

Abstract
Infectious bursal disease virus (IBDV), the causative agent of a highly contagious disease in chickens, carries a small nonstructural protein (NS). This protein has been implicated to play a role in the induction of apoptosis. In this study, we investigate the kinetics of viral replication during a single round of viral replication and examine the mechanism of IBDV-induced apoptosis. Our results show that it is caspase dependent and activates caspases 3 and 9. Nuclear factor kappa B (NF-κB) is also activated and is required for IBDV-induced apoptosis. The NF-κB inhibitor MG132 completely inhibited IBDV-induced DNA fragmentation, caspase 3 activation, and NF-κB activation. To study the function of the NS protein in this context, we generated the recombinant rGLS virus and an NS knockout mutant, rGLSNSΔ virus, using reverse genetics. Comparisons of the replication kinetics and markers for virally induced apoptosis indicated that the NS knockout mutant virus induces earlier and increased DNA fragmentation, caspase activity, and NF-κB activation. These results suggest that the NS protein has an antiapoptotic function at the early stage of virus infection.
Infectious bursal disease virus (IBDV) is the causative agent of a highly contagious disease in young chickens known as infectious bursal disease (IBD). The virus replicates in the cytoplasm of infected cells and targets the precursors of antibody-producing B cells in the bursa of Fabricius (BF). A hallmark of IBDV replication in the BF is the depletion of B cells and atrophy of the BF, resulting in immunosuppression (4). Therefore, IBD is economically very important to the poultry industry worldwide. There are several isolates of IBDV which induce characteristic bursal lesions. Classic and very virulent IBDV strains cause hemorrhagic inflammation of the BF, whereas variant strains (GLS and E/Del) cause rapid bursal atrophy without evoking an inflammation response, suggesting differences in the apoptotic processes and pathogenesis of the disease (26, 38, 39, 42).
IBDV is a member of the Birnaviridae family, and its genome consists of two segments of double-stranded RNA (12, 13). The smaller segment, B, encodes VP1, a 97-kDa multifunctional protein with polymerase and capping enzyme activities (40). The larger segment, A, contains a large open reading frame (ORF) encoding a 110-kDa precursor protein that is processed into mature VP2 and VP3 structural proteins by the viral protease, VP4 (2, 20, 21). In addition, segment A encodes a 17-kDa nonstructural (NS) protein (also known as VP5) from a small ORF overlapping the ORF encoding the N-terminal region of VP2 (32). The NS protein is highly basic, cysteine-rich, and conserved among all serotype I IBDV strains.
Apoptosis, or programmed cell death, is a controlled physiological process of host cells to remove unwanted cells, including virus-infected cells. As a defense mechanism in response to virus infection, infected host cells undergo apoptosis, which occurs at the early stage of viral infection, thus limiting viral propagation. To overcome host resistance, many viruses carry antiapoptotic factors to inhibit apoptosis. However, in some cases, viruses may trigger apoptosis at the late stage of viral replication to facilitate viral release and spread (17). The executioners in apoptosis are caspases, a family of cysteine-dependent, aspartate-directed proteinases. Caspases function as both effector caspases, such as caspases 3, 6, and 7, and initiator caspases, such as caspases 8 and 9 (43). Procaspases 8 and 9 are activated in the receptor-mediated and mitochondrial pathways, respectively, and then they successively activate the effector caspases downstream (16, 43). It is known that NF-κB is one of the central players in apoptosis regulation. In most cases, NF-κB acts as an antiapoptotic regulator, inhibiting cell death and assisting cell survival (3). However, in minor cases, the activation of NF-κB leads to apoptosis. NF-κB can be activated by both external and internal stresses. Generally, oxidative stresses are common NF-κB stimuli (6, 22, 31). In latent cells, NF-κB is withheld in the cytoplasm, being bound with its inhibitor, IκB. NF-κB can be released only after IκB is phosphorylated and degraded, possibly via the ubiquitin and proteasome pathway. As a result, the released NF-κB will be translocated to the nucleus, working as the transcriptional regulator of many related genes (7, 8, 10).
Previous studies have shown that IBDV induces apoptosis both in vitro and in vivo (26, 34, 42, 45). By transiently expressing the NS protein, VP5, in chicken embryo fibroblasts (CEF) and BSC-1 and Cos-1 cells, Lombardo and coworkers demonstrated that this protein accumulates within the host plasma membrane and induces cell lysis (30). Using a reverse genetic system, we generated an NS knockout mutant of IBDV-D78 and demonstrated that the NS protein plays a role in the induction of apoptosis and IBDV pathogenesis, since this virus did not induce bursal lesions in susceptible chickens after infection (46, 47). Jungmann and coworkers reported that apoptosis is induced by IBDV replication in productively infected cells as well as in antigen-negative cells in their vicinity, suggesting an indirect mechanism for the induction of apoptosis in vivo (24). However, to date, the mechanisms for IBDV-induced apoptosis remain unknown. In this report, we focus on a single round of IBDV replication in infected cells and examine the apoptotic kinetics and possible pathways of IBDV-induced apoptosis. Furthermore, we generated IBDV GLS and its NS knockout mutant by using reverse genetics, and we compared apoptosis and signaling pathways to elucidate the function of the NS protein in the pathogenesis of IBDV infection.
MATERIALS AND METHODS
Viruses, cells, and reagents.
The cell culture-adapted antigenic variant strain GLSTC was obtained from previously acknowledged sources (38). This virus was plaque purified twice in secondary CEF cells and propagated in Vero cells. Primary CEF cells were prepared from 10-day-old embryonated eggs (SPAFAS, Inc., Storrs, Conn.) as described previously (33, 46). Secondary CEF cells, maintained in a growth medium consisting of M199-F10 (50%-50% [vol/vol]) and 5% fetal bovine serum (FBS), were used in all experiments, including virus titration and plaque assays. Vero cells were maintained in M199 medium supplemented with 5% FBS at 37°C in a humidified 5% CO2 incubator and used for the transfection and propagation of virus stocks. Recombinant viruses of the GLSTC (tissue culture adapted) strain, rGLSA and rGLSNSΔ, were recovered using reverse genetics as described previously (29, 46). The chemical reagents used in this study are as follows. z-VAD-FMK (Z-Val-Ala-Asp-fluoromethylketone), MG132 (Z-Leu-Leu-Leu-CHO; Biomol Research Labs, Inc.), and z-FA-FMK (Z-Phe-Val-fluoromethylketone; Trevigen Inc.) were dissolved in dimethyl sulfoxide at a concentration of 10 mg/ml or 25 mg/ml. PDTC (pyrrolidinedithiocarbamate; Sigma) was prepared in nuclease-free water. Poly(I · C) was prepared with nuclease-free water according to the instructions provided by the company (Amersham Biotech Inc.). CEF cells were transfected with 2 to 12 μg of synthetic double-stranded RNA (dsRNA) [poly(I · C)], using Lipofectin reagent, as described previously (33, 46).
DNA fragmentation assay.
Mock-infected or virus-infected cells (2 × 106) grown in 25-cm2 tissue culture flasks were harvested at different time points, and low-molecular-weight DNAs were extracted. A DNA fragmentation assay was performed as described previously, with slight modifications (14). Briefly, the collected cells were washed in cold phosphate-buffered saline (PBS), resuspended in 300 μl of ice-cold lysis buffer (10 mM Tris [pH 7.5], 10 mM EDTA [pH 7.5], and 0.2% Triton X-100), and incubated on ice for 30 min. Lysates were centrifuged at 10,000 × g at 4°C for 10 min, and supernatants were subjected to phenol-chloroform extraction twice, once with buffered phenol-chloroform and once with chloroform-isoamyl alcohol (24:1 [vol/vol]), by using 1.5 ml of phase-lock gel (Eppendorf, Brinkmann Instruments). DNAs were ethanol precipitated with 500 mM NaCl. DNA samples were resuspended in 15 μl of sterile water and treated for 15 min at 37°C with RNase A at a final concentration of 1.0 μg/μl. All samples were run in a 2% agarose gel in 1× Tris-borate-EDTA buffer and stained with ethidium bromide.
Western blot analysis.
To detect viral protein expression levels, IBDV-infected CEF cells (in a six-well plate) were harvested at the indicated time points and pelleted by centrifugation. The cells were washed with ice-cold PBS, resuspended in 30 μl of PBS, and mixed with an equal volume of 2× sodium dodecyl sulfate sample loading buffer. For immunoblotting, the proteins were electrophoretically separated in a 12.5% sodium dodecyl sulfate-polyacrylamide gel. The primary antibody was a rabbit anti-IBDV polyclonal antibody diluted 1:200 in blocking solution. The detection of IBDV-specific protein was performed with an enhanced chemiluminescence Western blot detection system (Amersham Pharmacia Biotech Inc., NJ).
Growth curve for IBDV.
Infected cell cultures were freeze-thawed three times at different intervals, and the titers of infectious progeny were determined by a plaque assay. Briefly, the supernatants from infected cell cultures were diluted serially 10 times. Monolayers of secondary CEF cells grown in a six-well plate were inoculated with each dilution of supernatant. At 1 hour postinfection, the cells were overlaid with 3 ml of 0.9% SeaPlaque agarose (Difco) in minimum essential medium containing 5% FBS and 1% l-glutamine. After 3 days of incubation at 37°C, the overlays were removed, and the cells were fixed and stained with a solution containing 25% formalin, 10% ethanol, 5% acetic acid, and 1% crystal violet for 5 min at room temperature. After rinsing of the cells with distilled water, the plaques were counted.
NF-κB EMSA.
CEF cells grown in 75-cm2 tissue culture flasks were either mock infected or infected with GLSTC at a multiplicity of infection (MOI) of 10 PFU per cell. Total cell extracts were prepared, and electrophoretic mobility shift assays (EMSAs) were performed as previously described (35). Briefly, 2 × 106 infected CEF cells were trypsinized and washed once with cold PBS. The cells were resuspended in 20 μl high-salt detergent buffer containing 20 mM HEPES, pH 7.9, 350 mM NaCl, 20% (wt/vol) glycerol, 1% (wt/vol) NP-40, 1 mM MgCl2, 0.5 mM EDTA, 0.1 mM EGTA, 0.5 mM dithiothreitol (DTT), 0.1% phenylmethylsulfonyl fluoride (PMSF), and 1% protease inhibitor cocktail (Boehringer Mannheim, Indianapolis, IN) and incubated on ice for 30 min. The cell lysates were centrifuged for 5 min at 13,000 × g at 4°C. Equal amounts of protein (15 μg) were added to reaction mixtures that contained 20 μg bovine serum albumin (Sigma), 2 μg poly(dI · dC) (Amersham Biotech Inc.), 2 μl buffer D (20 mM HEPES, pH 7.9, 20% glycerin, 100 mM KCl, 0.5 mM EDTA, 0.25% NP-40, 2 mM DTT, 0.1% PMSF), 4 μl buffer F (20% Ficoll 400, 100 mM HEPES, 300 mM KCl, 10 mM DTT, 0.1% PMSF), and 0.2 pmol γ-32P-end-labeled probes in a final volume of 20 μl. For competition experiments, a 10-fold excess of unlabeled NF-κB consensus oligonucleotide or a double-stranded oligonucleotide for another transcription factor, AP-1, was added to reaction mixtures and incubated for 10 min at room temperature before γ-32P-end-labeled probes were added. After incubation at room temperature for another 25 min in the presence of γ-32P-end-labeled probes, all reaction mixtures were electrophoresed in 5% nondenaturing polyacrylamide gels at 55 mA for 20 min at 4°C. Subsequently, the gels were dried, prepared for autoradiography, and exposed to a phosphor screen (Molecular Dynamics) for storage at room temperature. The 32P-labeled bands were visualized using a phosphorimager (Storm; Molecular Dynamics, Sunnyvale, CA).
The double-stranded NF-κB oligonucleotides (5′-AGT TGA GGG GAC TTT CCC AGG C-3′ and the complementary strand, 3′-TCA ACT CCC CTG AAA GGG TCC G-5′) (Promega) were labeled using [γ-32P]ATP (3,000 Ci/mmol; Amersham) and T4 polynucleotide kinase (Promega) according to the instructions provided by the manufacturer. The AP-1 oligonucleotide contained the sequence 5′-GAT CGA ACT GAC CGC CCG CGG CCC GT-3′ and the complementary strand, 3′-GCG AAC TAC TCA GTC GGC CTT-5′.
Caspase 3, 8, and 9 activity assays.
Caspase activity assays were performed using the procedures described in fluorometric assay kits obtained from BioVision (Mountain View, CA). CEF cells (1 × 106) grown in a six-well plate were mock infected or infected with GLSTC virus at an MOI of 10 PFU per cell and incubated for various intervals. The cells were trypsinized and washed with cold PBS. The cell pellets were resuspended in 50 μl of chilled cell lysis buffer and incubated on ice for 10 min. The cell lysates were harvested by centrifugation at 10,000 × g for 10 min. The supernatants were collected and frozen at −70°C until samples from all the time points were harvested.
The fluorometric assay utilizes peptide substrates consisting of the consensus cleavage sequence for each caspase labeled with AFC (7-amino-4-trifuoromethyl coumarin), such as DEVD-AFC (synthetic caspase 3 substrate), IETD-AFC (synthetic caspase 8 substrate), and LEHD-AFC (synthetic caspase 9 substrate), which were used in the study. Activated caspases in apoptotic cells cleave the synthetic substrates to release free AFC, which is then quantified using a fluorometer. For each reaction, 50 μl of cell lysate and 50 μl of 2× reaction buffer containing 5 mM DTT were mixed and then incubated with 50 mM of the caspase substrate at 37°C for 90 min. Finally, the reactions were analyzed in a fluorescence plate reader (Luminescence LS 55 spectrometer; Perkin-Elmer Instruments, Shelton, CT) with a 400-nm excitation filter and a 505-nm emission filter. The amount of fluorescence detected was directly proportional to the amount of caspase activity. Results of all experiments are reported as means ± standard deviations (SD).
RESULTS
The kinetics of IBDV replication and apoptosis.
In the first set of experiments, we investigated IBDV-induced apoptosis and possible pathways during the early stage of viral infection in order to provide valuable indicators for analysis of the NS protein function. First, the replication and apoptotic kinetics during a single round of the viral life cycle were studied by infecting CEF cells with GLSTC virus (MOI = 10.0). Viral protein expression was detected by immunostaining, and the production of infectious viral progeny was quantified by plaque assay at different time points. As shown in Fig. 1A, the increased viral protein expression in infected cells could be detected at 8 h postinfection (p.i.). Accordingly, there was very little viral protein detected prior to 7 h p.i., but the viral yields increased 3-, 13-, and 17-fold at 9, 12, and 14 h, respectively, compared with the sample from 3 h p.i. (Fig. 1B). A DNA laddering assay was utilized to investigate IBDV-induced apoptosis, as shown in Fig. 1C. DNA fragmentation, which is a characteristic event in apoptotic cells, became detectable between 12 and 14 h p.i., suggesting that apoptosis occurred at the late stage of viral replication.
FIG. 1.

Kinetics of IBDV replication and induction of apoptosis in CEF cells during a single round of the viral life cycle. CEF cells were infected with GLSTC virus (MOI = 10.0) and harvested at the indicated time intervals. (A) Kinetics of viral protein expression. A virus-specific protein (VP3 [32 kDa]) in cell lysates was detected by Western blotting using an enhanced chemiluminescence system as described in Materials and Methods. (B) Synthesis of infectious viral progenies. Viral yields were quantified by plaque assay at the indicated times. Values of virus titers are shown as means ± SD from three independent experiments. (C) DNA laddering in IBDV-infected CEF cells. At the indicated time points, infected and mock-infected cells were harvested for a DNA fragmentation assay.
IBDV infection activates caspases 3 and 9 but not caspase 8.
To investigate if IBDV-induced apoptosis is caspase dependent, caspase 3 activity in CEF cells during IBDV infection was examined at different time points. Caspase 3 activity was detected by a fluorometric method based on cleavage of the DEVD-AFC substrate. Figure 2A shows that caspase 3 was activated at 9 h p.i. and that its activity increased over time, which correlates with the DNA laddering results. Caspases 8 and 9 are both pivotal initiator caspases in either extrinsic or intrinsic pathways. To determine which initiator caspase(s) is involved in IBDV-induced apoptosis, the activation of caspases 8 and 9 was examined. As shown in Fig. 2B, caspase 9 was activated during IBDV infection, but caspase 8 was not. As a positive control, dsRNA was used, which induces both caspases 8 and 9 (Fig. 2C).
FIG. 2.

Time courses of caspase 3, 8, and 9 activities in IBDV-infected cells. CEF cells were mock infected or infected with GLSTC virus at an MOI of 10 PFU per cell, and cell lysates were prepared at the indicated times postinfection. The enzymatic activities of caspases 3, 8, and 9 were quantified as described in Materials and Methods. (A) Caspase 3-like activity at the indicated times after infection. (B) Caspase 8- and 9-like activities at the indicated times after infection. P values were calculated using an unpaired t test. By conventional criteria, the differences in caspase 3 activities (A) and caspase 8 and 9 activities (2) between control and GLSTC-infected CEF cells at different time points were evaluated, and the results are marked (* or **). (C) As a positive control for caspase 8 and 9 activation, 2 μg or 12 μg of the synthetic dsRNA poly(I · C) was transfected into CEF cells by using Lipofectin. At 3 hours posttransfection, cell lysates were prepared, and the enzymatic activities of caspases 8 and 9 were determined.
The irreversible, cell-permeative, broad-spectrum caspase inhibitor z-VAD-FMK was used to confirm this result. As expected, this protein completely inhibited both caspase 3 and 9 activities during IBDV infection (Fig. 3A) and partially inhibited DNA laddering induced by IBDV (Fig. 3B). Taken together, our results indicate that IBDV infection activates caspases 3 and 9, but not caspase 8, at the late stage of viral replication.
FIG. 3.
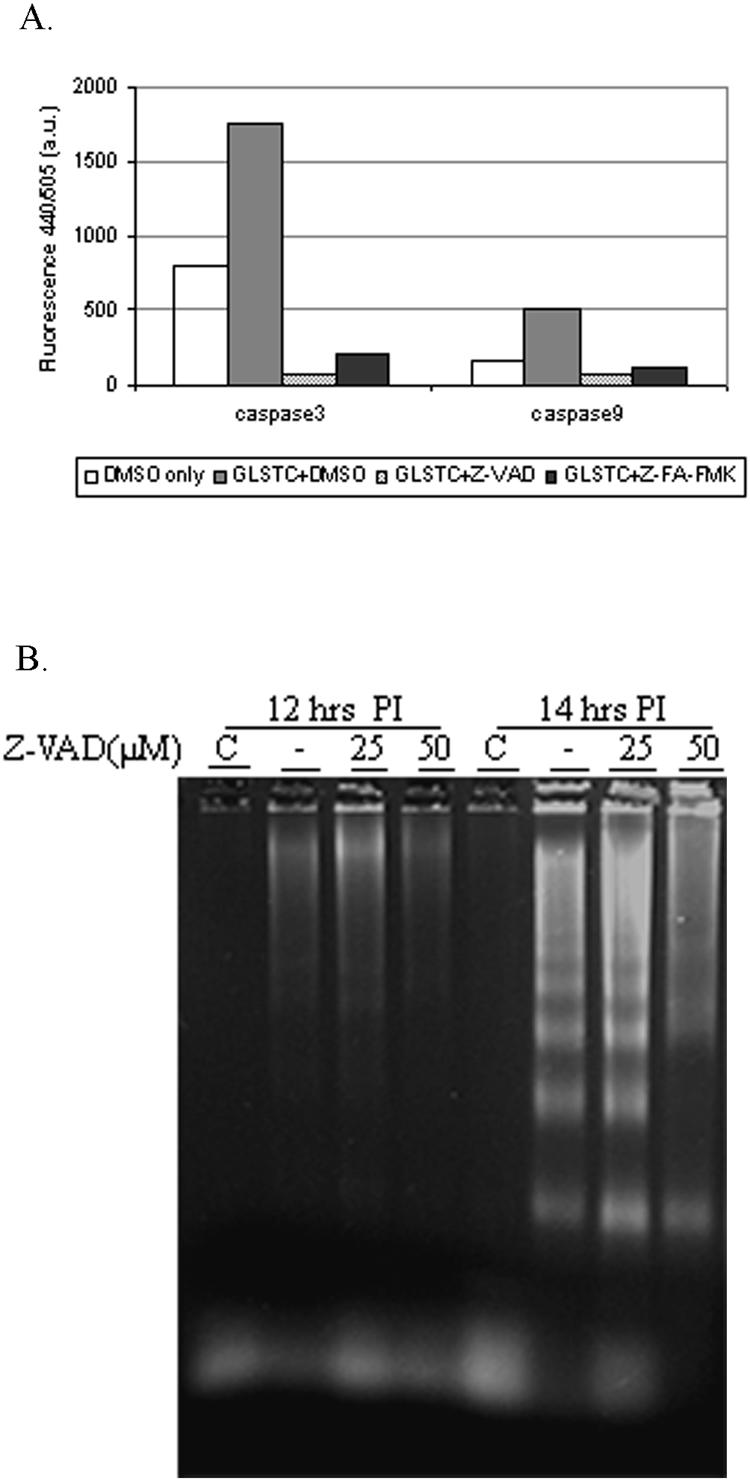
Effect of a broad-spectrum caspase inhibitor, z-VAD-FMK, on caspase 3 and caspase 9 activities and IBDV-induced DNA fragmentation. (A) CEF cells were mock infected (with dimethyl sulfoxide [DMSO]) or infected with GLSTC virus at an MOI of 10 PFU per cell in the presence of 50 μM of z-VAD-FMK or 50 μM of z-FA-FMK (negative inhibitor control). Infected cells were collected, lysed at 14 h postinfection, and used to assess caspase 3 and 9 enzymatic activities. The mean values from two independent experiments are shown. (B) DNA laddering assay performed at 12 and 14 h postinfection in the presence and absence of different concentrations of z-VAD-FMK.
NF-κB is activated after IBDV infection.
NF-κB, a very important player in the regulation of cell growth and survival (1), is activated in response to intrinsic and extrinsic signals, including virus infection (11). Therefore, we examined the activation of NF-κB by using an EMSA after virus infection. CEF cells were either mock infected or infected with GLSTC virus, and cell lysates were prepared at various time intervals. Cell lysates were incubated with a saturating amount of 32P-labeled oligonucleotide containing the NF-κB consensus binding sequence and were resolved in a nondenaturing polyacrylamide gel. Figure 4A shows that following IBDV infection, the NF-κB protein, capable of binding to the radiolabeled oligonucleotides, shifted to a higher-molecular-mass position, indicating its activation. NF-κB activation was detected at 6 h p.i. and further increased at approximately 16 h p.i. No NF-κB activity was detected in mock-infected cultures.
FIG. 4.

Time course of NF-κB gel shift activities in whole-cell lysates prepared from IBDV-infected cells. (A) CEF cells (1 × 106) were either mock infected or infected with GLSTC virus at an MOI of 10 PFU per cell. The cells were washed and trypsinized, and whole-cell lysates were obtained at the indicated time points. NF-κB gel shift activities were evaluated as described in Materials and Methods. (B) Specificity of NF-κB binding activity in EMSA. The whole-cell lysates utilized for panel A at 10 and 16 h postinfection were incubated with a 32P-labeled NF-κB consensus oligonucleotide probe alone (lanes N) or in the presence of a 10-fold excess of either unlabeled NF-κB oligonucleotide (lanes C) or an unlabeled oligonucleotide probe that binds to the transcription factor AP-1 (lanes A). A representative result from three independent experiments is shown.
To verify if the DNA binding was specific for NF-κB, a competitive EMSA was carried out. The binding reaction of cell lysate with the 32P-labeled NF-κB consensus oligonucleotide was performed in the presence of a 10-fold excess of either unlabeled NF-κB consensus oligonucleotide or an unlabeled oligonucleotide that binds to AP-1, another transcription factor. As shown in Fig. 4B, the gel shift activity was competitively inhibited by the unlabeled NF-κB consensus oligonucleotide but not by unlabeled AP-1, indicating that the binding of the 32P-labeled NF-κB oligonucleotide to NF-κB was specific.
Both a proteasome inhibitor and an antioxidant inhibit IBDV-induced NF-κB activation and apoptosis.
It has been shown that the degradation of IκB requires proteasome activity and that only after IκB is degraded can NF-κB be released and translocated to the nucleus to start the transcription of downstream genes (6, 8). Therefore, to determine if NF-κB activation is required for IBDV-induced apoptosis, a synthetic peptide inhibitor of the aldehyde proteasome pathway, MG132, was used (10, 36). CEF cells were infected with GLSTC virus (MOI = 10.0) in the presence or absence of different concentrations of MG132. The cell lysates and DNA extracts were subjected to EMSA and DNA laddering analysis at 14 or 16 h p.i. As shown in Fig. 5A, the treatment of CEF cells with MG132 completely eliminated NF-κB activity. As expected, MG132 at 50 μM also inhibited IBDV-induced DNA laddering (Fig. 5B). In addition, no cytopathic effect (CPE) was observed in the presence of MG132, whereas obvious CPE was observed between 12 and 14 h p.i. in the absence of MG132 (data not shown). The accumulation of reactive oxygen species (ROS) in stressed cells is a very common event that is associated with cell death pathways, and several studies suggest that an elevated ROS level enhances NF-κB activation. To determine if ROS accumulation is the upstream event of IBDV-induced NF-κB activation, the antioxidant PDTC was used (15, 35). CEF cells were infected with GLSTC virus in the presence of various concentrations of PDTC, and cell lysates were harvested for NF-κB EMSA and the DNA laddering assay at 14 h p.i. Our results show that PDTC also inhibits NF-κB activities (Fig. 5C). Moreover, both apoptosis and CPE were inhibited by PDTC, and DNA fragmentation was partially inhibited (Fig. 5D). Since caspase 3 is an important indicator of IBDV-induced apoptosis, caspase 3 activity was measured in the presence of MG132 at 14 h p.i. Figure 5E shows that MG132 also inhibits IBDV-induced caspase 3 activation.
FIG. 5.

Blocking of NF-κB activity and DNA fragmentation by the proteasome inhibitor MG132 and the antioxidant PDTC. EMSA and DNA laddering analyses were performed as described in Materials and Methods. CEF cells were infected with GLSTC virus (MOI = 10.0) in the presence and absence of 25 or 50 μM of MG132 and harvested at 14 and 16 h postinfection. Whole-cell lysates were assessed for NF-κB activity by EMSA (A). At 14 h postinoculation, low-molecular-weight DNAs were also extracted and subjected to a DNA laddering assay (B). The antioxidant PDTC was added at the indicated concentrations (C and D) to the cell culture at the same time that GLSTC virus was inoculated. Cells were harvested, and cell lysates were assessed for NF-κB activity and apoptosis by EMSA (C) and DNA laddering (D) analyses, respectively. A 1-kb Plus DNA ladder (lane M) was used as a marker. Caspase 3 activity was measured in the presence and absence of MG132 or PDTC at different concentrations and at 14 h postinfection with GLSTC virus (E).
Function of NS protein in IBDV-induced apoptosis.
Having demonstrated that both caspase 3 and 9 activation and NF-κB activation can be used as markers for IBDV-induced apoptosis, we wanted to determine the function of the NS protein in apoptosis during a single round of viral replication. For this experiment, we recovered the recombinant rGLSA virus and its NS knockout mutant, rGLSNSΔ, using reverse genetics (29, 33). When CEF cells were infected with these two viruses, no difference in viral protein synthesis (based on VP3 expression) was observed (Fig. 6A). However, rGLSNSΔ virus replicated slower and produced sevenfold fewer progenies than its counterpart, rGLSA virus, at 14 h p.i. (Fig. 6B). In addition, the CPE in rGLSNSΔ virus-infected cells was earlier and greater than that in rGLSA virus-infected CEF cells during a single round of viral replication (data not shown). We then examined the apoptosis induced by rGLSNSΔ virus after infection, as represented by DNA laddering, caspase 3 and 9 activation, and NF-κB activation. Since the NS-deficient virus does not grow to as high of a titer as the wild-type virus, we infected CEF cells with rGLSNSΔ virus at an MOI of 1 and with rGLSA virus at an MOI of 1 or 2. As shown in Fig. 6C, rGLSNSΔ virus induced stronger DNA laddering than rGLSA virus at 12 and 14 h p.i., even though the latter was inoculated at an MOI of 2. Similarly, rGLSNSΔ virus induced more NF-κB activation (Fig. 6D) and higher caspase 3 and 9 activities (Fig. 6E and F) than rGLSA virus at 9, 12, and 14 h p.i. Taken together, our results indicate that rGLSNSΔ virus induces increased apoptosis compared to rGLSA virus, suggesting that the NS protein is antiapoptotic during the early stage of viral replication.
FIG. 6.

Replication and apoptotic kinetics of rGLS and rGLSNSΔ viruses at early stage of virus infection. CEF cells (1 × 106) were inoculated with equal amounts of the rGLSNSΔ (NSΔ) and rGLSA (A) viruses at an MOI of 1 PFU per cell. Cell lysates were harvested and analyzed for the virus-specific protein VP3 by immunoblotting (A). Infected cells were freeze-thawed three times, and viruses were quantified by plaque assay at the indicated time points (B). The low-molecular-weight DNAs from cell lysates were extracted and analyzed by a DNA laddering assay (C) and by EMSA at 9, 12, and 14 h postinfection (D). Caspase 3 and 9 activities were also measured 12 h after virus infection at the indicated MOI per cell (E and F). All assays were performed as described in the legends to Fig. 1 to 5.
DISCUSSION
During virus infection, a variety of signal transduction pathways can be activated, leading to apoptosis of infected cells. For example, reovirus and Sindbis virus initiate apoptosis via both cellular receptor- and mitochondrion-mediated caspase-dependent pathways (11, 23, 25). During reovirus infection, both caspase 8 and caspase 9 are activated. Caspase 8 activation can be detected as early as 6 h postinfection, whereas caspase 9 can be detected at 12 h postinfection (25). Other viruses, such as dengue virus and bovine viral diarrhea virus, cause internal stresses, which damage mitochondrial membrane integrity or potentials so that apoptosis is initiated (22, 37).
For this study, the kinetics and signaling pathways of IBDV-induced apoptosis were studied in cell culture for the first time, based on a single round of replication. Three important characteristics of IBDV-induced apoptosis were revealed. First, as our data show, IBDV-induced apoptosis is caspase dependent. After infection of CEF cells with IBDV, DNA fragmentation can be detected, and both the effector caspase 3 and the initiator caspase 9 are significantly activated. The broad-spectrum caspase inhibitor z-VAD-FMK, known to inhibit caspases 8 and 9 and, subsequently, caspase 3 (41), also inhibits the activation of caspases 9 and 3 and partially inhibits DNA fragmentation induced by IBDV infection. Caspase 8 is not activated by IBDV infection, and apoptosis is detected in the late stage of a single round of viral replication, suggesting that IBDV-induced apoptosis may not be receptor mediated or that the receptor-mediated pathway is inhibited.
It has been shown that reovirus and flaviviruses induce apoptosis, which requires the activation of NF-κB (11, 22, 27). In this pathway, oxidative stress is considered one of the most important mediators of apoptosis, and NF-κB has been shown to function downstream of ROS in some situations (5, 22, 31). For infectious bronchitis virus and porcine transmissible gastroenteritis virus, the induction of apoptosis is caspase dependent and also involves cellular oxidative stress (14, 28). In this study, we show the second important characteristic for IBDV-induced apoptosis, which is the activation of NF-κB during the first round of the viral life cycle. Activation of NF-κB was detected at 8 h and peaked between 12 and 14 h postinfection. The addition of a proteasome inhibitor, MG132, dramatically inhibited NF-κB activation and also prevented infected cells from undergoing apoptosis. Similarly, the antioxidant PDTC inhibited IBDV-induced NF-κB activation and partially prevented cells from undergoing apoptosis. Although the precise apoptotic pathway that IBDV employs is not known, it is possible that the oxidative stress in infected cells may be the crucial step in IBDV-induced apoptosis, as shown for reovirus and flaviviruses.
The third important characteristic, as presented in this study, is that IBDV-induced apoptosis occurs at a late stage during virus infection in CEF cells. An increase in viral protein expression was observed at 8 h postinfection, whereas the viral yield was significantly higher at 14 h postinfection, which coincided with DNA fragmentation and cytopathic effects. It is evident that apoptosis occurs at a time when IBDV synthesis is complete and the progenies need to be released from infected cells.
For dsRNA and positive-strand RNA viruses, proteins are synthesized once viruses enter the cells, and then replication is initiated. There are many factors that can induce apoptosis, such as receptor binding, dsRNA, and stresses caused by virus replication. Responding to these invading foreign signals and products, infected host cells sacrifice themselves by going to programmed cell death before completing their life cycle, limiting viral replication (17). Presumably, virally induced apoptosis needs to be inhibited during the early stage of viral infection. Some viruses carry an antiapoptotic protein to counteract apoptosis. In reovirus, an attachment protein, sigma 1, determines the capacity to induce apoptosis (44). Recently, it was shown that a sigma 1-deficient reovirus caused significantly reduced caspase 3 activation and injury in the heart and brain tissues of infected mice compared to the wild type (19). Similarly, a nonstructural protein, NS5A, encoded by hepatitis C virus, was shown to have an antiapoptotic function (9). Infectious pancreatic necrosis virus, another member of the Birnaviridae family, encodes a 15-kDa nonstructural protein, VP5, which contains a Bcl-2 motif and also functions as an antiapoptotic protein (18). In the case of IBDV, the 17-kDa NS protein does not possess a Bcl-2 motif but contains a PEST motif. Proteins containing this type of motif are generally short-lived and expressed early and usually have a regulatory function.
In this study, by comparing IBDV-induced apoptotic characteristics of NS-deficient rGLSNSΔ virus and rGLSA virus, we provide important evidence suggesting that the NS protein functions as an antiapoptotic protein during the early stage of IBDV replication. First, the NS-deficient virus causes earlier and greater apoptotic effects than rGLSA virus, as shown by DNA laddering, caspase 3 activity, and NF-κB activation. Second, early apoptosis is accompanied by less production of viral progeny, and lastly, the viral protein translational level is not affected, as indicated by the Western blot results. Therefore, the NS protein performs the function of inhibiting apoptosis initiated by viral replication and prevents infected cells from undergoing cell death before the virus finishes its life cycle. Without this inhibition, apoptosis would occur earlier, when the virus still needs the host to complete its propagation. Thus, early apoptosis of infected cells results in reduced production of viral progeny. Consequently, the NS-deficient virus replicates slower and has a 1-log lower titer than rGLSA virus after several rounds of replication (data not shown).
The antiapoptotic function of the NS protein can also instigate attenuation of the virus in vivo. Earlier, we showed that the replication efficiency of IBDV in the BF modulates virulence in vivo (29). Bursa-derived wild-type IBDV replicates most efficiently in the BF, causing bursal lesions. On the other hand, the cell culture-adapted virus does not replicate efficiently due to mutations in the VP2 or VP1 protein, and it is attenuated. As our data indicate here, a loss of antiapoptotic function by the NS protein also decreases the replication efficiency, which can lead to further attenuation. In fact, this can explain why the NS-deficient rD78 mutant virus reported earlier (46), as well as rGLSNSΔ virus (data not shown), is attenuated in vivo and does not induce bursal lesions. These data may be in disagreement with our findings that the NS-deficient rD78 virus induced less apoptosis (by terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling assay) than the wild-type virus at 72 h p.i. However, this can be explained since the comparison of apoptosis was made under dissimilar conditions and not during the early stage of viral infection (between 9 and 12 h p.i., when the apoptosis of NS-deficient virus was greater than that of the wild type) (Fig. 6C), which consequently reduced the viral titer of NS-deficient rD78 virus throughout the growth period, and it seemed to be less apoptotic than the wild-type virus at 72 h p.i., when the cells were highly confluent.
Previously, it was shown that the NS protein accumulates within the host plasma membrane and induces cell lysis (30), and transient expression of the NS protein in vitro induces apoptosis (47). Since transient expression does not mimic virus infection, the results may be quite different from the point of view of the effect of viral infection on the host antiviral defense. Therefore, we speculate that the NS protein might be a regulatory protein which is antiapoptotic at the early stage of virus infection and targets the plasma membrane at the end of the viral life cycle. This results in cell death and dissemination of the IBDV progeny. A mutant rGLSNSΔ virus, lacking the last 20 residues of the NS protein (representing the cytoplasmic domain), was generated and exhibited similar kinetics to those of the rGLSNSΔ virus (data not shown). Therefore, further studies are needed to map the functional domain of the NS protein.
Acknowledgments
This project was supported by the National Research Initiative of the USDA Cooperative State Research, Education, and Extension Service under grant number 1997-02492 to V.N.V.
We thank Gerard H. Edwards for technical assistance.
REFERENCES
- 1.Ashkenazi, A., and V. M. Dixit. 1998. Death receptors: signaling and modulation. Science 281:1305-1358. [DOI] [PubMed] [Google Scholar]
- 2.Azad, A. A., S. A. Barrett, and K. J. Fahey. 1985. The characterization and molecular cloning of the double-stranded RNA genome of an Australian strain of infectious bursal disease virus. Virology 143:35-44. [DOI] [PubMed] [Google Scholar]
- 3.Barkett, M., and T. D. Gilmore. 1999. Control of apoptosis by Rel/NF-kappaB transcription factors. Oncogene 18:6910-6924. [DOI] [PubMed] [Google Scholar]
- 4.Becht, H. 1980. Infectious bursal disease virus. Curr. Top. Microbiol. Immunol. 90:107-121. [DOI] [PubMed] [Google Scholar]
- 5.Bonizzi, G., J. Piette, S. Schoonbroodt, R. Greimers, L. Havard, M.-P. Merville, and V. Bours. 1999. Reactive oxygen intermediate-dependent NF-κB activation by interleukin-1β requires 5-lipoxygenase or NADPH oxidase activity. Mol. Cell. Biol. 19:1950-1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brockman, J. A., D. C. Scherer, T. A. McKinsey, S. M. Hall, X. Qi, W. Y. Lee, and D. W. Ballard. 1995. Coupling of a signal response domain in IκBα to multiple pathways for NF-κB activation. Mol. Cell. Biol. 15:2809-2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown, K., S. Gerstberger, L. Carlson, G. Franzoso, and U. Siebenlist. 1995. Control of I kappa B-alpha proteolysis by site-specific, signal-induced phosphorylation. Science 267:1485-1488. [DOI] [PubMed] [Google Scholar]
- 8.Chen, Z., J. Hagler, V. J. Palombella, F. Melandri, D. Scherer, D. Ballard, and T. Maniatis. 1995. Signal-induced site-specific phosphorylation targets I kappa B alpha to the ubiquitin-proteasome pathway. Genes Dev. 9:1586-1597. [DOI] [PubMed] [Google Scholar]
- 9.Chung, Y. L., M. L. Sheu, and S. H. Yen. 2003. Hepatitis C virus NS5A as a potential viral Bcl-2 homologue interacts with Bax and inhibits apoptosis in hepatocellular carcinoma. Int. J. Cancer 107:65-73. [DOI] [PubMed] [Google Scholar]
- 10.Clifton, D. R., R. A. Goss, S. K. Sahni, D. van Antwerp, R. B. Baggs, V. J. Marder, D. J. Silverman, and L. A. Sporn. 1998. NF-kappa B-dependent inhibition of apoptosis is essential for host cell survival during Rickettsia rickettsii infection. Proc. Natl. Acad. Sci. USA 95:4646-4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Connolly, J. L., S. E. Rodgers, P. Clarke, D. W. Ballard, L. D. Kerr, K. L. Tyler, and T. S. Dermody. 2000. Reovirus-induced apoptosis requires activation of transcription factor NF-κB. J. Virol. 74:2981-2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delmas, B., F. S. B. Kibenge, J. A. Leong, E. Mundt, V. N. Vakharia, and J. L. Wu. 2005. Birnaviridae, p. 561-569. In C. M. Facquet, M. A. Mayo, J. Maniloff, U. Desselberger, and A. L. Ball (ed.), Virus taxonomy. Academic Press, London, United Kingdom.
- 13.Dobos, P., B. J. Hill, R. Hallett, D. T. Kells, H. Becht, and D. Teninges. 1979. Biophysical and biochemical characterization of five animal viruses with bisegmented double-stranded RNA genomes. J. Virol. 32:593-605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eleouet, J. F., S. Chilmonczyk, L. Besnardeau, and H. Laude. 1998. Transmissible gastroenteritis coronavirus induces programmed cell death in infected cells through a caspase-dependent pathway. J. Virol. 72:4918-4924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gong, G., G. Waris, R. Tanveer, and A. Siddiqui. 2001. Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF-kappa B. Proc. Natl. Acad. Sci. USA 98:9599-9604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Green, D. R., and J. C. Reed. 1998. Mitochondria and apoptosis. Science 281:1309-1312. [DOI] [PubMed] [Google Scholar]
- 17.Hay, S., and G. Kannourakis. 2002. A time to kill: viral manipulation of the cell death program. J. Gen. Virol. 83:1547-1564. [DOI] [PubMed] [Google Scholar]
- 18.Hong, J. R., H. Y. Gong, and J. L. Wu. 2002. IPNV VP5, a novel anti-apoptosis gene of the Bcl-2 family, regulates Mcl-1 and viral protein expression. Virology 295:217-229. [DOI] [PubMed] [Google Scholar]
- 19.Hoyt, C. C., S. M. Richardson-Burns, R. J. Goody, B. A. Robinson, R. L. Debiasi, and K. L. Tyler. 2005. Nonstructural protein sigma1s is a determinant of reovirus virulence and influences the kinetics and severity of apoptosis induction in the heart and central nervous system. J. Virol. 79:2743-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hudson, P. J., N. M. McKern, B. E. Power, and A. A. Azad. 1986. Genomic structure of the large RNA segment of infectious bursal disease virus. Nucleic Acids Res. 14:5001-5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jagadish, M. N., V. J. Staton, P. J. Hudson, and A. A. Azad. 1988. Birnavirus precursor polyprotein is processed in Escherichia coli by its own virus-encoded polypeptide. J. Virol. 62:1084-1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jan, J. T., B. H. Chen, S. H. Ma, C. I. Liu, H. P. Tsai, H. C. Wu, S. Y. Jiang, K. D. Yang, and M. F. Shaio. 2000. Potential dengue virus-triggered apoptotic pathway in human neuroblastoma cells: arachidonic acid, superoxide anion, and NF-κB are sequentially involved. J. Virol. 74:8680-8691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jan, J. T., and D. E. Griffin. 1999. Induction of apoptosis by Sindbis virus occurs at cell entry and does not require virus replication. J. Virol. 73:10296-10302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jungmann, A., H. Nieper, and H. Müller. 2001. Apoptosis is induced by infectious bursal disease virus replication in productively infected cells as well as in antigen-negative cells in their vicinity. J. Gen. Virol. 82:1107-1115. [DOI] [PubMed] [Google Scholar]
- 25.Kominsky, D. J., R. J. Bickel, and K. L. Tyler. 2002. Reovirus-induced apoptosis requires both death receptor- and mitochondrial-mediated caspase-dependent pathways of cell death. Cell Death Differ. 9:926-933. [DOI] [PubMed] [Google Scholar]
- 26.Lam, K. M. 1997. Morphological evidence of apoptosis in chickens infected with infectious bursal disease virus. J. Comp. Pathol. 116:367-377. [DOI] [PubMed] [Google Scholar]
- 27.Liao, C. L., Y. L. Lin, B. C. Wu, C. H. Tsao, M. C. Wang, C. I. Liu, Y. L. Huang, J. H. Chen, J. P. Wang, and L. K. Chen. 2001. Salicylates inhibit flavivirus replication independently of blocking nuclear factor kappa B activation. J. Virol. 75:7828-7839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu, C., H. Y. Xu, and D. X. Liu. 2001. Induction of caspase-dependent apoptosis in cultured cells by the avian coronavirus infectious bronchitis virus. J. Virol. 75:6402-6409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu, M., and V. N. Vakharia. 2004. VP1 protein of infectious bursal disease virus modulates the virulence in vivo. Virology 330:62-73. [DOI] [PubMed] [Google Scholar]
- 30.Lombardo, E., A. Maraver, I. Espinosa, A. Fernandez-Arias, and J. F. Rodriguez. 2000. VP5, the nonstructural polypeptide of infectious bursal disease virus, accumulates within the host plasma membrane and induces cell lysis. Virology 277:345-357. [DOI] [PubMed] [Google Scholar]
- 31.Manna, S. K., and B. B. Aggarwal. 1999. Lipopolysaccharide inhibits TNF-induced apoptosis: role of nuclear factor-kappaB activation and reactive oxygen intermediates. J. Immunol. 162:1510-1518. [PubMed] [Google Scholar]
- 32.Mundt, E., J. Beyer, and H. Müller. 1995. Identification of a novel viral protein in infectious bursal disease virus-infected cells. J. Gen. Virol. 76:437-443. [DOI] [PubMed] [Google Scholar]
- 33.Mundt, E., and V. N. Vakharia. 1996. Synthetic transcripts of double-stranded birnavirus genome are infectious. Proc. Natl. Acad. Sci. USA 93:11131-11136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ojeda, F., I. Skardova, M. I. Guarda, J. Ulloa, and H. Folch. 1997. Proliferation and apoptosis in infection with infectious bursal disease virus: a flow cytometric study. Avian Dis. 41:312-316. [PubMed] [Google Scholar]
- 35.Pahl, H. L., and P. A. Baeuerle. 1995. A novel signal transduction pathway from the endoplasmic reticulum to the nucleus is mediated by transcription factor NF-kappa B. EMBO J. 14:2580-2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rock, K. L., C. Gramm, L. Rothstein, K. Clark, R. Stein, L. Dick, D. Hwang, and A. L. Goldberg. 1994. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 78:761-771. [DOI] [PubMed] [Google Scholar]
- 37.Schweizer, M., and E. Peterhans. 1999. Oxidative stress in cells infected with bovine viral diarrhoea virus: a crucial step in the induction of apoptosis. J. Gen. Virol. 80:1147-1155. [DOI] [PubMed] [Google Scholar]
- 38.Snyder, D. B., D. P. Lana, P. K. Savage, F. S. Yancey, S. A. Mengel, and W. W. Marquardt. 1988. Differentiation of infectious bursal disease viruses directly from infected tissues with neutralizing monoclonal antibodies: evidence of a major antigenic shift in recent field isolates. Avian Dis. 32:535-539. [PubMed] [Google Scholar]
- 39.Snyder, D. B., V. N. Vakharia, and P. K. Savage. 1992. Naturally occurring-neutralizing monoclonal antibody escape variants define the epidemiology of infectious bursal disease viruses in the United States. Arch. Virol. 127:89-101. [DOI] [PubMed] [Google Scholar]
- 40.Spies, U., and H. Müller. 1990. Demonstration of enzyme activities required for cap structure formation in infectious bursal disease virus, a member of the birnavirus group. J. Gen. Virol. 71:977-981. [DOI] [PubMed] [Google Scholar]
- 41.Sun, X. M., M. MacFarlane, J. Zhuang, B. B. Wolf, D. R. Green, and G. M. Cohen. 1999. Distinct caspase cascades are initiated in receptor-mediated and chemical-induced apoptosis. J. Biol. Chem. 274:5053-5060. [DOI] [PubMed] [Google Scholar]
- 42.Tanimura, N., and J. M. Sharma. 1998. In-situ apoptosis in chickens infected with infectious bursal disease virus. J. Comp. Pathol. 118:15-27. [DOI] [PubMed] [Google Scholar]
- 43.Thornberry, N. A., and Y. Lazebnik. 1998. Caspases: enemies within. Science 281:1312-1316. [DOI] [PubMed] [Google Scholar]
- 44.Tyler, K. L., M. K. Squier, S. E. Rodgers, B. E. Schneider, S. M. Oberhaus, T. A. Grdina, J. J. Cohen, and T. S. Dermody. 1995. Differences in the capacity of reovirus strains to induce apoptosis are determined by the viral attachment protein sigma 1. J. Virol. 69:6972-6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vasconcelos, A. C., and K. M. Lam. 1994. Apoptosis induced by infectious bursal disease virus. J. Gen. Virol. 75:1803-1806.8021611 [Google Scholar]
- 46.Yao, K., M. A. Goodwin, and V. N. Vakharia. 1998. Generation of a mutant infectious bursal disease virus that does not cause bursal lesions. J. Virol. 72:2647-2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yao, K., and V. N. Vakharia. 2001. Induction of apoptosis in vitro by the 17-kDa nonstructural protein of infectious bursal disease virus: possible role in viral pathogenesis. Virology 285:50-58. [DOI] [PubMed] [Google Scholar]