

Abstract
The spike protein VP4 is a key component of the membrane penetration apparatus of rotavirus, a nonenveloped virus that causes childhood gastroenteritis. Trypsin cleavage of VP4 produces a fragment, VP5*, with a potential membrane interaction region, and primes rotavirus for cell entry. During entry, the part of VP5* that protrudes from the virus folds back on itself and reorganizes from a local dimer to a trimer. Here, we report that a globular domain of VP5*, the VP5* antigen domain, is an autonomously folding unit that alternatively forms well-ordered dimers and trimers. Because the domain contains heterotypic neutralizing epitopes and is soluble when expressed directly, it is a promising potential subunit vaccine component. X-ray crystal structures show that the dimer resembles the spike body on trypsin-primed virions, and the trimer resembles the folded-back form of the spike. The same structural elements pack differently to form key intermolecular contacts in both oligomers. The intrinsic molecular property of alternatively forming dimers and trimers facilitates the VP5* reorganization, which is thought to mediate membrane penetration during cell entry.
Keywords: dimer, rearrangement, rotavirus, trimer, VP5*
Introduction
Cell entry by rotavirus is mediated by a series of molecular rearrangements and interactions that translocate a 710 Å diameter subviral particle across a cellular membrane and into the cytoplasm. The two outermost proteins of the nonenveloped virion, VP7 and VP4, are the moving parts of the translocation apparatus. VP7, a glycoprotein, forms calcium-dependent trimers, which pack together in the smooth outer shell of the virion (Yeager et al, 1990; Dormitzer and Greenberg, 1992; Prasad and Chiu, 1994). The dissociation of VP7 trimers in low calcium may underlie the controlled disassembly of the outer capsid during entry. VP4 forms spikes (Figure 1A), which protrude from the VP7 shell in electron cryomicroscopy image reconstructions of trypsin-primed virions (Shaw et al, 1993; Yeager et al, 1994). Trypsin primes rotavirus for efficient infectivity by cleaving VP4 into two fragments: VP8* and VP5* (Estes et al, 1981). VP8* forms the ‘heads' of the spikes (Dormitzer et al, 2002) and binds sialic acid in some rotavirus strains (Ciarlet et al, 2002). Together with the N-terminus of VP8*, VP5* forms the spike ‘body' (Dormitzer et al, 2004). The body is linked by an asymmetric stalk to a ‘foot,' which is buried beneath the VP7 shell (Shaw et al, 1993; Yeager et al, 1994).
Figure 1.
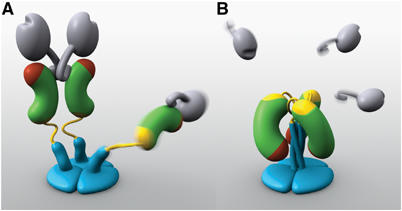
Models of two VP4 conformations. (A) The primed state. Two rigid subunits form the spike visible in electron cryomicroscopy image reconstructions of trypsin-primed virions. A third subunit is flexible. VP8* is gray, with an N-terminal tether and a globular head creased by the sialoside-binding site. The VP5* antigen domain is green bean-shape, with a red membrane interaction region and a yellow GH loop. An additional β-strand C-terminal to the antigen domain is also yellow. The spike body includes the VP5* antigen domain, part of the VP8* tether, and the GH loop. The foot is blue, as is a protruding region that rearranges into the coiled-coil. (B) The putative post-membrane penetration state. VP8* has dissociated; the yellow parts of each subunit have joined in a β-annulus; the α-helical triple coiled-coil has zipped up; and the VP5* antigen domain has folded back. The models were produced by Digizyme, Inc.
VP4 performs a series of molecular gymnastics during viral entry. Prior to trypsin cleavage, it is flexible (Crawford et al, 2001). The priming trypsin cleavage triggers its rearrangement into rigid spikes with approximate two-fold symmetry of their protruding parts (Shaw et al, 1993; Yeager et al, 1994; Figure 1A). After an unknown second triggering event, cleaved VP4 undergoes another rearrangement, in which two VP5* subunits fold back on themselves and join a third subunit to form a tightly associated trimer, shaped like a folded umbrella (Dormitzer et al, 2004; Figure 1b). VP8* probably dissociates from VP5* before or during the fold-back. The reorganization translocates the body domain's hydrophobic apex, which is a potential membrane interaction region, by at least 55 Å towards the molecule's base. The fold-back resembles those of enveloped virus membrane fusion proteins and may disrupt a cellular membrane.
The primary evidence for the trimeric, folded-back state of VP5* is a 3.2 Å resolution X-ray crystal structure of VP5CT (Figure 2A), a protease cleavage fragment of rhesus rotavirus (RRV) VP4 that consists of residues A248 to L525 or F528 (Dormitzer et al, 2004). Key structural elements holding the trimer together include a C-terminal triple α-helical coiled-coil, a nine-stranded β-annulus, and a ‘cap' of hydrophobic residues. Some electron cryomicroscopy evidence suggests that three VP4 subunits clustered at each peripentonal channel join to form the trimers. Although the molecular envelope reconstructed for the protruding part of the primed spike has approximate two-fold symmetry, the foot has apparent three-fold (or pseudo-hexameric) symmetry (Yeager et al, 1994). In addition, high pH treatment causes VP4 to coalesce into three foreshortened protrusions at each peripentonal channel (Pesavento et al, 2005). Therefore, the primed conformation of VP4 may include not only the rigid, two-fold clustered protruding spike but also a third protruding subunit, which remains flexible and, therefore, absent from averaged image reconstructions (Figure 1A).
Figure 2.
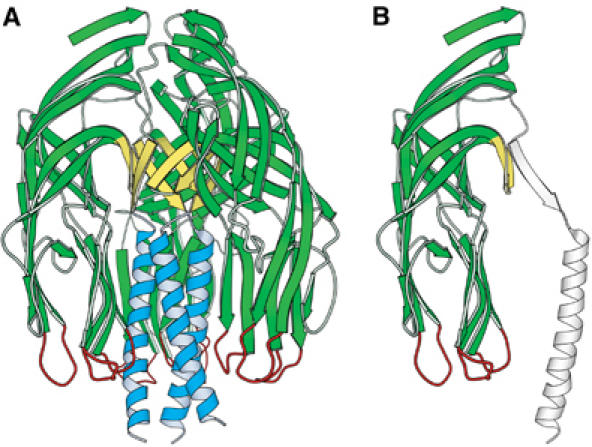
VP5CT and the VP5* antigen domain. (A) Ribbon diagram of the VP5CT trimer, colored to match Figure 1. VP5CT does not include the foot region. (B) Ribbon diagram of a single VP5CT subunit. The part that forms the VP5* antigen domain is green, yellow, and red; the remainder is drawn in outline.
Each subunit of the VP5CT trimer contains a globular domain between residues R247 and D479 (Figure 2B). This globular domain does not include the coiled-coil and is missing one of the β-strands that each subunit contributes to the β-annulus. It does include the potential membrane interaction region and all known antibody neutralization escape mutations, including mutations selected by heterotypically neutralizing antibodies, on VP5* (summarized in Dormitzer et al, 2004). This domain also includes the minimal in vitro-translated fragment immunoprecipitated by neutralizing monoclonal antibodies recognizing VP5* (Mackow et al, 1990). Therefore, we refer to this region as the ‘VP5* antigen domain.'
Because VP5CT is stable, soluble, and presents heterotypic neutralizing epitopes, it might be considered a promising antigen for use in a recombinant vaccine against rotavirus, a pathogen that kills approximately 440 000 children annually (Parashar et al, 2003). However, this fragment is produced very inefficiently by proteolysis from VP4 (Dormitzer et al, 2001). Direct expression of a fragment equivalent to VP5CT yields insoluble protein (unpublished data). Examination of the VP5CT structure suggests that its N-terminal and C-terminal parts, which make trimer contacts, may only fold properly on an intact VP4 precursor, accounting for the insolubility of the directly expressed protein. We hypothesized that an engineered domain that lacks the residues that form the coiled-coil and β-annulus might be expressed directly and efficiently as a soluble protein. We have expressed the VP5* antigen domain in insect cells and report biochemical and X-ray crystallographic analyses of the purified protein. These analyses demonstrate that this autonomously folding domain self-associates in both dimers and trimers, with many of the same structural elements making key intersubunit contacts in both states. This intrinsic molecular property, together with the ‘zipping-up' of a coiled-coil, underlies the molecular gymnastics of VP4 during cell entry.
Results and discussion
Expression and biochemical characterization
We expressed the RRV VP5* antigen domain with an N-terminal histidine tag in bacteria and insect cells. When expressed in Escherichia coli, the domain is insoluble (data not shown). When expressed in Sf9 insect cells from a recombinant baculovirus vector, the domain is soluble and readily purified by nickel affinity chromatography and size exclusion chromatography, with anion exchange chromatography interposed when higher purity is needed (Supplementary Figure 1A and B). This procedure yields up to 4 mg of purified protein from each liter of insect cell culture. The purified domain is soluble to 4 mg/ml. SDS–PAGE, mass spectrometry, and N-terminal sequencing reveal some heterogeneity due to inconsistent cleavage of the histidine tag by adventitious proteases (Supplementary Figure 1B). The solubility of the directly expressed VP5* antigen domain suggests that further structure-based engineering may yield an optimized version of the domain that can be produced efficiently enough to be a practical immunogen for inclusion in a subunit rotavirus vaccine.
Equilibrium analytical ultracentrifugation demonstrates self-association of the domain. The domain's hydrodynamic behavior can be modeled as a dynamic equilibrium between monomers and trimers with an association constant of between 18.7 and 62.5 (Supplementary Figure 1C). The domain's equilibrium distribution is similar at 4 and 22°C and at pH 5.6 and 8.0 (Supplementary Table). The hydrodynamic data cannot rule out the possibility that a small proportion of oligomers other than trimers are also present in solution, but the simple monomer–trimer equilibrium model is sufficient to fit the observed distributions closely.
Crystal structure of the VP5* antigen domain dimer
At 25°C and pH 5.6, the VP5* antigen domain crystallizes by the hanging drop method, using 2-methyl-2,4-pentanediol (MPD) as a precipitant. When frozen, these crystals diffract X-rays coherently to 1.5 Å interplanar spacing (although anisotropy limits the resolution of useable data to 1.6 Å). We determined the structure of these crystals by molecular replacement, using VP5CT as an initial phasing model (See Materials and methods and Table I).
Table 1.
X-ray diffraction data collection and refinement statistics
| Dimer | Trimer | |
|---|---|---|
| Data collection statistics | ||
| Resolution limit (Å) | 1.6 | 2.0 |
| Unique reflections | 73654 | 55552 |
| Redundancya | 4.70 (4.36) | 4.71 (4.56) |
| Completenessa (%) | 93.6 (69.9) | 94.9 (97.0) |
| I/σa | 28.36 (4.54) | 26.85 (4.25) |
| Rsyma,b (%) | 4.0 (29.4) | 4.6 (40.8) |
| Refinement statistics | ||
| Polypeptide chains | 2 | 3 |
| Protein atoms | 3535 | 5542 |
| Water molecules | 418 | 488 |
| MPD molecules | 4 | 0 |
| Tris molecules | 1 | 0 |
| Residues in allowed regions of Ramachandran plot (%) | 100 | 100 |
| Residues in most favored regions of Ramachandran plot (%) | 91.3 | 89 |
| RMSD bond lengths (Å) | 0.012 | 0.0078 |
| RMSD bond angles (deg) | 1.61 | 1.50 |
| Mean B value (Å2) | 32.0 | 40.1 |
| RMSD main chain B (Å2) | 2.20 | 1.52 |
| Resolution range (Å) | 45.5–1.6 | 50–2.0 |
| R-factorc | 20.3 | 22.2 |
| Free R-factorc | 22.5 | 25.2 |
| aValues for last shell given in parentheses. | ||
| bRsym=∑(I−〈I〉)/I. 〈I〉 is the average intensity over symmetry equivalent reflections. | ||
| cR-factor=(∑∣∣Fobs∣−∣Fcalc∣∣)/∑∣Fobs∣, where the summation is over the working set of reflections. For the free R-factor, the summation is over the test set of reflections (5% of the total reflections). | ||
This VP5* antigen domain crystal structure reveals a dimer. Figure 3A shows the dimer in the orientation of the spike on a trypsin-primed virion. In this orientation, the heads would be located above the diagram, and the foot would be buried under the VP7 shell below the diagram. The dimer has maximal dimensions of 78 Å (height), 45 Å (width), and 30 Å (depth), as measured on a Cα trace. The N- and C-termini of each subunit are at the bottom, and three loops (B′C′, D′E′, and F″G′) with hydrophobic tips are at the top. In the dimer, the F″G′ loop, which resembles the Semliki Forest virus fusion loop in primary sequence (Mackow et al, 1988), is distal to the approximate two-fold axis, and the B′C′ and D′E′ loops are adjacent to this axis. Like the protruding VP4 dyad on virions (Shaw et al, 1993; Yeager et al, 1994), the crystallized antigen domain dimer is slightly asymmetrical. A highly ordered molecule of MPD (not shown) is bound between the sheets of the β-sandwich of the green subunit in Figure 3A, but is absent from the blue subunit. In solution at pH 5.6 and 22°C, the antigen domain is in a dynamic equilibrium, with monomers and trimers as the main species (Supplementary Table). The change in solvent characteristics caused by MPD and glycerol in the crystallization mother liquor or the asymmetry introduced by the insertion of an ordered MPD molecule into some molecules may alter the equilibrium in favor of dimer formation.
Figure 3.
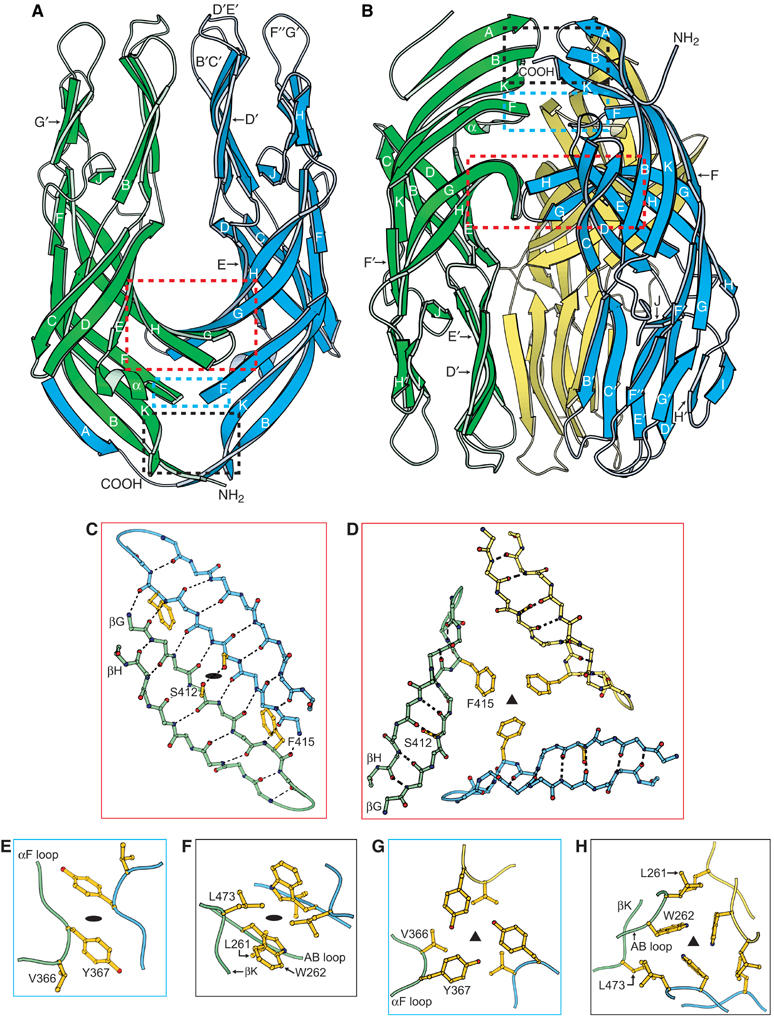
Crystal structures of the VP5* antigen domain dimer and trimer. (A) Ribbon diagram of the dimer. The orientation matches the model of the rigid spikes on the virion in Figure 1A. The red, blue, and black boxes show the regions detailed in panels (C), (E), and (F), respectively. The termini of the green subunit are indicated. Secondary structural elements, including the three hydrophobic loops of the blue subunit are labeled. (B) Ribbon diagram of the trimer. The orientation matches the model of the rearranged trimer in Figure 1B. The red, blue, and black boxes show the regions detailed in panels (D), (G), and (H), respectively. The termini of the blue subunit are indicated. (C, E, F) Atomic details of the key intersubunit contacts of the dimer. Black ovals indicate the approximate two-fold axis. In panel (F), the L261 side chain is below the W262 side chain. (D, G, H) Atomic details of the key intersubunit contacts of the trimer. Black triangles indicate the three-fold axis. In panel (H), the W262 rings are seen on-edge with the five-atom pyrrole ring closest to the viewer. Panels (C)–(H) are drawn as if looking down from the tops of the ribbon diagrams in panels (A) and (B). Panels (C) and (D), panels (E) and (G), and panels (F) and (H) are pairs, showing alternative packing of the same residues. The depicted side chains are discussed in the text. Dashed black lines indicate hydrogen bonds.
The contacts between subunits at the top of the dimer are insubstantial—salt bridges between E293 of one subunit and R341 of the other subunit and a hydrogen bond between the hydroxyl of S293 and its counterpart across the approximate two-fold axis (not shown). Instead, the dimer is primarily held together by extensive two-fold interactions near the bottom of the structure. These contacts include an intersubunit β-sheet (red box in Figure 3A and C) and a hydrophobic core (blue and black boxes in Figure 3A, E, and F). The central strands of the intersubunit β-sheet (strand G from each subunit) share eight backbone amide-to-backbone carbonyl hydrogen bonds and an additional hydrogen bond between the S412 side chain and its symmetry mate (Figure 3C). This interaction creates a continuous 10-stranded β-sheet (CDEHGGHEDC), which forms a saddle across the dimer interface (Figure 3A).
The hydrophobic core on the two-fold axis is below the new β-sheet. One dimer contact is made by the aromatic ring of Y367 stacking against its symmetry mate (Figure 3E). Below this, two W262 aromatic rings pack tangentially against each other, separated at their distal ends by a sandwiched pair of L473 side chains (Figure 3F). At the bottom of the structure, the L261 side chain contacts its symmetry mate (Figure 3F). An additional intersubunit contact, formed by the N-terminus of one subunit (blue in Figure 3A) crossing over to pack as strand A against the other subunit, is discussed below under ‘Comparison of VP5* subunit structures.' The dimer contacts bury 1846 Å2 (15.2%) of the surface of each subunit. Excluding the crossed-over N-terminus, 1371 Å2 (11.9%) are buried.
Fit of the dimer to the body of the primed VP4 spike
A 12 Å resolution electron cryomicroscopy image reconstruction of a trypsin-primed rotavirus virion provides a molecular envelope of the dimeric, protruding region of cleaved VP4 (Figure 4). Previously, we described the ‘in silico' trimming and reorientation of two subunits of the VP5CT trimer to model a potential fit of a hypothesized dimeric arrangement of the subunits to this molecular envelope (Dormitzer et al, 2004). In that fit, the trimmed subunits were arranged as a parallel dyad (although they did not contact each other), with the hydrophobic apices occupying the ‘shoulders' of the spike body and each F″G′ loop located distal to the approximate two-fold axis (Supplementary Figure 2). The dimeric crystal structure demonstrates that the VP5* antigen domain actually forms a dimer with parallel subunits in which the F″G′ loop of each subunit is distal to the two-fold axis (Figure 3A). The VP5* antigen domain dimer fits the electron cryomicroscopy envelope in the orientation reported for the models carved from the VP5CT structure (Figure 4). The gap between the subunits of the dimer in the crystal structure corresponds to the hole in the spike body, the hydrophobic apices correspond to the ‘shoulders,' and the C-termini extend into the stalk to connect with the buried foot domain.
Figure 4.
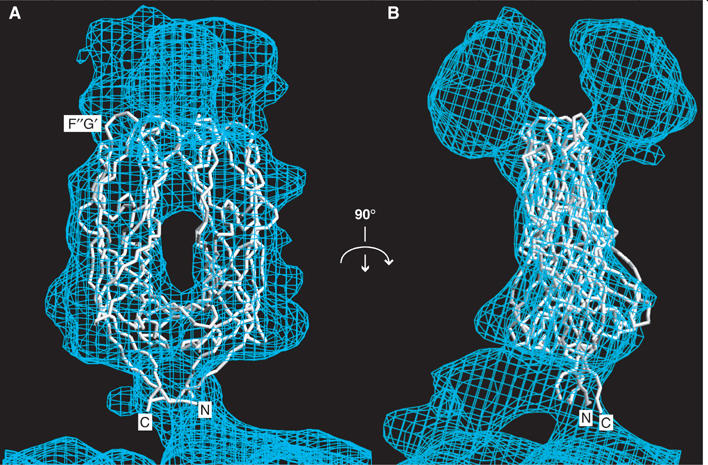
The VP5* antigen domain dimer fit to the molecular envelope of the primed spike. The molecular envelope of an approximately 12 Å resolution electron cryomicroscopy image reconstruction of a VP4 spike on a trypsin-primed SA11-4F rotavirus virion is contoured at 0.5 σ. (A) Depicted from the perspective of Figures 1A and 3A. The Cα trace includes residues T259-N477 of one subunit (on the left) and residues T259-S476 of the second subunit (on the right). The termini and the F″G′ loop of one subunit are indicated. (B) Rotated 90° about a vertical axis from panel (A).
Despite the good overall match of the dimeric crystal structure to the electron cryomicroscopy envelope, the fit is imperfect, with 25.6% of the atoms protruding beyond an envelope contoured at 0.5σ (Figure 4). In the previously reported fit of trimmed VP5CT subunits (Supplementary Figure 2, Dormitzer et al, 2004), which were not constrained in their orientation relative to each other by dimer contacts, 18.9% of the atoms protruded. Relative to the equivalent parts of the trimmed VP5CT subunits, the hydrophobic apices of the VP5* antigen domain dimer extend further beyond the spike shoulders. The dimer subunits are closer to the approximate dyad axis of the molecular envelope, leaving the lateral parts unfilled. Although not apparent from the perspectives in Figure 4, the flexible tip (with high thermal parameters) of each GH hairpin of the dimer protrudes from the molecular envelope. A shift in the GH hairpin (described under ‘Comparison of VP5* subunit structures') from its position in VP5CT to form the central strands of the intersubunit β-sheet in the dimer fills in some of the envelope at the base of the body on the approximate dyad axis. The fit of the dimeric crystal structure to the molecular envelope could be improved if the dimer were extended about the ‘joint' formed by its proximal two-fold contact so that the apices separated from each other. This distortion would produce a better match to the previously reported independent fit of each electronically trimmed subunit from VP5CT (Supplementary Figure 2).
The imperfect match between the dimeric crystal structure and the molecular envelope of the primed spike probably reflects the conformational effects of interactions of the VP5* antigen domain with VP8* and with the stalk and foot regions of VP5* on the virion. In the primed spike, approximately 60 N-terminal residues of each VP8* fragment tether each head to the body (Figure 1A; Dormitzer et al, 2004). Many of these residues probably insert between the two hydrophobic VP5* apices to form the distal dyad contact of the spike body. The alternative possibility, that the VP8* N-termini fill the lateral part of the electron cryomicroscopy envelope, is less likely, as antibody neutralization escape mutations are broadly distributed over the surface of the VP5* antigen domain, indicating surface exposure on the virion (Dormitzer et al, 2004). Insertion of the N-terminal residues of VP8* into the insubstantial distal two-fold contact of the dimeric crystal structure would force the VP5* apices apart, producing the extension about the proximal two-fold contact that improves the fit to the envelope. Because the VP8* N-terminus forms the distal two-fold contact and tethers the heads to the body, VP8* probably either dissociates from the folded back, trimeric form of VP5* or has a substantially altered association. Early dissociation of the heads could allow the VP5* antigen domains to flex about their proximal two-fold contact and achieve the conformation in the dimeric crystal structure prior to trimerizing and folding back.
Crystal structure of the VP5* antigen trimer
During storage at 8°C at pH 8.0, the VP5* antigen domain crystallizes in batch without added precipitant. These crystals, when frozen, diffract X-rays coherently to 2 Å interplanar spacing. We determined the structure of these crystals by molecular replacement, using VP5CT as an initial phasing model (see Materials and methods and Table I).
The structure reveals a VP5* antigen domain trimer (Figure 3B). Like the VP5CT trimer (Figure 2A), the VP5* antigen domain trimer is shaped like an umbrella. Unlike the VP5CT trimer, the VP5* antigen domain trimer lacks the coiled-coil or β-annulus that form the umbrella's ‘post' due to the C-terminal truncation of the recombinant construct. In the VP5* antigen domain trimer, 1286 Å2 (11.0%) of the surface of each subunit are buried. The additional three-fold contacts of VP5CT raise its buried surface area to 3956 Å2 (25.8%). The tips of the globular ‘shades' of the VP5* antigen domain umbrella approach each other more closely than those of VP5CT because they are not separated by a coiled-coil ‘post.' Except for the position of the GH loop (discussed under ‘Comparison of VP5* subunit structures'), the globular regions of the VP5* antigen domain and VP5CT trimers are essentially identical (RMSD of 0.76 Å for Cα of residues Y267-T410 and S423-L470).
The fold-back rearrangement of VP5* (Figure 1) inverts the antigen domain of the trimer relative to the dimer. Correspondingly, in Figure 3, the dimer and the trimer are both depicted with the probable position of the foot at the bottom, but the subunits of the trimer are inverted relative to those of the dimer. In Figure 3B, the trimer is presented with each subunit's termini at the top and hydrophobic apex at the bottom. The maximal dimensions of the trimer are 74 Å (height) and 33 Å (radius), as measured on a Cα trace.
The view of the blue subunit in Figure 3B displays features that are apparent at the 1.6 and 2.0 Å resolutions of the VP5* antigen domain structures, but not at the 3.2 Å resolution of the VP5CT structure (Dormitzer et al, 2004). Specifically, adjacent pairs of glycines interrupt the β-structure of the F′G loop, so that strand F′ from the VP5CT structure is divided into strands F′ and F″ (by G382 and G383) in the antigen domain structures, and strand G is divided into strands G′ and G (by G399 and G400). The more radial limb of the shortest loop at the bottom of the trimer structure (the loop that includes strand H′) had weak electron density in the VP5CT maps and was modeled as a coil (Figure 2A). In the new maps, these residues have strong density and form β-strand I (Figure 3B). Accordingly, VP5CT strand I becomes strand J, and VP5CT strand J becomes strand K in the antigen domain structures.
The antigen domain trimer is held together by a hydrophobic core centered on the three-fold axis near the top of the structure. The packing of this hydrophobic core is the same in the VP5* antigen domain trimer and in VP5CT. Three main levels of hydrophobic interactions are apparent (red, blue, and black boxes in Figure 3B). At the bottom level (red box in Figure 3A and D), the F415 aromatic ring of each subunit packs about the three-fold axis and forms the base of a large solvent filled cavity (volume 390 Å3; Supplementary Figure 3). This cavity is also present in VP5CT and communicates with the molecule's exterior through narrow channels. During entry, it could allow room for movements associated with molecular rearrangements. The ceiling of the cavity is formed by Y367 aromatic rings (blue box in Figure 3B and G), which pack about the three-fold axis, reinforced by interdigitating V366 side chains. Above this (black box in Figure 3B and H), a tight ‘propeller' of three W262 aromatic side chains is held together by interactions between the hydrogen bound to each pyrrole nitrogen and the π electrons of the adjacent indole ring. The propeller is reinforced by interdigitating L473 side chains. The packing of each L261 side chain against the L473 side chain of the adjacent subunit caps the hydrophobic core (Figure 3H).
The structure of the VP5* antigen domain trimer has several implications for VP4 rearrangements. First, near the bottom of the structure, the subunits do not interact. In VP5CT, the interactions between the lower portions of each domain and the coiled-coil are polar. Thus, the lack of a central coiled-coil in the VP5* antigen domain trimer does not expose hydrophobic patches. The overall hydrophilicity of the sides (but not the apex) of the globular domain allows for its free rotation through solvent during the fold-back translocation. Second, alternative packing of the same residues in the dimer and the trimer allows the formation of two well-ordered oligomers. Residues L261, W262, Y367, and L473 make key hydrophobic contacts about both the two-fold axis of the dimer (Figure 3E and F) and the three-fold axis of the trimer (Figure 3G and H). Other residues, such as V366 (Figure 3E and G) and F415 (Figure 3C and D), only make intersubunit contacts in the trimer.
Comparison of VP5* subunit structures
Superposition of a subunit from the VP5* antigen domain dimer on a subunit from the VP5CT trimer (Figure 5) demonstrates that most of the globular domain remains rigid during the two- to three-fold reorganization (RMSD 0.85 Å for Cα of residues D270-Q360, A372-T410, and S423-L470). However, the GH loop rotates by approximately 61° relative to the rest of the domain, displacing its tip (residue D417) by 18.4 Å (two-headed arrow in Figure 5). In the dimer, the GH loop forms the central strands of the intersubunit β-sheet (Figure 3C); in the VP5CT trimer, this loop forms six of nine strands of the intersubunit β-annulus (Figure 2A). The shift of the loop disrupts the intersubunit β-sheet of the dimer and allows the F415 aromatic rings to pack around the three-fold axis of the trimer (Figure 3D). In VP5CT, hydrogen bonds in the β-annulus between strand G and an additional β-strand formed by residues just N-terminal to the coiled-coil substitute for the disrupted bonds in the intersubunit β-sheet and clamp the subunits in the folded-back conformation (Dormitzer et al, 2004). The shift in the GH loop is accompanied by the partial uncoiling of an adjacent, short α-helix (labeled ‘α' in the green subunits of Figure 3A and B) between β-strands E and F. The GH loop of the VP5* antigen domain trimer does not form part of an intersubunit β-sheet or β-annulus, and its position may not model a conformation attained during the rearrangements that lead to cell entry.
Figure 5.
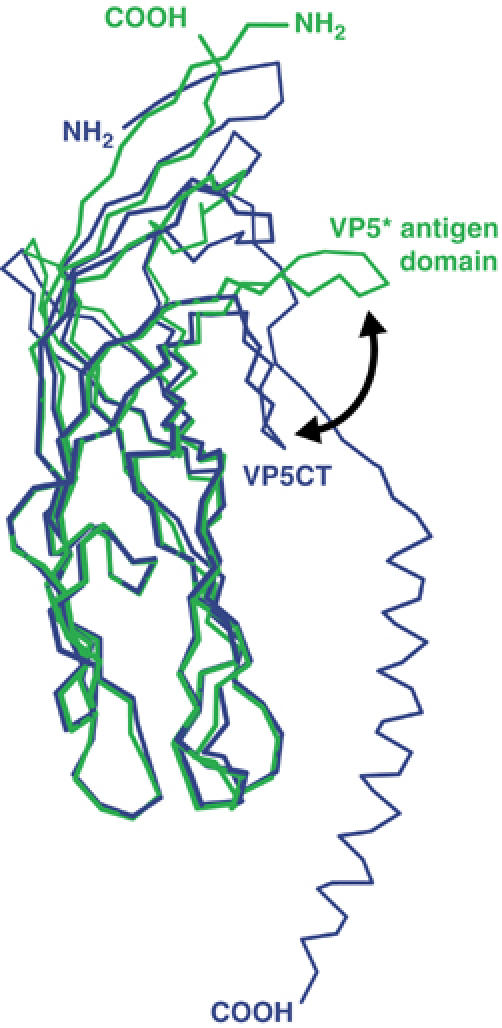
Superposition of the VP5* antigen domain and VP5CT. Residues S260-S476 of a VP5* antigen domain subunit in the dimer conformation are drawn as a green Cα trace. Residues I254-L522 of a VP5CT subunit are drawn as a blue Cα trace. The black arrow indicates the movement of the GH loop between the two states.
Comparison of the VP5* antigen domain dimer and VP5CT trimer subunits also reveals significant rearrangements of the domains' termini (Figure 5). The C-termini of the dimer and trimer point in opposite directions, reflecting the fold-back relative to the foot domain. The N-terminus of one subunit of the dimer (the blue subunit in Figure 3A) crosses over to form intersubunit β-strand A, which is hydrogen bonded to strand B of the other subunit. The equivalent residues of the other dimer subunit are either disordered or missing due to proteolysis. In the trimer, the N-terminus of each subunit folds back, so that β-strand A is hydrogen bonded to strand B of its own subunit (Figures 3B and 5). Whether strand A is exchanged or retained, the same residues on strands A and B hydrogen bond to each other in both dimer and trimer (E252–N268, I254–Q266, and V256–E264). The crossed-over strand of the dimer does not fit the molecular envelope of the primed VP4 spike (and is omitted from the Cα trace in Figure 4). Therefore, it is doubtful that the crossover occurs in the primed conformation of the spike on the virion. Trypsin cleavage of VP4 to produce an N-terminus at A248, but not an alternative N-terminus at residue N242, primes particles to infect cells and to mediate cell–cell ‘fusion from without' (Arias et al, 1996; Gilbert and Greenberg, 1998). The specificity of the priming cleavage suggests that the interactions of N-terminal residues of VP5* in the unprimed, primed, or folded-back states of the spike may have an essential role in controlling VP5* rearrangements.
Comparison to other viral entry proteins
The core of the VP5* antigen domain is an eight-stranded β-sandwich, composed of β-sheets BKF and CDEHG (Figure 3A and B). Functional appendages mounted on this core include three adjacent hydrophobic loops (B′C′, E′D′, and F″G′), which may interact with membranes, and the CD loop, which projects from the opposite end of the β-sandwich and may bind an integrin (Graham et al, 2003). The ability to form dimers or trimers alternatively is added to the core β-sandwich by residues in N- and C-terminal extensions and by the GH loop.
The fold of the core framework that bears these functional appendages is unique—the DALI structural similarity search algorithm (Holm and Sander, 1993) reveals no molecule in the Protein Data Bank with the same fold. The closest match is the reovirus σ1 knob (z score 4.1), which is very similar and probably homologous to the adenovirus fiber protein (Chappell et al, 2002). Rotavirus and reovirus are both members of the family Reoviridae. VP4 and σ1 both form viral spikes that function in attachment by binding cell surface proteins and sialic acid, albeit using different structural elements. The σ1 eight-stranded antiparallel β-barrel superimposes strikingly well on the VP5* antigen domain β-sandwich (Figure 6A), with an RMSD of 2.58 Å for 76 corresponding Cα atoms, 9.2% of which are identical. However, the two proteins have somewhat different folds. The σ1 β-barrel has a double Greek key fold (Figure 6B). Although the five C-terminal strands (E, F, G, H, and K) of the VP5* antigen domain β-sandwich share this connectivity, the three N-terminal strands (B, C, and D) are arranged in the opposite order (Figure 6B).
Figure 6.
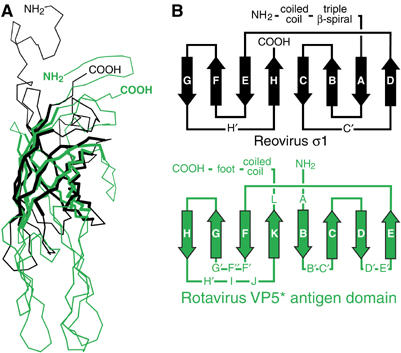
Comparison of the rotavirus VP5* antigen domain and the reovirus σ1 knob. (A) Superposition of Cα traces. Residues Y298 to T455 of reovirus σ1 (black; PDB identification code 1KKE) are superimposed on residues I254-L473 of a subunit of the rotavirus VP5* antigen domain trimer (green). The residues used to calculate the superposition matrix are drawn with thick lines. (B) Folding diagram of the β-barrel of reovirus σ1 and the β-sandwich of rotavirus VP5*. Arrows depict β-strands.
There are several possible explanations for the similarities and differences between the folds of the rotavirus VP5* antigen domain and the reovirus σ1 head. The two proteins could lack common ancestry, yet have converged on closely related folds. However, there is no known functional significance to the details of β-barrel folding that would drive the convergence to such similar structures in the spike proteins of two viruses in the same family. Another possibility is that small initial mutations in a common ancestral domain could have lead to a change in the pathway of protein folding, with a three-strand section of the VP5* β-sandwich or σ1 β-barrel flipping ‘inside out.' However, such refolding would necessitate solvent exposure of a number of hydrophobic side chains that pack in the interior of the β-structures. In a third scenario, genetic rearrangements could have altered the connectivity of the three N-terminal strands. Altered connectivity by this mechanism has been inferred by comparing the sequences of the palm domains of the RNA-dependant RNA polymerases (RdRps) of the tetraviruses and birnaviruses to the sequences of other RdRps of known structure (Gorbalenya et al, 2002). In the case of VP5* and σ1, such a rearrangement would require exchange of sequences encoding strands B and D of VP5* (or strands A and C of σ1).
A fourth possibility is suggested by the observation that the galectin-like sialic acid-binding domain of VP8*, which is N-terminal to the VP5* antigen domain, bears no structural relationship to the β-spirals in the sialic acid-binding region N-terminal to the σ1 head (Chappell et al, 2002). The overall organization of the rotavirus spike is consistent with the heads having arisen by the insertion of a host galectin-like carbohydrate-binding domain into an ancestral headless spike protein (Dormitzer et al, 2002). Thus, the three N-terminal strands of the β-structures may have separate origins in insertions that included a galectin-like domain or a β-spiral region added to the N-terminus of a common Greek-key ancestral core. Indeed, a systematic review of Greek-key β-barrels and β-sandwiches shows that the arrangement of the five staves with shared connectivity in VP5* and σ1 is conserved in many Greek-key β-structures (Zhang and Kim, 2000). Additional strands added to either terminus of this five-stranded core are more variably connected, suggesting that they are appended to a common folding intermediate.
Functional significance of the alternative oligomeric states
Several observations indicate that the VP5* antigen domain dimer and trimer model two functionally important states of the rotavirus spike protein. The extensive two-fold contacts in the dimeric crystal structure, including nine hydrogen bonds in the intersubunit β-sheet and a well-ordered hydrophobic core (Figure 3), provide evidence that the dimer is not simply an artefact of crystal packing. The fit of the dimer to the spikes on primed virions is confirmed by electron cryomicroscopy image reconstructions showing decoration of the ‘shoulders' of the spike by the antigen-binding fragment of a neutralizing monoclonal antibody that selects an escape mutation in the loop of the domain that fills the shoulders (Tihova et al, 2001). There are, however, subtle differences between the orientation of the domain in the crystal and on the virus, probably due to interactions with other parts of VP4 on the virion.
The reinforcement of the three-fold contacts observed in the crystal of VP5* antigen domain trimers by a triple coiled-coil and β-annulus in the more complete VP5CT fragment confirms the significance of the trimeric state of the antigen domain. Although analytical ultracentrifugation (Supplementary Figure 1C) indicates that the VP5* antigen domain is in dynamic equilibrium between monomers and trimers in solution, the additional contacts in VP5CT yield a very stable oligomer. VP5CT is more resistant to denaturation by guanidine hydrochloride than uncleaved VP4 (unpublished data) and its subunits do not dissociate in the presence of SDS (Dormitzer et al, 2001). This stability suggests that, during cell entry, the two- to three-fold rearrangement of VP5* is essentially irreversible.
The two- to three-fold reorganization and fold-back of VP5* is probably linked to membrane penetration. A two- to three-fold reorganization, associated with a fold-back rearrangement, mediates membrane fusion by class II-enveloped virus fusion proteins, such as the Semliki Forest virus and dengue fever virus envelope glycoproteins (Gibbons et al, 2004; Modis et al, 2004). The structural bases for the oligomeric reorganizations of the class II fusion proteins and VP5* are distinct. In the class II fusion proteins, different surfaces interact in the dimer and the trimer. In VP5*, many of the same residues at the N- and C-terminus and in the GH loop of the antigen domain have alternative packings with their symmetry mates in the dimer and the trimer. The formation of both a spike-like dimer and an umbrella-shaped trimer by the VP5* antigen domain through alternative packings demonstrates a molecular property that underlies the reorganization of a nonenveloped virus membrane penetration protein from a metastable primed spike to a trimeric final state.
Materials and methods
Subcloning
The coding sequence for RRV VP4 residues R247-D479 was amplified by PCR from a plasmid containing a cDNA copy of the full-length VP4 gene (GenBank accession code AY033150). This fragment was ligated into pET-28b (Novagen), which had been cleaved with NdeI and BamHI. A fragment excised with XbaI and XhoI encodes an N-terminal 6-histidine tag separated by a thrombin cleavage site from VP4 and was ligated into pFastBac1 (Invitrogen). A recombinant baculovirus expressing this fusion was generated using the Bac-to-Bac system (Invitrogen).
Protein expression and purification
Insect cell culture and baculovirus propagation were carried out as previously described (Willis et al, 1998). For protein production, suspension cultures of Sf9 cells, maintained in spinner flasks at 28°C, were infected with the recombinant baculovirus. Infected cells were harvested by centrifugation 72–86 h after infection and lysed by freezing and thawing in 20 mM Tris (pH 8.0), 100 mM NaCl, 10 mM imidazole, and 1 mM phenylmethylsulfonyl fluoride (PMSF). The lysate was clarified by centrifugation at 235 400 g for 1 h. Proteins in the clarified lysate were bound to nickel-charged HiTrap Chelating HP resin (Amersham Biosciences) and eluted with imidazole. Some samples were further processed by anion exchange chromatography on Mono Q HR 5/5 (Amersham Biosciences) after concentration and buffer exchange to less than 10 mM NaCl using an Amicon Ultra-4 centrifugal filter device (Millipore). Size exclusion chromatography on a Hi-Load 16/60 Superdex 75 or 200 column (Amersham Biosciences), equilibrated in 20 mM Tris (pH 8.0), 100 mM NaCl, and 1 mM EDTA (TNE), was used as a final purification step. Chromatography was performed using an AKTA system (Amersham Biosciences).
Analytical ultracentrifugation
Samples of the VP5* antigen domain were microdialyzed against TNE or ANE (20 mM Na acetate, pH 5.6, 100 mM NaCl, 1 mM EDTA). The diasylate was used as a blank and as diluent. Samples with protein concentrations of 75–300 μg/ml were centrifuged in an AN-60 Ti rotor at speeds of 10 000 and 14 000 r.p.m. in an Optima XL-A analytical ultracentrifuge (Beckman Coulter). Absorbance data were measured at 280 nm with 10 replicates. Attainment of equilibrium was confirmed by comparing absorbance curves at 6-h intervals. Origin software (Microcal Software, Inc.) was used to fit the data to the following model:
![]()
where Ar is the absorbance at radius x; A0,1 is the absorbance at the reference radius x0; H is a constant to account for protein specific volume, solvent density, angular velocity, and temperature; M is the monomer MW; N is the stoichiometry of the association; K is the association constant; and E is the baseline offset. A0,1 was constrained to be greater than 0. The monomer molecular weight was fixed at 27 644, based on MALDI mass spectrometry and SDS–PAGE (Supplementary Figure 1B). Protein-specific volumes and solvent densities were calculated based on published data (McRorie and Voelker, 1993). Each reported association constant (Supplementary Table) was determined by simultaneously fitting curves to protein distributions from six combinations of starting concentration and rotor speed.
Crystallization
At a concentration of 4 mg/ml in TNE, the VP5* antigen domain crystallized in batch at 8°C. The crystals belong to space group P212121 with unit cell parameters of a=93.12 Å, b=94.03 Å, and c=97.50 Å and contain one trimer in each asymmetric unit. These crystals were flash frozen by immersion in liquid nitrogen, using TNE with 25% glycerol as a freezing buffer. A second crystal form was obtained by the hanging drop vapor diffusion method after 72 h at 25°C with a well solution containing 100 mM sodium acetate, pH 5.6, 7% MPD, and 2% glycerol and with 2 μl drops containing a 1:1 mixture of 4 mg/ml protein in TNE and well solution. The crystals of this form belong to space group P21212 with unit cell parameters of a=163.36 Å, b=54.81 Å, and c=65.73 Å and contain one dimer in each asymmetric unit. These crystals were frozen after serial soaks in freezing buffers consisting of well solution with 12, 16, and 20% MPD.
Structure determination
X-ray diffraction data were collected at 100 K at the Advanced Light Source, beamline 8.2.2. Diffraction data were integrated and scaled using HKL2000 (HKL Research, Inc.). Starting phases for both crystal structures were obtained by molecular replacement using AMoRe (Navaza, 1994) in CCP4 (Collaborative Computational Project, 1994). Residues Y267-L470 from the VP5CT crystal structure (PDB identification code 1SLQ) were used as a search model. The structure was refined by multiple cycles of simulated annealing, energy minimization, and individual B-factor refinement using CNS (Brunger et al, 1998) alternating with rebuilding in O (Jones et al, 1991). Simulated annealing omit maps used in rebuilding were calculated using CNS (Brunger et al, 1998). Data collection and refinement statistics are reported in Table I.
Structure analysis and figure preparation
Structure geometry was analyzed using PROCHECK (Laskowski et al, 1993). Molecular alignments were obtained using LSQMAN (Kleywegt, 1996). Cavities were modeled using CAVENV (Collaborative Computational Project, 1994) and O (Jones et al, 1991), and their volumes were calculated using MAMA (Kleywegt and Jones, 1999). Buried surface area was calculated using AREAIMOL (Lee and Richards, 1971). The dimeric form of the VP5* antigen domain was fit to the molecular envelope of a VP4 spike from an approximately 12 Å resolution electron cryomicroscopy-based image reconstruction of trypsin-primed SA11-4F rotavirus particles, as previously described (Dormitzer et al, 2004). A structural similarity search was conducted using the DALI algorithm (Holm and Sander, 1993). Figures were made using MOLSCRIPT (Kraulis, 1991), O (Jones et al, 1991), Illustrator and Photoshop (Adobe Systems, Inc.), and Origin (Microcal Software, Inc.).
Coordinates
Atomic coordinates and structure factor amplitudes for the VP5* antigen domain dimer and trimer have been deposited in the Protein Data Bank (http://www.rcsb.org). The identification code for the dimer is 2B4H; the code for the trimer is 2B4I.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Table
Acknowledgments
We thank Marina Babyonyshev for skillful technical assistance, BV Venkataram Prasad for the electron cryomicroscopy image reconstruction of a rotavirus particle, Stephen C Harrison for scientific advice, the staff of ALS Beamline 8.2.2 for assistance in X-ray diffraction data collection, and Gaël McGill for three-dimensional modeling of VP4. This work was supported by NIH grant R01 AI 053174 and an Ellison Medical Foundation New Scholars in Global Infectious Diseases Award to PRD.
References
- Arias CF, Romero P, Alvarez V, Lopez S (1996) Trypsin activation pathway of rotavirus infectivity. J Virol 70: 5832–5839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Chappell JD, Prota AE, Dermody TS, Stehle T (2002) Crystal structure of reovirus attachment protein sigma1 reveals evolutionary relationship to adenovirus fiber. EMBO J 21: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciarlet M, Ludert JE, Iturriza-Gomara M, Liprandi F, Gray JJ, Desselberger U, Estes MK (2002) The initial interaction of rotavirus strains with N-acetyl-neuraminic (sialic) acid residues on the cell surface correlates with VP4 genotype, not species of origin. J. Virol 76: 4087–4095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project (1994) The CCP4 Suite: programs for protein crystallography. Acta Crystallogr D 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Crawford SE, Mukherjee SK, Estes MK, Lawton JA, Shaw AL, Ramig RF, Prasad BV (2001) Trypsin cleavage stabilizes the rotavirus VP4 spike. J Virol 75: 6052–6061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dormitzer PR, Greenberg HB (1992) Calcium chelation induces a conformational change in recombinant herpes simplex virus-1-expressed rotavirus VP7. Virology 189: 828–832 [DOI] [PubMed] [Google Scholar]
- Dormitzer PR, Greenberg HB, Harrison SC (2001) Proteolysis of monomeric recombinant rotavirus VP4 yields an oligomeric VP5* core. J Virol 75: 7339–7350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dormitzer PR, Nason EB, Prasad BV, Harrison SC (2004) Structural rearrangements in the membrane penetration protein of a non-enveloped virus. Nature 430: 1053–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dormitzer PR, Sun Z-YJ, Wagner G, Harrison SC (2002) The rhesus rotavirus VP4 sialic acid binding domain has a galectin fold with a novel carbohydrate binding site. EMBO J 21: 885–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estes MK, Graham DY, Mason BB (1981) Proteolytic enhancement of rotavirus infectivity: molecular mechanisms. J Virol 39: 879–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons DL, Vaney MC, Roussel A, Vigouroux A, Reilly B, Lepault J, Kielian M, Rey FA (2004) Conformational change and protein–protein interactions of the fusion protein of Semliki Forest virus. Nature 427: 320–325 [DOI] [PubMed] [Google Scholar]
- Gilbert JM, Greenberg HB (1998) Cleavage of rhesus rotavirus VP4 after arginine 247 is essential for rotavirus-like particle-induced fusion from without. J Virol 72: 5323–5327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya AE, Pringle FM, Zeddam JL, Luke BT, Cameron CE, Kalmakoff J, Hanzlik TN, Gordon KH, Ward VK (2002) The palm subdomain-based active site is internally permuted in viral RNA-dependent RNA polymerases of an ancient lineage. J Mol Biol 324: 47–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham KL, Halasz P, Tan Y, Hewish MJ, Takada Y, Mackow ER, Robinson MK, Coulson BS (2003) Integrin-using rotaviruses bind alpha2beta1 integrin alpha2 I domain via VP4 DGE sequence and recognize alphaXbeta2 and alphaVbeta3 by using VP7 during cell entry. J Virol 77: 9969–9978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, Sander C (1993) Protein structure comparison by alignment of distance matrices. J Mol Biol 233: 123–138 [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Kleywegt GJ (1996) Use of non-crystallographic symmetry in protein structure refinement. Acta Crystallogr D 52: 842–857 [DOI] [PubMed] [Google Scholar]
- Kleywegt GJ, Jones TA (1999) Software for handling macromolecular envelopes. Acta Crystallogr D 52: 842–857 [DOI] [PubMed] [Google Scholar]
- Kraulis J (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr 24: 946–950 [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the sterochemical quality of protein structures. J Appl Crystallogr 26: 283–291 [Google Scholar]
- Lee B, Richards FM (1971) The interpretation of protein structures: estimation of static accessibility. J Mol Biol 55: 379–400 [DOI] [PubMed] [Google Scholar]
- Mackow ER, Shaw RD, Matsui SM, Vo PT, Dang MN, Greenberg HB (1988) The rhesus rotavirus gene encoding protein VP3: location of amino acids involved in homologous and heterologous rotavirus neutralization and identification of a putative fusion region. Proc Natl Acad Sci USA 85: 645–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackow ER, Yamanaka MY, Dang MN, Greenberg HB (1990) DNA amplification-restricted transcription-translation: rapid analysis of rhesus rotavirus neutralization sites. (published erratum appears in Proc Natl Acad Sci USA 1990 June; 87(11): 4411). Proc Natl Acad Sci USA 87: 518–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRorie DK, Voelker PJ (1993) Self-associating systems in the analytical ultracentrifuge. Fullerton, California: Beckman Instruments [Google Scholar]
- Modis Y, Ogata S, Clements D, Harrison SC (2004) Structure of the dengue virus envelope protein after membrane fusion. Nature 427: 313–319 [DOI] [PubMed] [Google Scholar]
- Navaza J (1994) AmoRe: an automated package for molecular replacement. Acta Crystallogr A 50: 157–163 [Google Scholar]
- Parashar UD, Hummelman EG, Bresee JS, Miller MA, Glass RI (2003) Global illness and deaths caused by rotavirus disease in children. Emerg Infect Dis 9: 565–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesavento JB, Crawford SE, Roberts E, Estes MK, Prasad BV (2005) pH-induced conformational change of the rotavirus VP4 spike: implications for cell entry and antibody neutralization. J Virol 79: 8572–8580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad BV, Chiu W (1994) Structure of rotavirus. Curr Top Microbiol Immunol 185: 9–29 [DOI] [PubMed] [Google Scholar]
- Shaw AL, Rothnagel R, Chen D, Ramig RF, Chiu W, Prasad BV (1993) Three-dimensional visualization of the rotavirus hemagglutinin structure. Cell 74: 693–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tihova M, Dryden KA, Bellamy AR, Greenberg HB, Yeager M (2001) Localization of membrane permeabilization and receptor binding sites on the VP4 hemagglutinin of rotavirus: implications for cell entry. J Mol Biol 314: 985–992 [DOI] [PubMed] [Google Scholar]
- Willis SH, Peng C, Ponce de Leon M, Nicola AV, Rux AH, Cohen GH, Eisenberg RJ (1998) Expression and purification of secreted forms of HSV glycoproteins from baculovirus-infected insect cells. In Methods in Molecular Medicine: Herpes Simplex Virus Protocols, Brown SM, McLean AR (eds.), Vol. 10. Totowa, NJ: Humana Press Inc. [DOI] [PubMed] [Google Scholar]
- Yeager M, Berriman JA, Baker TS, Bellamy AR (1994) Three-dimensional structure of the rotavirus haemagglutinin VP4 by cryo-electron microscopy and difference map analysis. EMBO J 13: 1011–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeager M, Dryden KA, Olson NH, Greenberg HB, Baker TS (1990) Three-dimensional structure of rhesus rotavirus by cryoelectron microscopy and image reconstruction. J Cell Biol 110: 2133–2144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Kim S-H (2000) A comprehensive analysis of the Greek key motifs in protein β-barrels and β-sandwiches. Proteins: Stuct Funct Genet 40: 409–419 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Table