

Abstract
The cell division-independent growth of terminally differentiated cardiomyocytes is commonly associated with cardiovascular disease. We demonstrate that it is accompanied by a substantial rise in transcription by RNA polymerase (pol) III, which produces essential components of the biosynthetic apparatus, including 5S rRNA and tRNAs. This increase in transcription is achieved by changes in both the activity and level of the essential pol III-specific transcription factor TFIIIB. Erk and c-Myc, which directly activate TFIIIB in proliferating fibroblasts, also induce pol III transcription in growing cardiomyocytes. Furthermore, hypertrophic stimulation increases expression of the essential TFIIIB subunit Brf1, an effect not seen when fibroblasts proliferate. Erk mediates this induction of Brf1 expression and therefore contributes in at least two ways to pol III transcriptional activation during hypertrophy. Increased production of tRNA and 5S rRNA will contribute to the enhanced translational capacity required to sustain hypertrophic growth.
Keywords: c-Myc, Erk, hypertrophy, pol III transcription, TFIIIB
Introduction
In comparison to cell division, the mechanisms underlying cell growth (increase in mass) are poorly understood. Progress towards understanding the latter requires systems in which growth can be studied independently from proliferation. Mammalian cardiomyocytes terminally differentiate and lose the capacity to divide shortly after birth (Li et al, 1996; Soonpaa et al, 1996). Consequently, enlargement of existing cardiomyocytes accounts for the increase in heart size during postnatal development (Zak, 1974). This increase in cell size, in the absence of proliferation, is known as hypertrophy, and provides a useful model for studying mechanisms contributing directly to growth. Cardiomyocyte hypertrophy also occurs in adult hearts in response to a range of stimuli, allowing the heart to adjust its contractile capacity according to demand. However, sustained cardiomyocyte growth is frequently associated with cardiovascular disorders, and is an independent risk factor for heart disease (Levy et al, 1990).
A key hallmark of hypertrophy is an accumulation of protein, owing to accelerated rates of protein synthesis (Chien et al, 1991). Although this enhanced biosynthesis involves an increase in both translational efficiency and capacity, the latter appears particularly important for sustained hypertrophic growth, as associated with cardiac pathologies (reviewed by Hannan et al, 2003). Despite this, relatively few studies have attempted to elucidate the mechanisms responsible for this elevated capacity for protein synthesis.
Translational capacity is determined by the availability of components of the protein synthetic apparatus, including ribosomes, tRNAs and translation factors. Transcription by RNA polymerase (pol) I, which synthesises the large rRNAs, is increased during hypertrophy (McDermott et al, 1991; Hannan et al, 1995, 1996; Luyken et al, 1996), and this is essential for cardiomyocyte growth (Brandenburger et al, 2001). Pol III synthesises 5S rRNA, which is found in the ribosome in equimolar amounts with the pol I-produced rRNAs (Wool, 1979). Furthermore, pol III generates several additional small, untranslated RNAs essential for biosynthesis, including tRNAs (reviewed by White, 2005). Therefore, we hypothesised that, like pol I, pol III transcription will also be activated during myocardial hypertrophy.
Transcription by pol III is known to be tightly regulated in accordance with metabolic requirements in various other eukaryotic systems. The basis of this control has been well studied in proliferating mammalian cells, where the growth activators c-Myc and Erk have been shown to induce pol III transcription, whereas negative regulators of growth, such as RB, have the opposite effect (White et al, 1996; Felton-Edkins et al, 2003; Gomez-Roman et al, 2003). Each of these regulatory proteins targets the pol III-specific transcription factor TFIIIB (Chu et al, 1997; Larminie et al, 1997; Felton-Edkins et al, 2003; Gomez-Roman et al, 2003). However, these studies on mammalian pol III control have employed immortalised, proliferating cell lines, whereas the regulation of pol III transcription during the cell division-independent growth of terminally differentiated cells has not been addressed. Using cultured neonatal cardiomyocytes and animal models, we demonstrate that transcription by pol III is induced substantially during hypertrophic growth. This is associated with increased recruitment of TFIIIB and pol III to class III genes. Based on the data presented, we propose that regulation of pol III transcription is achieved through the concerted action of several proteins known to interact with TFIIIB, and through Erk-mediated induction of the essential TFIIIB subunit Brf1.
Results
Pol III transcription is activated during hypertrophic growth
Serum-starved cardiomyocytes were exposed to hypertrophic stimuli (foetal calf serum (FCS), endothelin-1 (ET-1) or phenylephrine (PE)) for 16 h. This caused elevated protein synthesis (Figure 1A), an increase in cell volume (Figure 1B) and re-expression of the foetal gene atrial natriuretic factor (ANF) (Figure 1C, top panel), which is a marker of hypertrophy (Chien et al, 1991). These effects occurred without significant induction of DNA synthesis or cellular proliferation (Figure 1A and B, respectively), and thus are indicative of a bona fide hypertrophic response. RT–PCR revealed a substantial increase in the level of 5S rRNA, U6 snRNA and precursors for tRNAs in response to hypertrophic stimulation (Figure 1C and D), whereas Northern blotting demonstrated a parallel increase in pol III transcripts from the B2 middle repetitive gene family (Figure 1E and F). The specificity of these effects is indicated by the constant level of the control mRNA encoding acidic ribosomal phosphoprotein (ARPP) P0 under all conditions (Figure 1C and E). B2 and tRNA precursor transcripts have short half-lives (Bladon et al, 1990; Winter et al, 2000) and the increase in their abundance suggests enhanced transcriptional output by pol III. Furthermore, whole-cell extracts prepared from FCS-stimulated cardiomyocytes transcribe pol III templates (including 5S rRNA, B2, tRNALeu and adenovirus VAI) more actively than extracts prepared from serum-starved cells (Figure 1G and H). We conclude that the pol III transcription machinery is activated during cardiomyocyte hypertrophy in culture. This is consistent with a previous report that levels of the pol III-synthesised transcript U7-2 (MRP) increase in response to spontaneous contraction of cardiomyocytes (Hannan and Rothblum, 1995).
Figure 1.
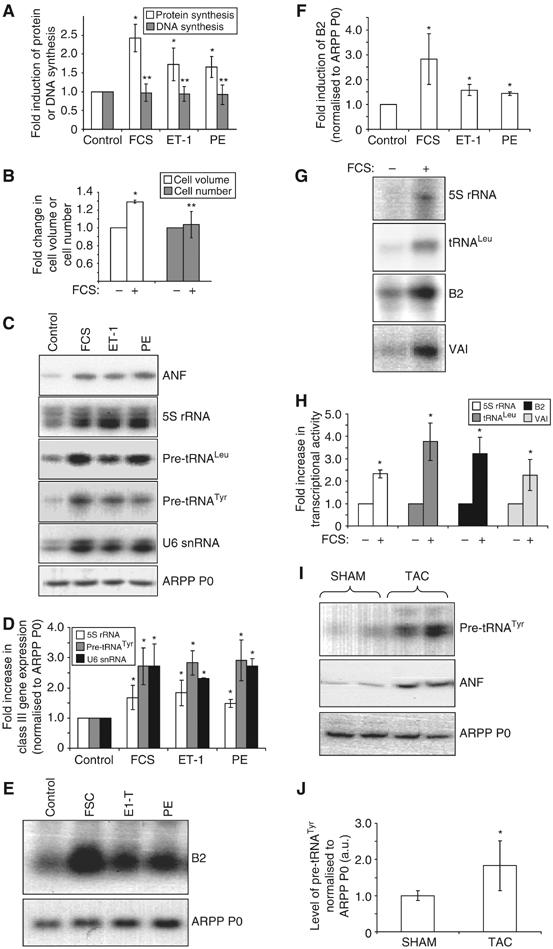
Hypertrophic stimuli activate pol III transcription. (A–F) Cultured cardiomyocytes were serum-starved for 24 h, then either maintained in serum-free media (control) or exposed to 10% FCS, 100 nM ET-1 or 100 μM PE for 16 h, as indicated. (A) Protein and DNA synthesis rates were determined by measuring incorporation of [35S]methionine/cysteine or [3H]thymidine, respectively. The data represent the mean of five separate experiments, each with three replicates per condition. (B) Myocyte volume and number were assayed using a Z2 Coulter counter. The fold increases represent the mean of three separate experiments, each with two replicates per condition. (C) Specific primers for the transcripts indicated were used to PCR amplify cDNAs generated from total RNA of cultured cardiomyocytes treated as specified. (D) RT–PCR transcript intensities were quantified and the level of class III transcripts normalised to ARPP P0. The average fold increases are represented. (E) Northern analysis was performed using total RNA from cardiomyocytes treated as indicated. Northern blots were probed for B2, then stripped and re-probed for ARPP P0. (F) B2 and ARPP P0 RNA levels were quantified by densitometry, and B2 normalised to ARPP P0. The average fold increases are represented. (G) Whole-cell extracts (20 μg), prepared from serum-starved or FCS-stimulated cardiomyocytes, were tested for their ability to transcribe 5S rRNA, tRNALeu, B2 and VAI templates in vitro. (H) Average fold increases in transcriptional activity are displayed graphically. (I) Activation of pol III transcription accompanies hypertrophic growth in situ. RNA was extracted from hearts derived from mice subjected to TAC or sham operation. RT–PCR was performed using pre-tRNATyr-, ANF- and ARPP P0-specific primers. RT–PCRs using RNA derived from two separate animals per condition are shown. (J) Six hearts were analysed as in (I) for each condition, and densitometry used to quantify transcript levels. The average level of pre-tRNATyr, normalised to ARPP P0, is shown. For all bar charts, error bars indicate the standard deviation from the mean (*significantly higher than control, P<0.05; **no significant difference from control).
To assess whether pol III transcription is enhanced during hypertrophic growth in situ, hearts were removed from adult mice in which pressure overload hypertrophy had been induced by transverse aortic constriction (TAC) for 2 weeks. Control mice were subjected to sham operation. RNA was extracted from these hearts and analysed by RT–PCR. TAC caused a clear increase in ANF mRNA levels, consistent with a hypertrophic response (Figure 1I). This was accompanied by a significant increase in the level of pre-tRNATyr transcripts (Figure 1I and J). Thus, pol III transcripts are not only induced during cardiomyocyte hypertrophy in culture but also in the adult heart.
Hypertrophic growth is accompanied by increased TFIIIB and pol III promoter occupancy in vivo
Expression of most class III genes depends on the productive assembly of the basal pol III-specific transcription factors TFIIIB and TFIIIC at the appropriate promoters: TFIIIC is responsible for initial promoter binding, and positions TFIIIB just upstream of the transcription start site; TFIIIB then recruits pol III, allowing transcription to commence (reviewed by Schramm and Hernandez, 2002). Chromatin immunoprecipitation (ChIP) assays were used to compare the association of these factors with class III genes before and after hypertrophic stimulation. They revealed that serum induces a marked increase in pol III binding to 5S rRNA and tRNA genes in vivo (Figure 2), consistent with the hypertrophy-associated increase in transcription. Hypertrophic growth is also accompanied by an increase in the level of TFIIIB bound, whereas TFIIIC occupancy is unaffected.
Figure 2.
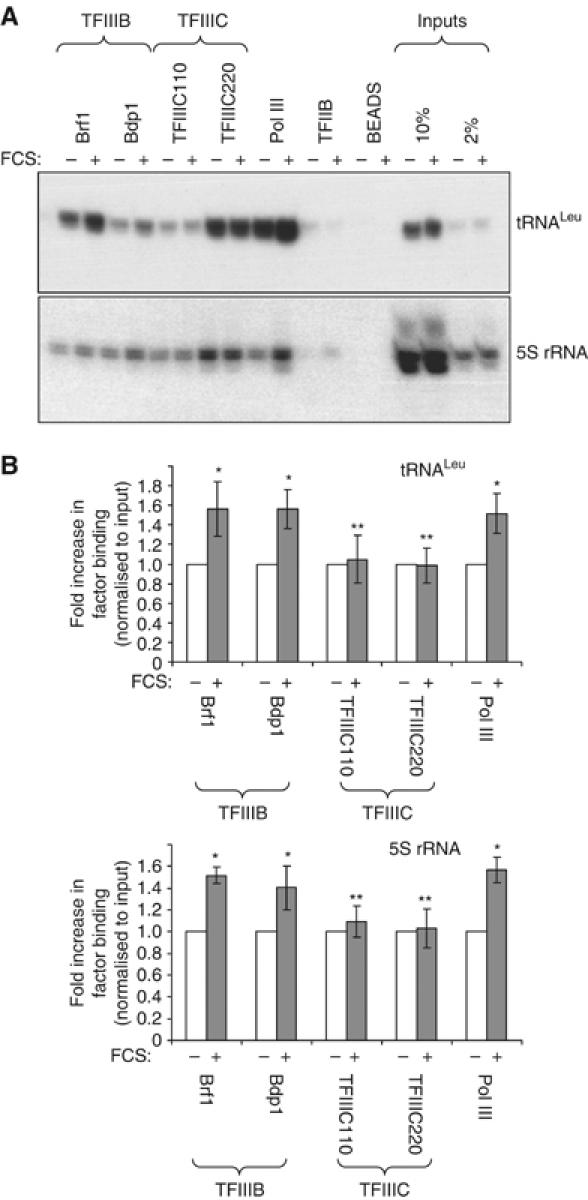
Hypertrophic stimulation enhances interaction of TFIIIB and pol III, but not TFIIIC, with class III genes in vivo. Cultured cardiomyocytes were treated with 10% FCS for 16 h to induce hypertrophic growth. Control cells were maintained in serum-free medium. ChIPs were performed with antibodies against TFIIIB (Brf1 and Bdp1), TFIIIC (110 and 220) and pol III (RPC155), as indicated. Negative control ChIPs used a TFIIB antibody and beads alone. (A) Association of each factor with 5S rRNA and tRNALeu genes, in unstimulated and hypertrophic cells, was analysed by PCR with gene-specific primers. Input genomic DNA (10 and 2% of that used in ChIPs) was analysed in parallel. (B) PCR products were quantified by densitometry and normalised to the appropriate input. The average fold increases in factor binding are represented (n=3; *significantly higher than control, P<0.05; **no significant difference from control).
Regulation of pol III transcription through changes in Brf1 abundance
TFIIIB is composed of three subunits: TBP, Brf1 and Bdp1. The activity of this complex is tightly regulated to modulate pol III output. In proliferating fibroblasts, this is achieved through protein–protein interactions with several regulators, rather than through alterations in TFIIIB abundance (Figure 3A; White, 2005). In contrast, a significant induction of Brf1 expression was found in response to the hypertrophic stimulation of cardiomyocytes (Figure 3B). This effect is specific to Brf1, as no change was found in the levels of the other TFIIIB subunits (Bdp1 and TBP), nor in various subunits of TFIIIC (110 and 220) or pol III (BN51 and C39). This induction of Brf1 is also apparent at the mRNA level (Figure 3C), and therefore may be a result of increased Brf1 gene transcription.
Figure 3.
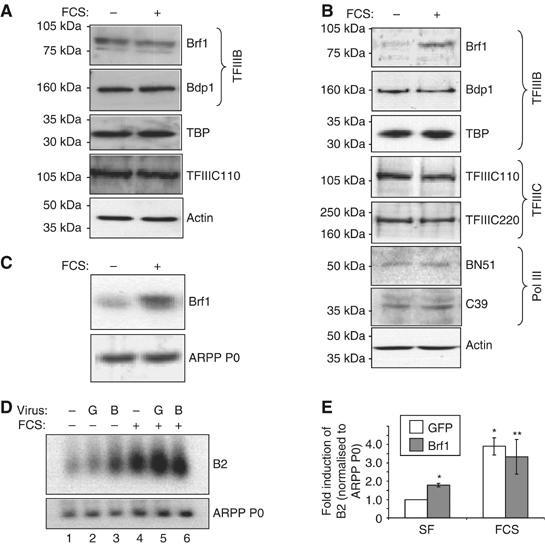
Brf1 levels limit pol III transcription in unstimulated cardiomyocytes, but increase specifically after hypertrophic stimulation. (A) Whole-cell lysates of rat1A fibroblasts that had been serum-starved for 24 h then either maintained in serum-free media or exposed to 10% FCS for 16 h were resolved by SDS–PAGE, and immunoblotted using antibodies against the proteins indicated. (B) The same procedure as outlined in (A) was used to analyse expression of the indicated proteins in cardiomyocytes. (C) Cultured cardiomyocytes were treated as in (A), then total RNA was extracted and used to generate cDNAs, which were amplified by PCR using primers specific for Brf1 or ARPP P0. (D) Cultured cardiomyocytes were infected with adenoviruses expressing HA-Brf1 (B) or GFP (G). Cells were serum-starved or stimulated with 10% FCS for 16 h before extracting whole-cell RNA. RNA was analysed by Northern blotting using B2- and ARPP P0-specific probes. (E) Northern blots were quantified by densitometry and B2 levels normalised to ARPP P0. The fold increases in normalised B2 expression relative to serum-starved, GFP control infected cells are shown. (n=8; *significantly different from SF GFP, P<0.05; **not significantly different from FCS GFP.)
To investigate whether increased Brf1 levels are able to contribute to the activation of transcription by pol III during hypertrophy, the effects of raising the concentration of this TFIIIB subunit were assessed. Cultured cardiomyocytes were infected with adenoviruses expressing GFP, as a negative control, or HA-tagged Brf1. Western analyses showed that cells infected with virus encoding HA-Brf1 had approximately 2.5-fold more Brf1 than cells infected with control virus (data not shown). This is similar to the magnitude of endogenous Brf1 induction in stimulated cardiomyocytes (see Figure 4D). In the absence of hypertrophic stimuli, raising Brf1 levels in this way caused a significant induction of B2 transcripts (almost two-fold) compared to control-infected cells (Figure 3D, compare lanes 2 and 3, and E). This suggests that Brf1 is limiting for pol III transcription under basal conditions in terminally differentiated cardiomyocytes. However, in serum-stimulated cells, B2 expression does not change significantly in response to exogenous Brf1 (Figure 3D and E), suggesting that the level of this subunit no longer restricts class III gene expression once hypertrophy has been induced. These data are consistent with a model whereby the hypertrophy-associated increase in the abundance of endogenous Brf1 contributes to the observed activation of pol III transcription in growing cardiomyocytes.
Figure 4.
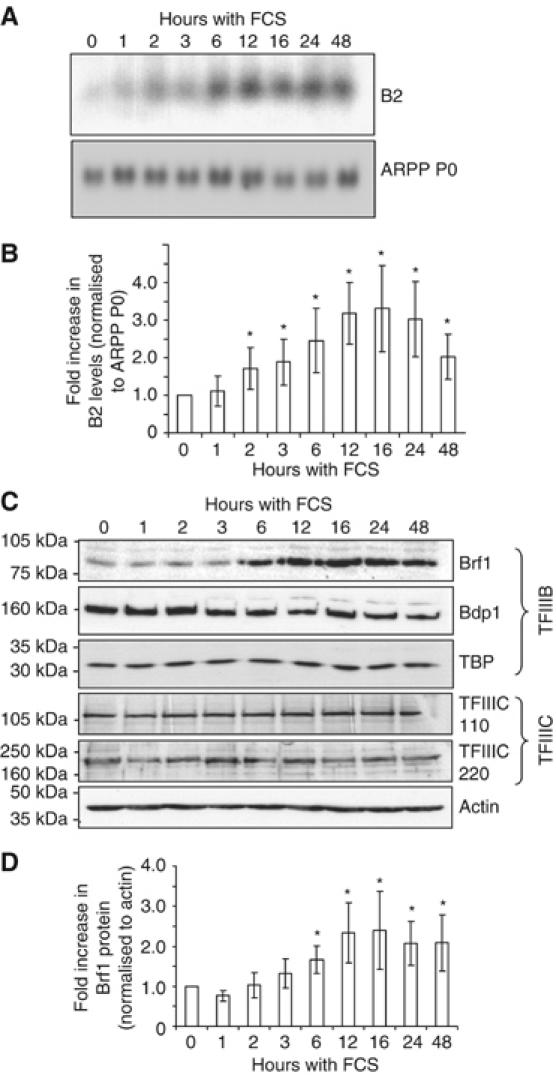
Pol III transcripts increase within 2 h of hypertrophic stimulation, followed by Brf1 induction within 6 h. Serum-starved cardiomyocytes were stimulated with 10% FCS for the times indicated (0–48 h). Whole-cell protein or RNA was then extracted. (A) B2 and ARPP P0 RNA levels were analysed by Northern blotting, and (B) quantified by densitometry (n=4; *significantly different from control, P<0.05). (C) Protein lysates were resolved by SDS–PAGE and Western blotting performed using antibodies against the indicated proteins. (D) The level of Brf1 at each time point was quantified and normalised to actin (n=4; *significantly different from control, P<0.05).
Known TFIIIB regulators respond to the hypertrophic stimulation of cardiomyocytes
Although Brf1 seems to be limiting for at least some pol III templates in unstimulated cardiomyocytes, increasing Brf1 alone is insufficient to induce class III gene expression to the same extent as FCS (Figure 3D and E). Furthermore, pol III transcription rises within 2 h of hypertrophic stimulation (Figure 4A and B), whereas Brf1 protein levels do not increase significantly until after 3–6 h (Figure 4C and D). Therefore, although maximum levels of Brf1 (2.4-fold higher than control cells) correlate with the highest level of class III gene expression (attained after 16 h), additional mechanisms may also contribute to pol III transcriptional regulation in cardiomyocytes.
In proliferating cells, both c-Myc and Erk bind to and activate TFIIIB, leading to increased class III gene expression (Felton-Edkins et al, 2003; Gomez-Roman et al, 2003). In contrast, hypophosphorylated RB sequesters TFIIIB in an inactive complex, thus restraining pol III transcription; this repression is relieved when RB becomes hyperphosphorylated during late G1 phase of the cell cycle (Scott et al, 2001). Each of these known pol III regulators has also been implicated in the control of hypertrophic cardiomyocyte growth (Sadoshima et al, 1997; Xiao et al, 2001; Bueno and Molkentin, 2002), and might therefore influence the pol III machinery in these terminally differentiated cells. In accordance with previous work (Starksen et al, 1986; Bogoyevitch et al, 1994; Sadoshima et al, 1997), Erk, c-Myc and RB were found to respond to the hypertrophic stimulation of cardiomyocytes (Figure 5A). Specifically, Erk is activated by phosphorylation and c-Myc expression is induced, whereas RB is inactivated by phosphorylation at several residues. These events were monitored over time (Figure 5B) and compared to the induction of class III gene expression (Figure 4A). Both Erk and c-Myc are activated within 1 h of hypertrophic stimulation, thus preceding the initial increase in pol III transcription. However, RB phosphorylation does not occur until 6–12 h following FCS addition (after cyclin D1 induction), and hence is unlikely to contribute to the initial activation of transcription by pol III. Nonetheless, RB phosphorylation, like Brf1 accumulation, does coincide with the peak of class III gene expression, suggesting that derepression of TFIIIB may contribute to maximum pol III output. The levels of c-Myc and phosphorylated RB decrease between 24 and 48 h. This may at least partly account for the reduction in pol III transcripts at this time.
Figure 5.
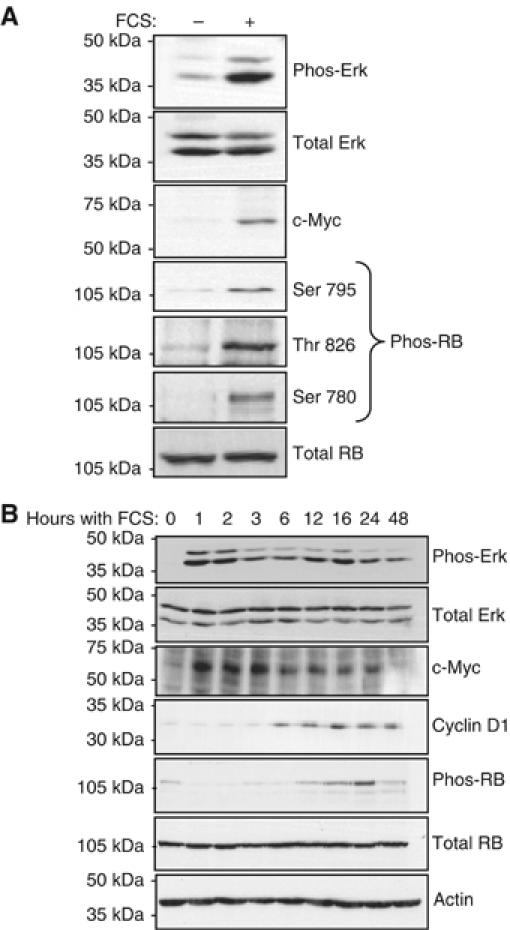
Known TFIIIB regulators respond to hypertrophic stimulation. (A) Whole-cell lysates were prepared from cultured cardiomyocytes treated as in Figure 3B, and analysed by Western blotting using antibodies against the proteins, or phosphoproteins (‘phos-'), specified. The phos-RB antibodies used specifically detect RB phosphorylated on the indicated sites. (B) Cardiomyocytes were cultured as described in Figure 4, and protein lysates analysed by Western blotting to detect the indicated proteins.
c-Myc and Erk activate pol III transcription in cardiomyocytes
The rapid activation of Erk and c-Myc by hypertrophic stimulation makes these proteins plausible candidates for the initial induction of class III gene expression. Therefore, their involvement in pol III activation in cardiomyocytes was investigated further. Cultured cardiomyocytes were transduced with adenoviruses expressing Lac Z, as a control or c-Myc. Figure 6A and B demonstrate that c-Myc triggers an increase in tRNA expression, as determined by RT–PCR. This confirms that pol III activity responds to c-Myc in this cell type.
Figure 6.
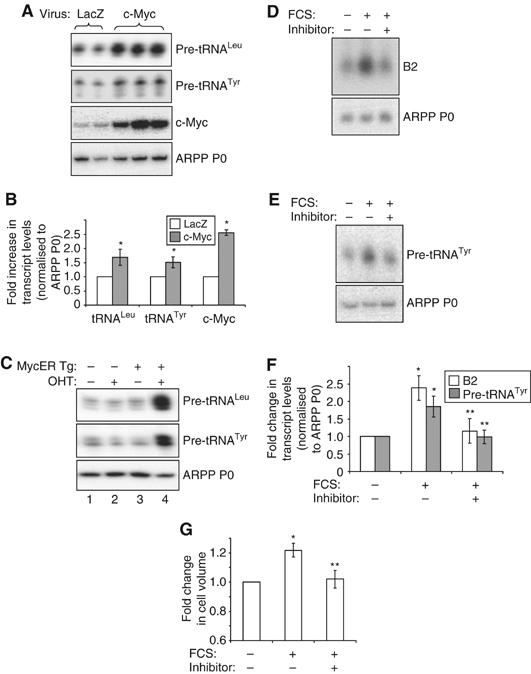
c-Myc regulates pol III transcription in cardiomyocytes. (A, B) Cultured cardiomyocytes were infected with adenovirus expressing Lac Z (control) or c-Myc, as indicated. Cells were grown in the absence of hypertrophic stimuli, then total RNA extracted and used to generate cDNAs by reverse transcription. cDNAs were amplified by PCR using primers specific for the indicated transcripts. (A) Representative PCRs are shown. (B) The average fold increases in transcript levels (normalised to ARPP P0), induced by c-Myc, are represented graphically (n=3; *significantly higher than LacZ-expressing control, P<0.05). (C) c-Myc induces pol III transcription in the heart. RNA was extracted from hearts derived from wild-type adult mice (lanes 1 and 2) or transgenic littermates expressing MycER specifically in cardiomyocytes (MycER Tg, lanes 3 and 4). Mice had either been exposed to 4-hydroxytamoxifen (OHT) (lanes 2 and 4) or vehicle (lanes 1 and 3) for 1 week before removal of hearts. RNA was analysed by RT–PCR using pre-tRNA- and ARPP P0-specific primers, as specified. (D–G) Cells were cultured in the absence of serum (SF) or stimulated with 10% FCS for 24 h. c-Myc inhibitor (‘+') or vehicle (‘−') were also included for 24 h where indicated. Whole-cell RNA was analysed by (D) Northern blotting or (E) RT–PCR. (F) tRNA and B2 levels were quantified by densitometry and normalised to ARPP P0, as represented graphically (n=3; *significantly higher than SF+vehicle, P<0.05; **significantly lower than FCS+vehicle, P<0.05). (G) Cell volume was assayed using a Z2 coulter counter and the average fold changes in cell size are shown. (n=3; *significantly higher than SF+vehicle, P<0.05; **significantly lower than FCS+vehicle, P<0.05).
To study the role of c-Myc in the adult myocardium, Xiao et al (2001) developed a mouse model in which an oestrogen-responsive Myc fusion protein (MycER) is expressed specifically in cardiomyocytes. In the absence of an appropriate ligand, the fusion is sequestered in the cytoplasm, but 4-hydroxytamoxifen induces MycER nuclear translocation. This model revealed that the activation of c-Myc in the heart is sufficient to induce several features of cardiac hypertrophy, including an increase in cell size without division (Xiao et al, 2001). We found that MycER activation also results in a substantial increase in the levels of pre-tRNAs (Figure 6C), demonstrating that c-Myc can activate pol III transcription in the intact heart. This enhancement of class III gene expression may contribute to the induction of hypertrophic growth by c-Myc.
To determine whether c-Myc is necessary for the activation of pol III transcription during hypertrophy, cardiomyocytes were stimulated with serum in the presence of a specific c-Myc inhibitor (Yin et al, 2003). Both RT–PCR and Northern analyses demonstrated that when c-Myc activity is inhibited, the serum induction of pol III transcripts is significantly diminished (Figure 6D–F). Myc inhibition also prevented the induction of hypertrophic growth by serum, as determined by cell volume measurements (Figure 6G). This provides evidence that c-Myc is essential for cardiomyocyte hypertrophy.
Erk activation is sufficient to induce the hypertrophic growth of cardiomyocytes (Bueno et al, 2000; Yue et al, 2000; Wang and Proud, 2002a). To determine whether the activation of Erk in the absence of hypertrophic stimuli could also induce transcription by pol III, cultured cardiomyocytes were infected with varying amounts of adenoviruses expressing the upstream Erk activator MEK1 (CAMEK) or β-galactosidase (β-gal), as a control. Western analysis confirmed that the CAMEK adenovirus effectively induces Erk phosphorylation, and hence activation (Figure 7A). This is accompanied by a substantial increase in the level of B2 (Figure 7B) and tRNA (Figure 7C) transcripts. Furthermore, blocking Erk activation with the MEK inhibitor PD98059 attenuates the serum-induced increase in tRNA (Figure 7D) and B2 (Figure 7E) expression. As reported previously (Yue et al, 2000; Wang and Proud, 2002a), Erk inhibition also impairs serum-induced hypertrophic growth, as determined by measuring protein synthesis (Figure 7F). Therefore, Erk activates both pol III transcription and protein synthesis in terminally differentiated cardiomyocytes.
Figure 7.
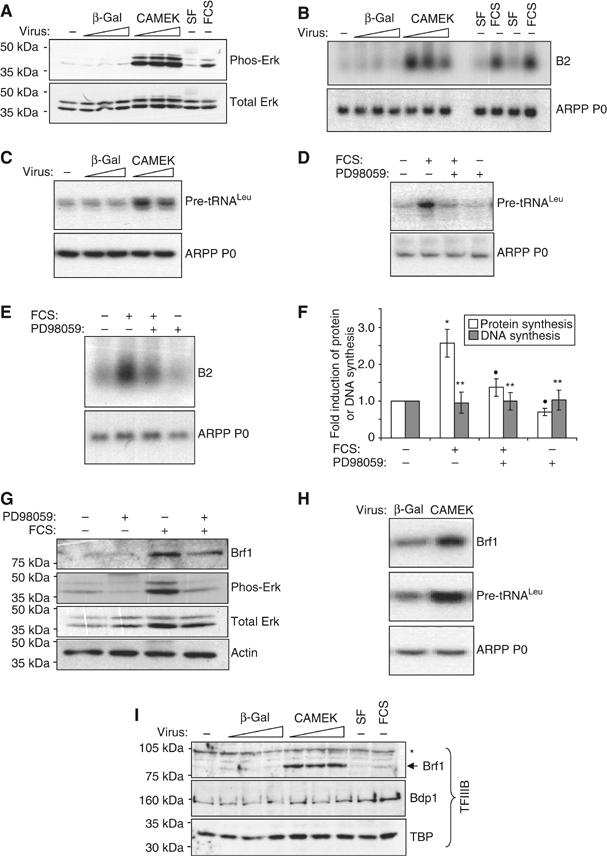
Erk induces pol III transcription and Brf1 expression in cardiomyocytes. (A) Cultured cardiomyocytes were infected (at a multiplicity of infection (m.o.i.) of 20, 50 or 80) with adenovirus expressing CAMEK or β-gal (negative control), as indicated. Cells were serum-starved for 48 h before harvesting. As a positive control for Erk activation, uninfected cells were incubated without (SF) or with 10% FCS for 16 h. Whole-cell protein was analysed by Western blotting using the antibodies indicated. (B) Total RNA from cells treated as in (A) was analysed by Northern blotting using the indicated probes. (C) Infections were carried out as in (A), using an m.o.i of 50 or 80, then total RNA analysed by RT–PCR using pre-tRNALeu- and ARPP P0-specific primers. (D–G) Cells were cultured in the absence of serum or stimulated with 10% FCS for 16 h. PD98059 or vehicle were also included for 16 h where indicated. Whole-cell RNA was analysed by (D) RT–PCR or (E) Northern blotting. (F) Protein and DNA synthesis rates were determined by measuring the incorporation of [35S]methionine/cysteine or [3H]thymidine, respectively. The fold increases represent the mean of four separate experiments, each with three replicates per condition (*significantly higher than SF+vehicle, P<0.05; •significantly lower than FCS+vehicle, P<0.05; **not significantly different from SF+vehicle). (G) Protein lysates were analysed by Western blotting using antibodies against the proteins indicated. (H) Total RNA was extracted from cardiomyocytes infected (at an m.o.i of 50) with adenovirus expressing CAMEK or β-gal (negative control), as indicated. Cells were serum-starved for 48 h before harvesting. RT–PCR was performed using the primers specified. (I) Whole-cell lysates, treated as in (A), were analysed by Western blotting using the indicated antibodies (*nonspecific band).
Erk induces Brf1 expression in cardiomyocytes
Erk directly phosphorylates the Brf1 subunit of TFIIIB in fibroblasts, and this facilitates the rapid induction of pol III transcription following mitogenic stimulation (Felton-Edkins et al, 2003). Our data suggest that the same is true in hypertrophic cardiomyocytes. However, in cardiomyocytes, expression of Brf1 itself is also induced under these conditions (Figure 3). We tested whether Erk mediates the induction of this limiting TFIIIB subunit.
Inhibiting Erk activity with PD98059 reduces the level of Brf1 expression in the presence of FCS by approximately 35% (Figure 7G), suggesting that signalling through the Erk pathway contributes to the induction of Brf1 during hypertrophic growth. Furthermore, Erk activation is sufficient to induce Brf1 expression at both the mRNA (Figure 7H) and protein (Figure 7I) levels in the absence of hypertrophic stimuli. Induction is clearly specific, as TBP and Bdp1 abundance do not respond to Erk activation (Figure 7I). Given that Brf1 is limiting for pol III transcription in the absence of hypertrophic stimuli, this induction of Brf1 is likely to have a significant impact on the activation of pol III transcription, and perhaps cell growth, by Erk in cardiomyocytes.
Discussion
Hypertrophic growth of cultured cardiomyocytes involves a substantial increase in the expression of various pol III products, including tRNAs and 5S rRNA. Analysis of mouse ventricular tissue suggests that induction of pol III transcription also accompanies hypertrophy in living animals, demonstrating the physiological relevance of the response. ChIP experiments revealed a specific increase in TFIIIB promoter occupancy on tRNA and 5S rRNA genes during hypertrophic growth, whereas the association of TFIIIC was unaffected. Several TFIIIB-directed mechanisms apparently contribute to this (Figure 8). Both Erk and c-Myc activate pol III transcription in these terminally differentiated cells, and are likely to mediate the initial induction of transcription by targeting TFIIIB directly. However, limiting levels of Brf1, and hence TFIIIB, may restrict the extent to which these molecules can activate pol III transcription initially. Further restriction of TFIIIB availability through interaction with hypophosphorylated RB is also probable. The Erk-mediated increase in Brf1 abundance by 6 h, and subsequent inactivation of RB by 12 h, would relieve these restrictions, thus allowing maximum class III gene expression. It is likely that the resulting increase in pol III transcripts contributes to enhanced cardiomyocyte translational capacity, and therefore assists hypertrophic growth.
Figure 8.
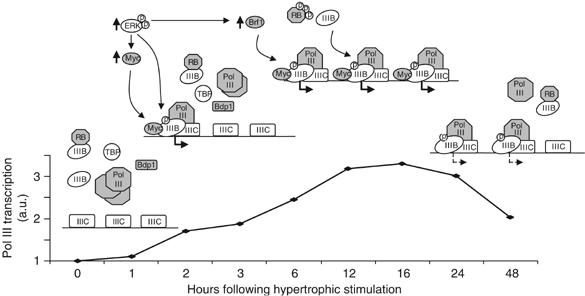
Model for activation of pol III transcription following induction of hypertrophic growth. Hypertrophic stimulation leads to the rapid activation of Erk and c-Myc, which are likely to target TFIIIB directly (as documented in other cell types), leading to the initial induction of pol III transcription. However, limiting levels of Brf1, and sequestration of TFIIIB by hypophosphorylated RB, may restrict the extent to which these molecules activate transcription at early time points. The increase in Brf1 abundance by 6 h, and the subsequent hyperphosphorylation of RB, would relieve these restrictions, thus allowing maximum pol III output. Decreased c-Myc levels and RB dephosphorylation might account for the downregulation of pol III transcription occurring 24–48 h after hypertrophic stimulation. Abbreviations used: TFIIIB, IIIB; TFIIIC, IIIC; c-Myc, Myc. Bold arrows below transcription complexes indicate active class III gene transcription; dashed arrows indicate transcription at a reduced rate.
There is considerable overlap between the mechanisms used to regulate TFIIIB activity in proliferating and in nondividing, terminally differentiated cells. For instance, both Erk and c-Myc target TFIIIB during proliferation (Felton-Edkins et al, 2003; Gomez-Roman et al, 2003), and our data suggest that these proteins are also involved in activating transcription by pol III during the cell division-independent growth of cardiomyocytes. Each of these proteins has previously been shown to promote the hypertrophic growth of cardiomyocytes (Glennon et al, 1996; Bueno et al, 2000; Yue et al, 2000; Xiao et al, 2001; Wang and Proud, 2002a), and some downstream targets likely to be involved in mediating this response have been identified. For example, known targets of the Erk pathway in these cells include the ribosomal protein S6 kinases, eIF4E-binding protein 1 and the translation elongation factor eEF2, which regulate translational efficiency (Wang et al, 2001; Iijima et al, 2002; Wang and Proud, 2002a, 2002b). Growth-associated genes induced by c-Myc in the myocardium include those encoding ribosomal proteins and other proteins involved in ribosome biogenesis (Xiao et al, 2001). Our data provide further evidence that Erk and c-Myc enhance cardiomyocyte biosynthesis, by demonstrating that they upregulate pol III transcription in these terminally differentiated cells.
Changes in RB phosphorylation are typically associated with cell division (Mittnacht, 2005). Although cardiomyocytes do not proliferate, RB nevertheless becomes phosphorylated on several sites following hypertrophic stimulation (Figure 5A; Sadoshima et al, 1997; Nozato et al, 2001). The consequences of this RB phosphorylation for hypertrophic growth have not been established; however, activation or inhibition of cyclin-dependent kinases (CDKs), which mediate RB phosphorylation, can induce or repress hypertrophic growth, respectively (Tamamori et al, 1998; Nozato et al, 2001; Busk et al, 2002; Tamamori-Adachi et al, 2002). We found that RB phosphorylation coincides with maximal class III gene expression in hypertrophic cardiomyocytes. As phosphorylation by CDKs has been shown to dissociate RB from TFIIIB (Scott et al, 2001), we suggest that the observed phosphorylation of RB will liberate TFIIIB molecules, thus maximising pol III output after 12–24 h of hypertrophic stimulation.
We also found a novel mode of TFIIIB regulation in cardiomyocytes, through enhanced Brf1 expression. In unstimulated cardiomyocytes, Brf1 levels are relatively low and limiting for pol III output. However, induction of hypertrophic growth is accompanied by a specific increase in the level of Brf1 mRNA and protein, such that this subunit no longer limits transcription. Although Brf1 levels are regulated in response to growth conditions in Saccharomyces cerevisiae (Sethy et al, 1995), this is the first indication that pol III transcription can be activated by a growth factor-dependent increase in Brf1 in mammals. Differentiation of F9 embryonal carcinoma cells is accompanied by a dramatic decrease in the rate of transcription by pol III (White et al, 1989), which can be partly attributed to a decline in Brf1 levels (Alzuherri and White, 1998). The restrictive abundance of Brf1 in unstimulated cardiomyocytes may reflect the differentiated nature of these cells, and necessitate the induction of this factor following hypertrophic stimulation, in contrast to the mitogenic stimulation of undifferentiated fibroblasts. Reducing Brf1 abundance may be a common means of restricting pol III output following terminal differentiation.
Erk contributes to the induction of Brf1 in cardiomyocytes: thus, Erk activation is sufficient to increase the level of Brf1 mRNA and protein, whereas Erk inhibition attenuates induction of Brf1. By raising expression of this limiting factor, Erk will activate the cell's capacity for pol III transcription. In contrast, Brf1 levels are not influenced by Erk activity in fibroblasts (Scott et al, 2001; Felton-Edkins et al, 2003). In fact, this is the first evidence for Erk-mediated changes in Brf1 abundance. The mechanistic basis of these cell-type-specific effects is currently unclear, and it remains to be determined whether this effect is unique to cardiomyocytes, or is more generally employed for controlling pol III output.
The activation of pol I transcription is essential for cardiomyocyte growth (Brandenburger et al, 2001). Given that pol III transcribes complementary components of the protein synthetic machinery, and that numerous regulatory mechanisms are involved in ensuring the appropriate expression of class III genes, it is highly probable that increased transcription by pol III will also be a critical feature of cardiomyocyte hypertrophy, and indeed cell growth in general. This will be tested by specifically preventing the hypertrophy-associated increase in pol III transcription, and analysing how this affects cell growth.
Materials and methods
Cell culture
Rat 1A fibroblasts were maintained as described previously (Felton-Edkins et al, 2003) and incubated in serum-free medium for 24 h before stimulation with 10% FCS. Cardiomyocytes were dissociated from the ventricles of neonatal Sprague–Dawley rats using multiple rounds of enzymatic digestion with 0.6 mg/ml pancreatin (Sigma) and 0.48 mg/ml collagenase (Worthington) in 20 mM HEPES (pH 7.35), 120 mM NaCl, 0.8 mM NaH2PO4, 5 mM glucose, 5.4 mM KCl, 0.8 mM MgSO4, 100 U/ml penicillin and 100 μg/ml streptomycin. To deplete the myocytes of fibroblasts, cells were preplated in DMEM/Medium-199 (M199) (4:1 (v/v)) supplemented with 10% (v/v) horse serum (HS), 5% (v/v) heat-inactivated FCS, penicillin (100 U/ml) and streptomycin (100 μg/ml). Myocytes were then plated onto six- or 24-well culture dishes precoated with 1% (w/v) gelatin at a density of 103 cells/mm2. After 24 h, confluent myocytes were beating spontaneously. Cells were then washed in the same media supplemented with 4% HS and without FCS, and then maintained in this medium until use. For experiments, serum was withdrawn for 24 h before treatment with 10% FCS, 100 nM ET-1, 100 μM PE (Sigma), 50 μM PD98059 or 64 μM c-Myc inhibitor (10058-F4; Calbiochem) for the indicated times.
Northern blotting
Total cellular RNA was extracted from cultured cardiomyocytes using TRI reagent, according to the manufacturer's protocol (Sigma). Northern blotting, using 10 μg of RNA, was performed as previously (Cairns and White, 1998). B2 and ARPP P0 probes have been described (White et al, 1989; Hurford et al, 1997).
Animal studies
For TAC, a fixed pressure overload was obtained by surgically constricting the transverse aorta, as described (Rockman et al, 1994). Sham-operated animals underwent the identical surgical procedure, except without aortic constriction. After 2 weeks, animals were euthanized and hearts snap-frozen for subsequent analyses. Transgenic MycER mice were generated and treated as previously (Xiao et al, 2001).
RT–PCR
RNA extracted from hearts or tissue (following homogenisation in TRI reagent) was used to generate cDNAs by reverse transcription, which were then PCR-amplified as previously (Sutcliffe et al, 2000). Primers and cycling parameters have been described for 5S rRNA, pre-tRNATyr, ARPP P0 (Winter et al, 2000), pre-tRNALeu and pre-tRNAArg (Daly et al, 2005). Other primers were as follows:
ANF: 5′-ATG GGC TCC TTC TCC ATC AC-3′ and 5′-TCT TCG GTA CCG GAA GCT G-5′ to give a 455 bp product;
Brf1: 5′-CTA CTT GGT TTG CCG AAC G-3′ and 5′-TCC TCT GTC CAG CGG TGT A-3′ to give a 490 bp product;
c-Myc: 5′-CCA GTG AGG ATA TCT GGA AG-3′ and 5′-TTC ACC ATG TCT CCT CCA AG-3′ to give a 180 bp product;
Cyclin D2: 5′-TTA CCT GGA CCG TTT CTT GG-3′ and 5′-TGC TCA ATG AAG TCG TGA GG-3′ to give a 240 bp product.
U6 snRNA: 5′-GCT CGC TTC GGC AGC ACA TAT AC-3′ and 5′-TAT CGA ACG CTT CAC GAA TTT GCG-3′ to give a 96 bp product.
PCR was performed using the following cycling parameters:
ANF: 94°C for 1 min, 20–25 cycles of (94°C for 45 s, 63°C for 45 s, 72°C for 90 s), 72°C for 2 min.
Brf1: 95°C for 5 min, 21–25 cycles of (95°C for 1 min, 61°C for 30 s, 72°C for 1 min), 72°C for 5 min.
c-Myc: 95°C for 2 min, 20–23 cycles of (95°C for 30 s, 55.3°C for 30 s, 72°C for 1 min), 72°C for 5 min.
Cyclin D2: 95°C for 3 min, 23 cycles of (95°C for 30 s, 55.3°C for 30 s, 72°C for 30 s), 72°C for 5 min.
U6 snRNA: 95°C for 3 min, 25 cycles of (95°C for 1 min, 60°C for 30 s, 72°C for 1 min), 72°C for 5 min.
Western blotting
Cells were washed twice in ice-cold PBS then scraped into lysis buffer (20 mM HEPES (pH 7.8), 150 mM NaCl, 25% glycerol, 50 mM NaF, 0.2 mM EDTA, 0.5% Triton X-100, 0.5% NP-40, 10 mM β-glycerophosphate, 1 mM sodium orthovanadate, 1 mM PMSF, 1 mM DTT, 0.5 μg/ml leupeptin, 1.0 μg/ml trypsin inhibitor, 0.5 μg/ml aprotinin and 40 μg/ml bestatin). After 10 min incubation on ice, and passage through a 26-G needle three times, the lysates were cleared by centrifugation at 4°C for 10 min. Protein (75 μg) was resolved by SDS–PAGE on 7.8% polyacrylamide gels, and specific proteins detected by Western blotting as described (White et al, 1995). RB antibody C-15, cyclin D1 antibody 72-13G, TBP antibody 58C9, c-Myc antibody 9E10 and actin antibody C11 were from Santa Cruz Biotechnology. Antibodies against Erk and phosphorylated forms of RB were from Cell Signalling Technology. Peptide antisera against Brf1 (128), Bdp1 (2663), TFIIIC220 (Ab7) and TFIIIC110 (4286) have been described previously (Shen et al, 1996; Cairns and White, 1998; Sutcliffe et al, 2000; Fairley et al, 2003). Peptide antisera against pol III subunits BN51 (2308) and C39 (2307) were raised by immunizing rabbits with synthetic peptides SEGNAAGRPARQGPD (human BN51 residues 2–16) and PPDADPVEIENRIIE (human C39 residues 10–24), respectively. Co-immunoprecipitation experiments demonstrate the specificity of these pol III antibodies (see Supplementary data).
Transcription assays
Whole-cell extracts were prepared as described by White et al (1995) and 20 μg used to transcribe plasmid DNA in vitro as previously (White et al, 1989), except pBR322 was not included and the incubations were for 1 h at 30°C. The plasmids used have been described (White et al, 1995).
ChIP assays
ChIP assays were performed as described previously (Gomez-Roman et al, 2003). Antibodies used in this assay against Brf1, Bdp1, TFIIIC110 and 220 are described above (under section Western blotting). Antibody C18 (Santa Cruz Biotechnology) was used against TFIIB. Antibody 1900 (Fairley et al, 2003) was used against pol III subunit RPC155. The specificity of this pol III antibody is demonstrated in the Supplementary Figure and in Fairley et al, 2005. Immunoprecipitated DNA was quantitated by PCR using the indicated primers (described above under section RT–PCR).
Cardiomyocyte volume and number analysis
Cardiomyocytes were dissociated from dishes using trypsin, then resuspended in DMEM/M199 (4:1) supplemented with 10% FCS, 100 U/ml penicillin and 100 μg/ml streptomycin. Cells were diluted in Isoton II (Beckman Coulter) to give 20 000 cells/ml, and analysed using a Coulter Z2 particle count and size analyser (Beckman Coulter) with a 100 μm aperture.
DNA and protein synthesis assays
Cardiomyocytes were grown on 24-well plates and treated as indicated (3 wells/condition). To monitor DNA synthesis, thymidine incorporation was measured as described by Scott et al (2001), except [3H]thymidine was added to cells 16 h before harvesting. To measure protein synthesis, 5 μCi of [35S]methionine/cysteine was added per ml of medium 1 h before harvesting. Rates of protein synthesis were assayed by measuring the incorporation of radiolabel into acid-insoluble material, as described previously (Welsh and Proud, 1992). Data were normalised for cell number.
Replication-deficient adenovirus production and infection
GFP-tagged replication-deficient adenovirus expressing HA-tagged human Brf1 was constructed using the method of He et al (1998). Briefly, an HindIII/XbaI fragment of pcDNA3HA.Brf1 was ligated into the HindIII and XbaI sites of pAdTrackCMV. Recombinant adenovirus was then generated by bacterial homologous recombination between the resulting pAdTrackCMV-HA.Brf1 and pAdEasy-1. Virus was amplified and purified in HEK293 cells, as described previously (He et al, 1998). Cardiomyocytes were infected at an m.o.i of 1.5 in 2 ml of fresh serum-free medium. Cells were incubated for 4 h, then serum-free medium was replaced with fresh medium. After 24 h, cells were serum-starved or stimulated with 10% FCS for 16 h. Infectivity was approximately 85%, as determined by GFP expression. Replication-deficient adenovirus expressing CAMEK (derived from rabbit MEK) was generated and used to infect cardiomyocytes as described previously (Bueno et al, 2000). Adenovirus expressing human c-Myc was prepared as described previously (MacLellan et al, 2000), and used to infect cardiomyocytes at an m.o.i. of 50. Cells were cultured in serum-free medium for an additional 48 h before harvest.
Quantification and statistical analysis of data
Data were quantified by densitometry using TotalLab (photoretix) version 1.11. Statistical analyses were performed using Student's t-tests (two-tailed distribution, unequal variance). A P-value of less than 0.05 was taken as a statistically significant difference between two groups.
Supplementary Material
Supplementary Methods
Supplementary Figure
Acknowledgments
We thank Jeffery Molkentin for the CAMEK-expressing adenovirus, and Arnie Berk for antibody Ab7. This work has been supported by Project Grants 062625 and 068710 from the Wellcome Trust, C1288 from Cancer Research UK and C19792 from the Biotechnology and Biological Sciences Research Council. WRM was supported by gifts from the Laubisch Fund as well as an NIH R01 HL62448.
References
- Alzuherri HM, White RJ (1998) Regulation of a TATA-binding protein-associated factor during cellular differentiation. J Biol Chem 273: 17166–17171 [DOI] [PubMed] [Google Scholar]
- Bladon TS, Frégeau CJ, McBurney MW (1990) Synthesis and processing of small B2 transcripts in mouse embryonal carcinoma cells. Mol Cell Biol 10: 4058–4067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogoyevitch MA, Glennon PE, Andersson MB, Clerk A, Lazou A, Marshall CJ, Parker PJ, Sugden P (1994) Endothelin-1 and fibroblast growth factors stimulate the mitogen-activated protein kinase signaling cascade in cardiac myocytes. J Biol Chem 269: 1110–1119 [PubMed] [Google Scholar]
- Brandenburger Y, Jenkins A, Autelitano DJ, Hannan RD (2001) Increased expression of UBF is a critical determinant of rRNA synthesis and hypertrophic growth of cardiac myocytes. FASEB J 15: 2051–2053 [DOI] [PubMed] [Google Scholar]
- Bueno OF, De Windt LJ, Tymitz KM, Witt SA, Kimball TR, Klevitsky R, Hewett TE, Jones SP, Lefer DJ, Peng C, Kitis RN, Molkentin JD (2000) The MEK1-Erk1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J 19: 6341–6350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueno OF, Molkentin JD (2002) Involvement of extracellular signal-regulated kinases 1/2 in cardiac hypertrophy and cell death. Circ Res 91: 776–781 [DOI] [PubMed] [Google Scholar]
- Busk PK, Bartkova J, Strøm CC, Wulf-Andersen L, Hinrichsen R, Christoffersen TEH, Latella L, Bartek J, Haunsø S, Sheikh SP (2002) Involvement of cyclin D in left ventricular hypertrophy in vivo and in vitro. Cardiovasc Res 56: 64–75 [DOI] [PubMed] [Google Scholar]
- Cairns CA, White RJ (1998) P53 is a general repressor of RNA polymerase III transcription. EMBO J 17: 3112–3123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien KR, Knowlton KU, Zhu H, Chien S (1991) Regulation of cardiac gene expression during myocardial growth and hypertrophy: molecular studies of an adaptive physiological response. FASEB J 5: 3037–3046 [DOI] [PubMed] [Google Scholar]
- Chu WM, Wang Z, Roeder RG, Schmid CW (1997) RNA polymerase III transcription repressed by RB through its interactions with TFIIIB and TFIIIC2. J Biol Chem 272: 14755–14761 [DOI] [PubMed] [Google Scholar]
- Daly NL, Arvanitis DA, Fairley JA, Gomez-Roman N, Morton JP, Graham SV, Spandidos DA, White RJ (2005) Deregulation of RNA polymerase III transcription in cervical epithelium in response to high-risk human papillomavirus. Oncogene 24: 880–888 [DOI] [PubMed] [Google Scholar]
- Fairley JA, Kantidakis T, Kenneth NS, Intine RV, Maraia RJ, White RJ (2005) Human La is found at RNA polymerase III-transcribed genes in vivo. Proc Natl Acad Sci USA 102: 18350–18355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairley JA, Scott PH, White RJ (2003) TFIIIB is phosphorylated, disrupted and selectively released from tRNA promoters during mitosis in vivo. EMBO J 22: 5841–5850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felton-Edkins ZA, Fairley JA, Graham EL, Johnston IM, White RJ, Scott PH (2003) The mitogen-activated protein (MAP) kinase Erk induces tRNA synthesis by phosphorylating TFIIIB. EMBO J 22: 2422–2432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glennon PE, Kaddoura S, Sale EM, Sale GJ, Fuller SJ, Sugden P (1996) Depletion of mitogen-activated protein kinase using an antisense oligodeoxynucleotide approach downregulates the phenylephrine-induced hypertrophic response in rat cardiac myocytes. Circ Res 78: 954–961 [DOI] [PubMed] [Google Scholar]
- Gomez-Roman N, Grandori C, Eisenman RN, White RJ (2003) Direct activation of RNA polymerase III transcription by c-Myc. Nature 421: 290–294 [DOI] [PubMed] [Google Scholar]
- Hannan RD, Jenkins A, Jenkins AK, Brandenburger Y (2003) Cardiac hypertrophy: a matter of translation. Clin Exp Pharm Physiol 30: 517–527 [DOI] [PubMed] [Google Scholar]
- Hannan RD, Luyken J, Rothblum LI (1995) Regulation of rDNA transcription factors during cardiomyocyte hypertrophy induced by adrenergic agents. J Biol Chem 270: 8290–8297 [DOI] [PubMed] [Google Scholar]
- Hannan RD, Luyken J, Rothblum LI (1996) Regulation of ribosomal DNA transcription during contraction-induced hypertrophy of neonatal cardiomyocytes. J Biol Chem 271: 3213–3220 [DOI] [PubMed] [Google Scholar]
- Hannan RD, Rothblum LI (1995) Regulation of ribosomal DNA transcription during neonatal cardiomyocyte hypertrophy. Cardiovasc Res 30: 501–510 [PubMed] [Google Scholar]
- He T-C, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B (1998) A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA 95: 2509–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurford RK, Cobrinik D, Lee M-H, Dyson N (1997) pRB and p107/p130 are required for the regulated expression of different sets of E2F responsive genes. Genes Dev 11: 1447–14639192872 [Google Scholar]
- Iijima Y, Laser M, Shiraishi H, Willey CD, Sundaravadivel B, Xu L, McDermott PJ, Kuppuswamy D (2002) c-Raf/MEK/Erk pathway controls protein kinase C-mediated p70S6K activation in adult cardiac muscle cells. J Biol Chem 277: 23065–23075 [DOI] [PubMed] [Google Scholar]
- Larminie CGC, Cairns CA, Mital M, Martin K, Kouzarides T, Jackson SP, White RJ (1997) Mechanistic analysis of RNA polymerase III regulation by the retinoblastoma protein. EMBO J 16: 2061–2071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP (1990) Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med 322: 1561–1566 [DOI] [PubMed] [Google Scholar]
- Li F, Wang X, Capasso JM, Gerdes AM (1996) Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J Mol Cell Cardiol 28: 1737–1746 [DOI] [PubMed] [Google Scholar]
- Luyken J, Hannan RD, Cheung JY, Rothblum LI (1996) Regulation of rDNA transcription during endothelin-1 induced hypertrophy of neonatal cardiomyocytes: hyperphosphorylation of upstream binding factor, an rDNA transcription factor. Circ Res 78: 354–361 [DOI] [PubMed] [Google Scholar]
- MacLellan WR, Xiao G, Abdellatif M, Schneider MD (2000) A novel Rb- and p300-binding protein inhibits transactivation by myoD. Mol Cell Biol 20: 8903–8915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott PJ, Carl LL, Conner KJ, Allo SN (1991) Transcriptional regulation of ribosomal RNA synthesis during growth of cardiac myocytes in culture. J Biol Chem 266: 4409–4416 [PubMed] [Google Scholar]
- Mittnacht S (2005) The retinoblastoma protein—from bench to bedside. Eur J Cell Biol 84: 97–107 [DOI] [PubMed] [Google Scholar]
- Nozato T, Ito H, Watanabe M, Ono Y, Adachi S, Tanaka H, Hiroe M, Sunamori M, Marumo F (2001) Overexpression of cdk inhibitor p16INK4a by adenovirus vector inhibits cardiac hypertrophy in vitro and in vivo: a novel strategy for the gene therapy of cardiac hypertrophy. J Mol Cell Cardiol 33: 1493–1504 [DOI] [PubMed] [Google Scholar]
- Rockman HA, Wachhorst SP, Mao L, Ross J Jr (1994) ANG II receptor blockade prevents ventricular hypertrophy and ANF gene expression with pressure overload in mice. Am J Physiol 266: H2468–H2475 [DOI] [PubMed] [Google Scholar]
- Sadoshima J, Aoki H, Izumo S (1997) Angiotensin II and serum differentially regulate expression of cyclins, activity of cyclin-dependent kinases, and phosphorylation of retinoblastoma gene product in neonatal rat cardiac myocytes. Circ Res 80: 228–241 [DOI] [PubMed] [Google Scholar]
- Schramm L, Hernandez N (2002) Recruitment of RNA polymerase III to its target promoters. Genes Dev 16: 2593–2620 [DOI] [PubMed] [Google Scholar]
- Scott PH, Cairns CA, Sutcliffe JE, Alzuherri HM, McLees A, Winter AG, White RJ (2001) Regulation of RNA polymerase III transcription during cell cycle entry. J Biol Chem 276: 1005–1014 [DOI] [PubMed] [Google Scholar]
- Sethy I, Moir RD, Librizzi M, Willis IM (1995) In vitro evidence for growth regulation of tRNA gene transcription in yeast. J Biol Chem 270: 28463–28470 [DOI] [PubMed] [Google Scholar]
- Shen Y, Igo M, Yalamanchili P, Berk AJ, Dasgupta A (1996) DNA binding domain and subunit interactions of transcription factor IIIC revealed by dissection with poliovirus 3C protease. Mol Cell Biol 16: 4163–4171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soonpaa MH, Kim KK, Pajak L, Franklin M, Field LJ (1996) Cardiomyocyte DNA synthesis and binucleation during murine development. Am J Physiol 271: H2183–H2189 [DOI] [PubMed] [Google Scholar]
- Starksen NF, Simpson P, Bishopric N, Coughlin SR, Lee WMF, Escobedo JA, Williams LT (1986) Cardiac myocyte hypertrophy is associated with c-myc protooncogene expression. Proc Natl Acad Sci USA 83: 8348–8350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutcliffe JE, Brown TRP, Allison SJ, Scott PH, White RJ (2000) Retinoblastoma protein disrupts interactions required for RNA polymerase III transcription. Mol Cell Biol 20: 9192–9202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamori M, Ito H, Hiroe M, Terada Y, Marumo F, Ikeda M (1998) Essential roles for G1 cyclin-dependent kinase activity in development of cardiomyocyte hypertrophy. Am J Physiol 275: H2036–H2040 [DOI] [PubMed] [Google Scholar]
- Tamamori-Adachi M, Ito H, Nobori K, Hayashida K, Kawauchi J, Adachi S, Ikeda MA, Kitajima S (2002) Expression of cyclin D1 and CDK4 causes hypertrophic growth of cardiomyocytes in culture: a possible implication for cardiac hypertrophy. Biochem Biophys Res Comm 296: 274–280 [DOI] [PubMed] [Google Scholar]
- Wang L, Gout I, Proud CG (2001) Cross-talk between the Erk and p70 S6 kinase (S6 K) signalling pathways: MEK-dependent activation of S6K2 in cardiomyocytes. J Biol Chem 276: 32670–32677 [DOI] [PubMed] [Google Scholar]
- Wang L, Proud CG (2002a) Ras/Erk signaling is essential for activation of protein synthesis by Gq protein-coupled receptor agonists in adult cardiomyocytes. Circ Res 91: 821–829 [DOI] [PubMed] [Google Scholar]
- Wang L, Proud CG (2002b) Regulation of the phosphorylation of elongation factor 2 by MEK-dependent signalling in adult rat cardiomyocytes. FEBS Lett 531: 285–289 [DOI] [PubMed] [Google Scholar]
- Welsh GI, Proud CG (1992) Regulation of protein synthesis in Swiss 3T3 fibroblasts. Rapid activation of the guanine-nucleotide-exchange factor by insulin and growth factors. Biochem J 284: 19–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RJ (2005) RNA polymerases I and III, growth control and cancer. Nat Rev Mol Cell Biol 6: 69–78 [DOI] [PubMed] [Google Scholar]
- White RJ, Gottlieb TM, Downes CS, Jackson SP (1995) Mitotic regulation of a TATA-binding-protein-containing complex. Mol Cell Biol 15: 1983–1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RJ, Stott D, Rigby PWJ (1989) Regulation of RNA polymerase III transcription in response to F9 embyonal carcinoma stem cell differentiation. Cell 59: 1081–1092 [DOI] [PubMed] [Google Scholar]
- White RJ, Trouche D, Martin K, Jackson SP, Kouzarides T (1996) Repression of RNA polymerase III transcription by the retinoblastoma protein. Nature 382: 88–90 [DOI] [PubMed] [Google Scholar]
- Winter AG, Sourvinos G, Allison SJ, Tosh K, Scott PH, Spandidos DA, White RJ (2000) RNA polymerase III transcription factor TFIIIC2 is overexpressed in ovarian tumours. Proc Natl Acad Sci USA 97: 12619–12624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wool IG (1979) The structure and function of eukaryotic ribosomes. Ann Rev Biochem 48: 719–754 [DOI] [PubMed] [Google Scholar]
- Xiao G, Mao S, Baumgarten G, Serrano J, Jordan MC, Roos KP, Fishbein MC, MacLellan WR (2001) Inducible activation of c-Myc in adult myocardium in vivo provokes cardiac myocyte hypertrophy and reactivation of DNA synthesis. Circ Res 89: 1122–1129 [DOI] [PubMed] [Google Scholar]
- Yin X, Giap C, Lazo JS, Prochownik EV (2003) Low molecular weight inhibitors of Myc–Max interaction and function. Oncogene 22: 6151–6159 [DOI] [PubMed] [Google Scholar]
- Yue TL, Gu JL, Wang C, Reith AD, Lee JC, Mirabile RC, Kreutz R, Wang Y, Maleef B, Parsons AA, Ohlstein EH (2000) Extracellular signal-regulated kinase plays an essential role in hypertrophic agonists, endothelin-1 and phenylephrine-induced cardiomyocyte hypertrophy. J Biol Chem 275: 37895–37901 [DOI] [PubMed] [Google Scholar]
- Zak R (1974) Development and proliferative capacity of cardiac muscle cells. Circ Res 34 and 35 (Suppl II): II-17–II-26 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods
Supplementary Figure