

Abstract
DNA topoisomerase II is an essential nuclear enzyme that modulates DNA processes by altering the topological state of double-stranded DNA. This enzyme is required for chromosome condensation and segregation; however, the regulatory mechanism of its activation is largely unknown. Here we demonstrate that topoisomerase IIα is activated in response to genotoxic stress. Concomitant with the activation, the expression of topoisomerase IIα is increased following DNA damage. The results also demonstrate that the proapoptotic kinase protein kinase C δ (PKCδ) interacts with topoisomerase IIα. This association is in an S-phase-specific manner and is required for stabilization and catalytic activation of topoisomerase IIα in response to DNA damage. Conversely, inhibition of PKCδ activity attenuates DNA damage-induced activation of topoisomerase IIα. Finally, aberrant activation of topoisomerase IIα by PKCδ is associated with induction of apoptosis upon exposure to genotoxic agents. These findings indicate that PKCδ regulates topoisomerase IIα and thereby cell fate in the genotoxic stress response.
DNA topoisomerase II is a nuclear enzyme that regulates DNA topology via transient double-strand breaks in the DNA helix (5, 38, 39). Topoisomerase II is involved in cell proliferation and has been implicated in indispensable cellular processes, such as replication, transcription, recombination, and chromosomal condensation and segregation (38). This enzyme also has an essential function as a structural component of mitotic chromosome and interphase nuclear scaffolds (9, 14). Given the fact that expression of topoisomerase II in proliferating cells is higher than that in quiescent cells, this enzyme is a clinically useful target to elicit cytotoxic effects in proliferating tumor cells. Indeed, a variety of anticancer agents target topoisomerase II (20) and interfere with its catalytic activity by trapping the enzyme in a form that is covalently bound to DNA. These stable enzyme-associated DNA complexes induce DNA damage and cell death. In addition to catalytically inactivating topoisomerase II, previous studies have shown that aberrant expression of topoisomerase II is associated with the induction of apoptosis (28, 37). While forced expression of topoisomerase IIα in cells triggered apoptotic cell death, nuclear localization of the enzyme was required for efficient apoptotic induction. By contrast, another report demonstrated that depletion of topoisomerase IIα conferred induction of apoptosis (1). Taken together, these results suggest that appropriate regulation of topoisomerase IIα expression is essential for cell viability and proliferation. In this regard, deregulated expression of topoisomerase IIα is associated with the commitment of apoptotic cell death; however, the mechanism remains unclear.
The protein kinase C (PKC) family of serine/threonine kinases is subdivided into (i) conventional PKCs (PKC α [PKCα], PKCβ, and PKCγ) that are calcium dependent and activated by diacylglycerol (DAG), (ii) novel PKCs (PKCδ, PKCɛ, PKCθ, and PKCμ) that are calcium independent and activated by DAG, and (iii) atypical PKCs (PKCζ and PKCλ) that are calcium independent and not activated by DAG (32). The ubiquitously expressed novel PKC, PKCδ, is tyrosine phosphorylated and activated by c-Abl and Lyn in the response to DNA damage (45, 48). PKCδ interacts with the nuclear DNA-dependent protein kinase catalytic subunit (DNA-PKcs) (2). Phosphorylation of DNA-PKcs by PKCδ inhibits the function of DNA-PKcs to form complexes with DNA and to phosphorylate downstream targets (2). In addition, cells deficient in DNA-PK are resistant to apoptosis induced by overexpression of the PKCδ catalytic domain. Other studies have demonstrated that the nuclear complex of c-Abl and Lyn includes the protein tyrosine phosphatase SHPTP1 (Src homology 2 domain [SH2]-containing protein tyrosine phosphatase 1) (21, 42) and that PKCδ phosphorylates and inactivates SHPTP1 in response to DNA damage (44). In cells that respond to genotoxic stress with apoptosis, PKCδ is cleaved by caspase-3 into a constitutively active catalytic fragment (PKCδCF) (10, 11). The finding that PKCδCF induced nuclear condensation and DNA fragmentation indicates that cleavage of PKCδ contributes to the apoptotic response (15). In this context, a recent study has demonstrated that PKCδ translocated to the nucleus and regulated Rad9 by phosphorylation in the apoptotic response to DNA damage (46). Furthermore, another study showed that cells derived from PKCδ-null transgenic mice were defective in mitochondrion-dependent apoptosis induced by various agents such as UV irradiation and hydrogen peroxide (27). These findings collectively support an essential role for PKCδ in the induction of apoptosis in the genotoxic stress response.
The present study demonstrates that PKCδ interacts with topoisomerase IIα. This interaction is required for stabilization and activation of topoisomerase IIα following DNA damage. The results also demonstrate that genotoxic stress-induced topoisomerase IIα activation confers the induction of PKCδ-mediated apoptosis.
MATERIALS AND METHODS
Cell culture.
Human MOLT-4, U-937, and HL-60 leukemia cells were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mM l-glutamine. Human U2-OS osteosarcoma cells, 293T embryonal kidney cells, mouse embryo fibroblasts (MEFs), and pkcδ−/− MEFs (29) were grown in Dulbecco's modified Eagle's medium containing 10% heat-inactivated fetal bovine serum and antibiotics. Cells were treated with 10 μM 1-β-d-arabinofuranosylcytosine (ara-C; Sigma-Aldrich), 500 μM cisplatin (CDDP; Sigma-Aldrich), 5 μM rottlerin (Sigma-Aldrich), 100 nM bistratene A (Sigma-Aldrich), 500 ng/ml nocodazole (Sigma-Aldrich), 5 μg/ml aphidicolin (WAKO), or 10 μM ICRF-193 (Zenyaku).
Plasmids.
PKCδ expression plasmids were described previously (44, 45). Plasmids for the expression of topoisomerase IIα glutathione S-transferase (GST) fusion proteins were constructed by PCR using topoisomerase IIα cDNA fused with green fluorescent protein (GFP) (GFP-TopoIIα) (30). To construct the GFP-topoisomerase IIα mutant in which the C-terminal domain was deleted (GFP-TopoIIαΔC), GFP-TopoIIα was digested with restriction enzymes PstI and SmaI. After the PstI site was blunt ended, the DNA was subjected to self-ligation, resulting in the truncated form of GFP-TopoIIα encoding amino acid residues 1 to 1145.
Cell transfections.
Cell transfections were performed as described previously (43, 47). The total DNA concentration was kept constant by including an empty vector.
Protein identification by mass spectrometry analysis.
293T cells were transfected with Flag vector or PKCδ tagged with a Flag epitope (Flag-PKCδ). At 48 h posttransfection, cells were lysed with 0.1% NP-40 lysis buffer (0.1% NP-40, 50 mM Tris-Cl, pH 7.6, 150 mM NaCl, 2 μg/ml aprotinin, 1 mM dithiothreitol [DTT], 10 mM NaF, 1 mM phenylmethylsulfonyl fluoride [PMSF], 10 μg/ml leupeptin, 1 μM pepstatin, and 1 mM Na3VO4). Lysates were centrifuged at 14,000 × g for 15 min, and the supernatants were subjected to immunoprecipitation with anti-Flag agarose (Sigma-Aldrich). Flag-PKCδ-associated complexes were then separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and visualized by silver staining. Protein bands were cut out from gels, digested with trypsin, and analyzed by matrix-assisted laser desorption ionization-time of flight mass spectrometry as described previously (12).
Immunoprecipitation. For coimmunoprecipitation of PKCδ and topoisomerase IIα, nuclear lysates from MOLT-4 cells were prepared as described previously (7). Precleared lysates (1 mg) were incubated with anti-topoisomerase IIα (Alexis Biochemicals) or anti-PKCδ (Santa Cruz Biotechnology) antibodies for 2 h at 4°C followed by 1 h of incubation with protein A-Sepharose beads (Amersham Biosciences). The immune complexes were washed three times with 0.1% NP-40 lysis buffer and then eluted by boiling for 5 min in 50 mM Tris-Cl, pH 6.8, containing 2% SDS, 6% 2-mercaptoethanol, 0.01% bromophenol blue, and 10% glycerol. The eluted samples were subjected to immunoblot analysis.
Immunoblot analysis.
Cells were lysed on ice for 30 min with 1% NP-40 lysis buffer (1% NP-40, 50 mM Tris-Cl, pH 7.6, 150 mM NaCl, 2 μg/ml aprotinin, 1 mM DTT, 10 mM NaF, 1 mM PMSF, 10 μg/ml leupeptin, 1 μM pepstatin, and 1 mM Na3VO4). Lysates were centrifuged at 14,000 × g for 15 min, and the supernatants were analyzed by immunoblotting. Cell lysates or immunoprecipitates were separated by SDS-PAGE and transferred to nitrocellulose filters. The filters were then incubated with anti-Flag, anti-GST (Nacalai Tesque), anti-GFP (Nacalai Tesque), anti-topoisomerase IIα (MBL or Topogen), anti-topoisomerase IIα and β (MBL), anti-PKCδ, anti-phospho-PKCδ (Thr505) (Cell Signaling Technology) or antitubulin (Sigma-Aldrich). The antigen-antibody complexes were visualized by chemiluminescence (Perkin-Elmer).
In vitro binding assays.
Cell lysates were incubated with purified proteins fused to GST in lysis buffer for 2 h at 4°C. The adsorbates were resolved by SDS-PAGE and analyzed by immunoblotting with anti-topoisomerase IIα or anti-PKCδ.
Preparation of nuclear extracts.
To prepare nuclear extracts for topoisomerase II catalytic activity assays, 3 × 107 to 5 × 107 cells were washed first with phosphate-buffered saline and then with 1 ml of buffer A (10 mM HEPES, pH 7.6, 15 mM KCl, 0.1 mM EDTA, 1 mM DTT, 0.5 mM PMSF, and 10 μg/ml leupeptin) and resuspended in buffer A containing 0.2% NP-40. After centrifugation, cells were resuspended in 1 ml of buffer A containing 0.25 M sucrose. Subsequently, samples were collected by centrifugation and resuspended in buffer D (50 mM HEPES, pH 7.6, 400 mM KCl, 0.1 mM EDTA, 10% glycerol, 1 mM DTT, 0.5 mM PMSF, and 10 μg/ml leupeptin). After the samples were mixed and gently rocked for 30 min at 4°C, they were centrifuged for 15 min at 1,400 rpm. Supernatants were used as nuclear extracts.
Topoisomerase IIα catalytic activity assays.
Topoisomerase II activity was assayed by the decatenation of kinetoplast DNA (KDNA) or relaxation of pBluescript. The decatenation assays were performed by incubating 0.2 μg KDNA (Nippon Gene) with nuclear extracts or recombinant topoisomerase IIα (Topogen) in assay buffer A (50 mM Tris-HCl, pH 8.0, 120 mM KCl, 10 mM MgCl2, 0.5 mM ATP, 0.5 mM DTT, and 30 μg/ml bovine serum albumin). After incubation for 20 min at 37°C, the reactions were stopped by the addition of stop buffer (5% Sarkosyl, 0.0025% bromophenol blue, and 25% glycerol). The reaction products were resolved on a 1% agarose gel containing 0.5 μg/ml ethidium bromide. The relaxation assays were performed by incubating 0.2 μg pBluescript with nuclear extracts or recombinant topoisomerase IIα in assay buffer B (30 mM Tris-HCl, pH 7.6, 60 mM KCl, 8 mM MgCl2, 3 mM ATP, 15 mM 2-mercaptoethanol, and 30 μg/ml bovine serum albumin). After incubation for 15 min at 37°C, the reactions were quenched by the addition of 0.1 volume of 10% SDS. The reaction products were resolved on a 1% native agarose gel.
siRNA transfections.
Small interfering RNA duplexes (siRNAs) targeting PKCδ were synthesized and purified by Invitrogen (Stealth RNAi). Stealth RNAi sequences were 5′-AUUAGCACAAUCUGGAUGACGCGCC-3′ for PKCδsiRNA1, 5′-AAACUCAUGGUUCUUGAUGUAGUGG-3′ for PKCδsiRNA2, 5′-AAAGAAGGUGGCGAUAAACUCAUGG-3′ for PKCδsiRNA3, and 5′-AACUCCGGUCUUCUUCUCGAAACCC-3′ for PKCδsiRNA4. siRNAs targeting topoisomerase IIα were synthesized and purified by QIAGEN. The siRNA sequences were 5′-AAGACUGUCUGUUGAAAGATT-3′ for topoisomerase IIαsiRNA1 and 5′-CAUAUUUUGCUCCGCCCAGTT-3′ for topoisomerase IIαsiRNA2. Scrambled siRNA was purchased from QIAGEN and used as a negative control. Transfection of siRNAs was performed using Lipofectamine 2000 (Invitrogen).
In vitro kinase assays.
In vitro kinase assays were performed as described previously (44).
RT-PCR analysis for topoisomerase IIα gene expression.
Total cellular RNA was extracted using the RNeasy kit (QIAGEN). First-strand cDNA synthesis and the following PCR were performed with 500 ng of total RNA using SuperScript one-step reverse transcriptase PCR (RT-PCR) system (Invitrogen) according to the manufacturer's protocol. For topoisomerase IIα gene expression, the nucleotide sequence of 5′-GCCCTCCTGCTACACATTTC-3′ was used as the sense primer, and 5′-AACACTTGGGCTTTACTTCACTT-3′ was used as the antisense primer. For β-actin gene expression, the nucleotide sequence of 5′-CAGGGCGTGATGGTGGGCA-3′ was used as the sense primer, and 5′-CAAACATCATCTGGGTCATCTTCTC-3′ was used as the antisense primer. The reaction products were resolved on a 2% agarose gel.
Cell cycle analysis.
DNA content was assessed by staining ethanol-fixed cells with propidium iodide and monitoring by using a FACScan (Becton Dickinson). Cell cycle phases were determined by using the CellQuest program (Becton Dickinson).
Assessment of apoptosis.
Apoptotic cells were detected by terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assays using the DeadEnd colorimetric TUNEL system (Promega).
RESULTS
Identification of topoisomerase IIα as a novel PKCδ-interacting protein.
To identify cellular proteins that interact with PKCδ, 293T cells were transiently transfected with PKCδ tagged with a Flag epitope. Anti-Flag immunoprecipitates were resolved by SDS-PAGE, and the coimmunoprecipitating proteins were analyzed by mass spectrometry. The results revealed that topoisomerase IIα was one of the PKCδ-interacting proteins (Fig. 1A). To assess whether endogenous PKCδ associates with endogenous topoisomerase IIα in cells, anti-PKCδ immunoprecipitates from MOLT-4 cell lysates were analyzed by immunoblotting with anti-topoisomerase IIα. PKCδ and topoisomerase IIα formed complexes in cells (Fig. 1B). In reciprocal experiments, immunoblot analysis of anti-topoisomerase IIα immunoprecipitates with anti-PKCδ confirmed the association of PKCδ and topoisomerase IIα (Fig. 1B). Similar results were obtained from U-937 and HL-60 cells (data not shown). To determine whether PKCδ associates with topoisomerase IIβ, lysates from MOLT-4 cells were subjected to immunoprecipitation with anti-PKCδ followed by immunoblotting with an anti-topoisomerase II antibody that recognizes both topoisomerase IIα (170 kDa) and IIβ (180 kDa). The results demonstrated that PKCδ interacts preferably with topoisomerase IIα and little, if any, with topoisomerase IIβ (Fig. 1C). To further define the association of PKCδ and topoisomerase IIα, cell lysates were incubated with purified GST, GST-PKCδ regulatory domain (RD) or GST-PKCδ catalytic fragment (CF) (Fig. 2A). Analysis of adsorbates with anti-topoisomerase IIα showed the binding of topoisomerase IIα to GST-PKCδCF, but not to GST or GST-PKCδRD (Fig. 2B). To map the PKCδ-interacting domain on topoisomerase IIα, cell lysates were incubated with GST fusion proteins containing topoisomerase IIα (amino acid residues 1 to 403, 388 to 864, 852 to 1233, or 1021 to 1531) (Fig. 2C). The results demonstrated that topoisomerase IIα (amino acid residues 1021 to 1531) contains the determinants responsible for binding to PKCδ (Fig. 2D). These findings indicate that PKCδCF interacts with the C-terminal region of topoisomerase IIα. To further confirm this in cells, we constructed a GFP-topoisomerase IIα mutant in which the C-terminal domain was deleted (GFP-TopoIIαΔC). GFP-TopoIIα or GFP-TopoIIαΔC was transfected into 293T cells. The finding that PKCδ interacts with full-length topoisomerase IIα, but not with topoisomerase IIα with the C-terminal domain deleted, indicates that the C-terminal region of topoisomerase IIα is required for binding to PKCδ in cells (Fig. 2E). To determine whether this interaction is direct, purified GST or GST-PKCδ was incubated with recombinant topoisomerase IIα. The finding that GST-PKCδ, and not GST, bound to recombinant topoisomerase IIα provides support that binding is direct (Fig. 2F). To assess further whether PKCδ phosphorylates topoisomerase IIα, purified GST-PKCδ was incubated with recombinant topoisomerase IIα. Analysis of the products by SDS-PAGE and autoradiography showed that topoisomerase IIα is a substrate for PKCδ (Fig. 2G). These data demonstrate that PKCδ directly binds to and phosphorylates topoisomerase IIα.
FIG. 1.
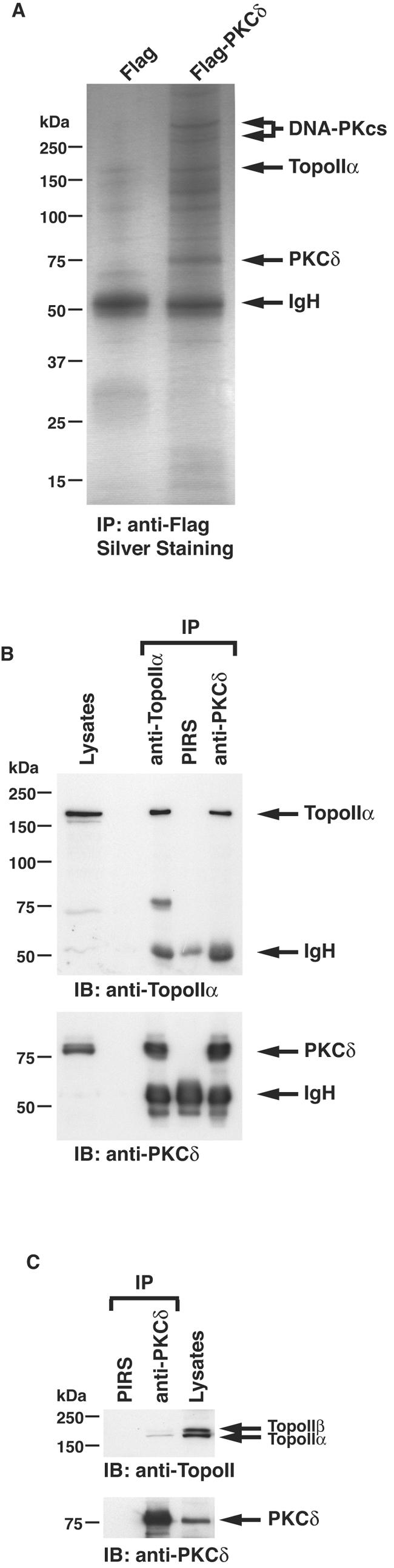
Association of PKCδ with topoisomerase IIα (TopoIIα). (A) Cell lysates from 293T cells transfected with Flag vector or Flag-PKCδ were immunoprecipitated (IP) with anti-Flag. Immunoprecipi- tates were resolved by SDS-PAGE and analyzed by silver staining. The polypeptides identified by mass spectrometric analyses are indicated to the right of the blot (IgH, immunoglobulin heavy chain). (B) Nuclear lysates from MOLT-4 cells were subjected to immunoprecipitation (IP) with preimmune rabbit serum (PIRS), anti-topoisomerase IIα (anti-TopoIIα), or anti-PKCδ. Cell lysates and immunoprecipitates were analyzed by immunoblotting (IB) with anti-topoisomerase IIα or anti-PKCδ. The finding that the binding stoichiometry of PKCδ with topoisomerase IIα was relatively high is mainly due to using nuclear lysates for immunoprecipitation. (C) Nuclear lysates from MOLT-4 cells were subjected to immunoprecipitation with PIRS or anti-PKCδ. Cell lysates and immunoprecipitates were analyzed by immunoblotting with anti-topoisomerase IIα and IIβ (anti-TopoII) or anti-PKCδ.
FIG. 2.


PKCδ directly interacts with and phosphorylates topoisomerase IIα. (A) Schematic representation of PKCδ. RD, regulatory domain; CF, catalytic fragment. (B) MOLT-4 cell lysates were incubated with GST, GST-PKCδRD, or GST-PKCδCF bound to glutathione beads. The adsorbates were analyzed by immunoblotting (IB) with anti-topoisomerase IIα (anti-TopoIIα) or anti-GST. GST-PKCδ fr., GST-PKCδ fragment. (C) Schematic representation of topoisomerase IIα. C-ter. domain, C-terminal domain. (D) MOLT-4 cell lysates were incubated with GST or GST-topoisomerase IIα fragments (GST-TopoIIα fr.) bound to glutathione beads. The adsorbates were analyzed by immunoblotting (IB) with anti-PKCδ or anti-GST. Each one of the bands specific for GST-topoisomerase IIα fragments is indicated by an asterisk. (E) 293T cells were transfected with GFP-TopoIIα or a truncated form of GFP-TopoIIα that encodes amino acid residues 1 to 1145 (GFP-TopoIIαΔC). Lysates were subjected to immunoprecipitation (IP) with anti-PKCδ or PIRS followed by immunoblot (IB) analysis with anti-GFP or anti-PKCδ. IgH, immunoglobulin heavy chain. (F) Recombinant topoisomerase IIα was incubated with glutathione beads containing GST or GST-PKCδ. The adsorbates were subjected to immunoblot (IB) analysis with anti-topoisomerase IIα (anti-TopoIIα) or anti-GST. (G) GST-PKCδ was incubated with or without recombinant topoisomerase IIα and [γ-32P]ATP. The reaction products were analyzed by SDS-PAGE and autoradiography or by Coomassie brilliant blue R-250 (CBB) staining.
PKCδ induces the catalytic activity of topoisomerase IIα.
To examine whether PKCδ is involved in topoisomerase IIα activity, purified topoisomerase IIα was incubated in the presence or absence of recombinant kinase-active PKCδ. We assayed catalytic activity of topoisomerase IIα by the decatenation of kinetoplast DNA. Decatenation activity was substantially enhanced in the presence of PKCδ (Fig. 3A). To determine whether stimulatory effects of PKCδ are confined to decatenation activity, we performed DNA relaxation assays. Purified topoisomerase IIα was incubated with supercoiled plasmids in the presence or absence of PKCδ. In concert with the decatenation assays, coincubation of PKCδ increased relaxation activity of topoisomerase IIα (Fig. 3B). To define PKCδ-mediated topoisomerase IIα activation in vitro more quantitatively, serially diluted PKCδ recombinant proteins were incubated with 0.25 U of topoisomerase IIα. Decatenation assays revealed that at least 20 ng of active PKCδ was necessary for full activation of topoisomerase IIα (Fig. 3C). To determine whether kinase activity is required for topoisomerase IIα activation, purified kinase-active or -inactive PKCδCF proteins were incubated with purified topoisomerase IIα. The kinase-active, but not kinase-inactive, PKCδ protein was capable of topoisomerase IIα activation, resulting in the decatenation of KDNA (Fig. 3D). These findings indicate that PKCδ induces catalytic activity of topoisomerase IIα in vitro. To assess involvement of PKCδ in topoisomerase IIα activation in vivo, MOLT-4 cells were treated with or without the PKCδ inhibitor rottlerin (18). Decatenation assays using nuclear lysates revealed that rottlerin treatment was associated with inhibition of topoisomerase IIα activity (Fig. 4A). Similar findings were obtained for U-937 and HL-60 cells (data not shown). To determine whether kinase activity of PKCδ is required for the activation of topoisomerase IIα, 293T cells were transfected with Flag vector, wild-type (wt) Flag-PKCδCF or the Flag-PKCδCF(K-R) mutant, which is catalytically inactive. Analysis of decatenation assays demonstrated that expression of the PKCδCF, but not the vector or the PKCδCF(K-R) mutant, induced topoisomerase IIα activity (Fig. 4B). Similar results were obtained with DNA relaxation assays (data not shown). To further establish the essential role for PKCδ in topoisomerase IIα regulation, pkcδ+/+ and pkcδ−/− MEFs were analyzed by the decatenation and DNA relaxation assays. The finding that topoisomerase IIα activity was diminished in pkcδ−/− MEFs provided further support for a pivotal role of PKCδ in topoisomerase IIα activation (Fig. 4C and D). To exclude the possibility that rottlerin is directly involved in the inhibition of topoisomerase IIα activity, pkcδ−/− MEFs were left untreated or treated with rottlerin for 1 h. Decatenation assays using nuclear lysates demonstrated that rottlerin treatment had no effect on topoisomerase IIα activity in pkcδ−/− MEFs (Fig. 4E).
FIG. 3.

Topoisomerase IIα is activated by PKCδ in vitro. (A) The indicated amount of purified topoisomerase IIα (TopoIIα) was incubated with (+) or without (−) kinase-active recombinant PKCδ (50 ng). Decatenation assays using reaction mixtures containing kinetoplast DNA were performed, and the reaction products were analyzed on a 1% agarose gel. Cat. KDNA, catenated KDNA; Decat. KDNA, decatenated KDNA. (B) Purified topoisomerase IIα was incubated with or without recombinant PKCδ. Relaxation assays using reaction mixtures containing pBluescript were performed, and the reaction products were analyzed on a 1% agarose gel. (C) Purified topoisomerase IIα (0.25 U) was incubated with or without (−) the indicated amount of kinase-active recombinant PKCδ. Decatenation assays using reaction mixtures containing KDNA were performed, and the reaction products were analyzed on a 1% agarose gel. (D) Purified topoisomerase IIα (0.25 U) was incubated with recombinant GST, GST-PKCδCF, or GST-PKCδCF(K-R). Decatenation assays using reaction mixtures containing KDNA were performed, and the reaction products were analyzed on a 1% agarose gel.
FIG. 4.

Topoisomerase IIα is activated by PKCδ in cells. (A) MOLT-4 cells were incubated in the presence (+) or absence (−) of rottlerin. Decatenation assays using nuclear lysates (Nuc. Lysates) were performed, and the reaction products were resolved on a 1% agarose gel. Cat. KDNA, catenated KDNA; Decat. KDNA, decatenated KDNA. (B) 293T cells were transfected with Flag vector, Flag-PKCδCF, or Flag-PKCδCF(K-R). Nuclear lysates were analyzed by decatenation assays (top blot). Cell lysates were subjected to immunoblot (IB) analysis with anti-Flag or antitubulin. (C and D) Nuclear lysates from pkcδ+/+ and pkcδ−/− MEFs were analyzed by the decatenation (C) and DNA relaxation (D) assays. (E) pkcδ−/− MEFs were left untreated or treated with rottlerin for 1 h. Nuclear lysates were analyzed by the decatenation assays. DMSO, dimethyl sulfoxide.
Association of PKCδ with topoisomerase IIα is required for topoisomerase IIα stabilization and activation.
Previous studies have demonstrated that expression of topoisomerase IIα is regulated in a cell cycle-dependent manner with levels that are low at G1, gradually increase from S to G2, and peak at M phase (19, 23, 35, 40). By contrast, topoisomerase IIβ expression is relatively constant throughout the cell cycle (19, 40). Furthermore, topoisomerase IIα is markedly degraded at the transition from M into G1 phase. Several studies have suggested that this degradation is associated with the ubiquitin-proteasome pathway (22, 31, 33). To examine PKCδ-mediated topoisomerase IIα activation in mitosis, MOLT-4 cells were synchronized in M phase by a microtubule inhibitor, nocodazole, in the presence or absence of rottlerin. Analysis of decatenation assays demonstrated that inhibition of PKCδ attenuated topoisomerase IIα activity in M phase (Fig. 5A). Similar results were obtained in U-937 cells (Fig. 5B). To verify and extend these findings, MOLT-4 cells or U-937 cells were synchronized at M phase in the presence or absence of PKCδ activator bistratene A (17). The finding that treatment with bistratene A increased the catalytic activity of topoisomerase IIα further supports the involvement of PKCδ in topoisomerase IIα activation in mitosis (Fig. 5C and data not shown). To define whether topoisomerase IIα regulation by PKCδ is cell cycle dependent, MOLT-4 cells synchronized at M phase were released by the removal of nocodazole from the cell culture medium. Consistent with previous studies, the activity of topoisomerase IIα gradually decreased after cells exited M phase and entered into G1 phase (Fig. 5D). Importantly, pretreatment of cells with rottlerin substantially attenuated topoisomerase II activity (Fig. 5D). Similar findings were obtained in U-937 cells (data not shown). To extend these analyses to S phase, MOLT-4 cells were synchronized in S phase by aphidicolin and then released by its removal. In concert with previous findings, topoisomerase IIα activation was induced from S to G2/M phase, reduced at G1 phase, and restored in S phase (Fig. 5E). Moreover, cell cycle-dependent topoisomerase IIα activation was in part abrogated by the pretreatment with rottlerin throughout the cell cycle (Fig. 5E). Similar results were obtained in U-937 cells (data not shown). To determine whether PKCδ activates topoisomerase IIα in a distinct cell cycle phase(s), cells were synchronized in M or S phase and then treated with or without rottlerin for 1 or 2 h. Decatenation assays demonstrated that treatment with rottlerin in M phase had no significant effect on topoisomerase IIα activity (Fig. 5F). By contrast, there was substantial attenuation of topoisomerase IIα activity in response to rottlerin in S phase (Fig. 5F). To further establish the involvement of PKCδ on topoisomerase IIα activation in S phase, pkcδ+/+ and pkcδ−/− MEFs were synchronized in S phase by aphidicolin. Analysis of decatenation assays demonstrated that topoisomerase IIα activity is substantially abrogated in pkcδ−/− MEFs (Fig. 5G). These findings indicate that PKCδ modulates topoisomerase IIα from S to G2/M phases. In concert with these results, association of PKCδ with topoisomerase IIα was confined to S phase (Fig. 6A, blot a). Moreover, pretreatment of cells with rottlerin was associated with down-regulation of this interaction and topoisomerase IIα expression (Fig. 6A, blots a and c). The finding that PKCδ was activated from late G1 to S phase further supports the S-phase-specific role for PKCδ in topoisomerase IIα regulation (Fig. 6A, blot d). Taken together, these results indicate that PKCδ is required for topoisomerase IIα stabilization and activation during S phase. To confirm that activity of PKCδ is required for interaction with topoisomerase IIα, MOLT-4 cells were left untreated or treated with rottlerin. The PKCδ-topoisomerase IIα complex formation was abrogated by treatment with rottlerin (Fig. 6B). In addition, treatment with rottlerin for longer periods (4 and 10 h) was associated with substantial attenuation of topoisomerase IIα expression (Fig. 6B). These results support a model in which PKCδ activation triggers stabilization and interaction with topoisomerase IIα. Previous studies have shown that PKCδ localizes to both the cytoplasm and the nucleus, and cytoplasmic PKCδ translocates to the nucleus upon exposure to various genotoxic agents (2, 6, 46, 48). In this regard, S-phase-specific interaction of PKCδ with topoisomerase IIα may be partially due to transient nuclear targeting of PKCδ in S phase. To examine this possibility, MOLT-4 cells were treated with or without aphidicolin. Subcellular fractionation assays demonstrated that the expression ratio of PKCδ in the nucleus and cytoplasm in asynchronous cells was comparable with that of S-phase-enriched cells (Fig. 6C). To define whether PKCδ modulates the mRNA levels of topoisomerase IIα, asynchronized and synchronized (S or G2/M) MOLT-4 cells were treated with or without rottlerin. RT-PCR assays revealed that the mRNA levels of topoisomerase IIα remained unchanged regardless of PKCδ activity in asynchronous, S-phase, and G2/M-phase cells (Fig. 7A). To further determine whether topoisomerase IIα stabilization by PKCδ is mainly due to posttranslational modification, 293T cells were stably transfected with GFP-tagged topoisomerase IIα (Fig. 7B). 293T/GFP-topoisomerase IIα cells were treated with aphidicolin to arrest cells in S phase in the presence or absence of rottlerin and then released by its removal. As shown with endogenous protein (Fig. 6A, blot c), inhibition of PKCδ by rottlerin down-regulated ectopic expression of topoisomerase IIα in both S and G2/M phases (Fig. 7C). These results demonstrate that PKCδ modulates topoisomerase IIα at a posttranslational level, and not at a transcriptional level.
FIG. 5.



PKCδ is involved in cell cycle-dependent activation of topoisomerase IIα. (A and B) MOLT-4 (A) and U-937 (B) cells were treated with nocodazole (+) in the presence (+) or absence (−) of rottlerin. Topoisomerase IIα activity was analyzed by decatenation assays (top blot). Lysates were analyzed by immunoblotting (IB) with anti-topoisomerase IIα (anti-TopoIIα) or anti-PKCδ. Nuc. Lysates, nuclear lysates; Cat. KDNA, catenated KDNA; Decat. KDNA, decatenated KDNA. (C) MOLT-4 cells were treated with nocodazole in the presence or absence of bistratene A or rottlerin. Nuclear lysates were analyzed by decatenation assays (top blot). Lysates were analyzed by immunoblotting with anti-topoisomerase IIα (anti-TopoIIα) or antitubulin (bottom blot). (D) MOLT-4 cells were treated with nocodazole for 16 h in the presence or absence of rottlerin and then released by nocodazole removal and harvested at the indicated times. Nuclear (Nuc.) lysates were prepared, and topoisomerase IIα activity was analyzed by decatenation assays (top blot). Lysates were analyzed by immunoblotting (IB) with anti-topoisomerase IIα or antitubulin (bottom blot). The cell cycle was monitored by using a FACscan and represented as the percentage of population in each cell cycle phase in the graph at the bottom of panel D. (E) MOLT-4 cells were treated with aphidicolin for 16 h in the presence or absence of rottlerin and then released by aphidicolin removal and harvested at the indicated times. Nuclear lysates were prepared, and topoisomerase IIα activity was analyzed by decatenation assays (top blot). Lysates were analyzed by immunoblotting with anti-topoisomerase IIα or antitubulin (bottom blot). The cell cycle was monitored by using a FACscan and represented as the percentage of population in each cell cycle phase in the graph. (F) MOLT-4 cells were treated with nocodazole or aphidicolin for 16 h and then with rottlerin for the indicated times. Topoisomerase IIα activity was analyzed by decatenation assays (top blot). Lysates were analyzed by immunoblotting with anti-topoisomerase IIα or antitubulin (bottom blot). The cell cycle was monitored by using a FACscan and represented as the percentage of population in each cell cycle phase in the graph. (G) pkcδ+/+ and pkcδ−/− MEFs were treated with aphidicolin for 16 h. Nuclear lysates were analyzed by the decatenation assays (top blot). Lysates were analyzed by immunoblotting with anti-topoisomerase IIα, anti-PKCδ, or antitubulin.
FIG. 6.

S-phase-specific interaction of PKCδ with topoisomerase IIα. (A) MOLT-4 cells were synchronized in G2/M phase by treatment with nocodazole in the presence or absence of rottlerin and then released into the cell cycle by its removal. Cells were harvested at the indicated times, and lysates were analyzed by immunoprecipitation (IP) with anti-PKCδ followed by immunoblotting (IB) with anti-topoisomerase IIα (anti-TopoIIα) (a) or anti-PKCδ (b). Cell lysates were also analyzed by immunoblotting with anti-topoisomerase IIα (c), anti-phospho-Thr505 PKCδ (d), anti-PKCδ (e), or antitubulin (f). The cell cycle was monitored by using a FACscan. DMSO, dimethyl sulfoxide; IgH, immunoglobulin heavy chain; P-Thr505 PKCδ, phospho-Thr505 PKCδ. (B) MOLT-4 cells were left untreated or treated with rottlerin for the indicated times. Cell lysates were subjected to immunoprecipitation with anti-PKCδ followed by immunoblot analysis with anti-topoisomerase IIα (anti-TopoIIα) or anti-PKCδ. Lysates were also analyzed by immunoblotting with anti-topoisomerase IIα, anti-PKCδ, or antitubulin. (C) MOLT-4 cells were left untreated or treated with aphidicolin to synchronize cells in S phase. Lysates from nuclear (N) and cytoplasmic (C) fractions were subjected to immunoblot (IB) analysis with anti-PKCδ, anti-topoisomerase IIα (anti-TopoIIα), or anti-IκBα.
FIG. 7.

PKCδ regulates a posttranslational modification of topoisomerase IIα. (A) MOLT-4 cells were left untreated or treated with aphidicolin or nocodazole for 16 h in the presence (+) or absence (−) of rottlerin. Total RNA was subjected to RT-PCR analysis using primer sets for topoisomerase IIα (TopoIIα) or β-actin. (B) 293T cells were stably transfected with GFP vector (293T/GFP) or GFP-topoisomerase IIα (293T/GFP-TopoIIα). Cell lysates were analyzed by immunoblotting (IB) with anti-GFP or antitubulin. (C) 293T/GFP-TopoIIα cells were treated with aphidicolin or nocodazole for 16 h in the presence or absence of rottlerin. After the cells were washed with phosphate-buffered saline twice, cells were mounted with Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories) and analyzed with a Nikon Eclipse TE2000-U microscope. The cell cycle was determined by using a FACscan. DMSO, dimethyl sulfoxide.
PKCδ stabilizes topoisomerase IIα expression in response to DNA damage.
To examine the status of topoisomerase IIα expression levels in response to DNA damage, MOLT-4 cells were treated with 1-β-d-arabinofuranocylcytosine. ara-C is incorporated into elongating DNA strands and causes arrest of DNA replication by functioning as a relative chain terminator and induces DNA double-strand breaks (26). Immunoblot analysis of cell lysates with anti-topoisomerase IIα demonstrated that ara-C treatment induced a transient increase of topoisomerase IIα expression (Fig. 8A). Similar findings were obtained with U-937 and HL-60 cells (Fig. 8C and data not shown). Moreover, as shown for ara-C, cisplatin treatment was also associated with up-regulation of topoisomerase IIα expression (Fig. 8B). These results indicate that certain types of genotoxic stress induce the expression of topoisomerase IIα. To assess the involvement of PKCδ in topoisomerase IIα expression, MOLT-4 cells were pretreated with rottlerin followed by ara-C treatment. Rottlerin pretreatment was associated with attenuation of ara-C-induced topoisomerase IIα expression (Fig. 8A). Similar results were obtained with CDDP treatment (Fig. 8B). These findings indicate that PKCδ is involved in the up-regulation of topoisomerase IIα expression in response to DNA damage. To assess whether the PKCδ-mediated increase of topoisomerase IIα expression following genotoxic stress is caused by a transcriptional modification, MOLT-4 cells were pretreated with or without rottlerin followed by treatment with ara-C. RT-PCR assays revealed that the mRNA levels of topoisomerase IIα remained unchanged regardless of PKCδ activity, in control and ara-C treated cells (Fig. 8D). To examine the involvement of PKCδ in posttranslational modulation of topoisomerase IIα upon exposure to genotoxic agents, 293T/topoisomerase IIα cells were treated with ara-C in the presence or absence of rottlerin. ara-C enhanced both endogenous and exogenous expression of topoisomerase IIα (Fig. 8E). Moreover, inactivation of PKCδ by rottlerin inhibited up-regulation of topoisomerase IIα expression (Fig. 8E). These results demonstrate that PKCδ induces topoisomerase IIα expression following ara-C treatment by a posttranslational, and not a transcriptional, regulation. To further define whether kinase activity of PKCδ is required for topoisomerase IIα expression, 293T cells were transfected with the Flag vector, wt Flag-PKCδCF, or Flag-PKCδCF(K-R) mutant. The finding that ectopic expression of the wt Flag-PKCδCF, but not the Flag vector or the Flag-PKCδCF(K-R) mutant, up-regulated topoisomerase IIα expression supports the role for PKCδ in kinase activity-dependent induction of topoisomerase IIα expression (Fig. 9A). To determine whether DNA damage induces the expression of topoisomerase IIα by a PKCδ-dependent mechanism, PKCδ was knocked down by transfection of U2-OS cells with siRNAs that target PKCδ (Fig. 9B). As shown in various cell types, treatment of U2-OS cells with ara-C also induced topoisomerase IIα expression (Fig. 9C). Importantly, knocking down PKCδ inhibited the up-regulation of topoisomerase IIα elicited by ara-C (Fig. 9C). To further define the direct role for PKCδ in DNA damage-induced topoisomerase IIα expression, pkcδ+/+ and pkcδ−/− MEFs were treated with ara-C. Immunoblot analysis with anti-topoisomerase IIα demonstrated that ara-C induced an increase of topoisomerase IIα expression in pkcδ+/+ MEFs (Fig. 9D). By contrast, there was no detectable induction of topoisomerase IIα in ara-C-treated pkcδ−/− MEFs (Fig. 9D). These findings indicate that topoisomerase IIα expression is up-regulated by a PKCδ-dependent mechanism in the response to genotoxic stress.
FIG. 8.

Inhibition of PKCδ by rottlerin abrogates DNA damage-induced expression of topoisomerase IIα. (A and B) MOLT-4 cells were pretreated with or without rottlerin for 1 h followed by treatment with ara-C (A) or cisplatin (CDDP) (B) for the indicated periods. Cell lysates were subjected to immunoblot (IB) analysis with anti-topoisomerase IIα (anti-TopoIIα) or anti-PKCδ. DMSO, dimethyl sulfoxide. (C) U-937 cells were treated and analyzed as described above for panel A. (D) MOLT-4 cells were left untreated or treated with ara-C for 4 h in the presence or absence of rottlerin. Total RNA was subjected to RT-PCR analysis using primer sets for topoisomerase IIα (TopoIIα) or β-actin. (E) 293T/GFP-TopoIIα cells were left untreated or treated with rottlerin for 1 h followed by treatment with ara-C for 4 h. Cell lysates were subjected to immunoblot analysis with anti-topoisomerase IIα or anti-PKCδ.
FIG. 9.

PKCδ-dependent induction of topoisomerase IIα expression in response to DNA damage. (A) 293T cells were transfected with Flag vector, Flag-PKCδCF, or Flag-PKCδCF(K-R). Cell lysates were analyzed by immunoblotting (IB) with anti-topoisomerase IIα (anti-TopoIIα), anti-Flag, or antitubulin. (B) U2-OS cells were left untransfected (Control) or transfected with the indicated siRNAs. Cell lysates were subjected to immunoblot (IB) analysis with anti-PKCδ or antitubulin. (C) U2-OS cells transfected with scrambled siRNA or PKCδsiRNA2 were left untreated or treated with ara-C for 4 h. Cell lysates were analyzed by immunoblotting with anti-topoisomerase IIα, anti-PKCδ, or antitubulin. (D) pkcδ+/+ and pkcδ−/− MEFs were left untreated or treated with ara-C for 4 h. Cell lysates were analyzed by immunoblotting with anti-topoisomerase IIα, anti-PKCδ, or antitubulin.
PKCδ activates topoisomerase IIα for the DNA damage response.
Previous studies have shown that PKCδ is activated in response to DNA damage (11). In this context and given the finding that PKCδ induces topoisomerase IIα expression following genotoxic stress, it is conceivable that PKCδ induces enzymatic activity of topoisomerase IIα in response to DNA damage. To address this issue, MOLT-4 cells were treated with ara-C in the presence or absence of rottlerin. Decatenation assays showed that topoisomerase IIα was activated by ara-C treatment (Fig. 10A). Importantly, pretreatment with rottlerin substantially attenuated ara-C-induced topoisomerase IIα activation (Fig. 10A). To verify these findings, we performed DNA relaxation assays. In concert with the decatenation assays, inhibition of PKCδ activation by rottlerin attenuated ara-C-induced topoisomerase IIα activity (Fig. 10B). Similar studies performed on U-937 and HL-60 cells confirmed these results (Fig. 10C and data not shown). Moreover, as found for ara-C, comparable results were obtained in the treatment of cells with CDDP (Fig. 10D and data not shown). To further assess whether DNA damage induces topoisomerase IIα activity by a PKCδ-dependent mechanism, PKCδ was knocked down by transfection of U2-OS cells with siRNAs. Treatment with ara-C induced topoisomerase IIα activity (Fig. 10E). Importantly, knocking down PKCδ inhibited the up-regulation of topoisomerase IIα activity elicited by ara-C (Fig. 10E). To confirm the involvement of PKCδ on topoisomerase IIα activation upon exposure to genotoxic agents, pkcδ+/+ and pkcδ−/− MEFs were left untreated or treated with ara-C. Analysis of decatenation assays demonstrated that topoisomerase IIα activity was substantially enhanced in pkcδ+/+, but not pkcδ−/−, MEFs (Fig. 10F). Taken together, these findings indicate that topoisomerase IIα is activated in response to certain genotoxic agents in a PKCδ-dependent mechanism.
FIG. 10.

PKCδ-dependent activation of topoisomerase IIα in response to DNA damage. (A and B) MOLT-4 cells were pretreated with or without rottlerin for 1 h followed by treatment with ara-C. Nuclear lysates (Nuc. Lysates) were analyzed by decatenation (A) and DNA relaxation (B) assays. DMSO, dimethyl sulfoxide; Cat. KDNA, catenated KDNA; Decat. KDNA, decatenated KDNA. (C) U-937 cells were treated as described above for panel A, and nuclear lysates were analyzed by the decatenation assays. (D) MOLT-4 cells were left untreated or treated with rottlerin for 1 h followed by the treatment with CDDP for the indicated times. Topoisomerase IIα activity was analyzed by decatenation assays. (E) U2-OS cells transfected with scrambled siRNA or PKCδsiRNA2 were left untreated or treated with ara-C for 4 h. Nuclear lysates were analyzed by the decatenation assays. (F) pkcδ+/+ and pkcδ−/− MEFs were left untreated or treated with ara-C for 4 h. Nuclear lysates were analyzed by the decatenation assays.
Activation of topoisomerase IIα is involved in PKCδ-mediated apoptosis in response to DNA damage.
Previous studies have shown that DNA damage elicited by ara-C efficiently induces apoptosis (26). Moreover, inhibition of PKCδ by (i) expression of dominant-negative PKCδ, (ii) rottlerin treatment, or (iii) knocking down PKCδ, attenuated ara-C-induced apoptosis (15, 46). These findings indicate that activation of PKCδ following genotoxic stress is associated with apoptosis execution; however, this mechanism is largely unknown. Importantly, the present study demonstrates that ara-C treatment induces expression and activation of topoisomerase IIα by a PKCδ-dependent mechanism. In this context, to examine the possibility that activation of topoisomerase IIα is involved in PKCδ-mediated apoptosis following DNA damage, cells were treated with ara-C in the presence or absence of a non-DNA-damaging catalytic inhibitor of topoisomerase IIα, ICRF-193 (36). Treatment of cells with ara-C was associated with apoptosis induction (Fig. 11A). In contrast, pretreatment with ICRF-193 substantially attenuated ara-C-induced apoptosis (Fig. 11A). These results suggest that ara-C-induced apoptosis is, at least in part, a topoisomerase IIα-dependent mechanism. To further define the role for topoisomerase IIα in ara-C-induced apoptosis, topoisomerase IIα was knocked down in U2-OS cells by transfection with topoisomerase IIαsiRNAs (Fig. 11B). Knocking down topoisomerase IIα attenuated the induction of apoptosis elicited by ara-C treatment (Fig. 11C). Moreover, as previously reported (46), pretreatment with rottlerin conferred a protective effect on ara-C-mediated apoptosis (Fig. 11C). By contrast, inhibition of PKCδ activity by rottlerin had little, if any, effect on attenuation of apoptosis when knocking down topoisomerase IIα (Fig. 11C). These findings collectively support the involvement of topoisomerase IIα as a positive regulator of apoptosis by PKCδ-mediated activation in response to DNA damage.
FIG. 11.

Topoisomerase IIα is required for PKCδ-induced apoptotic cell death in response to genotoxic stress. (A) MOLT-4 cells were treated with ara-C for the indicated times in the presence (open bar) or absence (closed bar) of ICRF-193. The percentages of apoptotic cells were determined by TUNEL assays. The results are represented as means ± standard deviations (error bars) obtained from four fields of 100 to 300 cells (each field) and three independent experiments. (B) U2-OS cells were transfected with the indicated siRNAs for 48 h and then left untreated (−) or treated (+) with ara-C for 4 h. Cell lysates were subjected to immunoblot (IB) analysis with anti-topoisomerase IIα (anti-TopoIIα) or antitubulin. Nuclear lysates were analyzed by decatenation assays (bottom blot). The cells had been transfected with scrambled siRNA (lanes S), topoisomerase IIα siRNA1 (lanes 1), or topoisomerase IIα siRNA2 (lanes 2). Nuc. Lysates, nuclear lysates; Cat. KDNA, catenated KDNA; Decat. KDNA, decatenated KDNA. (C) U2-OS cells transfected with the indicated siRNAs were left untreated (−) or treated (+) with rottlerin for 1 h followed by treatment with ara-C (+) for 24 h. The percentages of apoptotic cells were determined by TUNEL assays. The results are represented as means ± standard deviations (error bars) obtained from four fields of 100 to 300 cells, each performed over three independent experiments. Cell lysates were also analyzed by immunoblotting (IB) with anti-topoisomerase IIα (anti-TopoIIα) or antitubulin.
DISCUSSION
Activation of PKCδ in the DNA damage response.
Involvement of PKCδ in the DNA damage response is supported by the findings that both arrest of DNA replication and induction of DNA lesions are associated with PKCδ activation (44-46, 48). The available evidence indicates that full-length PKCδ is activated as an early event within 1 h of exposure to genotoxic agents. Activation of PKCδ by tyrosine phosphorylation upon exposure to genotoxic agents is mediated in part by the c-Abl kinase (48). Another study demonstrates that Lyn also phosphorylates PKCδ and that Lyn-mediated tyrosine phosphorylation of PKCδ contributes to PKCδ activation (45). PKCδ is also activated as a later event in the genotoxic stress response by caspase-3-mediated proteolytic cleavage (10, 11, 25). The cleaved C-terminal 40-kDa fragment contains the ATP-binding and kinase domains and is constitutively active (10, 11, 15). The finding that tyrosine phosphorylation of PKCδ is required for activation of caspase-3 and thereby PKCδ cleavage supports a link between both mechanisms of PKCδ activation (3). Whereas expression of PKCδCF induces apoptotic cell death (15), the precise events responsible for this response are unknown but may involve an interaction between PKCδCF and DNA-PKcs or Rad9 (2, 46). In addition, the present studies show for the first time that PKCδCF interacts with topoisomerase IIα. This interaction is necessary for PKCδ-mediated induction of apoptosis in response to certain types of genotoxic agents. Importantly, cleavage of PKCδ into the constitutively active catalytic fragment is irreversible and thus may function in prolonged stimulation of multiple proapoptotic pathways.
PKCδ interacts with topoisomerase IIα.
Previous studies have shown that PKCδ localizes to both the nucleus and cytoplasm (2, 6, 46, 48). While nuclear PKCδ associates constitutively with DNA-PKcs and Rad9 (2, 46), the nuclear targets of PKCδ are otherwise largely unknown. The present study demonstrates that nuclear PKCδ also associates with topoisomerase IIα. Our results show that the catalytic fragment of PKCδ directly binds to the C-terminal region of topoisomerase IIα. Notably, the finding that this interaction is confined to the S phase suggests a role for PKCδ in the gradual increase of topoisomerase IIα expression and activity during S phase. Moreover, the results demonstrate that, upon DNA damage in S phase by ara-C, activated PKCδ induces aberrant expression and activation of topoisomerase IIα. In this context, PKCδ-mediated apoptosis by ara-C is, at least in part, required for topoisomerase IIα activation. These findings collectively support a model in which modulation of topoisomerase IIα by PKCδ during S phase is essential for determination of cell fate.
PKCδ induces topoisomerase IIα expression and activation.
Activity of topoisomerase IIα is tightly regulated by posttranslational modifications, such as phosphorylation and ubiquitination. The topoisomerase II enzyme has been shown to be phosphorylated on multiple serine and threonine residues, the majority of which are located in the C-terminal region. Several lines of study have been conducted to examine whether topoisomerase IIα is phosphorylated in a cell cycle-dependent manner. The previous findings have indicated that this enzyme is hyperphosphorylated from S phase to G2/M phase. In concert with phosphorylation, expression and activation of topoisomerase IIα are induced and reach their maximal levels in M phase. Whereas there are a number of proteins involved in topoisomerase IIα regulation, our present study revealed PKCδ as a newly identified modulator of topoisomerase IIα expression and activation. Indeed, the present study demonstrates that PKCδ regulates the expression level of topoisomerase IIα. For example, inhibition of PKCδ was associated with down-regulation of topoisomerase IIα expression (Fig. 5A to G, 6A and B, and 7C). Induction of DNA damage-elicited topoisomerase IIα expression was also diminished by inhibition of PKCδ activity (Fig. 8A to C). Importantly, this regulation, at least in part, depends on the kinase activity of PKCδ (Fig. 9A). Taken together, these results suggest the possibility that PKCδ regulates topoisomerase IIα expression, resulting in the modulation of its activity. The present results also demonstrate that PKCδ is associated with induction of topoisomerase IIα stabilization and activation in S phase. The mechanism by which PKCδ achieves this is, at present, unclear. A potential explanation is that degradation of topoisomerase IIα is inhibited by PKCδ-mediated modification, while PKCδ phosphorylation of topoisomerase IIα could induce its activation. Importantly, the finding that PKCδ is activated and interacts with topoisomerase IIα during S phase further supports the S-phase-specific role for PKCδ in topoisomerase IIα modulation. In this context, other reports demonstrated that PKCδ is activated from late G1 to S phase (24, 34). Activation of PKCδ stimulated G1 phase cell cycle progression. Furthermore, activated PKCδ in S phase triggered caspase-dependent apoptotic cell death (34). Meanwhile, suppression of PKCδ activity was sufficient to inhibit DNA synthesis (24). Thus, taken together with the present findings, it is conceivable that PKCδ functions in cell cycle progression by modulating topoisomerase IIα in S phase. Obviously, further studies will be needed to define the precise role for PKCδ in cell cycle progression.
Our results also demonstrate that PKCδ promotes topoisomerase IIα expression and subsequent activation in response to certain types of genotoxic stress, such as ara-C treatment. Since ara-C is incorporated into DNA and causes DNA strand breaks, ara-C treatment arrests cells in S phase (13, 26). In this regard, the finding that activation of PKCδ following ara-C treatment caused more pronounced expression and activation of topoisomerase IIα further supports the model in which regulation of topoisomerase IIα by PKCδ is S phase specific.
Topoisomerase IIα is of functional importance in regulation of the PKCδ-dependent apoptotic response to DNA damage.
Whereas PKCδ is involved in the apoptotic response to DNA damage, the mechanism by which PKCδ induces DNA damage-elicited activation of the intrinsic apoptotic pathway is largely unknown. Certain insights have been derived from our previous finding that PKCδ regulates the interaction of human Rad9 (hRad9) with Bcl-2 and, consequently, the hRad9-mediated apoptotic response to DNA damage (46). Moreover, the results indicated that PKCδ translocates to the nucleus and thereby regulates hRad9 (46). Recently, we demonstrated that PKCδ regulates p53 by Ser46 phosphorylation (41). In that study, we show that p53-dependent apoptosis elicited by etoposide is attenuated by pretreatment of cells with rottlerin, indicating a pivotal role for PKCδ in induction of p53-mediated apoptosis. Thus, DNA damage-induced nuclear targeting of PKCδ could contribute to the induction of the intrinsic apoptotic pathway. In the present study, we further demonstrate that topoisomerase IIα functions as a novel nuclear effector of PKCδ-mediated apoptosis. We found that inappropriate expression of topoisomerase IIα by PKCδ is associated with genotoxic stress-elicited apoptosis. Moreover, the finding that PKCδ-mediated deregulation of topoisomerase IIα was S phase specific suggested the disruption of cell cycle checkpoints, resulting in the execution of apoptosis. In this regard, another study demonstrated that topoisomerase IIα-mediated cell death was triggered after progression through the G1-S phase transition but before the G2-M phase transition (28). Given the evidence for the involvement of topoisomerase IIα in the G2 checkpoint that regulates the entry into mitosis (8), aberrant activation of topoisomerase IIα during S phase could cause failure to arrest before mitosis. Unscheduled entry into mitosis could then trigger the intrinsic apoptotic pathway, which is characterized as “mitotic catastrophe” (4). Additional studies will be required to prove this scenario. However, as shown in the previous studies, the tight control of topoisomerase IIα is fundamental to the appropriate operation of the cell cycle (16, 19, 40). Thus, the findings in the present work support the model in which deregulation of topoisomerase IIα activity by PKCδ sensitizes the cells to genotoxic stress-induced catastrophic cell death.
Acknowledgments
We thank W. T. Beck for providing GFP-TopoIIα plasmid and T. Kasama for assisting in the mass spectrometry analysis.
This work was supported by grants from the Ministry of Education, Science and Culture of Japan (to K.Y., K.I.N., and Y.M.), the Nakajima Foundation (to K.Y.), Takeda Science Foundation (to K.Y.), Public Trust Haraguchi Memorial Cancer Research Fund (to K.Y.), Kanae Foundation for Life & Socio-medical Science (to K.Y.) and Kowa Life Science Foundation (to K.Y.).
REFERENCES
- 1.Akimitsu, N., K. Kamura, S. Tone, A. Sakaguchi, A. Kikuchi, H. Hamamoto, and K. Sekimizu. 2003. Induction of apoptosis by depletion of DNA topoisomerase IIα in mammalian cells. Biochem. Biophys. Res. Commun. 307:301-307. [DOI] [PubMed] [Google Scholar]
- 2.Bharti, A., S. K. Kraeft, M. Gounder, P. Pandey, S. Jin, Z. M. Yuan, S. P. Lees-Miller, R. Weichselbaum, D. Weaver, L. B. Chen, D. Kufe, and S. Kharbanda. 1998. Inactivation of DNA-dependent protein kinase by protein kinase Cδ: implications for apoptosis. Mol. Cell. Biol. 18:6719-6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blass, M., I. Kronfeld, G. Kazimirsky, P. M. Blumberg, and C. Brodie. 2002. Tyrosine phosphorylation of protein kinase Cδ is essential for its apoptotic effect in response to etoposide. Mol. Cell. Biol. 22:182-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Castedo, M., J. L. Perfettini, T. Roumier, K. Andreau, R. Medema, and G. Kroemer. 2004. Cell death by mitotic catastrophe: a molecular definition. Oncogene 23:2825-2837. [DOI] [PubMed] [Google Scholar]
- 5.Chen, A. Y., and L. F. Liu. 1994. DNA topoisomerases: essential enzymes and lethal targets. Annu. Rev. Pharmacol. Toxicol. 34:191-218. [DOI] [PubMed] [Google Scholar]
- 6.DeVries, T. A., M. C. Neville, and M. E. Reyland. 2002. Nuclear import of PKCδ is required for apoptosis: identification of a novel nuclear import sequence. EMBO J. 21:6050-6060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dignam, J. D., R. M. Lebovitz, and R. G. Roeder. 1983. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11:1475-1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Downes, C. S., D. J. Clarke, A. M. Mullinger, J. F. Gimenez-Abian, A. M. Creighton, and R. T. Johnson. 1994. A topoisomerase II-dependent G2 cycle checkpoint in mammalian cells. Nature 372:467-470. [DOI] [PubMed] [Google Scholar]
- 9.Earnshaw, W. C., B. Halligan, C. A. Cooke, M. M. Heck, and L. F. Liu. 1985. Topoisomerase II is a structural component of mitotic chromosome scaffolds. J. Cell Biol. 100:1706-1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Emoto, Y., H. Kisaki, Y. Manome, S. Kharbanda, and D. Kufe. 1996. Activation of protein kinase Cδ in human myeloid leukemia cells treated with 1-β-d-arabinofuranosylcytosine. Blood 87:1990-1996. [PubMed] [Google Scholar]
- 11.Emoto, Y., Y. Manome, G. Meinhardt, H. Kisaki, S. Kharbanda, M. Robertson, T. Ghayur, W. W. Wong, R. Kamen, R. Weichselbaum, and D. Kufe. 1995. Proteolytic activation of protein kinase C δ by an ICE-like protease in apoptotic cells. EMBO J. 14:6148-6156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Figeys, D., L. D. McBroom, and M. F. Moran. 2001. Mass spectrometry for the study of protein-protein interactions. Methods 24:230-239. [DOI] [PubMed] [Google Scholar]
- 13.Fram, R. J., and D. W. Kufe. 1982. DNA strand breaks caused by inhibitors of DNA synthesis: 1-β-d-arabinofuranosylcytosine and aphidicolin. Cancer Res. 42:4050-4053. [PubMed] [Google Scholar]
- 14.Gasser, S. M., T. Laroche, J. Falquet, E. Boy de la Tour, and U. K. Laemmli. 1986. Metaphase chromosome structure. Involvement of topoisomerase II. J. Mol. Biol. 188:613-629. [DOI] [PubMed] [Google Scholar]
- 15.Ghayur, T., M. Hugunin, R. V. Talanian, S. Ratnofsky, C. Quinlan, Y. Emoto, P. Pandey, R. Datta, Y. Huang, S. Kharbanda, H. Allen, R. Kamen, W. Wong, and D. Kufe. 1996. Proteolytic activation of protein kinase C δ by an ICE/CED 3-like protease induces characteristics of apoptosis. J. Exp. Med. 184:2399-2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goswami, P. C., J. L. Roti Roti, and C. R. Hunt. 1996. The cell cycle-coupled expression of topoisomerase IIα during S phase is regulated by mRNA stability and is disrupted by heat shock or ionizing radiation. Mol. Cell. Biol. 16:1500-1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Griffiths, G., B. Garrone, E. Deacon, P. Owen, J. Pongracz, G. Mead, A. Bradwell, D. Watters, and J. Lord. 1996. The polyether bistratene A activates protein kinase C-δ and induces growth arrest in HL60 cells. Biochem. Biophys. Res. Commun. 222:802-808. [DOI] [PubMed] [Google Scholar]
- 18.Gschwendt, M., H. J. Muller, K. Kielbassa, R. Zang, W. Kittstein, G. Rincke, and F. Marks. 1994. Rottlerin, a novel protein kinase inhibitor. Biochem. Biophys. Res. Commun. 199:93-98. [DOI] [PubMed] [Google Scholar]
- 19.Heck, M. M., W. N. Hittelman, and W. C. Earnshaw. 1988. Differential expression of DNA topoisomerases I and II during the eukaryotic cell cycle. Proc. Natl. Acad. Sci. USA 85:1086-1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kellner, U., M. Sehested, P. B. Jensen, F. Gieseler, and P. Rudolph. 2002. Culprit and victim—DNA topoisomerase II. Lancet Oncol. 3:235-243. [DOI] [PubMed] [Google Scholar]
- 21.Kharbanda, S., A. Bharti, D. Pei, J. Wang, P. Pandey, R. Ren, R. Weichselbaum, C. T. Walsh, and D. Kufe. 1996. The stress response to ionizing radiation involves c-Abl-dependent phosphorylation of SHPTP1. Proc. Natl. Acad. Sci. USA 93:6898-6901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim, H. D., A. Tomida, Y. Ogiso, and T. Tsuruo. 1999. Glucose-regulated stresses cause degradation of DNA topoisomerase IIα by inducing nuclear proteasome during G1 cell cycle arrest in cancer cells. J. Cell. Physiol. 180:97-104. [DOI] [PubMed] [Google Scholar]
- 23.Kimura, K., M. Saijo, M. Ui, and T. Enomoto. 1994. Growth state- and cell cycle-dependent fluctuation in the expression of two forms of DNA topoisomerase II and possible specific modification of the higher molecular weight form in the M phase. J. Biol. Chem. 269:1173-1176. [PubMed] [Google Scholar]
- 24.Kitamura, K., K. Mizuno, A. Etoh, Y. Akita, A. Miyamoto, K. Nakayama, and S. Ohno. 2003. The second phase activation of protein kinase C δ at late G1 is required for DNA synthesis in serum-induced cell cycle progression. Genes Cells 8:311-324. [DOI] [PubMed] [Google Scholar]
- 25.Koriyama, H., Z. Kouchi, T. Umeda, T. C. Saido, T. Momoi, S. Ishiura, and K. Suzuki. 1999. Proteolytic activation of protein kinase C δ and ɛ by caspase-3 in U937 cells during chemotherapeutic agent-induced apoptosis. Cell Signal. 11:831-838. [DOI] [PubMed] [Google Scholar]
- 26.Kufe, D. W., P. P. Major, E. M. Egan, and G. P. Beardsley. 1980. Correlation of cytotoxicity with incorporation of ara-C into DNA. J. Biol. Chem. 255:8997-9000. [PubMed] [Google Scholar]
- 27.Leitges, M., M. Mayr, U. Braun, U. Mayr, C. Li, G. Pfister, N. Ghaffari-Tabrizi, G. Baier, Y. Hu, and Q. Xu. 2001. Exacerbated vein graft arteriosclerosis in protein kinase Cδ-null mice. J. Clin. Investig. 108:1505-1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McPherson, J. P., and G. J. Goldenberg. 1998. Induction of apoptosis by deregulated expression of DNA topoisomerase IIα. Cancer Res. 58:4519-4524. [PubMed] [Google Scholar]
- 29.Miyamoto, A., K. Nakayama, H. Imaki, S. Hirose, Y. Jiang, M. Abe, T. Tsukiyama, H. Nagahama, S. Ohno, S. Hatakeyama, and K. I. Nakayama. 2002. Increased proliferation of B cells and auto-immunity in mice lacking protein kinase Cδ. Nature 416:865-869. [DOI] [PubMed] [Google Scholar]
- 30.Mo, Y. Y., K. A. Ameiss, and W. T. Beck. 1998. Overexpression of human DNA topoisomerase II α by fusion to enhanced green fluorescent protein. BioTechniques 25:1052-1057. [DOI] [PubMed] [Google Scholar]
- 31.Nakajima, T., K. Morita, N. Ohi, T. Arai, N. Nozaki, A. Kikuchi, F. Osaka, F. Yamao, and K. Oda. 1996. Degradation of topoisomerase IIα during adenovirus E1A-induced apoptosis is mediated by the activation of the ubiquitin proteolysis system. J. Biol. Chem. 271:24842-24849. [DOI] [PubMed] [Google Scholar]
- 32.Nishizuka, Y. 1988. The molecular heterogeneity of protein kinase C and its implications for cellular regulation. Nature 334:661-665. [DOI] [PubMed] [Google Scholar]
- 33.Salmena, L., V. Lam, J. P. McPherson, and G. J. Goldenberg. 2001. Role of proteasomal degradation in the cell cycle-dependent regulation of DNA topoisomerase IIα expression. Biochem. Pharmacol. 61:795-802. [DOI] [PubMed] [Google Scholar]
- 34.Santiago-Walker, A. E., A. J. Fikaris, G. G. Kao, E. J. Brown, M. G. Kazanietz, and J. L. Meinkoth. 2005. Protein kinase C δ stimulates apoptosis by initiating G1 phase cell cycle progression and S phase arrest. J. Biol. Chem. 280:32107-32114. [DOI] [PubMed] [Google Scholar]
- 35.Sugimoto, K., K. Yamada, M. Egashira, Y. Yazaki, H. Hirai, A. Kikuchi, and K. Oshimi. 1998. Temporal and spatial distribution of DNA topoisomerase II alters during proliferation, differentiation, and apoptosis in HL-60 cells. Blood 91:1407-1417. [PubMed] [Google Scholar]
- 36.Tanabe, K., Y. Ikegami, R. Ishida, and T. Andoh. 1991. Inhibition of topoisomerase II by antitumor agents bis(2,6-dioxopiperazine) derivatives. Cancer Res. 51:4903-4908. [PubMed] [Google Scholar]
- 37.Thielmann, H. W., and O. Popanda. 1998. Doxorubicin and gamma rays increase the level of DNA topoisomerase IIα in nuclei of normal and xeroderma pigmentosum fibroblasts. J. Cancer Res. Clin. Oncol. 124:355-366. [DOI] [PubMed] [Google Scholar]
- 38.Wang, J. C. 2002. Cellular roles of DNA topoisomerases: a molecular perspective. Nat. Rev. Mol. Cell. Biol. 3:430-440. [DOI] [PubMed] [Google Scholar]
- 39.Wang, J. C. 1996. DNA topoisomerases. Annu. Rev. Biochem. 65:635-692. [DOI] [PubMed] [Google Scholar]
- 40.Woessner, R. D., M. R. Mattern, C. K. Mirabelli, R. K. Johnson, and F. H. Drake. 1991. Proliferation- and cell cycle-dependent differences in expression of the 170 kilodalton and 180 kilodalton forms of topoisomerase II in NIH-3T3 cells. Cell Growth Differ. 2:209-214. [PubMed] [Google Scholar]
- 41.Yoshida, K. and H. Liu, and Y. Miki. 2006. Protein kinase C δ regulates Ser46 phosphorylation of p53 tumor suppressor in the apoptotic response to DNA damage. J. Biol. Chem. 281:5734-5740. [DOI] [PubMed] [Google Scholar]
- 42.Yoshida, K., S. Kharbanda, and D. Kufe. 1999. Functional interaction between SHPTP1 and the Lyn tyrosine kinase in the apoptotic response to DNA damage. J. Biol. Chem. 274:34663-34668. [DOI] [PubMed] [Google Scholar]
- 43.Yoshida, K., K. Komatsu, H. G. Wang, and D. Kufe. 2002. c-Abl tyrosine kinase regulates the human Rad9 checkpoint protein in response to DNA damage. Mol. Cell. Biol. 22:3292-3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yoshida, K., and D. Kufe. 2001. Negative regulation of the SHPTP1 protein tyrosine phosphatase by protein kinase C δ in response to DNA damage. Mol. Pharmacol. 60:1431-1438. [DOI] [PubMed] [Google Scholar]
- 45.Yoshida, K., Y. Miki, and D. Kufe. 2002. Activation of SAPK/JNK signaling by protein kinase Cδ in response to DNA damage. J. Biol. Chem. 277:48372-48378. [DOI] [PubMed] [Google Scholar]
- 46.Yoshida, K., H. G. Wang, Y. Miki, and D. Kufe. 2003. Protein kinase Cδ is responsible for constitutive and DNA damage-induced phosphorylation of Rad9. EMBO J. 22:1431-1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoshida, K., R. Weichselbaum, S. Kharbanda, and D. Kufe. 2000. Role for Lyn tyrosine kinase as a regulator of stress-activated protein kinase activity in response to DNA damage. Mol. Cell. Biol. 20:5370-5380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yuan, Z. M., T. Utsugisawa, T. Ishiko, S. Nakada, Y. Huang, S. Kharbanda, R. Weichselbaum, and D. Kufe. 1998. Activation of protein kinase C δ by the c-Abl tyrosine kinase in response to ionizing radiation. Oncogene 16:1643-1648. [DOI] [PubMed] [Google Scholar]