a novel PPARalpha/gamma agonist with beneficial effects on insulin resistance and carbohydrate and lipid metabolism in ob/ob mice and obese Zucker rats. J Lipid Res. 2002;43:1855–63. doi: 10.1194/jlr.m200127-jlr200. [DOI] [PubMed] [Google Scholar]
- 17.Palmer CN, Hsu MH, Griffin KJ, Raucy JL, Johnson EF. Peroxisome proliferator activated receptor-alpha expression in human liver. Mol Pharmacol. 1998;53:14–22. [PubMed] [Google Scholar]
- 18.Gonzalez FJ. Role of gene knockout and transgenic mice in the study of xenobiotic metabolism. Drug Metab Rev. 2003;35:319–35. doi: 10.1081/dmr-120026496. [DOI] [PubMed] [Google Scholar]
- 19.Gonzalez FJ, Kimura S. Study of P450 function using gene knockout and transgenic mice. Arch Biochem Biophys. 2003;409:153–58. doi: 10.1016/s0003-9861(02)00364-8. [DOI] [PubMed] [Google Scholar]
- 20.Henderson CJ, Wolf CR. Trans genic analysis of human drug-metabolizing enzymes: preclinical drug development and toxicology. Mol Interv. 2003;3:331–43. doi: 10.1124/mi.3.6.331. [DOI] [PubMed] [Google Scholar]
- 21.Gonzalez FJ. Cytochrome P450 humanised mice. Hum Genomics. 2004;1:300–6. doi: 10.1186/1479-7364-1-4-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gonzalez FJ. Role of cytochromes P450 in chemical toxicity and oxidative stress: studies with CYP2E1. Mutat Res. 2005;569:101–10. doi: 10.1016/j.mrfmmm.2004.04.021. [DOI] [PubMed] [Google Scholar]
- 23.Ereshefsky L, Riesenman C, Lam YW. Antidepressant drug interactions and the cytochrome P450 system. The role of cytochrome P450 2D6. Clin Pharmacokinet. 1995;29(Suppl 1):10–18. doi: 10.2165/00003088-199500291-00004. discussion 18–19. [DOI] [PubMed] [Google Scholar]
- 24.Quattrochi LC, Tukey RH. Nuclear uptake of the Ah (dioxin) receptor in response to omeprazole: transcriptional activation of the human CYP1A1 gene. Mol Pharmacol. 1993;43:504–8. [PubMed] [Google Scholar]
- 25.Yueh MF, Kawahara M, Raucy J. Cell-based high-throughput bioassays to assess induction and inhibition of CYP1A enzymes. Toxicol In Vitro. 2005;19:275–87. doi: 10.1016/j.tiv.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 26.Edwards DJ, Bellevue FH, 3rd, Woster PM. Identification of 6′,7′-dihydroxybergamottin, a cytochrome P450 inhibitor, in grapefruit juice. Drug Metab Dispos. 1996;24:1287–90. [PubMed] [Google Scholar]
- 27.Fukuda K, Ohta T, Oshima Y, Ohashi N, Yoshikawa M, Yamazoe Y. Specific CYP3A4 inhibitors in grapefruit juice: furocoumarin dimers as components of drug interaction. Pharmacogenetics. 1997;7:391–96. doi: 10.1097/00008571-199710000-00008. [DOI] [PubMed] [Google Scholar]
- 28.Moore LB, Goodwin B, Jones SA, Wisely GB, Serabjit-Singh CJ, et al. St. John’s wort induces hepatic drug metabolism through activation of the pregnane X receptor. Proc Natl Acad Sci USA. 2000;97:7500–2. doi: 10.1073/pnas.130155097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ingelman-Sundberg M. Pharmacogenetics of cytochrome P450 and its applications in drug therapy: the past, present and future. Trends Pharmacol Sci. 2004;25:193–200. doi: 10.1016/j.tips.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 30.Zanger UM, Raimundo S, Eichelbaum M. Cytochrome P450 2D6: overview and update on pharmacology, genetics, biochemistry. Naunyn Schmiedebergs Arch Pharmacol. 2004;369:23–37. doi: 10.1007/s00210-003-0832-2. [DOI] [PubMed] [Google Scholar]
- 31.Gonzalez FJ, Skoda RC, Kimura S, Umeno M, Zanger UM, et al. Characterization of the common genetic defect in humans deficient in debrisoquine metabolism. Nature. 1988;331:442–46. doi: 10.1038/331442a0. [DOI] [PubMed] [Google Scholar]
- 32.Johansson I, Lundqvist E, Bertilsson L, Dahl ML, Sjoqvist F, Ingelman-Sundberg M. Inherited amplification of an active gene in the cytochrome P450 CYP2D locus as a cause of ultrarapid metabolism of debrisoquine. Proc Natl Acad Sci USA. 1993;90:11825–29. doi: 10.1073/pnas.90.24.11825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rettie AE, Jones JP. Clinical and toxicological relevance of CYP2C9: drug-drug interactions and pharmacogenetics. Annu Rev Pharmacol Toxicol. 2005;45:477–94. doi: 10.1146/annurev.pharmtox.45.120403.095821. [DOI] [PubMed] [Google Scholar]
- 34.Bejjani BA, Lewis RA, Tomey KF, Anderson KL, Dueker DK, et al. Mutations in CYP1B1, the gene for cytochrome P4501B1, are the predominant cause of primary congenital glaucoma in Saudi Arabia. Am J Hum Genet. 1998;62:325–33. doi: 10.1086/301725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Libby RT, Smith RS, Savinova OV, Zabaleta A, Martin JE, et al. Modification of ocular defects in mouse developmental glaucoma models by tyrosinase. Science. 2003;299:1578–81. doi: 10.1126/science.1080095. [DOI] [PubMed] [Google Scholar]
- 36.Goodwin B, Redinbo MR, Kliewer SA. Regulation of cyp3a gene transcription by the pregnane x receptor. Annu Rev Pharmacol Toxicol. 2002;42:1–23. doi: 10.1146/annurev.pharmtox.42.111901.111051. [DOI] [PubMed] [Google Scholar]
- 37.Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ. Nuclear receptors and lipid physiology: opening the X-files. Science. 2001;294:1866–70. doi: 10.1126/science.294.5548.1866. [DOI] [PubMed] [Google Scholar]
- 38.Sueyoshi T, Negishi M. Phenobarbital response elements of cytochrome P450 genes and nuclear receptors. Annu Rev Pharmacol Toxicol. 2001;41:123–43. doi: 10.1146/annurev.pharmtox.41.1.123. [DOI] [PubMed] [Google Scholar]
- 39.Tzameli I, Moore DD. Role reversal: new insights from new ligands for the xenobiotic receptor CAR. Trends Endocrinol Metab. 2001;12:7–10. doi: 10.1016/s1043-2760(00)00332-5. [DOI] [PubMed] [Google Scholar]
- 40.Burbach KM, Poland A, Bradfield CA. Cloning of the Ah-receptor cDNA reveals a distinctive ligand-activated transcription factor. Proc Natl Acad Sci USA. 1992;89:8185–89. doi: 10.1073/pnas.89.17.8185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Swanson HI, Bradfield CA. The AH-receptor: genetics, structure and function. Pharmacogenetics. 1993;3:213–30. doi: 10.1097/00008571-199310000-00001. [DOI] [PubMed] [Google Scholar]
- 42.Baes M, Gulick T, Choi HS, Martinoli MG, Simha D, Moore DD. A new orphan member of the nuclear hormone receptor superfamily that interacts with a subset of retinoic acid response elements. Mol Cell Biol. 1994;14:1544–52. doi: 10.1128/mcb.14.3.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Forman BM, Tzameli I, Choi HS, Chen J, Simha D, et al. Androstane metabolites bind to and deactivate the nuclear receptor CAR-beta. Nature. 1998;395:612–15. doi: 10.1038/26996. [DOI] [PubMed] [Google Scholar]
- 44.Lehmann JM, McKee DD, Watson MA, Willson TM, Moore JT, Kliewer SA. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J Clin Invest. 1998;102:1016–23. doi: 10.1172/JCI3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kliewer SA, Moore JT, Wade L, Staudinger JL, Watson MA, et al. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell. 1998;92:73–82. doi: 10.1016/s0092-8674(00)80900-9. [DOI] [PubMed] [Google Scholar]
- 46.Blumberg B, Sabbagh W, Jr, Juguilon H, Bolado J, Jr, van Meter CM, et al. SXR, a novel steroid and xenobiotic-sensing nuclear receptor. Genes Dev. 1998;12:3195–205. doi: 10.1101/gad.12.20.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nolte RT, Wisely GB, Westin S, Cobb JE, Lambert MH, et al. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature. 1998;395:137–43. doi: 10.1038/25931. [DOI] [PubMed] [Google Scholar]
- 48.Nebert DW, Dalton TP, Okey AB, Gonzalez FJ. Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J Biol Chem. 2004;279:23847–50. doi: 10.1074/jbc.R400004200. [DOI] [PubMed] [Google Scholar]
- 49.Harper PA, Wong JY, Lam MS, Okey AB. Polymorphisms in the human AH receptor. Chem Biol Interact. 2002;141:161–87. doi: 10.1016/s0009-2797(02)00071-6. [DOI] [PubMed] [Google Scholar]
- 50.Handschin C, Meyer UA. Induction of drug metabolism: the role of nuclear receptors. Pharmacol Rev. 2003;55:649–73. doi: 10.1124/pr.55.4.2. [DOI] [PubMed] [Google Scholar]
- 51.Sonoda J, Xie W, Rosenfeld JM, Barwick JL, Guzelian PS, Evans RM. Regulation of a xenobiotic sulfonation cascade by nuclear pregnane X receptor (PXR) Proc Natl Acad Sci USA. 2002;99:13801–6. doi: 10.1073/pnas.212494599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xie W, Yeuh MF, Radominska-Pandya A, Saini SP, Negishi Y, et al. Control of steroid, heme, and carcinogen metabolism by nuclear pregnane X receptor and constitutive androstane receptor. Proc Natl Acad Sci USA. 2003;100:4150–55. doi: 10.1073/pnas.0438010100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xie W, Barwick JL, Simon CM, Pierce AM, Safe S, et al. Reciprocal activation of xenobiotic response genes by nuclear receptors SXR/PXR and CAR. Genes Dev. 2000;14:3014–23. doi: 10.1101/gad.846800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pascussi JM, Gerbal-Chaloin S, Fabre JM, Maurel P, Vilarem MJ. Dexamethasone enhances constitutive androstane receptor expression in human hepatocytes: consequences on cytochrome P450 gene regulation. Mol Pharmacol. 2000;58:1441–50. doi: 10.1124/mol.58.6.1441. [DOI] [PubMed] [Google Scholar]
- 55.Wei P, Zhang J, Egan-Hafley M, Liang S, Moore DD. The nuclear receptor CAR mediates specific xenobiotic induction of drug metabolism. Nature. 2000;407:920–23. doi: 10.1038/35038112. [DOI] [PubMed] [Google Scholar]
- 56.Honkakoski P, Zelko I, Sueyoshi T, Negishi M. The nuclear orphan receptor CAR-retinoid X receptor heterodimer activates the phenobarbital-responsive enhancer module of the CYP2B gene. Mol Cell Biol. 1998;18:5652–58. doi: 10.1128/mcb.18.10.5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kodama S, Koike C, Negishi M, Yamamoto Y. Nuclear receptors CAR and PXR cross talk with FOXO1 to regulate genes that encode drug-metabolizing and gluconeogenic enzymes. Mol Cell Biol. 2004;24:7931–40. doi: 10.1128/MCB.24.18.7931-7940.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang J, Huang W, Chua SS, Wei P, Moore DD. Modulation of acetaminophen-induced hepatotoxicity by the xenobiotic receptor CAR. Science. 2002;298:422–24. doi: 10.1126/science.1073502. [DOI] [PubMed] [Google Scholar]
- 59.Huang W, Zhang J, Chua SS, Qatanani M, Han Y, et al. Induction of bilirubin clearance by the constitutive androstane receptor (CAR) Proc Natl Acad Sci USA. 2003;100:4156–61. doi: 10.1073/pnas.0630614100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tzameli I, Pissios P, Schuetz EG, Moore DD. The xenobiotic compound 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene is an agonist ligand for the nuclear receptor CAR. Mol Cell Biol. 2000;20:2951–58. doi: 10.1128/mcb.20.9.2951-2958.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang H, Negishi M. Transcriptional regulation of cytochrome p450 2B genes by nuclear receptors. Curr Drug Metab. 2003;4:515–25. doi: 10.2174/1389200033489262. [DOI] [PubMed] [Google Scholar]
- 62.Moore LB, Parks DJ, Jones SA, Bledsoe RK, Consler TG, et al. Orphan nuclear receptors constitutive androstane receptor and pregnane X receptor share xenobiotic and steroid ligands. J Biol Chem. 2000;275:15122–27. doi: 10.1074/jbc.M001215200. [DOI] [PubMed] [Google Scholar]
- 63.Maglich JM, Parks DJ, Moore LB, Collins JL, Goodwin B, et al. Identification of a novel human constitutive androstane receptor (CAR) agonist and its use in the identification of CAR target genes. J Biol Chem. 2003;278:17277–83. doi: 10.1074/jbc.M300138200. [DOI] [PubMed] [Google Scholar]
- 64.Huang W, Zhang J, Wei P, Schrader WT, Moore DD. Meclizine is an agonist ligand for mouse constitutive androstane receptor (CAR) and an inverse agonist for human CAR. Mol Endocrinol. 2004;18:2402–8. doi: 10.1210/me.2004-0046. [DOI] [PubMed] [Google Scholar]
- 65.Huang W, Zhang J, Moore DD. A traditional herbal medicine enhances bilirubin clearance by activating the nuclear receptor CAR. J Clin Invest. 2004;113:137–43. doi: 10.1172/JCI200418385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Alvares K, Carrillo A, Yuan PM, Kawano H, Morimoto RI, Reddy JK. Identification of cytosolic peroxisome proliferator binding protein as a member of the heat shock protein HSP70 family. Proc Natl Acad Sci USA. 1990;87:5293–97. doi: 10.1073/pnas.87.14.5293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Klaunig JE, Babich MA, Baetcke KP, Cook JC, Corton JC, et al. PPAR-alpha agonist-induced rodent tumors: modes of action and human relevance. Crit Rev Toxicol. 2003;33:655–780. doi: 10.1080/713608372. [DOI] [PubMed] [Google Scholar]
- 68.Caldwell J. The current status of attempts to predict species differences in drug metabolism. Drug Metab Rev. 1981;12:221–37. doi: 10.3109/03602538108994030. [DOI] [PubMed] [Google Scholar]
- 69.Guengerich FP. Comparisons of catalytic selectivity of cytochrome P450 subfamily enzymes from different species. Chem Biol Interact. 1997;106:161–82. doi: 10.1016/s0009-2797(97)00068-9. [DOI] [PubMed] [Google Scholar]
- 70.Yu AM, Idle JR, Gonzalez FJ. Polymorphic cytochrome P450 2D6: humanized mouse model and endogenous substrates. Drug Metab Rev. 2004;36:243–77. doi: 10.1081/dmr-120034000. [DOI] [PubMed] [Google Scholar]
- 71.Corchero J, Granvil CP, Akiyama TE, Hayhurst GP, Pimprale S, et al. The CYP2D6 humanized mouse: effect of the human CYP2D6 transgene and HNF4alpha on the disposition of debrisoquine in the mouse. Mol Pharmacol. 2001;60:1260–67. doi: 10.1124/mol.60.6.1260. [DOI] [PubMed] [Google Scholar]
- 72.Hayhurst GP, Lee YH, Lambert G, Ward JM, Gonzalez FJ. Hepatocyte nuclear factor 4alpha (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol Cell Biol. 2001;21:1393–403. doi: 10.1128/MCB.21.4.1393-1403.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fonne-Pfister R, Bargetzi MJ, Meyer UA. MPTP, the neurotoxin inducing Parkinson’s disease, is a potent competitive inhibitor of human and rat cytochrome P450 isozymes (P450bufI, P450db1) catalyzing debrisoquine 4-hydroxylation. Biochem Biophys Res Commun. 1987;148:1144–50. doi: 10.1016/s0006-291x(87)80252-8. [DOI] [PubMed] [Google Scholar]
- 74.Siegle I, Fritz P, Eckhardt K, Zanger UM, Eichelbaum M. Cellular localization and regional distribution of CYP2D6 mRNA and protein expression in human brain. Pharmacogenetics. 2001;11:237–45. doi: 10.1097/00008571-200104000-00007. [DOI] [PubMed] [Google Scholar]
- 75.Miksys S, Rao Y, Hoffmann E, Mash DC, Tyndale RF. Regional and cellular expression of CYP2D6 in human brain: higher levels in alcoholics. J Neurochem. 2002;82:1376–87. doi: 10.1046/j.1471-4159.2002.01069.x. [DOI] [PubMed] [Google Scholar]
- 76.Gilham DE, Cairns W, Paine MJ, Modi S, Poulsom R, et al. Metabolism of MPTP by cytochrome P4502D6 and the demonstration of 2D6 mRNA in human foetal and adult brain by in situ hybridization. Xenobiotica. 1997;27:111–25. doi: 10.1080/004982597240802. [DOI] [PubMed] [Google Scholar]
- 77.McFayden MC, Melvin WT, Murray GI. Regional distribution of individual forms of cytochrome P450 mRNA in normal adult human brain. Biochem Pharmacol. 1998;55:825–30. doi: 10.1016/s0006-2952(97)00516-9. [DOI] [PubMed] [Google Scholar]
- 78.Voirol P, Jonzier-Perey M, Porchet F, Reymond MJ, Janzer RC, et al. Cytochrome P-450 activities in human and rat brain microsomes. Brain Res. 2000;855:235–43. doi: 10.1016/s0006-8993(99)02354-9. [DOI] [PubMed] [Google Scholar]
- 79.Llerena A, Edman G, Cobaleda J, Benitez J, Schalling D, Bertilsson L. Relationship between personality and debrisoquine hydroxylation capacity. Suggestion of an endogenous neuroactive substrate or product of the cytochrome P4502D6. Acta Psychiatr Scand. 1993;87:23–28. doi: 10.1111/j.1600-0447.1993.tb03325.x. [DOI] [PubMed] [Google Scholar]
- 80.Yu AM, Idle JR, Herraiz T, Kupfer A, Gonzalez FJ. Screening for endogenous substrates reveals that CYP2D6 is a 5-methoxyindolethylamine O-demethylase. Pharmacogenetics. 2003;13:307–19. doi: 10.1097/01.fpc.0000054094.48725.b7. [DOI] [PubMed] [Google Scholar]
- 81.Yu AM, Idle JR, Byrd LG, Krausz KW, Kupfer A, Gonzalez FJ. Regeneration of serotonin from 5-methoxytryptamine by polymorphic human CYP2D6. Pharmacogenetics. 2003;13:173–81. doi: 10.1097/01.fpc.0000054066.98065.7b. [DOI] [PubMed] [Google Scholar]
- 82.Guengerich FP. Cytochrome P-450 3A4: regulation and role in drug metabolism. Annu Rev Pharmacol Toxicol. 1999;39:1–17. doi: 10.1146/annurev.pharmtox.39.1.1. [DOI] [PubMed] [Google Scholar]
- 83.Kolars JC, Lown KS, Schmiedlin-Ren P, Ghosh M, Fang C, et al. CYP3A gene expression in human gut epithelium. Pharmacogenetics. 1994;4:247–59. doi: 10.1097/00008571-199410000-00003. [DOI] [PubMed] [Google Scholar]
- 84.McKinnon RA, Burgess WM, Hall PM, Roberts-Thomson SJ, Gonzalez FJ, Mc-Manus ME. Characterisation of CYP3A gene subfamily expression in human gastrointestinal tissues. Gut. 1995;36:259–67. doi: 10.1136/gut.36.2.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Glaeser H, Drescher S, van der Kuip H, Behrens C, Geick A, et al. Shed human enterocytes as a tool for the study of expression and function of intestinal drug-metabolizing enzymes and transporters. Clin Pharmacol Ther. 2002;71:131–40. doi: 10.1067/mcp.2002.121370. [DOI] [PubMed] [Google Scholar]
- 86.Obach RS, Zhang QY, Dunbar D, Kaminsky LS. Metabolic characterization of the major human small intestinal cytochrome p450s. Drug Metab Dispos. 2001;29:347–52. [PubMed] [Google Scholar]
- 87.Granvil CP, Yu AM, Elizondo G, Akiyama TE, Cheung C, et al. Expression of the human CYP3A4 gene in the small intestine of transgenic mice: in vitro metabolism and pharmacokinetics of midazolam. Drug Metab Dispos. 2003;31:548–58. doi: 10.1124/dmd.31.5.548. [DOI] [PubMed] [Google Scholar]
- 88.Yu AM, Fukamachi K, Krausz KW, Cheung C, Gonzalez FJ. Potential role for human cytochrome P450 CYP3A4 in estradiol homeostasis. Endocrinology. 2005;146:2911–19. doi: 10.1210/en.2004-1248. [DOI] [PubMed] [Google Scholar]
- 89.Wolbold R, Klein K, Burk O, Nussler AK, Neuhaus P, et al. Sex is a major determinant of CYP3A4 expression in human liver. Hepatology. 2003;38:978–88. doi: 10.1053/jhep.2003.50393. [DOI] [PubMed] [Google Scholar]
- 90.Sher A, Rahman MA. Enterohepatic recycling of estrogen and its relevance with female fertility. Arch Pharm Res. 2000;23:513–17. doi: 10.1007/BF02976582. [DOI] [PubMed] [Google Scholar]
- 91.Lieber CS. Cytochrome P-4502E1: its physiological and pathological role. Physiol Rev. 1997;77:517–44. doi: 10.1152/physrev.1997.77.2.517. [DOI] [PubMed] [Google Scholar]
- 92.Rubin E, Lieber CS. Alcoholism, alcohol, and drugs. Science. 1971;172:1097–102. doi: 10.1126/science.172.3988.1097. [DOI] [PubMed] [Google Scholar]
- 93.Kessova I, Cederbaum AI. CYP2E1: biochemistry, toxicology, regulation and function in ethanol-induced liver injury. Curr Mol Med. 2003;3:509–18. doi: 10.2174/1566524033479609. [DOI] [PubMed] [Google Scholar]
- 94.Bolt HM, Roos PH, Thier R. The cytochrome P-450 isoenzyme CYP2E1 in the biological processing of industrial chemicals: consequences for occupational and environmental medicine. Int Arch Occup Environ Health. 2003;76:174–85. doi: 10.1007/s00420-002-0407-4. [DOI] [PubMed] [Google Scholar]
- 95.Guengerich FP, Kim DH, Iwasaki M. Role of human cytochrome P-450 IIE1 in the oxidation of many low molecular weight cancer suspects. Chem Res Toxicol. 1991;4:168–79. doi: 10.1021/tx00020a008. [DOI] [PubMed] [Google Scholar]
- 96.Amet Y, Berthou F, Goasduff T, Salaun JP, Le Breton L, Menez JF. Evidence that cytochrome P450 2E1 is involved in the (omega-1)-hydroxylation of lauric acid in rat liver microsomes. Biochem Biophys Res Commun. 1994;203:1168–74. doi: 10.1006/bbrc.1994.2305. [DOI] [PubMed] [Google Scholar]
- 97.Fukuda T, Imai Y, Komori M, Nakamura M, Kusunose E, et al. Replacement of Thr-303 of P450 2E1 with serine modifies the regioselectivity of its fatty acid hydroxylase activity. J Biochem (Tokyo) 1993;113:7–12. doi: 10.1093/oxfordjournals.jbchem.a124006. [DOI] [PubMed] [Google Scholar]
- 98.Laethem RM, Balazy M, Falck JR, Laethem CL, Koop DR. Formation of 19(S)-, 19(R)-, and 18(R)-hydroxyeicosatetraenoic acids by alcohol-inducible cytochrome P450 2E1. J Biol Chem. 1993;268:12912–18. [PubMed] [Google Scholar]
- 99.DeBarber AE, Bleyle LA, Roullet JB, Koop DR. Omega-hydroxylation of farnesol by mammalian cytochromes p450. Biochim Biophys Acta. 2004;1682:18–27. doi: 10.1016/j.bbalip.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 100.Hayashi S, Watanabe J, Kawajiri K. Genetic polymorphisms in the 5′-flanking region change transcriptional regulation of the human cytochrome P450IIE1 gene. J Biochem (Tokyo) 1991;110:559–65. doi: 10.1093/oxfordjournals.jbchem.a123619. [DOI] [PubMed] [Google Scholar]
- 101.Persson I, Johansson I, Bergling H, Dahl ML, Seidegard J, et al. Genetic polymorphism of cytochrome P4502E1 in a Swedish population. Relationship to incidence of lung cancer. FEBS Lett. 1993;319:207–11. doi: 10.1016/0014-5793(93)80547-8. [DOI] [PubMed] [Google Scholar]
- 102.Wan YJ, Poland RE, Lin KM. Genetic polymorphism of CYP2E1, ADH2, and ALDH2 in Mexican-Americans. Genet Test. 1998;2:79–83. doi: 10.1089/gte.1998.2.79. [DOI] [PubMed] [Google Scholar]
- 103.Caro AA, Cederbaum AI. Oxidative stress, toxicology, and pharmacology of CYP2E1. Annu Rev Pharmacol Toxicol. 2004;44:27–42. doi: 10.1146/annurev.pharmtox.44.101802.121704. [DOI] [PubMed] [Google Scholar]
- 104.Iwahashi K, Ameno S, Ameno K, Okada N, Kinoshita H, et al. Relationship between alcoholism and CYP2E1 C/D polymorphism. Neuropsychobiology. 1998;38:218–21. doi: 10.1159/000026544. [DOI] [PubMed] [Google Scholar]
- 105.Konishi T, Calvillo M, Leng AS, Feng J, Lee T, et al. The ADH3*2 and CYP2E1 c2 alleles increase the risk of alcoholism in Mexican American men. Exp Mol Pathol. 2003;74:183–89. doi: 10.1016/s0014-4800(03)00006-6. [DOI] [PubMed] [Google Scholar]
- 106.Lee SS, Buters JT, Pineau T, Fernandez-Salguero P, Gonzalez FJ. Role of CYP2E1 in the hepatotoxicity of acet-aminophen. J Biol Chem. 1996;271:12063–67. doi: 10.1074/jbc.271.20.12063. [DOI] [PubMed] [Google Scholar]
- 107.Cheung C, Yu AM, Ward JM, Krausz KW, Akiyama TE, et al. The cyp2e1-humanized transgenic mouse: role of cyp2e1 in acetaminophen hepatotoxicity. Drug Metab Dispos. 2005;33:449–57. doi: 10.1124/dmd.104.002402. [DOI] [PubMed] [Google Scholar]
- 108.Moriguchi T, Motohashi H, Hosoya T, Nakajima O, Takahashi S, et al. Distinct response to dioxin in an aryl-hydrocarbon receptor (AHR)-humanized mouse. Proc Natl Acad Sci USA. 2003;100:5652–57. doi: 10.1073/pnas.1037886100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Xie W, Barwick JL, Downes M, Blumberg B, Simon CM, et al. Humanized xenobiotic response in mice expressing nuclear receptor SXR. Nature. 2000;406:435–39. doi: 10.1038/35019116. [DOI] [PubMed] [Google Scholar]
- 110.Saini SP, Mu Y, Gong H, Toma D, Uppal H, et al. Dual role of orphan nuclear receptor pregnane X receptor in bilirubin detoxification in mice. Hepatology. 2005;41:497–505. doi: 10.1002/hep.20570. [DOI] [PubMed] [Google Scholar]
- 111.Sonoda J, Chong LW, Downes M, Barish GD, Coulter S, et al. Pregnane X receptor prevents hepatorenal toxicity from cholesterol metabolites. Proc Natl Acad Sci USA. 2005;102:2198–203. doi: 10.1073/pnas.0409481102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lee SS, Pineau T, Drago J, Lee EJ, Owens JW, et al. Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol Cell Biol. 1995;15:3012–22. doi: 10.1128/mcb.15.6.3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Peters JM, Cattley RC, Gonzalez FJ. Role of PPAR alpha in the mechanism of action of the nongenotoxic carcinogen and peroxisome proliferator Wy-14643. Carcinogenesis. 1997;18:2029–33. doi: 10.1093/carcin/18.11.2029. [DOI] [PubMed] [Google Scholar]
- 114.Aoyama T, Peters JM, Iritani N, Nakajima T, Furihata K, et al. Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor alpha (PPARalpha) J Biol Chem. 1998;273:5678–84. doi: 10.1074/jbc.273.10.5678. [DOI] [PubMed] [Google Scholar]
- 115.Chu S, Huang Q, Alvares K, Yeldandi AV, Rao MS, Reddy JK. Transformation of mammalian cells by over-expressing H2O2-generating peroxisomal fatty acyl-CoA oxidase. Proc Natl Acad Sci USA. 1995;92:7080–84. doi: 10.1073/pnas.92.15.7080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Reddy JK, Rao MS. Oxidative DNA damage caused by persistent peroxisome proliferation: its role in hepatocarcinogenesis. Mutat Res. 1989;214:63–8. doi: 10.1016/0027-5107(89)90198-x. [DOI] [PubMed] [Google Scholar]
- 117.Reddy JK, Lalwai ND. Carcinogenesis by hepatic peroxisome proliferators: evaluation of the risk of hypolipidemic drugs and industrial plasticizers to humans. Crit Rev Toxicol. 1983;12:1–58. doi: 10.3109/10408448309029317. [DOI] [PubMed] [Google Scholar]
- 118.Blumcke S, Schwartzkopff W, Lobeck H, Edmondson NA, Prentice DE, Blane GF. Influence of fenofibrate on cellular and subcellular liver structure in hyperlipidemic patients. Atherosclerosis. 1983;46:105–16. doi: 10.1016/0021-9150(83)90169-7. [DOI] [PubMed] [Google Scholar]
- 119.Gariot P, Pointel JP, Barrat E, Drouin P, Debry G. [Morphometric study of hepatic peroxisomes in hyperlipoproteinemic patients treated with fenofibrate] Biomed Pharmacother. 1984;38:101–6. [PubMed] [Google Scholar]
- 120.De La Iglesia FA, Lewis JE, Buchanan RA, Marcus EL, McMahon G. Light and electron microscopy of liver in hyperlipoproteinemic patients under long-term gemfibrozil treatment. Atherosclerosis. 1982;43:19–37. doi: 10.1016/0021-9150(82)90096-x. [DOI] [PubMed] [Google Scholar]
- 121.Hanefeld M, Kemmer C, Kadner E. Relationship between morphological changes and lipid-lowering action of p-chlorphenoxyisobutyric acid (CPIB) on hepatic mitochondria and peroxisomes in man. Atherosclerosis. 1983;46:239–46. doi: 10.1016/0021-9150(83)90115-6. [DOI] [PubMed] [Google Scholar]
- 122.Frick MH, Elo O, Haapa K, Heinonen OP, Heinsalmi P, et al. Helsinki Heart Study: primary-prevention trial with gemfibrozil in middle-aged men with dyslipidemia. Safety of treatment, changes in risk factors, and incidence of coronary heart disease. N Engl J Med. 1987;317:1237–45. doi: 10.1056/NEJM198711123172001. [DOI] [PubMed] [Google Scholar]
- 123.Elcombe CR, Mitchell AM. Peroxisome proliferation due to di(2-ethylhexyl) phthalate (DEHP): species differences and possible mechanisms. Environ Health Perspect. 1986;70:211–19. doi: 10.1289/ehp.8670211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hsu MH, Savas U, Griffin KJ, Johnson EF. Identification of peroxisome proliferator-responsive human genes by elevated expression of the peroxisome proliferator-activated receptor alpha in HepG2 cells. J Biol Chem. 2001;276:27950–58. doi: 10.1074/jbc.M100258200. [DOI] [PubMed] [Google Scholar]
- 125.Yu S, Cao WQ, Kashireddy P, Meyer K, Jia Y, et al. Human peroxisome proliferator-activated receptor alpha (PPARalpha) supports the induction of peroxisome proliferation in PPARalpha-deficient mouse liver. J Biol Chem. 2001;276:42485–91. doi: 10.1074/jbc.M106480200. [DOI] [PubMed] [Google Scholar]
- 126.Lawrence JW, Li Y, Chen S, DeLuca JG, Berger JP, et al. Differential gene regulation in human versus rodent hepatocytes by peroxisome proliferator-activated receptor (PPAR) alpha. PPAR alpha fails to induce peroxisome proliferation-associated genes in human cells independently of the level of receptor expresson. J Biol Chem. 2001;276:31521–27. doi: 10.1074/jbc.M103306200. [DOI] [PubMed] [Google Scholar]
- 127.Cheung C, Akiyama TE, Ward JM, Nicol CJ, Feigenbaum L, et al. Diminished hepatocellular proliferation in mice humanized for the nuclear receptor peroxisome proliferator-activated receptor alpha. Cancer Res. 2004;64:3849–54. doi: 10.1158/0008-5472.CAN-04-0322. [DOI] [PubMed] [Google Scholar]
- 128.Jiang Z, Dalton TP, Jin L, Wang B, Tsuneoka Y, et al. Toward the evaluation of function in genetic variability: characterizing human SNP frequencies and establishing BAC-transgenic mice carrying the human CYP1A1 CYP1A2 locus. Hum Mutat. 2005;25:196–206. doi: 10.1002/humu.20134. [DOI] [PubMed] [Google Scholar]
- 129.Ueno T, Tamura S, Frels WI, Shou M, Gonzalez FJ, Kimura S. A transgenic mouse expressing human CYP1A2 in the pancreas. Biochem Pharmacol. 2000;60:857–63. doi: 10.1016/s0006-2952(00)00389-0. [DOI] [PubMed] [Google Scholar]
- 130.Hwang DY, Chae KR, Shin DH, Hwang JH, Lim CH, et al. Xenobiotic response in humanized double transgenic mice expressing tetracycline-controlled transactivator and human CYP1B1. Arch Biochem Biophys. 2001;395:32–40. doi: 10.1006/abbi.2001.2542. [DOI] [PubMed] [Google Scholar]
- 131.Morgan K, French SW, Morgan TR. Production of a cytochrome P450 2E1 transgenic mouse and initial evaluation of alcoholic liver damage. Hepatology. 2002;36:122–34. doi: 10.1053/jhep.2002.33720. [DOI] [PubMed] [Google Scholar]
- 132.Li Y, Yokoi T, Kitamura R, Sasaki M, Gunji M, et al. Establishment of transgenic mice carrying human fetus-specific CYP3A7. Arch Biochem Biophys. 1996;329:235–40. doi: 10.1006/abbi.1996.0214. [DOI] [PubMed] [Google Scholar]
- 133.Imaoka S, Hayashi K, Hiroi T, Yabusaki Y, Kamataki T, Funae Y. A transgenic mouse expressing human CYP4B1 in the liver. Biochem Biophys Res Commun. 2001;284:757–62. doi: 10.1006/bbrc.2001.5055. [DOI] [PubMed] [Google Scholar]
- 134.Chen JY, Levy-Wilson B, Goodart S, Cooper AD. Mice expressing the human CYP7A1 gene in the mouse CYP7A1 knock-out background lack induction of CYP7A1 expression by cholesterol feeding and have increased hypercholesterolemia when fed a high fat diet. J Biol Chem. 2002;277:42588–95. doi: 10.1074/jbc.M205117200. [DOI] [PubMed] [Google Scholar]
- 135.Goodart SA, Huynh C, Chen W, Cooper AD, Levy-Wilson B. Expression of the human cholesterol 7alpha-hydroxylase gene in transgenic mice. Biochem Biophys Res Commun. 1999;266:454–59. doi: 10.1006/bbrc.1999.1799. [DOI] [PubMed] [Google Scholar]
- 136.Miyake JH, Doung XD, Strauss W, Moore GL, Castellani LW, et al. Increased production of apolipoprotein B-containing lipoproteins in the absence of hyperlipidemia in transgenic mice expressing cholesterol 7alpha-hydroxylase. J Biol Chem. 2001;276:23304–11. doi: 10.1074/jbc.M101853200. [DOI] [PubMed] [Google Scholar]
- 137.Li X, Nokkala E, Yan W, Streng T, Saarinen N, et al. Altered structure and function of reproductive organs in transgenic male mice overexpressing human aromatase. Endocrinology. 2001;142:2435–42. doi: 10.1210/endo.142.6.8211. [DOI] [PubMed] [Google Scholar]
- 138.Meir K, Kitsberg D, Alkalay I, Szafer F, Rosen H, et al. Human sterol 27-hydroxylase (CYP27) overexpressor transgenic mouse model. Evidence against 27-hydroxycholesterol as a critical regulator of cholesterol homeostasis. J Biol Chem. 2002;277:34036–41. doi: 10.1074/jbc.M201122200. [DOI] [PubMed] [Google Scholar]
- 139.Zhang W, Moorthy B, Chen M, Muthiah K, Coffee R, et al. A Cyp1a2-luciferase transgenic CD-1 mouse model: responses to aryl hydrocarbons similar to the humanized AhR mice. Toxicol Sci. 2004;82:297–307. doi: 10.1093/toxsci/kfh260. [DOI] [PubMed] [Google Scholar]
- 140.Zhang W, Purchio AF, Chen K, Wu J, Lu L, et al. A transgenic mouse model with a luciferase reporter for studying in vivo transcriptional regulation of the human CYP3A4 gene. Drug Metab Dispos. 2003;31:1054–64. doi: 10.1124/dmd.31.8.1054. [DOI] [PubMed] [Google Scholar]
- 141.Robertson GR, Field J, Goodwin B, Bierach S, Tran M, et al. Transgenic mouse models of human CYP3A4 gene regulation. Mol Pharmacol. 2003;64:42–50. doi: 10.1124/mol.64.1.42. [DOI] [PubMed] [Google Scholar]