

Abstract
Inhibition of the mammalian soluble epoxide hydrolase (sEH) is a promising new therapy in the treatment of disorders resulting from hypertension and vascular inflammation. A spectrophotometric assay (4-nitrophenyl-trans-2,3-epoxy-3-phenylpropyl carbonate, NEPC) is currently used to screen libraries of chemicals; however this assay lacks the required sensitivity to differentiate the most potent inhibitors. A series of fluorescent α-cyanoester and α-cyanocarbonate epoxides that produce a strong fluorescent signal on epoxide hydrolysis by both human and murine sEH were designed as potential substrates for an in vitro inhibition assay. The murine enzyme showed a broad range of specificities, whereas the human enzyme showed the highest specificity for cyano(6-methoxy-naphthalen-2-yl)methyl trans-[(3-phenyloxiran-2-yl)methyl] carbonate. An in vitro inhibition assay was developed using this substrate and recombinant enzyme. The utility of the fluorescent assay was confirmed by determining the IC50 values for a series of known inhibitors. The new IC50 values were compared with those determined by spectrophotometric NEPC and radioactive tDPPO assays. The fluorescent assay ranked these inhibitors on the basis of IC50 values, whereas the NEPC assay did not. The ranking of inhibitor potency generally agreed with that determined using the tDPPO assay. These results show that the fluorescence-based assay is a valuable tool in the development of sEH inhibitors by revealing structure–activity relationships that previously were seen only by using the costly and labor-intensive radioactive tDPPO assay.
Keywords: Soluble epoxide hydrolase, α-Cyanoester, α-Cyanocarbonate, Kinetic assay, Fluorescent substrate
The soluble epoxide hydrolase (sEH, EC 3.3.2.3)1 is a member of the α/β-hydrolase fold family of enzymes [1] and catalyzes the hydrolysis of an epoxide to its corresponding diol through the catalytic addition of a water molecule [2]. The endogenous substrates for the sEH include epoxides of arachidonic acid [3,4] and linoleic acid [5,6]. Arachidonic acid epoxides (epoxyeicosatrienoic acid epoxides, EETs) are known modulators of blood pressure [7,8] and vascular permeability [9,10]. It has been shown that sEH inhibition not only lowers blood pressure in rodent models but also offers protection against hypertension-related renal damage [4,11,12].
For the past 6 years, we have investigated the therapeutic role of inhibition of mammalian sEH. Our inhibitor structures have evolved from simple symmetrical ureas to compounds containing multiple pharmacophores that possess increased potency, aqueous solubility, and bioavailability in rodent models [13–18]. Evaluation of these inhibitors typically begins with in vitro assays using purified recombinant sEH prior to in vivo studies. Traditionally, we have used racemic 4-nitrophenyl-trans-2,3-epoxy-3-phenylpropyl carbonate (NEPC) as a substrate for a continuous kinetic assay to determine inhibitor potency [14,19]. As the potency of our inhibitors increased (lower IC50), we found that the NEPC absorption-based assay was not sensitive enough to separate structurally different inhibitors on the basis of IC50 values. We addressed this issue previously by using a radioactive-based assay to measure these IC50 values [15]. Although this assay was able to separate the most potent inhibitors, allowing structure–activity analysis, it has the disadvantage of being time-consuming and costly. Therefore, with a view to assay hundreds of possible sEH inhibitors, it was necessary to investigate alternative assay strategies.
Spectroscopic assays present the advantages, compared with radioactive- and chromatographic-based assays, of being straightforward in design and execution and of using common laboratory instrumentation. One major limitation of the spectroscopic assay is that it requires a substrate whose optical output is modulated on reaction with the enzyme. The hydrolysis of an epoxide by sEH typically results in a diol whose spectral properties are similar to those of the parent epoxide; therefore, a substrate that has additional mechanisms in place for generating an optical signal is required. This has been addressed in the literature recently by oxidative cleavage of diols to the corresponding aldehyde using periodate [20–23]. The resultant aldehydes can then be coupled to suitable chromophores or fluorophores and quantified using absorption or fluorescence methods. Although these methods detect epoxide hydrolase activity effectively, they require high concentrations of substrate (millimolar) and rely on external chemical modification steps. An alternate strategy was presented by our previously described substrate, NEPC, which undergoes an intramolecular cyclization liberating the highly colored 4-nitrophenolate anion following hydrolysis of the epoxide moiety [19].
We recently reported that α-cyanoesters [24,25] and α-cyanoethers [26] are effective fluorescent substrates for the detection and quantitation of esterase and P450 activities, respectively. In the case of the α-cyanoesters, O-deacylation liberates a cyanohydrin intermediate that rapidly decomposes to the highly fluorescent 6-methoxy-2-naphthaldehyde (1). These substrates are highly sensitive, are hydrolytically stable, and show large changes in their UV and fluorescence spectra on hydrolysis. We would like to report the development of a series of fluorescent substrates for mammalian sEH, which combines the decomposition mechanism of NEPC with the latent fluorophore of the α-cyanoesters (Fig. 1). In addition, we have designed a continuous kinetic assay for the evaluation of sEH inhibitors. The substrates described in this work will be useful tools in the further development of potent sEH inhibitors.
Fig. 1.
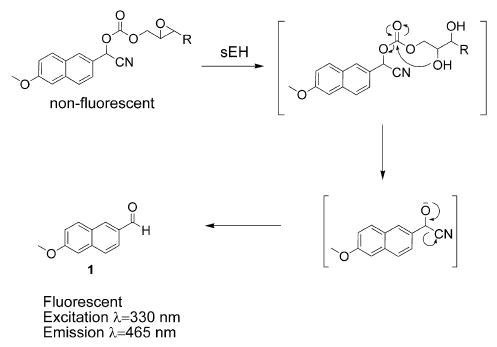
Proposed substrates for sEH. Following hydrolysis of the oxirane, the resultant diol undergoes an intramolecular cyclization, eliminating the cyanohydrin of aldehyde 1. The cyanohydrin rapidly decomposes to fluorescent aldehyde 1.
Materials and methods
Reagents
All reagents and solvents were purchased from Al-drich Chemical (Milwaukee, WI, USA) unless otherwise noted and were used without further purification. Tri-ethylamine (TEA) was distilled over CaH2 prior to use. 4-Chlorocinnamyl alcohol was synthesized as per Charette et al. [27]. Hept-3-enoic acid (85%) was purchased from TCI Chemicals (Portland, OR, USA) and was used as received. Although not explicitly labeled as such, 1H NMR indicated greater than 85% trans-isomer when compared with an authentic sample [28]. 6-Methoxy-2-naphthaldehyde (1) was purchased from Avocado Research Chemicals (Heysham, UK). 2-Hydroxy-2-(2-methoxynaphthalen-6-yl)-acetonitrile was synthesized as per Shan and Hammock [25]. (±)-NEPC was synthesized as per Dietze et al. [19].
Synthesis
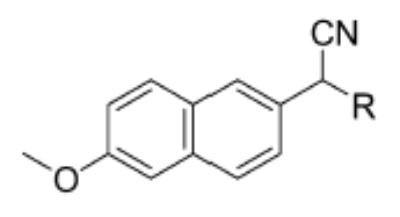
The fluorescent substrates used in this study (Table 1) were synthesized as shown in Scheme 1. All substrates were synthesized as racemic mixtures, each containing a trans-epoxide functionality. All chemical reactions were conducted under a nitrogen atmosphere using anhydrous solvents unless otherwise noted. Reaction progress was monitored using thin-layer chromatography (TLC) with 0.2-mm glass plates precoated with silica gel 60 F254 (Merck, Darmstadt, Germany). Chemical detection was based on the quenching of fluorescence from 254 nm ultraviolet light. Flash chromatography was performed with 32–63 μm silica gel (Sorbent Technologies, Atlanta, GA, USA). NMR spectra were recorded on a Varian Mercury 300 (Varian, Palo Alto, CA, USA) in CDCl3 using tetramethylsilane (TMS) as an internal reference unless otherwise noted. NMR peaks are reported in parts per million (ppm, δ) relative to TMS. High-resolution mass determinations were performed on a Micromass LCT, an orthogonal acceleration time-of-flight (oa-TOF) mass spectrometer, in both positive and negative modes (Micromass, Manchester, UK). Mass spectral calibration was performed using poly-d-l-alanine (P9003, Sigma, St. Louis, MO, USA). For brevity, representative synthetic procedures are listed below. For additional synthetic procedures and spectral characterization, refer to the accompanying supplemental information.
Table 1.
Substrates described in Scheme 1
| Substrate | Structure | Human sEH specific activity (nmol/min/mg) | Mouse sEH specific activity (nmol/min/mg) |
|---|---|---|---|
| 2 |
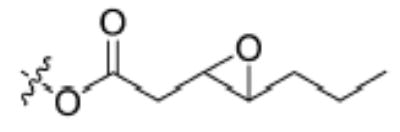
|
532 ± 24 | 275 ± 4 |
| 3 |
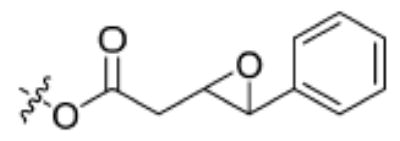
|
714 ± 23 | 214 ± 63 |
| 4 |
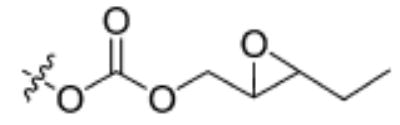
|
408 ± 14 | 125 ± 32 |
| 5 |
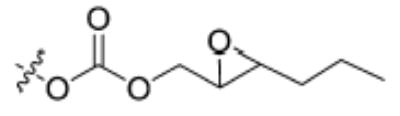
|
1022 ± 35 | 255 ± 45 |
| 6 |
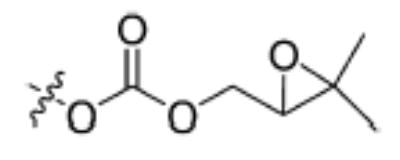
|
11.7 ± 0.2 | 8.4 ± 0.1 |
| 7 |
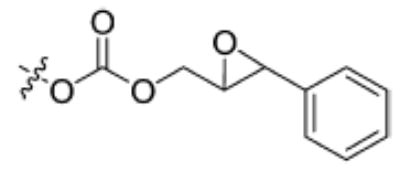
|
2689 ± 44 | 781 ± 129 |
| 8 |
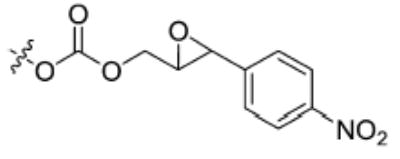
|
376 ± 6 | 859 ± 11 |
| 9 |
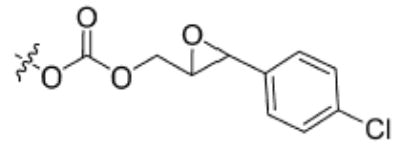
|
359 ± 6 | 1040 ± 93 |
Note. Substrates were assayed at 30 °C in BisTris–HCl buffer (25 mM, 0.1 mg/ml BSA, pH 7.0) with [S] = 10 μM, [murine sEH] = 0–3.6 μg/well, and [human sEH] = 0–12 μg/well. Data are the slopes ± standard errors determined by linear regression methods.
Scheme 1.
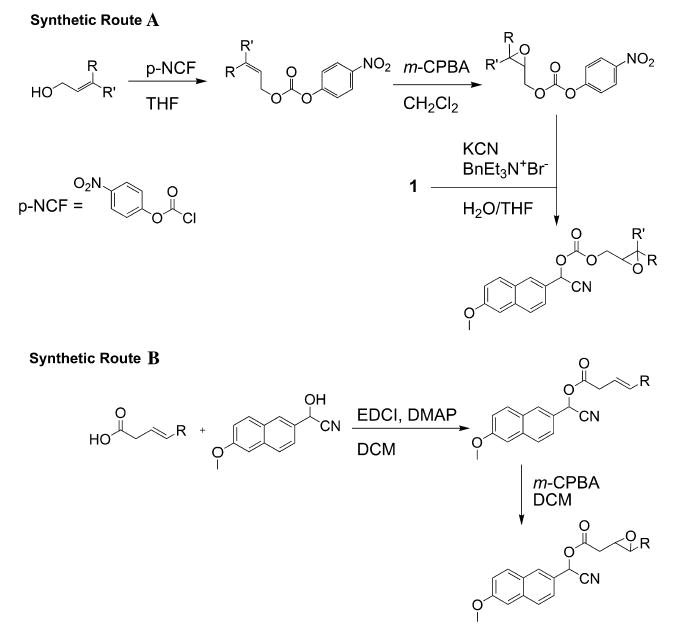
Synthetic routes used for the synthesis of the α-cyanocarbonates (route A) and α-cyanoesters (route B) used in this study.
Cyano(6-methoxy-naphthalen-2-yl)methyl trans-[(3-phenyloxiran-2-yl)methyl] carbonate (7)
A solution of KCN (206 mg) in H2O (0.50 ml) was added dropwise via syringe pump to a solution of aldehyde 1 (531 mg, 2.85 mmol) and (±)-NEPC (1.0 g, 3.17 mmol) in tetrahydrofuran (THF, 25 ml) over 1 h. The reaction was allowed to stir at 0 °C for 2 h and then was warmed to room temperature and stirred for an additional 2 h. Ethyl acetate (50 ml) was added to the reaction. The bright yellow precipitate was filtered, and the filtrate was washed with K2CO3(aq) (1 M) until the aqueous layer was colorless. The organic layer was dried over MgSO4, filtered, and evaporated. The residue was chromatographed twice on SiO2 (5% ethyl acetate in hexanes) to give the product as a clear viscous oil (589 mg, 53%). 1H (300 MHz) δ: 7.91 (br s, 1H), 7.76 (d, J = 8.8 Hz, 1H), 7.72 (d, J = 9.3 Hz, 1H), 7.52 (dd, J = 6.6, 1.9 Hz, 1H), 7.30–7.10 (m, 7H), 7.15 (d, J = 2.4 Hz, 1H), 6.38 (br s, 1H), 4.57–4.52 (m, 1H), 4.30–4.20 (m, 1H), 3.89 (s, 3H), 3.80–3.78 (m, 1H), 3.30–3.23 (m, 1H). 13C (75 MHz) δ: 158.8, 153.2, 135.6, 135.4, 129.8, 128.4, 128.1, 128.0, 125.6, 125.5, 124.7, 119.9, 115.6, 105.5, 68.5, 68.2, 67.0, 58.3, 56.1, 56.0, 55.2. HRMS (m/z): calculated for C46H39N2O10 [2M + H]+: 779.2605, found: 779.2589.
Cyano-(6-methoxynaphthalen-2-yl)methyl trans-2-pentenylacetate
trans-Hept-3-enoic acid (236 μl, 1.73 mmol), hydroxy-(6-methoxy-naphthalen-2-yl)-acetonitrile (355 mg, 1.65 mmol), EDCI (330 mg, 1.73 mmol), and DMAP (42 mg) all were dissolved in dichloromethane (15 ml) and stirred for 36 h. The reaction was washed with K2CO3(aq) (1 M, 2 × 20 ml) and dried over MgSO4. The solvent was evaporated, and residue was chromatographed on SiO2 (3:1 toluene:hexanes) to give the product as a yellow oil (339 mg, 66%). 1H (300 MHz) δ: 7.91 (d, J = 1.6 Hz, 1H), 7.79 (d, J = 8.4 Hz, 1H), 7.76 (d, J = 8.7 Hz, 1H), 7.50 (dd, J = 8.6, 1.8 Hz, 1H), 7.20 (dd, J = 8.9, 2.9 Hz, 1H), 7.14 (d, J = 2.5 Hz, 1H), 6.55 (s, 1H), 5.63–5.43 (m, 2H), 3.92 (s, 3H), 3.20–3.05 (m, 2H), 2.00 (q, J = 7.2 Hz, 2H), 1.36 (m, 2H), 0.86 (t, J = 7.3 Hz, 3H). 13C (75 MHz) δ: 170.2, 158.8, 135.9, 135.2, 129.8, 128.2, 128.0, 127.8, 126.5, 124.9, 120.0, 119.9, 116.2, 105.6, 63.0, 55.3, 37.4, 34.4, 22.1, 13.5.
Cyano(2-methoxynaphthalen-6-yl)methyl trans-2-(3-propyloxiran-2-yl)acetate (2)
Cyano-(6-methoxynaphthalen-2-yl)methyl trans-2-pentenylacetate (723 mg, 2.23 mmol) was dissolved in dichloromethane (10 ml) and treated with m-chloroperbenzoic acid (m-CPBA, 516 mg, 3.0 mmol). The reaction was stirred for 3 days. The reaction was washed with K2CO3 (1 M, 3 × 10 ml), and the organic layer was dried over MgSO4. The solvent was removed, and the residue was chromatographed on SiO2 (15% ethyl acetate in hexanes) to give the product as a clear oil (87 mg, 13%). 1H (300 MHz) δ: 7.91 (d, J = 1.6 Hz, 1H), 7.79 (d, J = 8.4 Hz, 1H), 7.76 (d, J = 8.7 Hz, 1H), 7.50 (m, 1H), 7.20 (dd, J = 8.9, 2.9 Hz, 1H), 7.14 (d, J = 2.5 Hz, 1H), 6.58 (s, 1H), 3.93 (s, 3H), 3.10–3.00 (m, 1H), 2.77–2.58 (m, 3H), 1.57–1.36 (m, 4H), 0.92 (t, J = 7.2 Hz, 3H). 13C (75 MHz) δ: 168.7, 168.6, 158.9, 135.3, 129.8, 128.2, 128.1, 127.9, 126.2, 126.2, 124.9, 119.9, 116.0, 116.0, 105.6, 63.3, 58.3, 55.3, 53.1, 53.1, 37.2, 37.2, 33.5, 19.0, 13.8. HRMS (m/z): calculated for C40H43N2O8 [2M + H]+: 679.3019, found: 679.3014.
Recombinant enzyme production and purification
Recombinant MsEH and HsEH were produced in a baculovirus expression system and purified by affinity chromatography [29–31]. The enzyme preparations were at least 97% pure, as evidenced by SDS–PAGE and scanning densitometry. No detectable esterase or glutathione transferase activities, which can interfere with sEH assays, were detected [19]. Protein concentration was quantified by using the Pierce BCA assay using Fraction V bovine serum albumin (BSA) as the calibrating standard.
Standardization curves
Fluorescence was measured in relative fluorescent units (RFU), and absorbance was measured in optical density (OD). Therefore, a conversion of the measured signal into moles of fluorophore or moles of chromophore was necessary for quantitative assessment. For this purpose, stock solutions of aldehyde 1 in dimethyl sulfoxide (DMSO) and 4-nitrophenol in ethanol at known concentrations were prepared. The stock solutions were diluted (1:25 stock:buffer for aldehyde 1 and 1:12.5 for 4-nitrophenol) with buffer (BisTris–HCl, 25 mM, pH 6.5, 7.0, 7.5, and 8.0 containing 0.1 mg/ml BSA) and were added to the same buffer in a black polystyrene 96-well microtiter plate (Greiner Bio-One, Long-wood, FL, USA) or in a clear polystyrene 96-well plate (ThermoLabsystems, Franklin, MA, USA), giving a final assay volume of 200 μl and a final DMSO or ethanol concentration of 1 or 2%, respectively (v/v). Fluorescence measurements were taken at 30 °C with an excitation wavelength of 330 nm (bandwidth = 20 nm) and an emission wavelength of 465 nm (bandwidth = 20 nm) using a SpectraFluor Plus (Tecan, Research Triangle Park, NC, USA) fluorescent plate reader with the following settings: manual gain, 60; integration time, 40 μs; number of flashes, 3. The OD readings of 4-nitrophenol were taken with a SpectraMax 340PC384 microplate spectrophotometer (Molecular Devices, Sunnyvale, CA, USA) at 405 nm. The relationship between moles of aldehyde 1 and RFU, as well as between moles of 4-nitrophenol and OD, was plotted, and the resulting calibration curve was used to determine the concentration of the respective reporter molecule in the subsequent experiments. All determinations were performed with at least three replicates, and linear regressions were performed with at least five datum points.
Aqueous solubility determination
Solubility determinations were performed in clear 96-well styrene flat-bottom microtiter plates using a SpectraMax 340PC384 microplate spectrophotometer. Stock solutions of the fluorescent substrates in DMSO were prepared at the following concentrations: 10, 7.5, 5, 2.5, 1.5, 1.0, 0.75, 0.5, 0.25, 0.1, and 0.05 mM. These solutions were further diluted 1:25 (v:v) in buffer (Bis-Tris–HCl, 25 mM, pH 7.0, containing 0.1 mg/ml BSA). Then 50 μl of the diluted stock solutions was added to 150 μl of assay buffer in a clear 96-well plate (final content of stock solution per well = 1%), and the absorptions of these mixtures at 800 nm were read. The measurements were performed in triplicate at 30 °C.
Aqueous stability determination
Stock solutions of substrate 7 (0.5 and 2.0 mM in DMSO) and NEPC (2.0 and 2.5 mM in ethanol) were prepared. These were diluted 1:25 (v:v) in BisTris–HCl buffer (25 mM, 0.1 mg/ml BSA, pH 6.5, 7.0, 7.5, and 8.0) except for the 2.5 mM NEPC stock solution, which was diluted 1:12.5 in buffer. Then 50 μl of the resultant dilutions was added to 150 μl of the corresponding buffer in a black or clear 96-well microtiter plate. The fluorescent substrate’s hydrolysis was monitored using a SpectraFluor Plus fluorescent plate reader by measuring the appearance of aldehyde 1 (excitation filter 330 nm, emission filter 465 nm) for 10 min and recording the fluorescence emission every 30 s at 30 °C. NEPC hydrolysis was monitored by measuring the absorbance at 405 nm for 10 min using a SpectraMax 340PC384 microplate spectrophotometer. All determinations were conducted with at least three replicates.
Specific activity determination
A 1 mM fluorescent substrate stock solution in DMSO was prediluted 1:25 in BisTris–HCl buffer (25 mM, 0.1 mg/ml BSA, pH 7.0). Then 50 μl of the resultant solution was added to 150 μl of previously incubated (5 min at 30 °C) enzyme solution in a black 96-well microtiter plate ([S]final = 10 μM). The enzyme solutions consisted of recombinant sEH from mouse or human in buffer (BisTris–HCl, 25 mM, pH 7.0, containing 0.1 mg/ml BSA) at various concentrations. The amount of substrate turned over was measured by determination of fluorescence as described above. Initial velocities were calculated by linear regression and plotted against the corresponding protein concentrations. Specific enzyme activities were determined by linear regression of the linear portion of these curves and contained at least five datum points. These determinations were performed in quadruplicate. Assays were run under conditions where product formation was linearly dependent both on the concentration of enzyme and on the time for the course of the assay.
Vmax/Km determination
These assays were performed as per the determination of specific enzyme activity (vide supra). Constant enzyme concentrations of recombinant sEH (murine sEH: 6.9 nM; human sEH: 2.9 nM) were tested for their activity with various substrate concentrations. For this purpose, stock solutions of substrate in DMSO were prepared with the following concentrations: 2.5, 1.5, 1.0, 0.75, 0.5, 0.25, 0.1, 0.05, and 0 mM. They were pre-diluted as described above and subsequently were added to preincubated enzyme/buffer mixtures at 30 °C as described above. Initial velocities of substrate turnover were plotted versus corresponding substrate concentrations, and Vmax/Km values were determined by linear regression of the linear portion of these curves and contained at least five datum points. These determinations were performed in quadruplicate.
Kinetic assay optimization
The following procedure was used to determine the optimal concentrations of enzyme and substrate for a kinetic assay. Various amounts of human sEH in Bis-Tris–HCl buffer (25 mM, pH 7.0, containing 0.1 mg/ml BSA, range of human sEH content = 0–71 ng/well) were tested with various final substrate concentrations (0–10 μM) in a matrix where well A1 contained the highest enzyme and substrate concentrations and well H12 contained neither enzyme nor substrate. Enzyme activity was monitored as described above, including preincubation of the enzyme. The chosen concentration of enzyme and substrate was then reevaluated in quadruplicate.
Inhibitor assay
IC50 values were determined using compound 7 as a substrate. Enzymes (0.88 nM for murine and 0.96 nM for human sEH) were incubated with inhibitors ([I] = 0.5–10,000 nM) for 5 min in BisTris–HCl buffer (25 mM, pH 7.0, containing 0.1 mg/ml of BSA) at 30 °C prior to substrate introduction ([S] = 5 μM). Enzyme activity was measured as described above by monitoring the appearance of aldehyde 1. Assays were performed in triplicate. By definition, IC50 values are concentrations of inhibitor that reduce enzyme activity by 50%. IC50 values were determined by regression of at least five datum points, with a minimum of two datum points in the linear region of the curve on either side of the IC50 values. The curve was generated from at least three separate runs, each in triplicate. Results are given as means and standard deviations.
Results and discussion
Design and synthesis
The substrates shown in Table 1 were designed to follow the same mechanistic path of degradation as reported by Dietze et al. [19] for NEPC. It was necessary to find a fluorescent reporter group whose fluorescence was modulated on cleavage of the ester or carbonate linker. Although umbelliferone phenylepoxy-esters have been reported to be good fluorescent substrates for sEH, they are hydrolytically unstable [19]. It has been shown that α-cyanoester derivatives of aldehyde 1 are essentially nonfluorescent, whereas the aldehyde produced by cleavage of the ester is highly fluorescent (vide supra). It was hypothesized that replacement of the 4-nitrophenoxide portion of NEPC with an α-cyanoester or a carbonate would result in an acceptable fluorescent substrate for sEH. Because it has been shown previously [19] that there is little difference between (+)-NEPC and (−)-NEPC, we opted for a racemic synthesis. In addition, to maintain consistency with Dietze and coworkers’ study [19], we chose to incorporate only trans-epoxides into the substrates described. On hydrolysis of the epoxide functionality, the resultant diol undergoes an intramolecular cyclization with the carbonyl of either the ester or the carbonate, followed by elimination of the cyanohydrin. The cyanohydrin rapidly decomposes to give the highly fluorescent 6-methoxy-2-naphthaldehyde. Although aliphatic alcohols do not possess the leaving group ability required for the proposed substrates [19], we felt that the steric bulk of the cyanohydrin leaving group would facilitate the decomposition of the substrate following epoxide hydrolysis.
The substrates that possess carbonate linkages were synthesized as shown in Scheme 1 (route A). The synthesis began with a trans-allylic alcohol that either was commercially available or, in the case of p-chlorocinnamyl alcohol, was known in the literature [27]. The starting allylic alcohols were reacted with p-nitrophenylchloroformate to give the corresponding carbonate in an acceptable yield. The carbonates were then smoothly converted to the corresponding epoxides using the mild oxidant m-CPBA. Initially, we thought that p-nitrophenyl epoxycarbonates would react with 2-hydroxy-2-(2-methoxynaphthalen-6-yl)-acetonitrile to form the desired α-cyanocarbonate substrates; however, we found that this procedure resulted solely in decomposition of the cyanohydrin. Therefore, we formed the cyanohydrin in situ via reaction of 6-methoxy-2-naphthaldehyde with KCN under phase transfer conditions. This methodology allowed the facile synthesis of the carbonate-based substrates described in Table 1 with yields ranging from 6 to 34% (overall) and purity greater than 95% (via 1H NMR).
The desired α-cyanoesters were synthesized by first coupling commercially available carboxylic acids to 2-hydroxy-2-(2-methoxynaphthalen-6-yl)-acetonitrile using the well-known coupling agent 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI) [32] (Scheme 1, route B). Subsequent epoxidation of the resulting α-cyanoester was accomplished with m-CPBA. It should be noted that this oxidation step occurs in very low yield (yields were <20%) and requires a lengthy reaction time.
Substrate selectivity
Having the required substrates in hand, we first investigated the selectivity of each substrate for both murine and human sEH. For this purpose, activity was measured for varying concentrations of enzymes while having a constant concentration of substrate ([S] = 10 μM). Specific activities for both enzymes were determined via linear regression of the linear portion of the curves of activity versus [E]. These results are summarized in Table 1. Carbonate 6 showed the lowest specific activity among the substrates tested for both human and murine sEH. This observation is easily explained by considering the steric congestion around the epoxide and is consistent with earlier observations that sEH converts disubstituted epoxides faster than it does trisubstituted epoxides (see [33] and references therein). The murine enzyme showed little preference for alkyl carbonates over alkyl esters, as evidenced by the similarity between specific activities determined for carbonates 4 and 5 and ester 2. In contrast, the human enzyme showed a preference for carbonate 5 over carbonate 4 and ester 2. Although the murine enzyme showed higher specific activities for aryl carbonates 7, 8, and 9 over the alkyl carbonates and esters, the picture is not so clear for the human enzyme. The human enzyme showed lower specific activities for carbonates 8 and 9 than for esters 2 and 3 and carbonates 4, 5, and 7. It is interesting to note that Dietze et al. [19] found that aryl-epoxy esters were four times more active than alkyl-epoxy esters in the murine epoxide hydrolase. The highest specific activity for the murine enzyme was obtained with carbonate 9, whereas the highest specific activity for the human enzyme was obtained with carbonate 7. Because human sEH is the ultimate target for inhibitor development, we chose to focus exclusively on the use of phenylepoxy-carbonate 7 as a fluorescent substrate in this study.
Aqueous solubility, stability, and Michaelis–Menten parameters
To determine the limit of solubility of 7 in buffer, solutions of various concentrations of the substrate were diluted with BisTris–HCl buffer (25 mM, pH 7.0, containing 0.1 mg/ml of BSA) in a clear 96-well microtiter plate. Substrate precipitation was seen at concentrations above the solubility limit and was detected by an increase in absorption at 800 nm. We determined that the limit of solubility for carbonate 7 was between 15 and 20 μM, which is at least twofold lower than the solubility of NEPC [13,19].
One of the shortcomings we have found in using NEPC to assay inhibitor potency is its large background hydrolysis. Because 4-nitrophenolate is a good leaving group, it was hypothesized that replacing this group with the 6-methoxynaphthyl cyanohydrin would yield a substrate that had increased hydrolytic stability. Fig. 2 illustrates the hydrolysis of both NEPC and carbonate 7 at different pH values and concentrations. Comparing the hydrolysis of NEPC to the hydrolysis of substrate 7 at equal concentrations shows that compound 7 is approximately three to four times more stable in aqueous solution at pH 7.0 than is NEPC. In addition, the hydrolytic stability of NEPC and compound 7 was investigated using optimized assay concentrations of substrate (50 and 5 μM, respectively). As can be seen in Fig. 2, compound 7 gives an overall lower background signal than does NEPC, due in part to the much lower concentration of substrate.
Fig. 2.
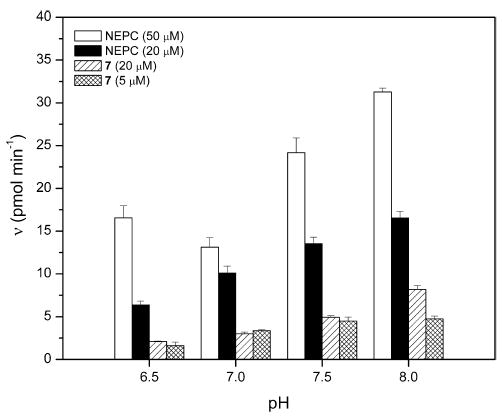
Hydrolytic stability of fluorescent substrate versus pH. [NEPC] = 50 μM (open bar) and 20 μM (filled bar), and [7] = 20 μM (hatched bar) and 5 μM (cross-hatched bar), in BisTris–HCl buffer (25 mM, 0.1 mg/ml BSA) at the indicated pH values. Data are means ± standard deviations of at least three replicates.
After determining the hydrolytic stability and aqueous solubility of substrate 7, we then attempted to determine the Michaelis–Menten parameters, KM and kcat. The rate of appearance of aldehyde 1 as a function of concentration of substrate 7 in both murine and human systems is shown in Fig. 3. Because of the substrate’s low solubility in the studied system, the curves plateau at [S] ≈ 10 μM and do not follow a hyperbolic shape as expected (r2 < 0.97 for both enzymes), thereby making it impossible to calculate both kinetic constants accurately. However, using the linear portion of the graph, we determined the Vmax/KM values for compound 7 to be 14.3 ± 1.3 and 12.5 ± 1.0 pmol/min/μM with the murine and human enzymes, respectively. Converting these to kcat/KM values, 0.368 and 0.172/s/μM for the human and murine systems, respectively, allows comparison with those for NEPC as determined by Morisseau et al. [13]. NEPC was shown to have kcat/KM values of 2.6 and 3.3/s/μM for the human and murine systems, respectively. From these data, we can see that NEPC is hydrolyzed roughly 10–20 times faster by the murine and human sEH than is substrate 7. However, this is offset by the lower background hydrolysis and much higher sensitivity of the fluorescent reporter group.
Fig. 3.
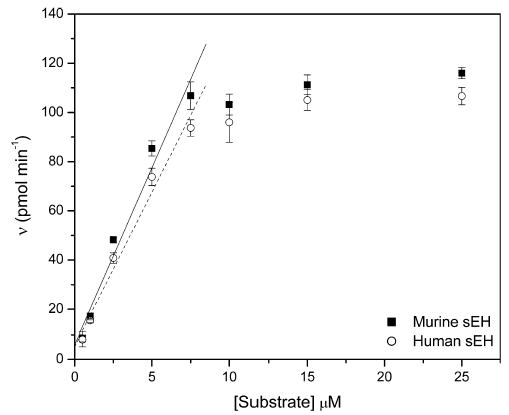
Vmax/Km determination for substrate 7. [Murine sEH] = 6.9 nM and [human sEH] = 2.9 nM. Linear portion of the data is indicated by a solid line for the murine sEH and by a broken line for the human sEH. Data are reported as average velocities ± standard deviations (n = 4).
Inhibitor assays
To choose the appropriate substrate/enzyme concentration combination for a kinetic assay, we looked at the hydrolysis of substrate 7 by both the human and murine recombinant enzymes. Using a black 96-well microtiter plate, a matrix of varying enzyme and substrate concentrations was prepared and evaluated. From this experiment, we chose concentrations of substrate and enzyme that satisfied the following stipulations: (i) substrate turnover was at least 10 times above background hydrolysis, (ii) enzyme concentrations were as small as possible, (iii) the substrate turnover rate was linear for a period of 10 min, and (iv) the enzyme concentration was low enough to ensure that the hydrolysis of the epoxide was rate limiting. We found that useful concentrations of enzyme and substrate were 1 nM and 5 μM, respectively. These concentrations were applicable to both murine and human enzyme systems. A substrate concentration of 5 μM is probably well below the KM of substrate 7. It was observed that at low concentrations of enzyme, there was a lag time of approximately 2 min, which is consistent with the observations made by Dietze and coworkers when developing the NEPC substrate [19]. In spite of the observed lag time and the fact that this assay could not be run under saturation conditions, we hypothesized that we could still use it to rank those inhibitors that could not be ranked using the NEPC absorption-based assay.
If substrate 7 is to be used in a kinetic assay, it is imperative that the cyclization of the diol is not the rate-limiting step. Attempts to compare the rate of substrate consumption to the rate of aldehyde appearance using HPLC methods were unsuccessful due to decomposition of compound 7 during the HPLC run. Analysis of the data obtained from the specific activity determination revealed that the rate of hydrolysis of compound 7 approached saturation with increasing concentrations of human and murine enzymes. In both cases, the maximal hydrolysis rate of substrate 7 was approximately 400 pmol/min. Because the rate of epoxide hydrolysis should increase linearly with enzyme concentration, the observed saturation can be attributed to the fact that epoxide hydrolysis occurs faster than diol cyclization at high concentrations of enzyme. Based on the data shown in Table 1, an enzyme concentration of 1 nm corresponds to a hydrolysis rate of approximately 30 and 9 pmol/min for the human and murine enzymes, respectively. At this low concentration of enzyme, epoxide hydrolysis is the rate-limiting step of the process.
To investigate the usefulness of this new assay, a series of sEH inhibitors were evaluated for inhibition potency. The inhibitors chosen (Fig. 4) were published previously and represent a wide range of potencies [14,15,17]. Table 2 compares the experimentally determined IC50 values using three different assay techniques: the absorption-based NEPC [19], radioactive tDPPO [15], and the currently described fluorescent assay. In the murine enzyme system, the fluorescent assay gives a relative pattern of inhibition for this series of compounds that is comparable to that obtained previously with the other assays. In contrast, the relative ranking of CHU and DCU in the human enzyme system differs between the radioactive- and optic-based assays. This highlights the difficulties encountered when relating IC50 values to inhibitor potency because it is apparent from Table 2 that IC50 values can be dependent on the method of determination. Because of the relatively large concentration of enzymes used, the NEPC assay does not allow us to differentiate the best inhibitors (CDU and AUDA) from each other on the basis of IC50 values. The high sensitivity of the fluorescent assay allows the use of a 100-fold lower concentration of enzyme, contributing to the differentiation of the more potent inhibitors. Compared with the tDPPO assay, which is also able to differentiate the best inhibitors, the new fluorescent assay yields a fourfold higher relative separation of CDU and AUDA for human sEH. Considering these results, the fluorescent assay described in this article has an effective range for IC50 values that is two orders of magnitude lower than NEPC.
Fig. 4.
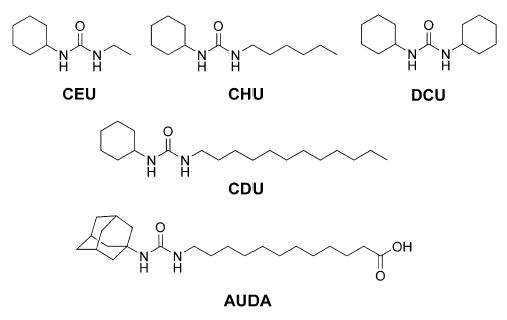
Inhibitors used to validate new assay.
Table 2.
Comparison of IC50 values for selected sEH inhibitors
| Mouse sEH IC50 (nM)a |
Human sEH IC50 (nM)a |
|||||
|---|---|---|---|---|---|---|
| Substrate | NEPCb | tDPPOb,c | 7 | NEPCb | tDPPOb,c | 7 |
| [S] (μM) | 50 | 50 | 5 | 50 | 50 | 5 |
| [Enzyme] (nM) | 100 | 8 | 1 | 200 | 16 | 1 |
| CEU | 51,700 ± 700 | n.d. | >100,000 | 42,000 ± 2000 | n.d. | 7500 ± 130 |
| CHU | 110 ± 30 | 55 ± 3 | 75 ± 8 | 70 ± 20 | 221 ± 2 | 25 ± 0.1 |
| DCU | 90 ± 10 | 81.8 ± 0.7 | 240 ± 20 | 160 ± 10 | 63 ± 0.3 | 52 ± 1 |
| CDU | 50 ± 10 | 9.8 ± 0.4 | 17 ± 3 | 100 ± 10 | 85.2 ± 0.5 | 7.0 ± 0.2 |
| AUDA | 50 ± 10 | 18 ± 1 | 27 ± 1 | 100 ± 10 | 69 ± 2 | 3.2 ± 0.1 |
Values are reported as averages ± standard deviations of at least three replicates.
Data determined previously (see text).
tDPPO = [3H] trans-1,3-diphenylpropene oxide.
Conclusion
We have developed a fluorescent assay for mammalian sEH inhibition studies. This assay is based on the enzymatic hydrolysis of a readily synthesized β-epoxy-carbonate that results in the production of a fluorescent aldehyde. This new assay has a sensitivity that is 100 times greater than that in the previously used spectrophotometric assay. This assay will be an invaluable tool for the development of new sEH inhibitors and will further our investigations into the relationship among sEH inhibition, hypertension, and vascular inflammation.
Supplementary Material
Acknowledgments
The authors thank James Sanborn for many helpful discussions and Jozsef Lango for assistance with mass spectral determinations. We also thank Rong Zhang for project initiation. Paul Jones was supported by an NIH Institutional Pre- and Postdoctoral Training grant (T32 DK07355) and by an NIH/NHLBI Ruth L. Kirschstein–NRSA grant (F32 HL078096). Nicola Wolf was supported by the Bavarian Research Foundation (Bayerische Forschungsstiftung). This work was supported in part by NIEHS grant ES02710, NIEHS Super-fund grant P42 ES04699, NHLBI STTR grant R41 HL078016 and NIH/NINDS grant R03 NS050841.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.ab.2005.03.041.
Footnotes
Abbreviations used: sEH, soluble epoxide hydrolase; EET, epoxyeicosatrienoic acid epoxides; NEPC, 4-nitrophenyl-trans-2,3-epoxy-3-phenylpropyl carbonate; TEA, triethylamine; TLC, thin-layer chromatography; TMS, tetramethylsilane; ppm, parts per million; oa-TOF, orthogonal acceleration time-of-flight; THF, tetrahydrofuran; m-CPBA, m-chloroperbenzoic acid; BSA, bovine serum albumin; RFU, relative fluorescent units; OD, optical density; DMSO, dimethyl sulfoxide; EDCI, 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride; tDPPO, [3H] trans-1,3-diphenylpropene oxide; P450, cytochrome P450.
References
- 1.Arand M, Grant DF, Beetham JK, Friedberg T, Oesch F, Hammock BD. Sequence similarity of mammalian epoxide hydrolases to the bacterial haloalkane dehalogenase and other related proteins: implication for the potential catalytic mechanism of enzymatic epoxide hydrolysis. FEBS Lett. 1994;338:251–256. doi: 10.1016/0014-5793(94)80278-5. [DOI] [PubMed] [Google Scholar]
- 2.Oesch F. Mammalian epoxide hydrases: inducible enzymes catalysing the inactivation of carcinogenic and cytotoxic metabolites derived from aromatic and olefinic compounds. Xenobiotica. 1973;3:305–340. doi: 10.3109/00498257309151525. [DOI] [PubMed] [Google Scholar]
- 3.Zeldin DC, Kobayashi J, Falck JR, Winder BS, Hammock BD, Snapper JR, Capdevila JH. Regio- and enantiofacial selectivity of epoxyeicosatrienoic acid hydration by cytosolic epoxide hydrolase. J Biol Chem. 1993;268:6402–6407. [PubMed] [Google Scholar]
- 4.Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, Parker C, Graham L, Engler MM, Hammock BD, Zeldin DC, Kroetz DL. Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ Res. 2000;87:992–998. doi: 10.1161/01.res.87.11.992. [DOI] [PubMed] [Google Scholar]
- 5.Moghaddam MF, Grant DF, Cheek JM, Greene JF, Williamson KC, Hammock BD. Bioactivation of leukotoxins to their toxic diols by epoxide hydrolase. Nat Med. 1997;3:562–566. doi: 10.1038/nm0597-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greene JF, Williamson KC, Newman JW, Morisseau C, Hammock BD. Metabolism of monoepoxides of methyl linoleate: bioactivation and detoxification. Arch Biochem Biophys. 2000;376:420–432. doi: 10.1006/abbi.2000.1753. [DOI] [PubMed] [Google Scholar]
- 7.Capdevila JH, Falck JR, Harris RC. Cytochrome P450 and arachidonic acid bioactivation: molecular and functional properties of the arachidonate monooxygenase. J Lipid Res. 2000;41:163–181. [PubMed] [Google Scholar]
- 8.Fleming I. Cytochrome P450 enzymes in vascular homeostasis. Circ Res. 2001;89:753–762. doi: 10.1161/hh2101.099268. [DOI] [PubMed] [Google Scholar]
- 9.Oltman CL, Weintraub NL, VanRollins M, Dellsperger KC. Epoxyeicosatrienoic acids and dihydroxyeicosatrienoic acids are potent vasodilators in the canine coronary microcirculation. Circ Res. 1998;83:932–939. doi: 10.1161/01.res.83.9.932. [DOI] [PubMed] [Google Scholar]
- 10.Fisslthaler B, Popp R, Kiss L, Potente M, Harder DR, Fleming I, Busse R. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature. 1999;401:493–497. doi: 10.1038/46816. [DOI] [PubMed] [Google Scholar]
- 11.Imig JD, Zhao X, Capdevila JH, Morisseau C, Hammock BD. Soluble epoxide hydrolase inhibition lowers arterial blood pressure in angiotensin II hypertension. Hypertension. 2002;39:690–694. doi: 10.1161/hy0202.103788. [DOI] [PubMed] [Google Scholar]
- 12.Zhao X, Yamamoto T, Newman JW, Kim IH, Watanabe T, Hammock BD, Stewart J, Pollock JS, Pollock DM, Imig JD. Soluble epoxide hydrolase inhibition protects the kidney from hypertension-induced damage. J Am Soc Nephrol. 2004;15:1244–1253. [PubMed] [Google Scholar]
- 13.Morisseau C, Du G, Newman JW, Hammock BD. Mechanism of mammalian soluble epoxide hydrolase inhibition by chalcone oxide derivatives. Arch Biochem Biophys. 1998;356:214–228. doi: 10.1006/abbi.1998.0756. [DOI] [PubMed] [Google Scholar]
- 14.Morisseau C, Goodrow MH, Dowdy D, Zheng J, Greene JF, Sanborn JR, Hammock BD. Potent urea and carbamate inhibitors of soluble epoxide hydrolases. Proc Natl Acad Sci USA. 1999;96:8849–8854. doi: 10.1073/pnas.96.16.8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morisseau C, Goodrow MH, Newman JW, Wheelock CE, Dowdy DL, Hammock BD. Structural refinement of inhibitors of urea-based soluble epoxide hydrolases. Biochem Pharmacol. 2002;63:1599–1608. doi: 10.1016/s0006-2952(02)00952-8. [DOI] [PubMed] [Google Scholar]
- 16.Nakagawa Y, Wheelock CE, Morisseau C, Goodrow MH, Hammock BG, Hammock BD. 3-D QSAR analysis of inhibition of murine soluble epoxide hydrolase (MsEH) by benzoylureas, arylureas, and their analogues. Bioorg Med Chem Lett. 2000;8:2663–2673. doi: 10.1016/s0968-0896(00)00198-x. [DOI] [PubMed] [Google Scholar]
- 17.McElroy NR, Jurs PC, Morisseau C, Hammock BD. QSAR and classification of murine and human soluble epoxide hydrolase inhibition by urea-like compounds. J Med Chem. 2003;46:1066–1080. doi: 10.1021/jm020269o. [DOI] [PubMed] [Google Scholar]
- 18.Kim IH, Morisseau C, Watanabe T, Hammock BD. Design, synthesis, and biological activity of 1,3-disubstituted ureas as potent inhibitors of the soluble epoxide hydrolase of increased water solubility. J Med Chem. 2004;47:2110–2122. doi: 10.1021/jm030514j. [DOI] [PubMed] [Google Scholar]
- 19.Dietze EC, Kuwano E, Hammock BD. Spectrophotometric substrates for cytosolic epoxide hydrolase. Anal Biochem. 1994;216:176–187. doi: 10.1006/abio.1994.1023. [DOI] [PubMed] [Google Scholar]
- 20.Doderer K, Lutz-Wahl S, Hauer B, Schmid RD. Spectrophotometric assay for epoxide hydrolase activity toward any epoxide. Anal Biochem. 2003;321:131–134. doi: 10.1016/s0003-2697(03)00399-3. [DOI] [PubMed] [Google Scholar]
- 21.Doderer K, Schmid RD. Fluorometric assay for determining epoxide hydrolase activity. Biotechnol Lett. 2004;26:835–839. doi: 10.1023/b:bile.0000025887.36874.33. [DOI] [PubMed] [Google Scholar]
- 22.Mateo C, Archelas A, Furstoss R. A spectrophotometric assay for measuring and detecting an epoxide hydrolase activity. Anal Biochem. 2003;314:135–141. doi: 10.1016/s0003-2697(02)00646-2. [DOI] [PubMed] [Google Scholar]
- 23.Badalassi F, Wahler D, Klein G, Crotti P, Reymond JL. A versatile periodate-coupled fluorogenic assay for hydrolytic enzymes. Angewandte Chemie—Intl Ed. 2000;39:4067–4070. doi: 10.1002/1521-3773(20001117)39:22<4067::aid-anie4067>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 24.Wheelock CE, Wheelock AM, Zhang R, Stok JE, Morisseau C, Le Valley SE, Green CE, Hammock BD. Evaluation of α-cyanoesters as fluorescent substrates for examining interindividual variation in general and pyrethroid-selective esterases in human liver microsomes. Anal Biochem. 2003:208–222. doi: 10.1016/s0003-2697(03)00002-2. [DOI] [PubMed] [Google Scholar]
- 25.Shan G, Hammock BD. Development of sensitive esterase assays based on α-cyano-containing esters. Anal Biochem. 2001;299:54–62. doi: 10.1006/abio.2001.5388. [DOI] [PubMed] [Google Scholar]
- 26.Zhang R, Kang KD, Shan G, Hammock BD. Design, synthesis, and evaluation of novel P450 fluorescent probes bearing s-cyanoether. Tetrahedron Lett. 2003:4331–4334. [Google Scholar]
- 27.Charette AB, Molinaro C, Brochu C. Catalytic asymmetric cyclopropanation of allylic alcohols with titanium–TADDOLate: scope of the cyclopropanation reaction. J Am Chem Soc. 2001;123:12168–12175. doi: 10.1021/ja0108382. [DOI] [PubMed] [Google Scholar]
- 28.Ragoussis N. Modified Knoevenagel condensations: synthesis of (E)-3-alkenoic acids. Tetrahedron Lett. 1987;28:93–96. [Google Scholar]
- 29.Grant DF, Storms DH, Hammock BD. Molecular cloning and expression of murine liver soluble epoxide hydrolase. J Biol Chem. 1993;268:17628–17633. [PubMed] [Google Scholar]
- 30.Beetham JK, Tian T, Hammock BD. cDNA cloning and expression of a soluble epoxide hydrolase from human liver. Arch Biochem Biophys. 1993;305:197–201. doi: 10.1006/abbi.1993.1411. [DOI] [PubMed] [Google Scholar]
- 31.Wixtrom RN, Silva MH, Hammock BD. Affinity purification of cytosolic epoxide hydrolase using derivatized epoxy-activated Sepharose gels. Anal Biochem. 1988;169:71–80. doi: 10.1016/0003-2697(88)90256-4. [DOI] [PubMed] [Google Scholar]
- 32.Dhaon MK, Olsen RK, Ramasamy K. Esterification of N-protected α-amino acids with alcohol/carbodiimide/4-(dimethylamino)pyridine: racemization of aspartic and glutamic acid derivatives. J Organic Chem. 1982;47:1962–1965. [Google Scholar]
- 33.Morisseau C, Hammock BD. Epoxide hydrolases: mechanisms, inhibitor designs, and biological roles. Annu Rev Pharmacol Toxicol. 2005;45:311–333. doi: 10.1146/annurev.pharmtox.45.120403.095920. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.