

Abstract
One unresolved issue in gut immunity is how the mucosal T lymphocytes are activated and which antigen presenting cell (APC) is critical for regulating this process. We have identified a unique population of APCs that is exclusively localized in the lamina propria (LP). These APCs constitutively expressed the costimulatory molecule CD70 and exhibited antigen presenting functions. After oral Listeria monocytogenes infection, the expansion and differentiation of antigen-specific T cells occurred in the gut mucosa in situ and blockade of CD70 costimulation abrogated the mucosal T cell expansion and effector functions. Thus, a potent CD70-dependent stimulation via specialized tissue-specific APC is required for the expansion and differentiation of gut mucosal T cells after an oral infection.
Introduction
Lymphocytes in the intestinal mucosa present several features that are distinct from their counterparts in the secondary lymphoid organs. Unlike T cells in the spleen and lymph nodes (LNs), the gut intraepithelial lymphocytes (IELs) exhibit a constitutively activated phenotype1-4. In contrast to peripheral T cells, the IELs are refractory to T cell receptor (TCR)-CD3 stimulation, but proliferate vigorously and secrete pro-inflammatory cytokines after CD2 stimulation5,6. In addition, the gut mucosal T cells express distinctive adhesion molecules and their costimulatory requirements are different from peripheral T cells7-9. Further, systemic administration of soluble protein antigen without adjuvant induces differentiated cytotoxic T cells in the mucosa but not in the spleen and LNs10. Finally, induction of interferon-γ (IFN-γ) gene expression in peripheral T cells and mucosal T cells occurs through the use of different cisregulatory elements and requires the recruitment of different transactivating factors11. One possible reason for these observations is that the T cells in the mucosa may be stimulated with distinct types of antigen presenting cells (APCs) and are thus imprinted differently during their differentiation.
Different subsets of dendritic cells (DCs) have been identified in the intestinal lymphoid organs. In addition to CD11c+CD11b+CD8α-DEC205- and CD11c+CD11b-CD8α+DEC205+ DC found in the spleen and LN, Peyer's patch (PP) and mesenteric lymph nodes (MLNs) also contain a unique population of CD11c+CD11b-CD8α-DEC205- DC12-14. Another subset of CD11cloB220+Gr1+CD8α+ plasmacytoid DC has also been described in the MLN and PP15,16. Functionally, the PP DCs (particularly the CD11b+ and plasmacytoid subsets) differ from the splenic and LN DC in their ability to induce cytokine production. For example, in contrast to the splenic DC-stimulated T cells, which produce IFN-γ, PP DC-primed T cells secrete predominantly interleukin-10 (IL-10)16,17. Further, after ligation of the costimulatory molecule RANK, the PP DCs produce IL-10, whereas splenic DCs produce IL-1218. As a consequence, treatment of mice with RANK-ligand during oral administration of soluble antigen enhances tolerance induction18 Thus, whereas splenic DCs provide a stimulatory environment, PP DCs generally provide an inhibitory milieu. In contrast to PP and MLN, the gut mucosal DC subsets remain poorly characterized14, although the presence of potentially tolerogenic subsets in LP has been suggested19.
One important feature of APCs is the expression of MHC and costimulatory molecules. The TNF-related costimulatory ligand, CD70 is expressed by activated murine DC in vitro as well as in vivo during a protracted Leishmania infection20,21. CD70 is normally only expressed transiently in vivo and its persistent expression in transgenic (Tg) mice results in profound activation and expansion of T cells, leading ultimately to the exhaustion of the naïve T cell pool22,23. However, constitutive CD70 expression by immune cells in vivo has not been reported. Here we have identified a unique population of APCs of non-hematopoietic origin that occur exclusively in the lamina propria (LP) and constitutively express CD70. We show that CD70+ APCs play a pivotal role in the expansion and differentiation of T cells in the gut mucosa.
Results
LP contains a unique CD70+ cell population.
Because the constitutively activated nature of T cells in the intestinal mucosa is reminiscent of the persistent activation of T cells seen in CD70 Tg mice23, we tested if CD70 expressing cells are present in the intestinal mucosa in naïve mice. Cells isolated from the intestinal mucosa and lymphoid organs were tested for CD70 expression by flow cytometry. We found a substantial portion of CD70+ cells in the gut LP, but not in other tissues such as the spleen, PP, MLN and bone marrow (BM) (Fig. 1a). The CD70+ cells were distributed along the entire length of the intestine including the colon (Fig. 1b). Immunohistological examination confirmed their exclusive distribution in the LP (Fig. 1c).
Figure 1.
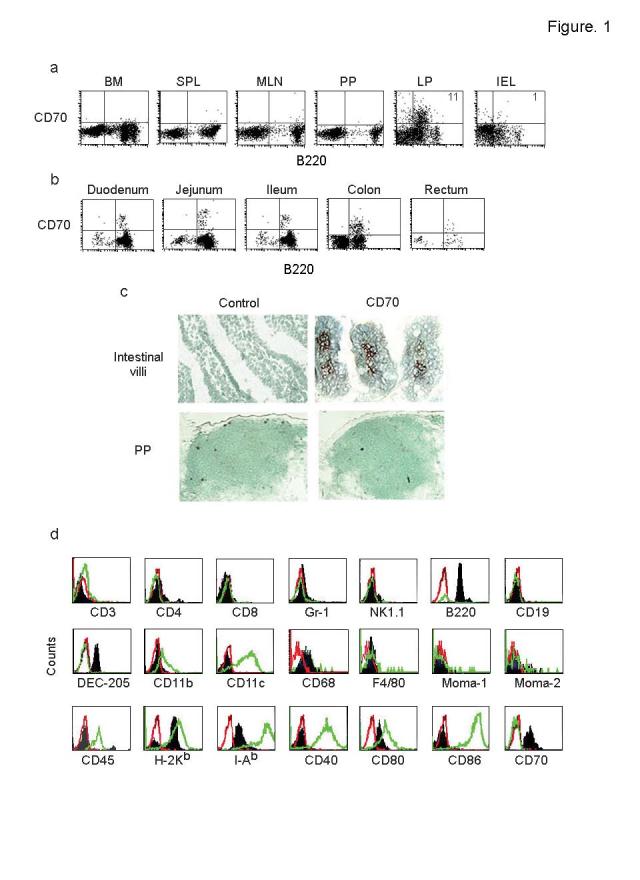
Presence of a unique CD70+ cell population in the gut lamina propria. (a) Single cell suspensions from indicated organs of wild-type mice were examined for the presence of CD70+ cells by flow cytometry. Percentage of cells expressing CD70 is indicated in the right upper quadrant. Representative results from >10 mice from 3 independent experiments are shown. (b) Isolated LP cells from different sections of the intestine were examined for the presence of CD70+ cells by flow cytometry. (c) Intestinal (upper panels) and PP sections (bottom panels) were stained with CD70 or isotype control antibody and examined histologically. Brown staining indicates CD70 positivity. (d) Isolated cells from LP and BMDCs were stained with indicated antibodies. Overlay histograms of isotype control (red open histograms), CD70-gated cells from the gut LP (dark filled histograms) and CD11c gated BMDC (green open histograms) are shown. Results are representative of 3 independent experiments.
In mice, surface CD70 is expressed by in vitro activated B cells, mature dendritic cells and to a lesser extent, T cells20,24. Thus, we examined the phenotype of the CD70 expressing cells and compared them to the BM-derived dendritic cells (BMDC) (Fig. 1d) as well as to the CD11c+ cells present in the LP (Supplementary Fig. 1a online). The CD70+ cells were negative for CD3, CD4 and CD8 expression, indicating they are not of T cell origin. They also did not express the NK1.1 or Gr-1 molecules. They had low expression of B220, but they were not B cells because they were CD19 negative. They were also not plasma cells because B220loCD70+ cells were present in the B cell-deficient MuMT mice as well as in the B and T cell-deficient RAG-1-/- mice (Supplementary Fig. 1d,e online). Although they had low expression of CD68 and stained for □-naphthyl acetate esterase (Supplementary Fig. 1b online), they were negative for the macrophage markers CD11b, F4/80, MOMA-1 and MOMA-2. We also tested for the DC markers CD11c, CD11b and DEC 205 and MHC expression12-14. Unlike BMDCs, the CD70+ cells did not express CD11c or CD11b. However, they expressed DEC 205, MHC class I and class II. Among the costimulatory molecules, CD70+ cells expressed CD80 but not CD40 or CD86 (Fig. 1d). Although CD40 antibody-treated DC have been reported to express CD7020,24, we did not find CD70 expression in BMDC after LPS stimulation. Unlike BMDCs, the MHC class II and costimulatory molecule expression could not be augmented in CD70+ cells by Toll-like receptor (TLR) ligands such as LPS, CpG oligonucleotides or TNF (data not shown). When cultured, CD70+ cells were loosely adherent and hetreogenous in shape and architecture but they did not exhibit typical dendrites (Supplementary Fig. 1c online). Thus, the gut LP contains a distinct CD70+ cell population with an unusual phenotype.
Non-hematopoietic origin of CD70+ cells. Because of the unusual phenotype of the CD70+ cells, we tested if they are of hematopoietic lineage. Although they stained weakly with B220 antibody, which is thought to recognize the isoform of CD45 expressed on B cells, they did not express the pan-leukocyte marker CD45 (Fig. 1d). Moreover, B220lo CD70+ cells were also detected in CD45-deficient mice (Fig. 2a). Thus, the CD70+ cells apparently express an unidentified protein other than CD45 that possesses an epitope recognized by the B220 antibody. We also confirmed that they are not of hematopoietic lineage in bone marrow reconstitution experiments. Because the CD70+ cells were negative for the commonly used congenic markers (CD45 and Thy1), we used bone marrow cells from human HLA-A2.1 Tg mice in C57 background as donor cells. In these mice, the HLA Tg is driven by the endogenous HLA-A2.1 promoter25 and thus all class I positive cells should express the molecule. In the Tg mice, CD70+ cells in the LP uniformly expressed the HLA-A2.1 (Fig. 2b). For reconstitution, lethally irradiated C57 mice were transfused with bone marrow cells from HLA-A2.1 Tg mice and after 6 weeks, their gut mucosal cells were examined for reconstitution and CD70 expression. Although the B220hi B cells expressed HLA-A2.1 uniformly indicating effective reconstitution, the B220loCD70+ cells did not express the Tg (Fig. 2b), showing that the B220loCD70+ cells do not originate from bone marrow-derived stem cells. Thus, the CD70+ cells constitute a distinct class of tissue-specific cells possessing some phenotypic features of professional antigen presenting cells that could have been missed by adhering to the conventional phenotypic markers of dendritic cells. However, the CD70+ cells did not stain with α smooth muscle actin, suggesting that they are not myofibroblasts, which are predominant in the gut mucosa26. Thus their exact lineage remains to be determined.
Figure 2.

Non-hematopoitic origin of CD70+ cells. (a) LP cells from CD45-/- mice were examined for the presence of B220loCD70+ cells by flow cytometry (bottom panel). Their splenocytes (top panel) were also tested to confirm the lack of CD45+ cells. The results are representative of 3 independent experiments. (b) C57 mice were lethally irradiated and injected with bone marrow cells from HLA-A2.1 transgenic mice and after 6 weeks, the B220hi B cells and B220lo CD70+ cells were examined for HLA-A2 Tg expression in the chimera (right panel). Tg expression by B220hi B cells and B220lo CD70+ cells in wild-type C57 or HLA-A2.1 Tg mice is shown in the left and middle panels respectively. Data are representative of results from 4 mice each.
Antigen presenting function of CD70+ cells
Because the CD70+ cells exhibited some phenotypic features of APCs, we tested if they were capable of presenting antigens. For these studies, we did not use CD70 antibody to isolate CD70+ cells to avoid affecting their function through CD70. Because they are B220+ but CD19-, we enriched for these cells from isolated LP cells of C57BL/6 mice (H-2b) by first eliminating CD19+ B cells and then selecting B220+ cells. The isolated cells were >90% enriched as assessed by CD70 expression (Supplementary Fig. 2a online). These cells efficiently stimulated allogenic H-2d T cells from Balb/c mice (Fig. 3a).
Figure 3.
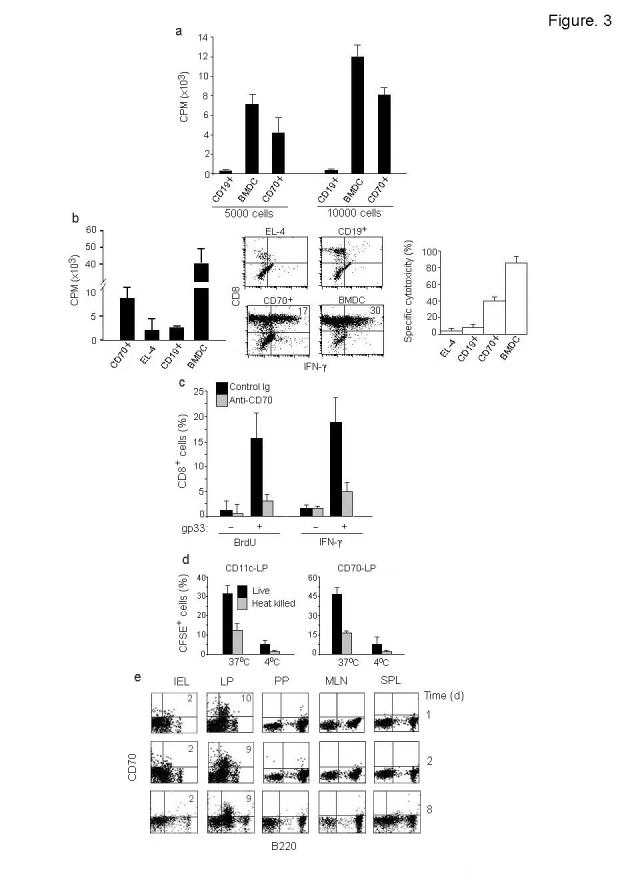
Antigen presenting function of CD70+ APC. (a) Immuno-magnetically enriched CD70+ cells, CD19+ B cells or LPS matured BMDC from C57BL/6 mice were cultured with T cells from BALB/c mice for 5 days and tested for 3H thymidine incorporation. Mean + s.d. of triplicate wells from 2 independent experiments is shown. (b) Purified CD8+ T cells from P14 mice were stimulated with gp33 peptide-pulsed CD70+ cells, B cells, EL-4 cells or BMDC and tested after 4 days for 3H thymidine incorporation (left) or after 6 days for IFN-γ production (middle) and cytotoxicity (right) using peptide pulsed EL-4 targets at an E:T ratio of 10:1. Mean + s.d. of triplicates from 2 independent experiments is shown. (c) P14 CD8 T cells were stimulated with gp33 peptide-pulsed CD70+ cells and incubated in the presence or absence of control or CD70 blocking antibody and BrdU incorporation and IFN-γ production determined after 4 days. Mean + s.d. of triplicates from 2 independent experiments is shown. (d) isolated LP cells from wild-type mice were incubated with either live or heat killed CSFE-labeled Lmdd (10 CFU/cell) at either 37°C or 4°C for 4 h, washed extensively, stained with CD11c or CD70 antibody and examined by flow cytometry. Results represent mean + s.d. of 3 experiments (e) Cells from different organs were tested for the presence of CD70+ cells at indicated times after oral Lm infection. Representative results from 3 independent experiments are shown.
We also tested the ability of CD70+ cells to stimulate a primary T cell response. Isolated CD70+ cells were pulsed with the LCMV glycoprotein peptide (gp33-41) and used to stimulate naïve CD8 T cells from the P14 TCR Tg mice (specific for gp33-41). The CD70+ cells stimulated and induced effector functions as determined by proliferation, IFN-γ production and cytotoxicity assays (Fig. 3b). Moreover, the P14 T cell activation could be inhibited by CD70 blocking antibody as determined by bromodeoxyuridine (BrdU) incorporation and IFN-γ production assays (Fig. 3c). The CD70 antibody did not directly affect T cells numbers or function (data not shown) and thus, the antibody appears to block the APC function of CD70+ cells. Although these results suggest that the CD70+ cells have the ability to stimulate a primary T cell response, given the method used for their isolation, we could not rule out the possibility of contamination with a small number of plasmacytoid DCs in these experiments.
Phagocytic function of CD70+ cells
We compared the CD70+ cells with CD11c+ cells present in LP for their ability to phagocytose Listeria monocytogenes (Lm). To determine capture of Lm, LP cells were infected with 10 cfu/cell of CFSE-labeled Lm. Because CFSE gets diluted with bacterial replication, we used a strain of D-alanine-deficient listeria (Lmdd) that is incapable of replication in the absence of externally supplemented D-alanine27. After 4 h of infection at 37°C and extensive washing, cells were stained with CD70 or CD11c and examined for green fluorescence by flow cytometry. Both CD11c+ cells and CD70+ cells, but not CD8 T cells, (data not shown) were able to take up Lm efficiently (Fig. 3d). The phagocytosis was not due to non-specific binding, because little increase in fluorescence was seen if the cultures were incubated at 4°C. The presence of CSFE-labeled Listeria inside CD70+ cells was also confirmed by fluorescence microscopy (Supplementary Fig. 2c online). We also confirmed that CD70+ cells could phagocytose Lm in vivo after oral infection (Supplementary Fig. 2b online).
Mucosal DC are thought to pickup antigen in the gut mucosa and migrate to MLNs to prime T cells12. To determine if CD70+ APC can migrate to PP and /or MLNs, and also to test if CD70 is induced on immune cells after oral Lm infection as shown in influenza virus infection24, we tested different organs for the presence of CD70 expressing cells 1, 2 and 8 days after Lm infection. Although CD70 expressing cells were consistently seen in the gut LP, no CD70 expressing cell was found in any other organs, including the draining MLN and PP (Fig. 3e). Thus, CD70 expression after Lm infection in vivo is confined to CD70+ APC. Although these results also suggest that the CD70+ APC remain confined to LP even during an oral infection, migration of small numbers of cells may be hard to detect by flow cytometry.
T cells expand in the gut mucosa
The presence of a unique population of APCs in the gut LP suggest that these cells might be involved in T cell stimulation in situ. To test this hypothesis, we used CD8+ T cells from P14 TCR Tg mice crossed to T-GFP mice (P14–T-GFP). In T-GFP mice, GFP uniformly expressed in naïve and early-activated CD8+ T cells is selectively turned off once the cells differentiate into phenotypically and functionally mature cytotoxic effector T cells28,29. Thus, P14–T-GFP system allows the identification of antigen-specific cells by gp33 tetramer staining as well as simultaneously assessing their differentiation status by GFP expression. Purified CD8+ T cells from P14–T-GFP mice (>97% GFP+ L-selectin+ CD69-; Supplementary Fig. 2d online and data not shown) were adoptively transferred to C57 recipients and 4 days later, the mice were infected orally with rLmgp33. Different organs were tested for the presence of gp33 tetramer+ donor derived CD8+ T cells by flow cytometry on days 3 and 8 after infection. We chose day 3 as an early time point because substantial expansion of activated T cells can be demonstrated on day 3 and 4 after antigen challenge in adoptive transfer experiments30,31. In the adoptively transferred uninfected mice, 4 days after transfer, the tetramer+ cells constituted less than 2% of CD8 T cells in the spleen, MLN, PP, IEL and LP (data not shown). In infected mice, an expanded population of tetramer positive CD8+ T cells was present only in the IEL and LP (>10%) after 3 days (Fig. 4a). This expansion was not due to non-specific expansion of memory cells because a similar expansion of tetramer+ cells also occurred in adoptive transfer experiments using CD8+ T cells from P14 mice in the RAG-1-deficient background (data not shown). Although the majority of tetramer positive cells in the secondary lymphoid organs were GFP+, those in LP and IEL were uniformly GFP- (Fig. 4b), indicating that the tetramer positive cells had differentiated into effector T cells in the gut mucosa. However, 8 days after infection, expansion of GFP- tetramer+ P14 cells was readily detectable in the lymphoid organs, suggesting that they also differentiated into effector cells.
Figure 4.

Expansion and differentiation of T cells occurs in the intestinal mucosa after oral Lm infection. (a) Wild-type mice were adoptively transferred with CD8+ cells from P14–T-GFP mice, gavaged with rLmgp33 and at indicated times after infection, cells from different organs were examined by flow cytometry for the presence of Db Gp33 tetramer+ cells. Dot plots of CD8-gated cells are shown and percentages of GFP+ and GFP- cells indicated. (b) Summary of the data (mean + s.d.) from 2 experiments done as in a with 3 mice each. (c) C57 mice were adoptively transferred with CSFE labeled CD8+ T cells from P14 mice and were either left alone (uninfected, left panel) or gavaged with rLmgp33 (right panel) and after 3 days, cells from different organs were stained with anti-CD8 antibody and Db Gp33 tetramer. Histograms of CFSE fluorescence on CD8 and tetramer-gated cells from one mouse (out of 3 examined) are shown. (d) Shows percent dividing Db Gp33 tetramer+ cells in different organs on day 3 post infection, determined by incorporation of BrdU administered 2 h before harvesting. Data (mean + s.d) from 2 independent experiments with 4 mice each are shown. (e) C57 mice were gavaged with rLmgp33 and the frequencies of gp33-specific IFN-γ producing CD8+ T cells in different organs determined after 5 days. Data (mean + s.d.) from 7 mice in 2 independent experiments are shown.
We also detected cell division in various organs after infection by adoptively transferring CSFE-labeled CD8+ T cells from P14 Tg mice into C57 recipients and gavage them with rLmgp33. On day 3 after infection, only a few tetramer+ cells in PP and MLN were dividing, whereas almost all tetramer+ cells in the LP and IEL had lost CFSE fluorescence, indicating that they had undergone multiple divisions (Fig. 4c). Collectively, these results suggest that the major expansion and differentiation of antigen-specific T cells early after an oral infection occurs in the intestinal mucosa where the CD70+ APC reside.
To test if the increased numbers of transferred cells seen on day 3 after infection in the IEL and LP was due to accumulation of dividing cells emigrating from MLN and PP or represent in situ proliferation, mice were adoptively transferred with P14 CD8+ T cells and infected with rLmgp33. Three days later, BrdU was injected intravenously and after 2h, the mice were killed and BrdU+ cells in different organs determined by flow cytometry. A substantial portion of gp33 tetramer+ cells in the LP were BrdU+, whereas only a small number of cells in MLN and PP incorporated BrdU (Fig. 4d). Thus, antigen-specific T cells were actively replicating in the intestinal LP.
Because the unnatural numbers of cytotoxic T lymphocyte (CTL) precursors that were present in the adoptive transfer system may not accurately represent the normal situation, we tested if endogenous CTLs also expand first in the mucosa after oral Lm infection. C57 mice were gavaged with rLmgp33 and the frequency of gp33 peptide-specific IFN-γ producing CD8+ T cells in various organs compared after 3 days or 5 days of infection in an ELISPOT assay. Unlike after adoptive transfer, substantial numbers of IFN-γ producing antigen-specific T cells were not seen in any organ at 3 days after infection (data not shown). However, gp33-specific IFN-γ producing CD8+ T cells could be demonstrated in IEL and LP, but not other organs 5 days after infection (Fig. 4e). Thus, although the kinetics with adoptive transfer were faster because of excessive precursor numbers, the T cell activation pattern reflected the natural infection. Taken together, our results show that at early time points after an oral Lm infection, the antigen-specific T cells expand and differentiate in situ in the gut mucosa.
LTA-/- mice fail to expand T cells
Although our results suggest that T cells expand and differentiate in the mucosa, they do not directly address where the initial priming of T cells occurs. Our finding that the tetramer+ cells in PP and MLNs did not lose GFP expression on day 3 after infection does not rule out priming in PP and MLNs because in T-GFP mice, early activated T cells continue to express GFP and it is only the fully differentiated effector cells that lose GFP expression28,29. Therefore, to determine if PP and MLN are required to generate a T cell response during Lm infection, we compared the expansion of adoptively transferred P14 CD8+ T cells after rLmgp33 infection in wild-type and lymphotoxin α-deficient (LTA-/-) mice which lack PP and LNs32. Although the spleen in both groups contained equivalent numbers of tetramer+ cells, the expansion of tetramer+ cells seen in the IEL in wild-type mice was completely absent in LTA-/- mice (Fig. 5a). However, LTA-/- mice had similar numbers of CD70+ APC as wild-type mice (Fig. 5b). Although intestinal homing defect has been noted for B cells in the LTA-/- mice33, the total T cell number as well as the T cell subset composition is unaltered in these mice, suggesting that the homing defect does not apply to T cells34. We also did not find differences in the total CD8+ T cell numbers between wild-type and LTA-/- mice even after Lm infection (Fig. 5c) and thus, the homing defects cannot explain the inability of T cells to expand in LTA-/- mice. Taken together, these results suggest that PP and/or MLN is required for the initial T cell priming following Lm infection.
Figure 5.

LTA-/- mice do not generate a gut mucosal T cell response. (a) Wild-type and LTA-/- mice were adoptively transferred with P14-T-GFP CD8+ cells and infected with rLmgp33 as in Fig. 4, and their IELs and spleen were examined for the presence of Db Gp33 tetramer+ cells on day 3 after infection. Percentages of tetramer positive cells is indicated in the representative histograms in the left panel and summary of the data (mean + s.d.) from 5 mice of each genotype in 2 independent experiments is shown in the right panel. (b) LP cells from wild-type and LTA-/- mice were stained with CD70 antibody. Frequencies of CD70+ cells (mean + s.d.) from 6 mice in 2 independent experiments is shown. (c) Summary of data (mean + s.d) on CD8 T cell numbers in IEL+LP in the experiment detailed in a is shown.
CD70 blockade abrogates mucosal T cell expansion
The profound expansion of effector-memory T cells seen in CD70 Tg mice is dependent on continuous CD27-CD70 interaction, and CD70 blocking antibody treatment can effectively reverse this to wild-type levels24. Thus, to test the importance of CD70-mediated costimulation via the CD70+ APC in antigen-specific T cell expansion, we administered CD70 antibody on the day of P14–T-GFP CD8+ T cell adoptive transfer and repeated on the day of infection with oral Lm, 4 days later. This treatment abrogated P14–T-GFP CD8+ T cell expansion on day 3 after infection in the LP and IEL (Fig. 6a). The effect of CD70 antibody treatment was specific to IEL and LP and did not affect the limited T cell expansion in the PP and MLN (Fig. 6b).
Figure 6.
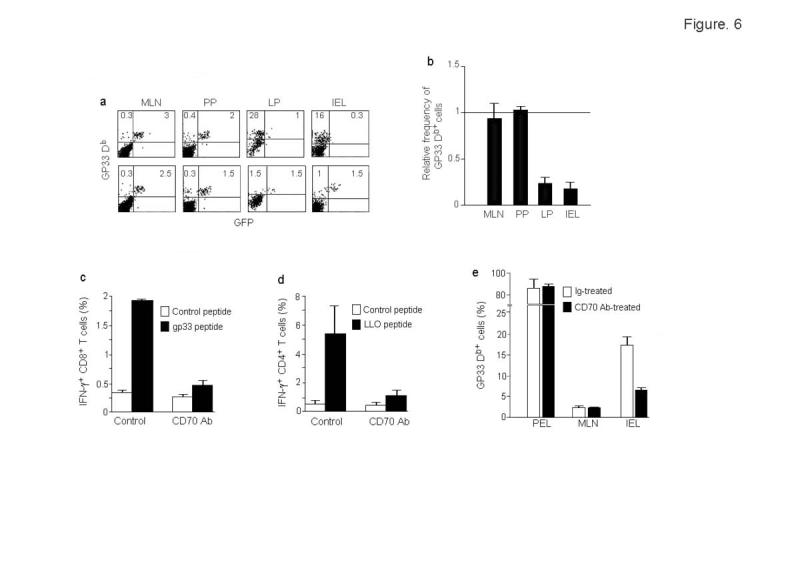
CD70 antibody treatment abrogates T cell expansion in LP and IEL. (a) Mice were adoptively transferred with P14-T-GFP CD8+ T cells and infected with rLmgp33 as in Fig. 4. On the day of adoptive transfer and on the day of infection, the mice were also injected iv with 100 μg of control hamster IgG or CD70 antibody. On day 3-post infection, cells from indicated organs were examined for the presence of Db Gp33 tetramer+ cells. Percentages of GFP+ and GFP- tetramer+ cells are indicated. (b) Summary of data (mean + s.d. from 6 mice in 2 independent experiments done as in a. Percentage of all tetramer+ CD8 T cells in CD70 antibody-treated mice were divided by that in control mice to obtain the relative frequency of tetramer positivity. (c, d) C57 mice were infected with rLmgp33 and treated with control or CD70 mAb and on day 7 post infection, LP T cells were tested for IFN-γ production by intracellular staining after 6h stimulation with either an irrelevant EBV peptide, the CD8 T cell epitopic peptide gp33 or the CD4 T cell epitopic peptide LLO190-201-pulsed BMDC. IFN-γ production (mean + s.d.) by CD8 and CD4-gated cells from 4 mice is shown in (c) and (d), respectively (e) Transferred mice treated with CD70 mAb or a control antibody were challenged with gp33 peptide in IFA ip and cells from different sites tested for tetramer+ CD8 T cells 3 days later. PEL, peritoneal exudates lymphocytes. Results (mean + s.d.) from 2 independent experiments with 3 mice each are shown.
To test if CD70 blockade can also affect the function of T cells in a non-adoptive transfer system, we measured the IFN-γ response to a CD8 as well as a dominant CD4 epitope in the presence or absence of CD70 antibody treatment. C57 mice infected orally with Lm were treated with control Ig or CD70 mAb and after 7 days, LP cells were stimulated with BMDC pulsed with the CD8+ T cell peptide gp33 or the immunodominant CD4+ T cell peptide, LLO190-20135. The IFN-γ response was nearly abrogated with CD70 antibody treatment, showing that CD70+ APC can stimulate CD8+ as well as CD4+ T cells in the gut mucosa (Fig. 6c,d).
In the above experiment, we could not formally prove that CD70 antibody had no effect on the T cell expansion at systemic sites, because only small numbers of tetramer+ cells could be detected even without antibody treatment in the PP and MLN 3 days after oral Lm infection. To more conclusively determine if CD70 antibody effect is specific to the gut or is also seen at peripheral sites, we repeated the adoptive transfer experiments in control and CD70 antibody-treated animals with a systemic rather than oral route of antigen challenge. After adoptive transfer of P14-T-GFP CD8+ T cells, the animals were injected intraperitoneally (ip) with gp33 peptide in Freund's incomplete adjuvant and 3 days later, the expansion of tetramer+ cells monitored in different organs. Under these conditions, although over 80% of CD8+ T cells accumulating in the peritoneal cavity were tetramer+, CD70 antibody treatment had no effect on their numbers. In contrast, CD70 blockade diminished the expansion of tetramer+ cells in the gut mucosa (Fig. 6e) showing that the CD70 antibody effect is confined to the gut where CD70+ APC are present.
The above results suggested that the expansion and differentiation of antigen-specific T cells in the gut at early time points is dependent on stimulation by CD70+ APCs. However, after oral Lm infection, the bacteria also spread to PP and MLN, reaching peak titers on day 2 and to spleen and liver, reaching peak titers on day 4 after infection36. Thus, T cells can presumably be activated by APCs in the lymphoid organs independently of the gut mucosa. Consistent with this, appreciable numbers of tetramer+ cells were detected at the systemic sites after 8 days of infection (Fig. 4a). To understand the relevance of CD70+ APC at later time points, we tested the effect of CD70 blockade on the T cell expansion at different sites 8 days after infection. CD70 antibody was administered to adoptively transferred mice every 2 days and the number of tetramer+ cells and their BrdU incorporation was tested after 8 days of infection. Even under these conditions, CD70 blockade selectively reduced the tetramer+ cell expansion in LP but not in PP or MLN (Supplementary Fig. 3 online). These results suggest that in general, a potent stimulation by CD70+ APC is required for T cell expansion in the gut mucosa.
Discussion
Here we showed that the expansion and differentiation of T cells after an oral infection occurs in the intestinal mucosa in situ and that restimulation via a unique tissue-specific APC is critical for this process. These findings may help understand how a robust immune response can be generated in the generally suppressive environment of the gut mucosa.
Several subsets of professional antigen presenting DCs have been described, based on the phenotype and function14,37-39. Although they are of non-hematopoietic lineage, the CD70+ APC share some of the features of professional APCs, including the expression of MHC class II, DEC-205, and costimulatory molecules CD70 and CD86. Moreover, they appear to be able to take up bacteria and present antigens to T cells in vitro as well as in vivo. However unlike the other DC subsets, the CD70+ APC is restricted in distribution to the LP compartment. Thus, CD70+ APCs appear to constitute a distinct class of tissue-specific APCs. Further studies should clarify the exact lineage of these cells.
The intestinal immune system has to delicately balance the induction of tolerance to harmless commensal bacteria and dietary antigens with the induction of active immunity to pathogens. Probably because the former outnumber the latter, the gut has a predisposition for tolerance induction14. DCs in PP and MLN elicit suppressive cytokines to the same stimuli that elicit stimulatory cytokines in splenic DCs17,18. Moreover, intestinal epithelial cells as well as intra-epithelial lymphocytes (IEL) produce copious amounts of the potentially immunosuppressive cytokine TGF-β40-42. Despite this apparently suppressive environment, IELs are constitutively activated in terms of their phenotype1-4. Moreover, the T cell responses to infections are more robust and more prolonged in the gut mucosa than in the periphery43-46. Our results suggest that a strong costimulation provided by CD70+ APC may be necessary to activate T cells in the generally suppressive intestinal milieu.
The issue of where and how intestinal T cells are activated has remained controversial. Based on the oligoclonality of T cell repertoire in the IEL that resembles T cell blasts in afferent lymph in the thoracic duct, it was originally proposed that T cells are primed to antigen in PP or MLN, then migrate to blood via thoracic duct before accumulating in LP and IEL47,48. However, studies in mice that lack PP, such as following in utero treatment with lymphotoxin β receptor-Ig (LTBR-Ig) fusion protein or in LTB-/- or LTA/LTB heterozygous mice have shown that PP are not indispensable for either mucosal IgA responses or tolerance induction49,50. Because impaired IgA responses were observed in the LTA-/- and LTBR-/- mice that lack both PP and MLN, it was then suggested that MLN might act as an alternative site for induction of mucosal immunity32,49,51-53. Even this view has been challenged by a study showing that the mucosal IgA deficiency in LTA-/- mice could be reversed by transfer of bone marrow cells from wild-type mice, suggesting that the presence of LT rather than intact PP or MLN is required for mucosal IgA production33.
The site of antigen priming for T cell immunity has not been studied in PP and MLN-deficient situations. Our results showing that LTA-/- mice, lacking PP and LN are incapable of expanding T cells in the mucosa, despite possessing CD70+ APC capable of providing stimulation suggest that priming occurs in PP and/or MLN for the gut mucosal T cell response. This is also consistent with previous studies showing that the initial priming of antigen-specific CTL occurs in the MLN30,31 as well as with a study showing that LTA-/- mice are more susceptible to Lm infection compared to wild-type mice54. Although defects in adhesion molecules and chemokine expression in the LTA-/- mice have been reported to inhibit intestinal migration of B cells33,34, T cell migration appear to be unaffected in these mice34. Thus, the defect in T cell expansion seen in LTA-/- mice is a consequence of lack of priming rather than defective homing of CD8 T cells.
Collectively our results suggest that although T cells may be primed in the PP and/or MLN, restimulation via the tissue-specific CD70+ APC is critical for the expansion and differentiation of T cells in the gut mucosa. Because CD70-mediated costimulation is critical during the mucosal stimulation, our findings may also provide a tool to augment mucosal response to an oral vaccine or to interfere with excessive mucosal immune response in situations like inflammatory bowel disease.
Methods
Mice. C57BL/6, BALB/c, MuMT, and RAG-1-/- mice were purchased from the Jackson Laboratory (Bar Harbor, Maine). P14 TCR Tg mice in RAG-1-/- background were purchased from the Taconic Farms, Germantown, NY. T-GFP Tg mice28, backcrossed to C57BL/6 mice for 7 generations were bred into P14 TCR-Tg mice, specific for the LCMV gp33-41 peptide55, that had been extensively backcrossed to C57BL/6 mice (N10, gift of Dr. Rafi Ahmed, Emory University, Atlanta, GA) to derive P14-T-GFP mice. LTA-/- mice32 was a gift from Dr. David Chaplin, University of Birmingham, Alabama. All mice were maintained under specific pathogen-free (SPF) conditions in microisolater cages and were used when they were 6-10 weeks of age. All animal experiments had been approved by the Institutional Review Board of the CBR Institute for Biomedical Research.
Adoptive transfer. Naïve CD8+ T cells were purified from splenocytes of P14 or P14-T-GFP mice by negative selection using the murine T cell CD8 subset isolation kit (R&D systems, Minneapolis, MN) according to the manufacturer's instructions. The isolated cells were >90% pure and >95% of them expressed GFP and high levels of L-selectin and were CD69-. C57BL/6 recipient mice were injected iv with 8×106 purified CD8+ T cells. In some experiments, the CD8 T cells from P14 mice were labeled with 1 μM carboxyfluorescein diacetate-succinimidyl ester (CSFE) before transfer.
Infection with Listeria monocytogenes. Mice were gavaged with 5×109 CFU of recombinant Lm encoding the LCMV glycoprotein29 (rLmgp33, gift of Dr. H. Shen, University of Pennsylvania School of Medicine, Philadelphia, PA) and at indicated times post infection, their spleen, MLN, PP, and intestine were harvested. In some experiments, isolated gut mucosal (LP+IEL) cells were infected with a D-Alanine-deficient Lm strain (Lmdd, gift of Dr. F. Frankel, University of Pennsylvania)27 that was labeled with CFSE.
Isolation of lymphocytes from PP, LP and IEL. Intestinal lymphocytes were isolated as described56. Briefly, PP were excised and the intestine was flushed with 50ml PBS and cut longitudinally along its entire length. After 3 washes, the intestine was cut into small pieces, transferred to a 50 ml tube in 20 ml HBSS containing 5% FBS and 0.1 mM EDTA and shaken with a magnetic stirrer for 20 min. The supernatant was harvested, the EDTA treatment repeated 2 more times and IEL isolated from the pooled supernatants. Finally, the intestinal pieces were digested with 200 U/ml of collagenase (type VIII, Sigma) with shaking at 37°C for 30 min. LP lymphocytes were isolated from the supernatants. For phenotyping and isolation of CD70+ cells, IEL and LP were used without further purification. In all other experiments to detect T cells, both IEL and LP lymphocytes were purified on a discontinuous percoll gradient. Briefly, the cells were washed 2 times in PBS and suspended in 8 ml of 44% Percoll and layered on 5 ml of 67% Percoll and centrifuged for 20 min at 600Xg. The interface cells were collected, washed and stained with antibodies for flow cytometric analysis.
Flow cytometry. MHC Db/gp33 tetramers were obtained from Beckman Coulter Immunomics, San Diego, CA. Anti-mouse DEC 205-FITC was from Research Diagnostics, Flanders, New Jersey. FITC, PE or Cy-5 conjugated antibodies to mouse CD4, CD8, CD11a, CD11b, CD11c, CD19, CD40, CD70, CD80, CD86, B220, Gr-1, NK1.1, H-2Kb, and I-Ab, were from BD PharMingen, San Diego, CA. MOMA-1, MOMA-2, F4/80 and CD68 antibodies were from Acris Antibodies, GmbH (Hiddenhausen, Germany). Blocking anti-CD70 antibody has been described23. Immunostaining and flow cytometric analysis were done as described29 using a FACScan flow cytometer (BD Biosciences, Mountain View, CA).
Generation of bone marrow-derived DC (BMDC). Bone marrow cells were cultured with 20 ng/ml each of rGM-CSF and IL4 (R&D systems) for 7 days and matured by culturing with 1μg/ml of LPS (Sigma) for 1 more day.
Immunomagnetic isolation and allogenic and peptide stimulation. Gut mucosal cells (LP) from C57BL/6 mice were first stained with CD19-coated Miltenyi beads and the B cells were depleted using a Miltenyi miniMACS system. The cells were subsequently stained with B220-coated Miltenyi beads to positively select B220+ CD70+ cells. The isolated cells were >90% pure as assessed by staining with CD70 antibody. To generate an alloresponse, different numbers of CD70+ or BMDC from C57BL/6 mice were irradiated (4000 rads) and cultured for 5 days with 105 T cells (purified using R&D systems T cell enrichment columns) from BALB/c mice in triplicate in 96 well plates, pulsed with 3H thymidine (0.5 μCi/well) for 6 hours, harvested and counted for thymidine incorporation using a Packard Topcount harvester and microplate reader. To generate a peptide-specific response, isolated CD70+ cells, CD19+ B cells, EL-4 cells and BMDC were pulsed with gp33-41 peptide (KAVYNFATC, 5 μg/ml, synthesized at BioSource, CA), washed and used to stimulate purified CD8 T cells from P14 mice. Proliferation was measured by 3H thymidine incorporation after 4 days and intracellular IFN-γ production and cytotoxicity was measured after 6 days using peptide pulsed EL-4 cells as described earlier28. To measure listeria-specific CD4 T cell response, LP cells from Lm-infected C57 mice were stimulated with the BMDC pulsed with the immunodominant CD4 T cell epitopic peptide, LLO190-20135 (NEKYAQAYPNVS, 5μg/ml) for 6 h in the presence of brefeldin A.
ELISPOT assay. ELISPOT assays were done in Millipore HA plates (Millipore, Bedford, MA) coated with anti-mouse IFN-γ (AN-18, BD Pharmingen). Single cell suspensions were plated in wells containing gp33-41 peptide-pulsed or unpulsed MC57G cells. Cytokine production was detected after 24 h using biotinylated antibody against IFN-γ (R4-6A2, BD Pharmingen) followed by alkaline phosphatase-conjugated anti-biotin monoclonal antibody (Vector Laboratories, Burlingame, Calif.) and precipitating alkaline phosphatase substrate, nitroblue tetrazolium-5-bromo-4-chloro-3-indolylphosphate (Pierce, Rockford, Ill.) Spots were counted using a digital imager and Immunospot software (version 3, Cellular Technology, Ltd, Cleveland, OH). The number of spots was normalized with respect to the number of CD8β+ T cells (determined in parallel by flow cytometry) to give a final result as spot-forming cells per 104 cells.
Immunohistology. Intestinal tissue was embedded in O.C.T. medium and snap-frozen in liquid nitrogen. Ten micron cryostat sections were reacted with CD70 mAb followed by goat anti-rat HRP and Nova Red substrate (Vector Laboratories, Burlingame, CA) and counter stained with 1% Methyl Green for examination with a light microscope.
BrdU incorporation assay. In vivo BrdU labeling was done as described by Meyer et al57. Two hours before sacrifice, mice were injected ip with 1 ml of BrdU (1-bromo-2' deoxyuridine and 5-fluoro-2' deoxyuridine) labeling reagent (Zymed, South San Francisco, CA) per 100 g body weight. Lymphocytes harvested from various organs were stained externally with anti-mouse CD8 Cy-5 and gp33 tetramer PE, fixed, permeabilized and stained internally with anti-BrdU/DNase FITC (Becton-Dickinson, San Jose, CA) as described earlier29. For vitro BrdU assay, 22 μl of labeling reagent was added to 200 μl of culture medium in 96 well and after overnight incubation, cells were stained for BrdU as described above.
Esterase staining. Intracellular esterase staining was done using α-naphthyl acetate esterase staining kit from Sigma Diagnostics according to the manufacturer's instructions. After counterstaining with hematoxylin, dark sating cells were enumerated by light microscopy by counting cells in 5 high power fields.
Antibody treatment. Mice were given anti–murine CD70 (clone 3B9) or control hamster mAb (100 μg per intraperitoneal injection) every 2 days.
Supplementary Material
Acknowledgements
We thank H. Shen for providing the rLmgp33 listeria strain and F. Frankel for providing Lmdd strain and A. Schlesinger for help with ELISPOT assays. This work was supported by NIH grant AI46566 to NM.
References
- 1.Klein JR. Ontogeny of the Thy-1-, Lyt-2+ murine intestinal intraepithelial lymphocyte. Characterization of a unique population of thymus-independent cytotoxic effector cells in the intestinal mucosa. J Exp Med. 1986;164:309–14. doi: 10.1084/jem.164.1.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huleatt JW, Lefrancois L. Antigen-driven induction of CD11c on intestinal intraepithelial lymphocytes and CD8+ T cells in vivo. J Immunol. 1995;154:5684–93. [PubMed] [Google Scholar]
- 3.Beagley KW, Husband AJ. Intraepithelial lymphocytes: origins, distribution, and function. Crit Rev Immunol. 1998;18:237–54. doi: 10.1615/critrevimmunol.v18.i3.40. [DOI] [PubMed] [Google Scholar]
- 4.Goodman T, Lefrancois L. Intraepithelial lymphocytes. Anatomical site, not T cell receptor form, dictates phenotype and function. J Exp Med. 1989;170:1569–81. doi: 10.1084/jem.170.5.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pirzer UC, Schurmann G, Post S, Betzler M, Meuer SC. Differential responsiveness to CD3-Ti vs. CD2-dependent activation of human intestinal T lymphocytes. Eur J Immunol. 1990;20:2339–42. doi: 10.1002/eji.1830201025. [DOI] [PubMed] [Google Scholar]
- 6.Targan SR, Deem RL, Liu M, Wang S, Nel A. Definition of a lamina propria T cell responsive state. Enhanced cytokine responsiveness of T cells stimulated through the CD2 pathway. J Immunol. 1995;154:664–75. [PubMed] [Google Scholar]
- 7.Zhou Z, Pollok KE, Kim KK, Kim YJ, Kwon BS. Functional analysis of T-cell antigen 4-1BB in activated intestinal intra-epithelial T lymphocytes. Immunol Lett. 1994;41:177–84. doi: 10.1016/0165-2478(94)90129-5. [DOI] [PubMed] [Google Scholar]
- 8.Wang HC, Klein JR. Multiple levels of activation of murine CD8(+) intraepithelial lymphocytes defined by OX40 (CD134) expression: effects on cell-mediated cytotoxicity, IFN-gamma, and IL-10 regulation. J Immunol. 2001;167:6717–23. doi: 10.4049/jimmunol.167.12.6717. [DOI] [PubMed] [Google Scholar]
- 9.Kim SK, Schluns KS, Lefrancois L. Induction and visualization of mucosal memory CD8 T cells following systemic virus infection. J Immunol. 1999;163:4125–32. [PubMed] [Google Scholar]
- 10.Kim SK, et al. Generation of mucosal cytotoxic T cells against soluble protein by tissue-specific environmental and costimulatory signals. Proc Natl Acad Sci U S A. 1998;95:10814–9. doi: 10.1073/pnas.95.18.10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gonsky R, et al. Mucosa-specific targets for regulation of IFN-gamma expression: lamina propria T cells use different cis-elements than peripheral blood T cells to regulate transactivation of IFN-gamma expression. J Immunol. 2000;164:1399–407. doi: 10.4049/jimmunol.164.3.1399. [DOI] [PubMed] [Google Scholar]
- 12.Huang FP, et al. A discrete subpopulation of dendritic cells transports apoptotic intestinal epithelial cells to T cell areas of mesenteric lymph nodes. J Exp Med. 2000;191:435–44. doi: 10.1084/jem.191.3.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iwasaki A, Kelsall BL. Localization of distinct Peyer's patch dendritic cell subsets and their recruitment by chemokines macrophage inflammatory protein (MIP)-3alpha, MIP-3beta, and secondary lymphoid organ chemokine. J Exp Med. 2000;191:1381–94. doi: 10.1084/jem.191.8.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mowat AM. Anatomical basis of tolerance and immunity to intestinal antigens. Nat Rev Immunol. 2003;3:331–41. doi: 10.1038/nri1057. [DOI] [PubMed] [Google Scholar]
- 15.Castellaneta A, Abe M, Morelli AE, Thomson AW. Identification and characterization of intestinal Peyer's patch interferon-alpha producing (plasmacytoid) dendritic cells. Hum Immunol. 2004;65:104–13. doi: 10.1016/j.humimm.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 16.Bilsborough J, George TC, Norment A, Viney JL. Mucosal CD8alpha+ DC, with a plasmacytoid phenotype, induce differentiation and support function of T cells with regulatory properties. Immunology. 2003;108:481–92. doi: 10.1046/j.1365-2567.2003.01606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iwasaki A, Kelsall BL. Freshly isolated Peyer's patch, but not spleen, dendritic cells produce interleukin 10 and induce the differentiation of T helper type 2 cells. J Exp Med. 1999;190:229–39. doi: 10.1084/jem.190.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williamson E, Bilsborough JM, Viney JL. Regulation of mucosal dendritic cell function by receptor activator of NF-kappa B (RANK)/RANK ligand interactions: impact on tolerance induction. J Immunol. 2002;169:3606–12. doi: 10.4049/jimmunol.169.7.3606. [DOI] [PubMed] [Google Scholar]
- 19.Martin P, et al. Characterization of a new subpopulation of mouse CD8alpha+ B220+ dendritic cells endowed with type 1 interferon production capacity and tolerogenic potential. Blood. 2002;100:383–90. doi: 10.1182/blood.v100.2.383. [DOI] [PubMed] [Google Scholar]
- 20.Futagawa T, et al. Expression and function of 4-1BB and 4-1BB ligand on murine dendritic cells. Int Immunol. 2002;14:275–86. doi: 10.1093/intimm/14.3.275. [DOI] [PubMed] [Google Scholar]
- 21.Akiba H, et al. Critical contribution of OX40 ligand to T helper cell type 2 differentiation in experimental leishmaniasis. J Exp Med. 2000;191:375–80. doi: 10.1084/jem.191.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arens R, et al. Constitutive CD27/CD70 interaction induces expansion of effector-type T cells and results in IFNgamma-mediated B cell depletion. Immunity. 2001;15:801–12. doi: 10.1016/s1074-7613(01)00236-9. [DOI] [PubMed] [Google Scholar]
- 23.Tesselaar K, et al. Lethal T cell immunodeficiency induced by chronic costimulation via CD27-CD70 interactions. Nat Immunol. 2003;4:49–54. doi: 10.1038/ni869. [DOI] [PubMed] [Google Scholar]
- 24.Tesselaar K, et al. Expression of the murine CD27 ligand CD70 in vitro and in vivo. J Immunol. 2003;170:33–40. doi: 10.4049/jimmunol.170.1.33. [DOI] [PubMed] [Google Scholar]
- 25.Le AX, et al. Cytotoxic T cell responses in HLA-A2.1 transgenic mice. Recognition of HLA alloantigens and utilization of HLA-A2.1 as a restriction element. J Immunol. 1989;142:1366–71. [PubMed] [Google Scholar]
- 26.Powell DW, et al. Myofibroblasts. I. Paracrine cells important in health and disease. Am J Physiol. 1999;277:C1–9. doi: 10.1152/ajpcell.1999.277.1.C1. [DOI] [PubMed] [Google Scholar]
- 27.Thompson RJ, Bouwer HG, Portnoy DA, Frankel FR. Pathogenicity and immunogenicity of a Listeria monocytogenes strain that requires D-alanine for growth. Infect Immun. 1998;66:3552–61. doi: 10.1128/iai.66.8.3552-3561.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manjunath N, et al. A transgenic mouse model to analyze CD8(+) effector T cell differentiation in vivo. Proc Natl Acad Sci U S A. 1999;96:13932–7. doi: 10.1073/pnas.96.24.13932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manjunath N, et al. Effector differentiation is not prerequisite for generation of memory cytotoxic T lymphocytes. J Clin Invest. 2001;108:871–8. doi: 10.1172/JCI13296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim SK, Reed DS, Heath WR, Carbone F, Lefrancois L. Activation and migration of CD8 T cells in the intestinal mucosa. J Immunol. 1997;159:4295–306. [PubMed] [Google Scholar]
- 31.Vezys V, Olson S, Lefrancois L. Expression of intestine-specific antigen reveals novel pathways of CD8 T cell tolerance induction. Immunity. 2000;12:505–14. doi: 10.1016/s1074-7613(00)80202-2. [DOI] [PubMed] [Google Scholar]
- 32.De Togni P, et al. Abnormal development of peripheral lymphoid organs in mice deficient in lymphotoxin. Science. 1994;264:703–7. doi: 10.1126/science.8171322. [DOI] [PubMed] [Google Scholar]
- 33.Kang HS, et al. Signaling via L TbetaR on the lamina propria stromal cells of the gut is required for IgA production. Nat Immunol. 2002;3:576–82. doi: 10.1038/ni795. [DOI] [PubMed] [Google Scholar]
- 34.Newberry RD, McDonough JS, McDonald KG, Lorenz RG. Postgestational lymphotoxin/lymphotoxin beta receptor interactions are essential for the presence of intestinal B lymphocytes. J Immunol. 2002;168:4988–97. doi: 10.4049/jimmunol.168.10.4988. [DOI] [PubMed] [Google Scholar]
- 35.Geginat G, Schenk S, Skoberne M, Goebel W, Hof H. A novel approach of direct ex vivo epitope mapping identifies dominant and subdominant CD4 and CD8 T cell epitopes from Listeria monocytogenes. J Immunol. 2001;166:1877–84. doi: 10.4049/jimmunol.166.3.1877. [DOI] [PubMed] [Google Scholar]
- 36.Marco AJ, et al. Penetration of Listeria monocytogenes in mice infected by the oral route. Microb Pathog. 1997;23:255–63. doi: 10.1006/mpat.1997.0144. [DOI] [PubMed] [Google Scholar]
- 37.Shortman K, Liu YJ. Mouse and human dendritic cell subtypes. Nat Rev Immunol. 2002;2:151–61. doi: 10.1038/nri746. [DOI] [PubMed] [Google Scholar]
- 38.Ardavin C. Origin, precursors and differentiation of mouse dendritic cells. Nat Rev Immunol. 2003;3:582–90. doi: 10.1038/nri1127. [DOI] [PubMed] [Google Scholar]
- 39.Wilson HL, O'Neill HC. Murine dendritic cell development: difficulties associated with subset analysis. Immunol Cell Biol. 2003;81:239–46. doi: 10.1046/j.1440-1711.2003.t01-1-01165.x. [DOI] [PubMed] [Google Scholar]
- 40.MacDonald TT. Effector and regulatory lymphoid cells and cytokines in mucosal sites. Curr Top Microbiol Immunol. 1999;236:113–35. doi: 10.1007/978-3-642-59951-4_7. [DOI] [PubMed] [Google Scholar]
- 41.Shires J, Theodoridis E, Hayday AC. Biological insights into TCRgammadelta+ and TCRalphabeta+ intraepithelial lymphocytes provided by serial analysis of gene expression (SAGE) Immunity. 2001;15:419–34. doi: 10.1016/s1074-7613(01)00192-3. [DOI] [PubMed] [Google Scholar]
- 42.Hayday A, Theodoridis E, Ramsburg E, Shires J. Intraepithelial lymphocytes: exploring the Third Way in immunology. Nat Immunol. 2001;2:997–1003. doi: 10.1038/ni1101-997. [DOI] [PubMed] [Google Scholar]
- 43.Pope C, et al. Organ-specific regulation of the CD8 T cell response to Listeria monocytogenes infection. J Immunol. 2001;166:3402–9. doi: 10.4049/jimmunol.166.5.3402. [DOI] [PubMed] [Google Scholar]
- 44.Offit PA, Cunningham SL, Dudzik KI. Memory and distribution of virus-specific cytotoxic T lymphocytes (CTLs) and CTL precursors after rotavirus infection. J Virol. 1991;65:1318–24. doi: 10.1128/jvi.65.3.1318-1324.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Belyakov IM, et al. Mucosal immunization with HIV-1 peptide vaccine induces mucosal and systemic cytotoxic T lymphocytes and protective immunity in mice against intrarectal recombinant HIV-vaccinia challenge. Proc Natl Acad Sci U S A. 1998;95:1709–14. doi: 10.1073/pnas.95.4.1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Masopust D, Jiang J, Shen H, Lefrancois L. Direct analysis of the dynamics of the intestinal mucosa CD8 T cell response to systemic virus infection. J Immunol. 2001;166:2348–56. doi: 10.4049/jimmunol.166.4.2348. [DOI] [PubMed] [Google Scholar]
- 47.Regnault A, Cumano A, Vassalli P, Guy-Grand D, Kourilsky P. Oligoclonal repertoire of the CD8 alpha alpha and the CD8 alpha beta TCR-alpha/beta murine intestinal intraepithelial T lymphocytes: evidence for the random emergence of T cells. J Exp Med. 1994;180:1345–58. doi: 10.1084/jem.180.4.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arstila T, et al. Identical T cell clones are located within the mouse gut epithelium and lamina propia and circulate in the thoracic duct lymph. J Exp Med. 2000;191:823–34. doi: 10.1084/jem.191.5.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamamoto M, et al. Alternate mucosal immune system: organized Peyer's patches are not required for IgA responses in the gastrointestinal tract. J Immunol. 2000;164:5184–91. doi: 10.4049/jimmunol.164.10.5184. [DOI] [PubMed] [Google Scholar]
- 50.Spahn TW, et al. Induction of oral tolerance to cellular immune responses in the absence of Peyer's patches. Eur J Immunol. 2001;31:1278–87. doi: 10.1002/1521-4141(200104)31:4<1278::aid-immu1278>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 51.Banks TA, et al. Lymphotoxin-alpha-deficient mice. Effects on secondary lymphoid organ development and humoral immune responsiveness. J Immunol. 1995;155:1685–93. [PubMed] [Google Scholar]
- 52.Davis IA, Knight KA, Rouse BT. The spleen and organized lymph nodes are not essential for the development of gut-induced mucosal immune responses in lymphotoxin-alpha deficient mice. Clin Immunol Immunopathol. 1998;89:150–9. doi: 10.1006/clin.1998.4601. [DOI] [PubMed] [Google Scholar]
- 53.Futterer A, Mink K, Luz A, Kosco-Vilbois MH, Pfeffer K. The lymphotoxin beta receptor controls organogenesis and affinity maturation in peripheral lymphoid tissues. Immunity. 1998;9:59–70. doi: 10.1016/s1074-7613(00)80588-9. [DOI] [PubMed] [Google Scholar]
- 54.Eugster HP, et al. Multiple immune abnormalities in tumor necrosis factor and lymphotoxin-alpha double-deficient mice. Int Immunol. 1996;8:23–36. doi: 10.1093/intimm/8.1.23. [DOI] [PubMed] [Google Scholar]
- 55.Pircher H, Burki K, Lang R, Hengartner H, Zinkernagel RM. Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature. 1989;342:559–61. doi: 10.1038/342559a0. [DOI] [PubMed] [Google Scholar]
- 56.Lefrancois L, Lycke N. In: Current Protocol Immunology. R C, editor. John Wiley & Sons; New York: 1996. pp. 3.19.1–16. [Google Scholar]
- 57.Meyer AL, et al. Rapid depletion of peripheral antigen-specific T cells in TCR-transgenic mice after oral administration of myelin basic protein. J Immunol. 2001;166:5773–81. doi: 10.4049/jimmunol.166.9.5773. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.