

Abstract
Cell cycle arrest in response to DNA damage depends upon coordinated interactions between DNA repair and checkpoint pathways. Here we examine the role of DNA repair and checkpoint genes in responding to unprotected telomeres in budding yeast cdc13-1 mutants. We show that Exo1 is unique among the repair genes tested because like Rad9 and Rad24 checkpoint proteins, Exo1 inhibits the growth of cdc13-1 mutants at the semipermissive temperatures. In contrast Mre11, Rad50, Xrs2, and Rad27 contribute to the vitality of cdc13-1 strains grown at permissive temperatures, while Din7, Msh2, Nuc1, Rad2, Rad52, and Yen1 show no effect. Exo1 is not required for cell cycle arrest of cdc13-1 mutants at 36° but is required to maintain arrest. Exo1 affects but is not essential for the production of ssDNA in subtelomeric Y′ repeats of cdc13-1 mutants. However, Exo1 is critical for generating ssDNA in subtelomeric X repeats and internal single-copy sequences. Surprisingly, and in contrast to Rad24, Exo1 is not essential to generate ssDNA in X or single-copy sequences in cdc13-1 rad9Δ mutants. We conclude that Rad24 and Exo1 regulate nucleases with different properties at uncapped telomeres and propose a model to explain our findings.
CHECKPOINT controls are evolutionarily conserved mechanisms that inhibit cell cycle progression when DNA is damaged (Hartwell and Weinert 1989; Lowndes and Murguia 2000; Zhou and Elledge 2000; Nyberg et al. 2002). They play important roles in the processes of meiosis and immune system development, contribute to the integrity of the neuronal system, help to maintain genetic stability, and prevent cancer (Zhou and Elledge 2000). Checkpoint pathways are thought of as signal transduction cascades that comprise stimuli, sensors, signalers, and targets (Zhou and Elledge 2000, 2003; Nyberg et al. 2002).
Recently two checkpoint sensor protein complexes have been shown to bind damaged DNA (Kondo et al. 2001; Melo et al. 2001; Rouse and Jackson 2002; Zou et al. 2002). In budding yeast, one complex comprises Rad17, Mec3, and Ddc1, forming a heterotrimeric, proliferating cell nuclear antigen (PCNA)-like ring structure, called the 9-1-1 complex (named after the mammalian and Schizosaccharomyces pombe orthologs Rad9, Rad1, and Hus1). Loading of this complex is dependent on an alternative replication factor C (RFC) complex made of Rad24 and the four small Rfc subunits (Green et al. 2000). The second, Mec1/Ddc2 complex, binds DNA independently of Rad24, Rad17, Mec3, and Ddc1. Both Rad17 and Mec1 complexes are essential for signaling cell cycle arrest in response to many types of DNA damage, suggesting that they are each necessary to stimulate the signal transduction cascade that results in cell cycle arrest. Another checkpoint protein, Rad9, is required to load neither the Rad17 nor the Mec1 complex and it may therefore act as a downstream signal transduction molecule or as a component of a third checkpoint complex (Gilbert et al. 2001; Melo et al. 2001). The nature of the interactions between checkpoint sensor proteins and damaged DNA is now being elucidated (Ellison and Stillman 2003; Majka and Burgers 2003; Zou and Elledge 2003).
A large body of evidence indicates that single-stranded DNA (ssDNA) is an important stimulus for cell cycle arrest in eukaryotes (Garvik et al. 1995; Huang et al. 1996; Lee et al. 1998; Usui et al. 2001; Vaze et al. 2002; Zou and Elledge 2003). Interestingly, checkpoint proteins not only recognize ssDNA but affect the rate at which ssDNA arises, suggesting that they have direct roles in regulating accumulation of ssDNA (Lydall and Weinert 1995).
Cells that are defective in Cdc13, a telomere-binding protein, accumulate large amounts of ssDNA specifically near telomeres (Garvik et al. 1995; Nugent et al. 1996; Booth et al. 2001). rad9Δ and rad24Δ checkpoint mutants are completely defective in cell cycle arrest in response to cdc13-1-induced defects, yet Rad24 contributes to ssDNA production, while Rad9 inhibits ssDNA production (Lydall and Weinert 1995). These observations can be explained by a model in which Rad24 is required for the activity of a 5′ to 3′ exonuclease that degrades the telomeres of Cdc13 mutants and in which Rad9 inhibits this putative exonuclease (Booth et al. 2001). One model is that the PCNA-like 9-1-1 complex loaded onto DNA by Rad24/Rfc possesses intrinsic exonuclease activity. This is plausible because there is evidence that members of the 9-1-1 complex possess 3′ to 5′ exonuclease activity in vitro (Freire et al. 1998; Naureckiene and Holloman 1999; Bessho and Sancar 2000; Lindsey-Boltz et al. 2001). However, the relevance of 3′ to 5′ exonuclease activities in vitro to the generation of ssDNA by 5′ to 3′ nuclease acitivity in vivo at the telomeres of cdc13-1 mutants is unclear (Booth et al. 2001). An alternative model is that the 9-1-1 complex loaded by Rad24 is required to anchor an as yet unidentified 5′ to 3′ exonuclease to DNA (Majka and Burgers 2003).
One nuclease that has the potential to be regulated by RAD24 in cdc13-1 mutants is Exo1. Exo1 is involved in the 5′ to 3′ resection of DSBs (Tsubouchi and Ogawa 2000; Tomita et al. 2003), in mismatch repair (Szankasi and Smith 1995; Tishkoff et al. 1997; Lewis et al. 2002), and in meiotic recombination (Khazanehdari and Borts 2000; Kirkpatrick et al. 2000). Interestingly, Exo1, like Rad24, contributes to generating ssDNA near the telomeres of cdc13-1 mutants (Maringele and Lydall 2002). A model showing how checkpoint proteins Ddc1, Mec3, Rad9, Rad17, and Rad24 might regulate Exo1 or other nuclease activities (ExoX) at uncapped telomeres of cdc13-1 mutants is shown in Figure 1. By carefully characterizing the role of EXO1, RAD9, and RAD24, in regulating ssDNA accumulation and cell cycle arrest of cdc13-1 mutants, we show that although Exo1 has a critical role in generating ssDNA in cdc13-1 mutants, RAD24 appears to regulate a nuclease other than Exo1.
Figure 1.—

A model of checkpoint regulation of Exo1 activity at cdc13-1 telomeres. This model implies that Rad24 and the small Rfc subunits (2, 3, 4, 5) load the checkpoint sliding clamp (Ddc1, Mec3, and Rad17) onto telomeres of cdc13-1 mutants at 36°, and this sliding clamp tethers Exo1 to DNA. Other nuclease activities (ExoX) may also exist. Rad9 inhibits nuclease activity.
MATERIALS AND METHODS
Yeast strains:
All strains used were in the W303 background and unless otherwise indicated contained RAD5, rather than the rad5-535 mutation (Fan et al. 1996). Standard genetic procedures of transformation and tetrad analysis were followed (Adams et al. 1997). Yeast strains were cultured and serial dilutions tested for growth on plates as previously described (Maringele and Lydall 2002).
Growth at different temperatures:
Incubators were set at the temperatures indicated, and in all cases plates at different temperatures were incubated in parallel. The temperatures within incubators oscillated around the set temperature by perhaps 1° or more. Comparatively close temperatures (27.3° and 28.2°) were used because we routinely observe that cdc13-1 rad9Δ mutants form colonies less well than cdc13-1 rad24Δ mutants do at semipermissive temperatures (see Figure 2R).
Figure 2.—

Deletion in EXO1 permits growth of cdc13-1 mutants at high temperatures. (A–S) Small aliquots of fivefold dilution series of the strains indicated, and growing at 20°, were transferred to plates and incubated at the temperatures shown for 3 days before being photographed. Some plates (E, J, O, and S) were incubated for three cycles of 36° for 4 hr followed by incubation at 23° for 4 hr and colonies were then allowed to form at 23° for 6 or 3 days. The relevant genotypes are indicated on the left, and strain numbers are shown in parentheses.
Synchronous cultures, viability, cell cycle position, and ssDNA measurements:
bar1 cdc13-1 cdc15-2 strains were released from G1 arrest at 23° and placed at 36°, and cell viability and cell cycle position were monitored as previously described (Lydall and Weinert 1997a). DNA was isolated from cells, and the fraction of ssDNA was measured by quantitative amplification of ssDNA (QAOS) as previously described (Booth et al. 2001; Jia et al. 2004). In all cases the ssDNA in unknown samples and in standards was measured in triplicate. The primers used to detect ssDNA in the X and Y′ repeats are described in Table 1 and in supplementary material at http://www.genetics.org/supplemental/. The primers used to detect ssDNA at PDA1, YER186C, and YER188W were as previously described (Booth et al. 2001; Jia et al. 2004).
TABLE 1.
Primers used to detect ssDNA in telomeric repeats
| Primer | Telomeric repeat |
Sequence | Type of primer |
|---|---|---|---|
| M 315 | AAGGAGCGCAGCGCCTGTACCA | Tag | |
| M 513 | X-repeat | AAGGAGCGCAGCGCCTGTACCACATTTTAATATCT | Tagging primer |
| M 512 | X-repeat | ATTGAGTGGATAGTAGATGGTGAAAAAGTGGTATAACG | Reverse primer |
| M 510 | X-repeat | TCATTCGGCGGCCCCAAATATTGTATAACTGCCC | Probe |
| M 520 | TGCCCTCGCATCGCTCTCGAA | Tag | |
| M 521 | Y′5000 | TGCCCTCGCATCGCTCTCGAAACAAAGTCAGTGA | Tagging primer |
| M 517 | Y′5000 | GTCCTGGAACGTTGTCACGAAAAAGC | Reverse primer |
| M 516 | Y′5000 | TGCTAGGCCGAACGACAGCTCTACGATGCGTACTT | Probe |
| M 316 | TGCCCTCGCATCGCTCTCACA | Tag | |
| M 243 | Y′600 | TGCCCTCGCATCGCTCTCACAGCCCTATCAG | Tagging primer |
| M 237 | Y′600 | GAGATCAGCTTGCGCTGGGAGTTACC | Reverse primer |
| M 526 | Y′600 | ACAGGAATGCCGTCCAATGCGGCACTTTAGA | Probe |
Yeast genome sequences used for primers are formatted differently; tag sequences (not present in the yeast genome) are in regular type, the yeast sequences in tagging primers and tag are in boldface type, reverse primers are in italics, and probes are underlined.
Microcolony assays:
Yeast strains dividing exponentially at 23° were arrested in G1 with α-factor for 2.5 hr. Arrested cells were briefly sonicated, spread as single cells on plates, and incubated for 15 hr at 36° before being photographed at 200× magnification.
RESULTS
EXO1 contributes to the temperature-sensitive phenotype of cdc13-1 strains:
To identify nucleases responsible for generating ssDNA near the telomeres of cdc13-1 mutants we combined mutations in genes encoding known nucleases and other DNA repair proteins with cdc13-1 and tested the ability of double mutants to grow at a range of temperatures. Removal of gene products that contribute to ssDNA production at telomeres of cdc13-1 mutants may increase the ability of cdc13-1 mutants to grow at semipermissive temperatures because lower levels of ssDNA at telomeres should result in less-pronounced cell cycle arrest. In contrast, removal of gene products that inhibit ssDNA production at telomeres of cdc13-1 mutants should decrease the ability of cdc13-1 mutants to grow at semipermissive temperatures because higher levels of ssDNA at telomeres should result in greater cell cycle arrest. For example, removal of the DNA repair gene YKU70 reduces the maximum permissive temperature (MPT) of cdc13-1 strains (Nugent et al. 1998; Polotnianka et al. 1998). In contrast, deletion of checkpoint genes like RAD9 and RAD24 increases the MPT of cdc13-1 mutants (Weinert and Hartwell 1993), presumably because checkpoint-defective cells can no longer signal that ssDNA is present at telomeres and/or because lower levels of ssDNA are present.
We examined the effect of EXO1 on cdc13-1 strains since EXO1 encodes a 5′ to 3′ exonuclease that contributes to, but is not essential for, cell cycle arrest of cdc13-1 strains (Maringele and Lydall 2002). We also tested the other four nucleases in the Exo1 class encoded by RAD27 (which is the FLAP endonuclease of budding yeast), RAD2, DIN7, and YEN1 (Fiorentini et al. 1997; Johnson et al. 1998). In addition we tested Mre11, Rad50, and Xrs2 (components of the MRX complex), which have been shown to function redundantly with Exo1 in generating ssDNA at DSBs in budding yeast (Tsubouchi and Ogawa 2000). Finally, we tested RAD52, which is required for virtually all homologous recombination pathways in budding yeast (Paques and Haber 1999), and NUC1, which encodes a mitochondrial exonuclease.
Figure 2 shows that EXO1 is unique among the repair genes tested because, like the RAD9 checkpoint gene, it inhibited the growth of cdc13-1 mutants at the semipermissive temperatures of 27.3° and 28.2° (i.e., cdc13-1 exo1Δ mutants formed colonies at 27.3°, whereas cdc13-1 EXO1 strains did not). In contrast, mutations in MRE11, RAD50, XRS2, and RAD27 made cdc13-1 strains grow poorly, such that even at 20° the double mutants grew slowly as has previously been noted (Nugent et al. 1998). These experiments show that the MRX complex genes and RAD27 function to maintain the vitality of cdc13-1 mutants whereas EXO1 functions to decrease the vitality of cdc13-1 mutants. RAD2, NUC1, YEN1, and DIN7 were neutral and did not affect the growth of cdc13-1 mutants. Grandin et al. (2001) have observed that cdc13-1 mec3Δ survivor strains that amplify telomeric repeats can grow at higher temperatures. However, this observation is not relevant to the better growth of cdc13-1 exo1Δ strains because neither these nor any of the other cdc13-1 strains we generated at 20° or 23° had amplified telomeric DNA, to generate survivors (see supplementary Figure 1 at http://www.genetics.org/supplemental/).
In comparison to checkpoint-defective cdc13-1 rad9Δ cells, cdc13-1 exo1Δ mutants were better able to form colonies at 28.2° (Figure 2D) and grew similarly to cdc13-1 rad24Δ cells (Figure 2R). Since rad9Δ and rad24Δ mutants are completely defective in checkpoint-dependent arrest after cdc13-1-induced damage, we have assumed that the differences in growth between the cdc13-1 rad9Δ and cdc13-1 rad24Δ strains at semipermissive temperatures are due to the more rapid accumulation of single-stranded DNA near the telomeres of cdc13-1 rad9Δ mutants (Lydall and Weinert 1995, 1997a). The growth of cdc13-1 exo1Δ mutants is consistent with this hypothesis since EXO1, like RAD24, is important for production of ssDNA near telomeres of cdc13-1 mutants (Maringele and Lydall 2002).
Interestingly, cdc13-1 exo1Δ mutants maintained high viability after three 4-hr cycles at 36° (SFigure 2, E and S) and could form large colonies more rapidly than cdc13-1 EXO1+ RAD+ cells (Figure 2S). This result is consistent with the idea that EXO1-dependent ssDNA, accumulating at the telomeres of cdc13-1 mutants over a 4-hr period at 36°, induces significant growth delay. It is notable that the phenotype of cdc13-1 rad24Δ mutants is different from cdc13-1 exo1Δ mutants in this assay. They retained reasonable viability, similar to cdc13-1 cells, but cdc13-1 rad24Δ colonies were smaller than cdc13-1 exo1Δ colonies after 3 days growth at 23° (Figure 2S).
EXO1 is required for rapid death of cdc13-1 rad9Δ strains:
To test whether Rad9 and Rad24 regulated Exo1, as suggested by Figure 1, we first created combinations of cdc13-1, exo1Δ, rad9Δ, and rad24Δ mutations and measured growth at a range of temperatures (Figure 3). Figure 3B shows that an exo1Δ mutation, like a rad24Δ mutation, improves the growth of cdc13-1 rad9Δ strains at 28.2°, suggesting that Exo1, like Rad24, is required for the accumulation of ssDNA at telomeres in cdc13-1 rad9Δ mutants.
Figure 3.—

Exo1 contributes to death of cdc13-1 rad9Δ mutants at high temperatures. (A–F) A fivefold dilution series of yeast strains indicated, and growing at 20°C, were transferred to plates and incubated at 23° and 28.2° for 2 days. In addition, some plates (C and F) were incubated for three cycles of 36° for 4 hr followed by incubation at 23° for 4 hr and colonies were then allowed to form at 23° for 5 days. (G) Strains DLY1468 (RAD+, squares), DLY1470 (rad9Δ, diamonds) and DLY1472 (rad24Δ, circles), which carried bar1 cdc13-1 cdc15-2 and the other mutations specified, were released from G1 arrest to 36°, and the ability of these cells to form colonies was determined. A single, representative experiment is shown. (H) Strains DLY 1431(exo1Δ RAD+, squares), DLY 1433 (exo1Δ rad9Δ, diamonds), and DLY 1434 (exo1Δ rad24Δ, circles) were treated as in G. A single, representative experiment is shown.
If Exo1 contributes to the production of ssDNA in cdc13-1 mutants, then it may also, like Rad24, contribute to the cell death that occurs when cdc13-1 rad9Δ mutants are cultured at 36° for short periods (Lydall and Weinert 1995). To test this, yeast cells growing on plates were subjected to three 4-hr periods at the restrictive temperature of 36°, separated by 4-hr periods of recovery at the permissive temperature 23°. Colonies were then allowed to form at 23°. Figure 3C shows that cdc13-1 rad9Δ exo1Δ cells formed considerably more colonies than cdc13-1 rad9Δ cells did after this protocol. In fact, cdc13-1 rad9Δ exo1Δ cells formed similar numbers of colonies as cdc13-1 RAD+ EXO1+ cells and slightly more than cdc13-1 rad9Δ rad24Δ cells did, with an estimated viability of 20–100%. Figure 3, D–F, shows that cdc13-1 rad9Δ rad24Δ exo1Δ strains behaved similarly to cdc13-1 rad9Δ exo1Δ strains in this assay.
To confirm that Exo1 has a major role in the cell death that occurs in cdc13-1 rad9Δ mutants we measured the ability of cdc13-1 mutants cultured in liquid at 36° to form colonies when returned to 23°. Figure 3, G and H, confirms that most of the reproductive cell death that occurs in cdc13-1 rad9Δ mutants cultured at 36° does not occur if EXO1 is deleted. Taken together the data in Figures 2 and 3 were consistent with the hypothesis that Exo1 is, like Rad24, responsible for generating ssDNA at the telomeres of cdc13-1 and cdc13-1 rad9Δ mutants and that this ssDNA activates checkpoint control pathways and contributes to cell death.
exo1Δ mutants escape from arrest caused by cdc13-1-induced DNA damage:
Cells with low levels of ssDNA, or with mutated cell signaling molecules, escape cell cycle arrest more readily than cells with high levels of ssDNA, and this phenomenon has been termed adaptation (Toczyski et al. 1997; Lee et al. 1998; Vaze et al. 2002). In asynchronous cultures cdc13-1 exo1Δ mutants arrested cell division less rapidly and completely than cdc13-1 EXO1+ cells did (Maringele and Lydall 2002). To determine whether this impaired cell cycle arrest was due to inefficient arrest, or due to arrest followed by escape from arrest, bar1 and cdc15-2 mutations were used to quantify the fraction of cdc13-1 mutants that had failed to arrest, or escaped arrest, during a single cell cycle (Lydall and Weinert 1997b).
BAR1 encodes a protease that degrades the mating pheromone α-factor. A bar1 mutation allows efficient G1 arrest of cells with comparatively low levels of α-factor. CDC15 is required for mitotic exit. At 36° cdc15-2 mutants arrest cell division in late mitosis with separated chromosomes and an elongated spindle. A population of bar1 cdc13-1 cdc15-2 mutants arrested in G1 with α-factor at 23° and released from G1 arrest by removing the α-factor and culturing at 36° will go through most of the events of a single cell cycle but not reenter G1. Checkpoint-proficient cells arrest at the metaphase/anaphase checkpoint due to cdc13-1-induced damage. Checkpoint-deficient cells do not arrest at metaphase but enter anaphase and arrest at late mitosis due to the cdc15-2 mutation. The fraction of checkpoint-defective cdc13-1 cdc15-2 cells that enter anaphase at 36° can be readily measured by examining the position of nuclear DNA within the population (Lydall and Weinert 1997b).
Figure 4A shows that checkpoint-proficient (RAD+) cdc13-1 cdc15-2 cells start to reach medial nuclear division (or metaphase/anaphase) 80 min after release from G1 arrest and that by 120 min >80% of the cells are arrested at medial nuclear division. Arrest at medial nuclear division is efficient since no cells reach late nuclear division (Figure 4B). As expected, checkpoint-defective strains, containing either rad9Δ or rad24Δ mutations, transiently appeared at medial nuclear division only before entering mitosis (Figure 4A) and accumulated at late nuclear division (the cdc15-2 arrest point, Figure 4B).
Figure 4.—
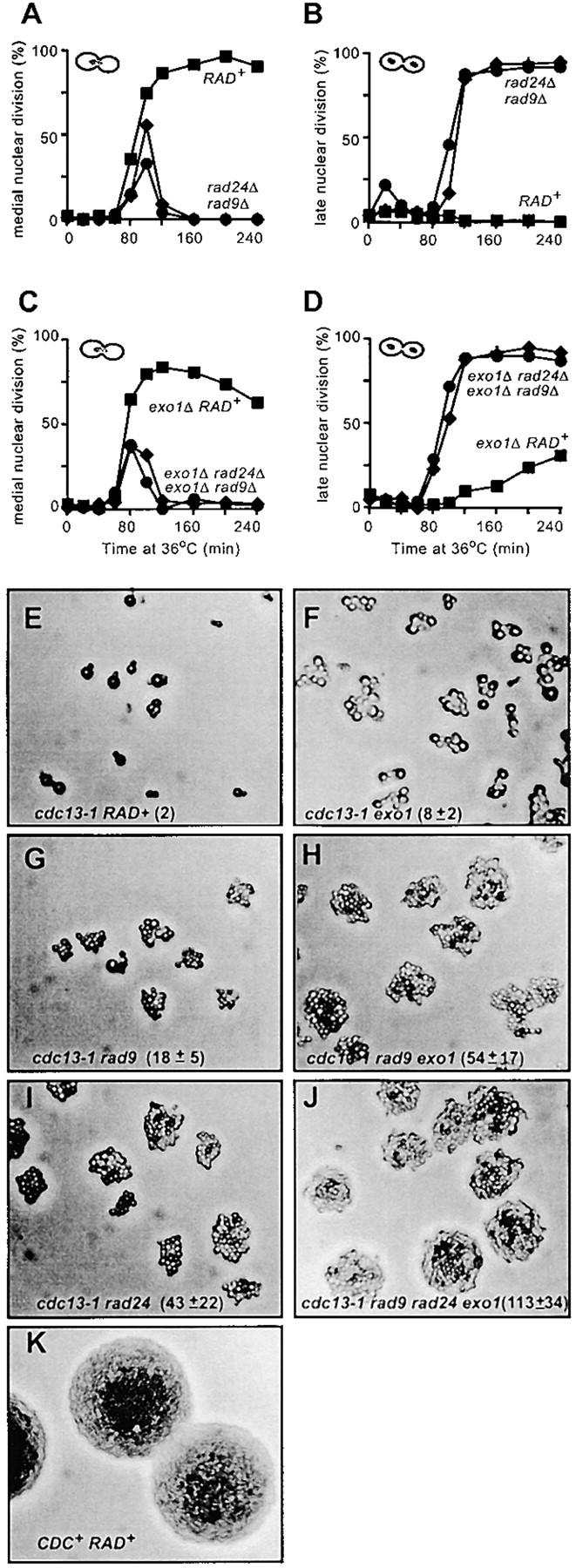
Deletion in EXO1 permits escape of cdc13-1 mutants from arrest at 36°. (A–D) The cell cycle positions of the yeast strains described in Figure 3, G and H, were monitored after staining nuclei with DAPI. A single, representative experiment is shown. (E–K) Yeast strains containing cdc13-1 (DLY1108), cdc13-1 exo1Δ (DLY1296), cdc13-1 rad9Δ (DLY1255), and cdc13-1 rad9Δ exo1Δ (DLY 1692). cdc13-1 rad24Δ (DLY1257), cdc13-1 rad9Δ rad24Δ exo1Δ (DLY1695), and CDC+ (DLY640) were released from G1 arrest and allowed to form microcolonies for 15 hr at 36° before being photographed at 200× magnification. In the W303 genetic background, cdc13-1 mutants form asymmetric dumbbells after long periods of growth at 36° (E), whereas in other genetic backgrounds the dumbbells remain symmetrical. The cell numbers within microcolonies were estimated from the photographs shown and are indicated, along with standard deviations (E–K). Small microcolonies are largely flat, and all cells are within the focal plane; however, as colony size increases, cells begin to grow out of the focal plane and are not visible.
An exo1Δ mutation allows a fraction of cdc13-1 cells arrested at medial nuclear division to escape arrest. Figure 4C shows that cdc13-1 cdc15-2 exo1Δ strains were largely arrested at medial nuclear division after 120 min at 36°, like cdc13-1 RAD+ strains at 36° (compare Figure 4A with 4C). However, cdc13-1 cdc15-2 exo1Δ mutants slowly escaped arrest and accumulated at late nuclear division, such that by 240 min ∼30% of cdc13-1 cdc15-2 exo1Δ cells had reached late nuclear division (the cdc15-2 arrest point, Figure 4D). Virtually no cdc13-1 RAD+ cells reached late nuclear division in this (Figure 3B) or other experiments (Lydall and Weinert 1995). Arrest of cdc13-1 cdc15-2 exo1Δ mutants at medial nuclear division at 36° was completely dependent on RAD9 and RAD24 (Figure 4, C and D), indicating that RAD9- and RAD24-dependent checkpoint pathways are responsible for the initial arrest of cdc13-1 exo1Δ mutants. These single cell cycle experiments suggest that an exo1Δ mutation allows cdc13-1 mutants that have arrested cell division to escape arrest.
If an exo1Δ mutation allows cdc13-1 mutants to escape cell cycle arrest and enter anaphase, then cdc13-1 exo1Δ mutants might be able to complete cell division and to divide. If so then after long periods of growth at 36° cdc13-1 exo1 mutants should form larger microcolonies than cdc13-1 strains do. To test this, we arrested single MATa cdc13-1 cells in G1 using the mating pheromone α-factor and incubated them on plates for 15 hr at 36°. Figure 4, E–J, shows the effect of exo1Δ, rad9Δ, and rad24Δ mutations on the ability of cdc13-1 strains to divide and form microcolonies at 36°. It is clear that an exo1Δ mutation increased the size of cdc13-1 microcolonies. Figure 4E shows that cdc13-1 cells mainly arrested cell division with two buds when cultured at 36°. In contrast, cdc13-1 exo1Δ mutants formed larger microcolonies, with most of the single cells eventually forming colonies of 5–10 cells after 15 hr at 36° (Figure 4F).
It is notable that most individual cdc13-1 exo1Δ cells were larger than the checkpoint defective cdc13-1 rad9Δ cells in microcolonies grown under identical conditions (compare Figure 4F with 4G). This observation is consistent with the existence of a checkpoint that extends each cell cycle of cdc13-1 exo1Δ mutants (see Figure 4, C and D), and that while arrested at this checkpoint cdc13-1 exo1Δ cells enlarge in size before escaping arrest.
Exo1 inhibits the growth of cdc13-1 rad9Δ colonies:
We have previously shown that cdc13-1 rad24Δ and cdc13-1 rad9Δ rad24Δ mutants form larger microcolonies than cdc13-1 rad9Δ cells do at 36° (Lydall and Weinert 1997a). Figure 4, H and I, shows that Exo1, like Rad24, appears to inhibit the division of cdc13-1 rad9Δ cells since cdc13-1 rad9Δ exo1Δ triple mutants form large-sized colonies, like cdc13-1 rad24Δ cells. This is consistent with the hypothesis that RAD24- and EXO1-dependent ssDNA production limits the division of cdc13-1 rad9Δ cells at 36°.
If Rad24 and Exo1 contribute to the same pathway to limit the division of cdc13-1 rad9Δ cells at 36°, then cdc13-1 rad9Δ rad24Δ exo1Δ quadruple mutants should form colonies similar in size to those of cdc13-1 rad9 rad24Δ or cdc13-1 rad9Δ exo1Δ triple mutants. This logic explains why a cdc13-1 rad9Δ rad17Δ rad24Δ mec3Δ mutant forms microcolonies similar in size to those of cdc13-1 rad9Δ rad17Δ and other similar triple mutants (Lydall and Weinert 1997a). However, if Rad24 and Exo1 contribute to independent pathways to limit division, then cdc13-1 rad9Δ rad24Δ exo1Δ quadruple mutants may form larger colonies than the corresponding triple mutants do. Figure 4J shows that cdc13-1 rad9Δ rad24Δ exo1Δ mutants do indeed form larger colonies than the corresponding triple mutants do. This suggests that Rad24 and Exo1 play different roles in limiting the division of cdc13-1 rad9Δ cells at 36° and is consistent with different growth of cdc13-1 exo1Δ vs. cdc13-1 rad24Δ mutants under other conditions, e.g., Figure 2S.
Msh2 does not contribute to cell cycle arrest of cdc13-1 mutants:
Exo1 binds Msh2, a core component of eukaryotic mismatch repair pathways, and plays an important role in mismatch repair (Szankasi and Smith 1995; Tishkoff et al. 1997; Marti et al. 2002). To test whether Msh2, like Exo1, regulated cellular responses to the cdc13-1 defect, we examined the effect of deleting MSH2 (Luhr et al. 1998). Although some early colony growth experiments suggested that Msh2 played a role in responding to the cdc13-1 defect, we concluded after more experiments that Msh2 plays no direct role in recruiting Exo1 to cdc13-1-defective telomeres (see Supplementary Figure 2 at http://www.genetics.org/supplemental/).
Exo1 is required for production of ssDNA at X repeats and single-copy subtelomeric sequences in cdc13-1 mutant cells:
To assess directly the role of Exo1 in generating ssDNA in cdc13-1 mutants we used synchronous cultures to examine ssDNA production at three repetitive loci found on numerous telomeres and three single-copy loci near the right telomere of chromosome V. Previously we showed that in cdc13-1 mutants ssDNA is generated in a telomere-to-centromere direction, with Rad9 inhibiting ssDNA production, and Rad24 being required for ssDNA production, and that ssDNA exists at least 30 kb from the telomere in cdc13-1 rad9 mutants (Lydall and Weinert 1995; Booth et al. 2001; Jia et al. 2004). We have also demonstrated that ssDNA accumulation depends on release from G1 (M. K. Zubko and D. Lydall, unpublished data).
Figure 5A indicates the two major telomere types found in budding yeast. All telomeres contain X repeats. Approximately half of the telomeres possess X repeats directly adjacent to the TG repeats within 1 kb of the chromosome terminus and are termed X-type telomeres. The other class of telomeres contains one or more Y′ repeats between the X and TG repeats, which are termed Y-type telomeres (Pryde et al. 1997). Subtelomeric Y′ repeats are highly dynamic, and their location and number vary between yeast strains (Louis et al. 1994). According to the Saccharomyces Genome Database, chromosome V of the sequenced S288C strain contains a single Y′; single X; and the single-copy genes YER188W, YER186C, and PDA1 at the indicated distances from the chromosome end. The W303 strains used in this study are reasonably closely related to S288C strains (Winzeler et al. 2003), but it is possible that there are different types or numbers of subtelomeric repeats, at this chromosome end, in W303 strains, and also that differences have arisen between strains while undergoing genetic crosses (Horowitz et al. 1984). For these reasons the distances of the loci from the end of chromosome V should be considered approximate and are shown in parentheses.
Figure 5.—
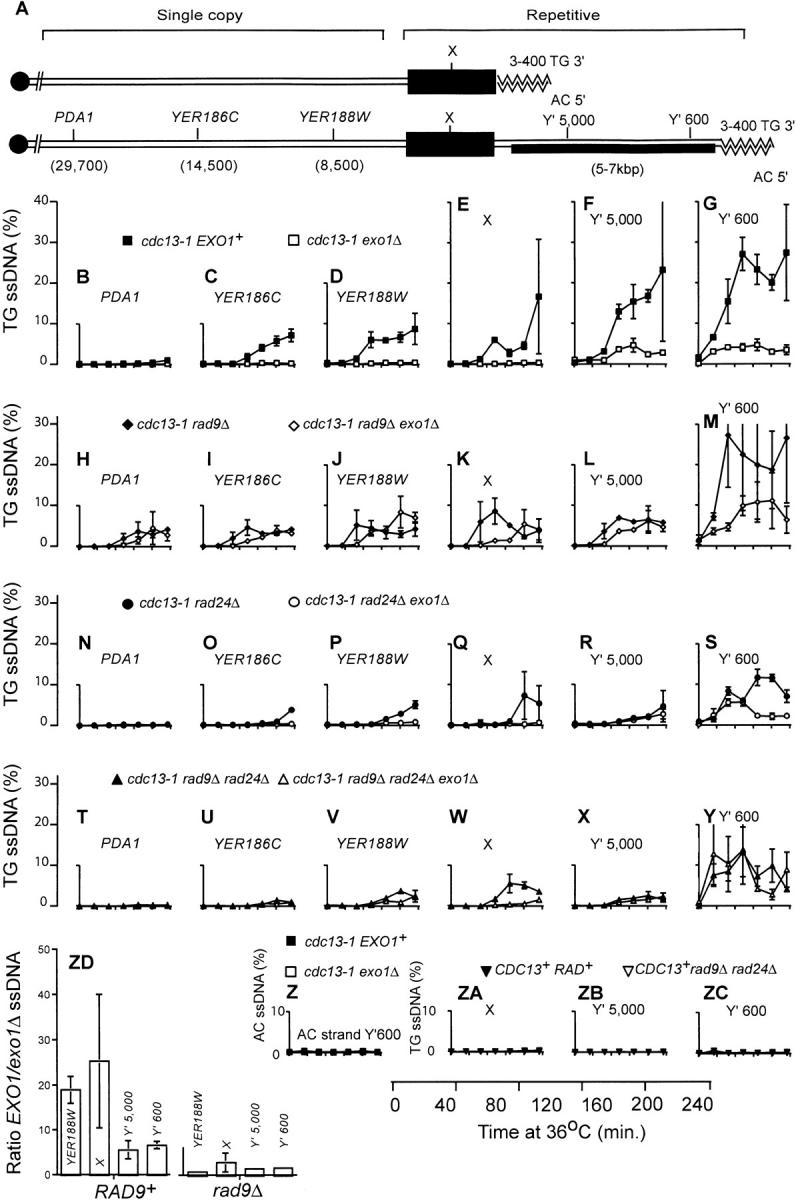
Exo1 is required to generate ssDNA in X and single-copy telomeric sequences in cdc13-1 mutants. (A) A schematic model of the two classes of telomere in budding yeast. One class contains an X repeat, but no Y′ repeats, and the other class contains one or more Y′ repeats, in addition to the X repeats. The bottom half of A is a representation of the right telomere of the sequenced chromosome V present in the Saccharomyces Genome Database. It comprises a 3- to 400-bp TG/AC repeat, a Y′ repeat, a 374-bp X repeat, and the YER188W, YER186C, and PDA1 single-copy loci. Using primers and probes directed to repetitive and single-copy loci we were able to detect the appearance of ssDNA in repetitive elements (at numerous telomeres, including 5R) and also specifically 8500, 14,500 and 29,700 bp from the right telomere of chromosome V. Yeast strains were released from G1 arrest to 36°, and the amount of ssDNA was measured by quantitative amplification of ssDNA (QAOS; Booth et al. 2001). In most cases the data points indicate the average amount of ssDNA measured in two independent strains of identical genotype, with error bars indicating the difference observed between the two strains. When the amount of ssDNA in a genotype had been previously measured (Booth et al. 2001), a single new experiment was performed with error bars representing the standard error of the mean of three independent measurements. In cases where a single strain of a particular genotype was identified, two independent synchronous cultures of that strain were performed and the difference in values between the two experiments is indicated by the error bars. (B–G) Yeast stains containing cdc13-1 (DLY1468 and DLY1469, solid squares) and cdc13-1 exo1Δ (DLY1431 and DLY1432, open squares) mutations. (H–M) Yeast strains containing cdc13-1 rad9Δ (DLY1470 and DLY1471, solid diamonds) and cdc13-1 rad9Δ exo1Δ (DLY1433 and DLY1476, open diamonds) mutations.(N–S) Yeast strains containing cdc13-1 rad24Δ (DLY1472, single experiment, solid circles) and cdc13-1 rad24Δ exo1Δ (DLY1434, duplicate experiments, open circles) mutations.(T–Y) Yeast strains containing cdc13-1 rad9 rad24Δ (DLY1474, single experiment, solid triangles) and cdc13-1rad9Δ rad24Δ exo1Δ (DLY1435 and 1477, open triangles) mutations. (Z) Yeast strains and symbols are as in G, and ssDNA was measured on the AC strand. (ZA–ZC) Yeast strains containing CDC13+ cdc15-2 RAD+ (DLY 1363, single experiment, solid downward-pointing triangles) and CDC13+ rad9Δ rad24Δ cdc15-2 (DLY1414, single experiment, open downward-pointing triangle) mutations. (ZD) A histogram showing the ratio between the amount of ssDNA observed at four telomeric loci in cdc13-1 EXO1+ vs. cdc13-1 exo1Δ strains and corresponding rad9Δ strains. Ratios shown are average ratios of ssDNA in EXO1+ vs. exo1Δ strains at 120-, 160-, 200-, and 240-min time points. The ratios were determined from the data plotted in D–G and J–M. The error bars show the standard error of the mean.
The accumulation of ssDNA in cdc13-1 EXO1+ and cdc13-1 exo1Δ mutants was measured by quantitative amplification of ssDNA (QAOS; Figure 5, B–G). In QAOS a tagging primer anneals to ssDNA but not to dsDNA at low temperature, and then primer extension creates a complementary, tagged, ssDNA-dependent molecule, which is detected by quantitative real-time PCR (Booth et al. 2001). QAOS can accurately measure ssDNA in single-copy yeast genes at levels >0.2%. Figure 5B shows that neither cdc13-1 nor cdc13-1 exo1Δ mutants generated significant levels of ssDNA at the PDA1 locus, 30 kb from the VR telomere, consistent with earlier experiments (Booth et al. 2001; Jia et al. 2004). However, closer to the telomere, at the YER186C locus (14,500 bp from the telomere), it is clear that cdc13-1 EXO1+ cells generated significant levels of ssDNA while cdc13-1 exo1Δ strains did not (Figure 5C). We rarely observe ssDNA at single-copy sequences rising much above 10% and have been unable to determine whether this is due to degradation of ssDNA in cdc13-1 mutants in vivo or during DNA preparation or because telomeres are only partially susceptible to nuclease activity in vivo; see discussion in Booth et al. (2001). At YER186C cdc13-1 EXO1+ strains began to accumulate ssDNA 120 min after releasing a G1 culture to 36° and reached a level of 5–6% ssDNA by 240 min, whereas cdc13-1 exo1Δ strains did not generate ssDNA above 1%. Closer to the telomere, at YER188W and in the repetitive X sequence, a similar pattern to that at YER186C was seen, with very little ssDNA being observed in cdc13-1exo1Δ cells, but significant levels being observed in cdc13-1 EXO1+ cells (Figure 5, D and E). These data demonstrate that Exo1 is essential for generating the vast majority of ssDNA at X repeats and single-copy sequences at telomeres of cdc13-1 mutants at 36°.
At repetitive Y′ repeats, found on approximately half of the telomeres, ssDNA levels increased significantly in cdc13-1exo1Δ cells (Figure 5, F and G; Maringele and Lydall 2002). We measured ssDNA ∼600 and 5000 bp from the telomeric ends of the Y′ repeats. Approximately 600 bp from the end of the telomere, ssDNA reached ∼5% 40 min after releasing G1-arrested strains to 36° and remained close to this level for the remaining 200 min. These levels were lower than those seen in cdc13-1 EXO1+ cells, which reached 20–30%. At the Y′5000 locus ssDNA levels were similar to those at Y′600, but the kinetics of appearance were slower, with ssDNA not accumulating beyond 1% until 80–120 min after release from G1 arrest (Figure 5F). This suggests that a 5′ to 3′ nuclease degrades the telomere beginning at the telomeric end.
If ssDNA in cdc13-1 mutants initiates at the chromosome terminus and extends toward the centromere, as suggested by the data here (Figure 5, B–G) and obtained earlier (Booth et al. 2001), then the termini of X-type telomeres appear to have different properties to the termini of Y′-type telomeres. At X-type telomeres, which contain no Y′ repeats and represent approximately half the telomeres in budding yeast, the X repeats lie within 1 kb of the chromosome end at a similar position to the Y′600 locus of Y′-type telomeres (Figure 5A). Yet, on average, the amount of ssDNA observed at X repeats in cdc13-1 exo1Δ mutants is considerably less than even half the amount of ssDNA observed at the Y′600 or Y′5000 loci. The left part of Figure 5ZD shows the ratio of ssDNA observed in cdc13-1 EXO1 vs. cdc13-1 exo1Δ mutants at YER188W, X, Y′5000 and Y′600 repeats. At YER188W and X repeats EXO1+ cells contain ∼20-fold more ssDNA that exo1Δ mutants do. However, in the Y′ repeats the differential is reduced to ∼6-fold. Thus, Exo1 is more important for generating ssDNA at X and single-copy telomeric sequences than in Y′ repeats of cdc13-1 mutants.
In summary, the data in Figure 5, B–G, are consistent with ssDNA in cdc13-1 mutants being generated by two, or more, 5′ to 3′ exonucleases. Exo1 is critical for the production of ssDNA in X repeats and of single-copy sequences on the right telomere of chromosome V (Maringele and Lydall 2002). A significant amount of ssDNA in Y′ repeats is also dependent on EXO1 but, in addition, an EXO1-independent nuclease(s) appears able to generate ssDNA in the repetitive Y′ sequences.
Rad9 inhibits Exo1 and other nucleases:
Most of the ssDNA and cell death that occur in cdc13-1 rad9Δ mutants are dependent on Rad24 (Lydall and Weinert 1995). Since an exo1Δ mutation rescues the rapid loss of viability observed in cdc13-1 rad9Δ mutants (Figure 3) it seemed likely that Exo1 would be required, like Rad24, for rapid generation of ssDNA in cdc13-1 rad9Δ mutants. To test this directly, we examined ssDNA production in cdc13-1 rad9Δ and cdc13-1 exo1Δ rad9Δ strains (Figure 5, H–M). Surprisingly, the effect of deleting EXO1 on ssDNA production in cdc13-1 rad9Δ mutants was considerably less than the effect of deleting RAD24. cdc13-1 rad9Δ exo1Δ mutants clearly generated significant levels of ssDNA at all telomeric loci tested (Figure 5, H–M), whereas a cdc13-1 rad9Δ rad24Δ strain generated considerably less ssDNA, particularly at loci further from the telomere (Figure 5, T–Y).
At all loci examined the accumulation of ssDNA is marginally slower in cdc13-1 exo1Δ rad9Δ strains than in cdc13-1 rad9Δ strains. At PDA1, a locus that becomes significantly single stranded only in cdc13-1 rad9Δ mutants but not in cdc13-1 RAD+ mutants, cdc13-1 exo1Δ rad9Δ mutants clearly generate significant levels of ssDNA reaching ∼5% (Figure 5, B and H). In cdc13-1 rad9Δ exo1Δ mutants the kinetics of ssDNA accumulation appear to be ∼40–80 min delayed in comparison with cdc13-1 rad9Δ EXO1+ cells. This is apparent at PDA1, YER186C, YER188W, the X, and the Y′5000 loci, where the ssDNA reaches a level >1% ∼40 min later (Figure 5, H–L). Therefore, it appears that Rad9 inhibits EXO1-dependent nuclease activity to some extent.
Comparison between Figure 5, B–E, and 5, H–K, demonstrates that while Exo1 is critical for generation of ssDNA in the X sequences and the single-copy sequences that lie internal to these in cdc13-1strains (Figure 5, B–E), Exo1 is much less important in this process in cdc13-1 rad9Δ strains (Figure 5, H–K). Figure 5ZD illustrates this because it shows the EXO1 independence of ssDNA production in cdc13-1 rad9Δ cells at both repetitive and single-copy sequences (right part of the figure) compared with the corresponding RAD9+ cells. The ssDNA that appears in cdc13-1 rad9Δ exo1Δ strains is clearly Exo1 independent, and it might be generated by a different nuclease, one that is normally inhibited by Rad9. Furthermore, these data suggest that Rad9 contributes to the integrity of some type of barrier or domain structure in cdc13-1 strains that ensures that ssDNA generation in X and single-copy telomeric sequences is largely dependent on Exo1.
Exo1 and Rad24 control nucleases with different properties:
Exo1 and Rad24 are each required for the efficient generation of ssDNA in cdc13-1 mutants (Figure 5; Lydall and Weinert 1995; Booth et al. 2001). A simple model to explain these data is that the Rad24, RFC-like protein (Lydall and Weinert 1997a; Green et al. 2000) is required to load or in some other manner to regulate the activity of Exo1. If so, then cdc13-1 exo1Δ rad24Δ triple mutants should behave like cdc13-1 exo1Δ and cdc13-1 rad24Δ double mutants. SFigure 5, N–S, shows that the patterns of ssDNA accumulation in cdc13-1 rad24Δ and cdc13-1 rad24Δ exo1Δ mutants are different. cdc13-1 rad24Δ exo1Δ mutants behave like cdc13-1 exo1Δ mutants and generate very little ssDNA in the X repeat and the single-copy sequences that lie internal to these. In contrast, cdc13-1 rad24Δ strains generate small but significant amounts of ssDNA at YER186C, YER188W, and the X repeat at late time points (Figure 5, O–Q). One explanation for these data is that Exo1 is essential for ssDNA production in the X and single-copy sequences and that Rad24 is only partially required for the activity of Exo1. However, examination of ssDNA accumulation in cdc13-1 rad9Δ mutants suggests that this simple explanation is insufficient. Rad24 is required for most of the ssDNA produced in cdc13-1 rad9Δ mutants (Figure 5, T–Y; Lydall and Weinert 1995; Booth et al. 2001) whereas Exo1 is not (Figure 5, H–M). This suggests that Rad9 plays a major role in inhibiting a RAD24-dependent, but EXO1-independent, nuclease that generates ssDNA in cdc13-1 mutants.
Exo1- and Rad24-independent nuclease activity in cdc13-1 mutants:
To determine if Exo1 is responsible for the small amount of ssDNA that accumulates near telomeres of cdc13-1 rad9Δ rad24Δ mutants, we examined the ssDNA accumulation in cdc13-1 rad9Δ rad24Δ exo1Δ quadruple mutants (Figure 5, T–Y). We found significant levels of ssDNA appearing in the Y′ sequences of cdc13-1 rad9Δ rad24Δ exo1Δ mutants (Figure 5Y). However, in the X sequences and those internal to the X sequences most but not all of the ssDNA that formed in cdc13-1 rad9Δ rad24Δ mutants was dependent on Exo1 (Figure 5, T–W). The observation that cdc13-1 rad24Δ exo1Δ strains generate significant levels of ssDNA at the Y′600 locus (Figure 5S) also demonstrates that Exo1- and Rad24-independent mechanisms must exist to generate ssDNA in the Y′ sequences of cdc13-1 mutants.
Finally, experimental controls show that no detectable ssDNA accumulates on the strand that ends with the 5′ AC repeats at the telomere, in either cdc13-1 or cdc13-1 exo1Δ mutants (Figure 5Z), and this is consistent with earlier studies on cdc13-1 mutants (Garvik et al. 1995). Figure 5, ZA–ZC, shows that all the ssDNA generated in the Y′ and X sequences is dependent on the cdc13-1 defect.
DISCUSSION
Telomeres contain various types of repetitive DNA structures and a large number of telomere-binding proteins that function to protect the telomere from repair and checkpoint pathways (Blackburn 2001; Cervantes and Lundblad 2002; Lydall 2003; Ferreira et al. 2004; Harrington 2004). In this article we have begun to dissect the interactions that occur between Rad9 and Rad24 checkpoint products and the Exo1 DNA repair protein at unprotected telomeres of budding yeast cdc13-1 mutants.
We establish that Exo1 is unique among products of 10 different DNA repair genes tested because like the Rad9 and Rad24 checkpoint proteins, it inhibits the growth of cdc13-1 mutants at semipermissive temperatures of ∼27°. In contrast, components of the MRX complex, Mre11, Rad50, and Xrs2, along with the FLAP endonuclease Rad27, have opposite properties to Exo1, and they contribute to the vitality of cdc13-1 strains at the permissive temperature of 23°. Other nucleases and DNA repair proteins encoded by RAD52, RAD2, MSH2, NUC1, YEN1, and DIN7 played no detectable role at the telomeres of cdc13-1 mutants because they neither inhibit nor enhance growth of cdc13-1 mutants at 23°.
There is evidence for overlapping functions between the MRX complex and Exo1 in DNA repair (Tsubouchi and Ogawa 2000; Moreau et al. 2001; Lee et al. 2002; Lewis et al. 2002). Indeed, the MRX complex functions as a nuclease to create ssDNA at telomeres created de novo (Diede and Gottschling 2001). It is possible that MRX plays a role in generating ssDNA in cdc13-1 mutants and represents ExoX or ExoY in Figure 6, but we have been unable to test this directly because cdc13-1 mrxΔ double mutants grow extremely poorly even at 20° (Figure 2). It is likely that the protective role of MRX at telomeres (Nugent et al. 1998; Maringele and Lydall 2002), or its role in recruiting telomerase (Tsukamoto et al. 2001), explains the poor growth of cdc13-1 mrxΔ double mutants. It is clear that Exo1 has very different properties to the components of the MRX in the context of the cdc13-1- and yku70Δ-induced telomere damage complex (this work and Nugent et al. 1998; Maringele and Lydall 2002).
Figure 6.—

A model for the interaction between nucleases and checkpoint proteins at cdc13-1-induced damage. According to this model, ssDNA formation begins at the chromosome end (where Cdc13p binds). Exo1, ExoX (which is Rad17, Rad24, Mec3, and Ddc1 dependent), and ExoY contribute to ssDNA production. Rad9 inhibits exonuclease activity by contributing to a nuclease progression barrier centered on X repeats. Exo1 and ExoX are both critical for generating ssDNA beyond the Rad9-dependent barrier. However, if the barrier is missing, due to the absence of Rad9, then ExoX becomes more important than Exo1 for generating ssDNA at single-copy sequences near telomeres. ExoY generates ssDNA in the absence of Exo1 and ExoX.
Exo1 is a mismatch repair-associated exonuclease, and some studies in mammalian cells suggest that mismatch repair pathways contribute to DNA damage checkpoint pathways (Davis et al. 1998; Yan et al. 2001). Indeed, recent experiments show that human Msh2 binds to human checkpoint PI3 type kinase, ATR (orthologue of budding yeast Mec1; Wang and Qin 2003). However, other studies have questioned the role of mismatch repair in checkpoint control (Aquilina et al. 1999; Strathdee et al. 2001). Furthermore, mismatch repair pathways regulate the growth of cells growing without telomerase (Rizki and Lundblad 2001). Our analyses lead us to conclude that Msh2, a core component of the mismatch repair machinery, plays no essential role in either recruiting either Exo1 or other nucleases or signaling cell cycle arrest, in cdc13-1 mutants.
Analysis of ssDNA production in cdc13-1 yeast strains containing combinations of exo1Δ rad9Δ and rad24Δ mutations shows that regulation of ssDNA production by nucleases and checkpoint pathways is complex. Our data support a model in which at least three independent nucleases attack the telomeres of cdc13-1 mutants at 36° (Figure 6). Exo1 is the primary nuclease active at the telomeres of cdc13-1 mutants (this work) and at telomeres of yku70Δ mutants at 37° (Maringele and Lydall 2002). ExoX and ExoY are as yet unidentified and play a lesser role in generating ssDNA. Their properties are described in Table 2 and below.
TABLE 2.
Nuclease activities at unprotected telomeres
| Important for telomeric ssDNA production in
|
||||
|---|---|---|---|---|
| Single-copy sequences in cdc13-1 cells | Single-copy sequences in cdc13-1 rad9Δ cells |
Y′ repeats in cdc13-1 cells | Y′ repeats in yku70Δ cells | |
| Exo1 | Yes | No | Partially | Yes |
| ExoX (Rad24-dependent) | Yes | Yes | Partially | No |
| ExoY | ? | ? | ? | |
We consider Exo1 the primary nuclease for generating ssDNA in cdc13-1 mutants because Exo1 is critical for generating ssDNA in X repeats and single-copy sequences internal to X. In addition, Exo1 is important for generating high levels of ssDNA in the Y′ repeats of cdc13-1 mutants. However, when Rad9 is missing, other nucleases, in particular a Rad24-dependent nuclease, designated ExoX, can generate ssDNA in single-copy sequences of cdc13-1 mutants. ExoX can be proposed because cdc13-1 rad9Δ rad24Δ strains (deficient in ExoX, due to the absence of Rad24, but proficient in Exo1) generate very little ssDNA internal to the X repeats, whereas cdc13-1 rad9Δ exo1Δ strains (deficient in Exo1 but proficient in ExoX) are able to generate high levels of ssDNA at these loci. The putative Rad24-dependent ExoX is, like Exo1, important for generating maximum levels of ssDNA in the Y′ repeats of cdc13-1 mutants. ExoY is another putative nuclease that generates ssDNA near the telomeres of cdc13-1 exo1Δ rad24Δ mutants. Alternatively, ExoY could be the same nuclease as ExoX but with an activity partially dependent on Rad24.
ExoX is not yet identified. ExoX may be the intrinsic nuclease activity of the checkpoint sliding clamp, Rad17, Mec3, and Ddc1 or may be another, so far unidentified 5′ to 3′ nuclease tethered to DNA by this sliding clamp. Alternatively, ExoX and/or ExoY may be some other combination of repair activities, e.g., combined helicase and endonuclease activities, or MRX activity. Further experiments will be necessary to define ExoX.
Our experiments show that Exo1 is critical for generating ssDNA at X repeats and single-copy subtelomeric sequences when Rad9 is present in cdc13-1 mutants, but Exo1 is less critical in Y′ repeats or in X repeats when Rad9 is missing (Table 2). Interestingly, Pryde and Louis have shown that there is a domain of transcriptional repression centered on the X repeat at telomeres; i.e., Y′ repeats are less transcriptionally silenced than X repeats (Pryde and Louis 1999). It seems plausible that this domain of transcriptional repression might share properties with a nuclease inhibition domain since it is located in a similar position. Other experiments suggest that Rad9 inhibits nuclease activity in cdc13-1 mutants by both kinase-dependent (Rad53 and Mec1) and kinase-independent mechanisms (Jia et al. 2004). Further experiments will be required to elucidate how Rad9 inhibits nucleases at uncapped telomeres.
We began our studies with the assumption that cdc13-1 rad9Δ mutants became rapidly inviable at 36° because of the rapid accumulation of ssDNA. exo1Δ and rad24Δ mutations each suppress the rapid loss of viabilty observed in cdc13-1 rad9Δ mutants, but cdc13-1 rad9Δ exo1Δ mutants, in contrast to cdc13-1 rad9Δ rad24Δ mutants, still generate high levels of ssDNA. This puzzle may be explained if Exo1 contributes directly to the loss of viability of cdc13-1 rad9Δ cells through enzymatic activities other than its 5′ to 3′ exonuclease activity. For example, Exo1 possesses FLAP endonuclease activity (Lee and Wilson 1999; Tran et al. 2002), and this activity could be responsible for forming cytotoxic lesions in cdc13-1 rad9Δ mutants. Other recent experiments show that Mec1 and Rad53 also contribute to the loss of viability of cdc13-1 rad9Δ strains and yet, like Exo1, they do not greatly affect the rate of accumulation of ssDNA (Jia et al. 2004).
Finally, analysis of cell cycle arrest in cdc13-1 exo1Δ mutants allows us to address the role of telomeric ssDNA in cell cycle arrest. Our data suggest that cdc13-1exo1Δ strains generate ssDNA in Y′ repeats, but not internally to these. After 4 hr incubation ∼30% of cdc13-1exo1Δ strains escape arrest without apparently removing or “repairing” the ssDNA (Figure 5, F and G). We assume, but have no direct evidence, that these cdc13-1 cells dividing in the presence of ssDNA at telomeres have downregulated checkpoint signal transduction pathways, as has been described at double-strand breaks (Leroy et al. 2003). The amount of ssDNA present in cdc13-1 exo1Δ strains can be estimated at ∼15 kb, on the basis that there are ∼40 Y′ repeats in G2 cells (64 telomeres), each with an average size of 6 kb, and 64 telomeric TG repeats with an average size of 350 bp, and 5% (13 kb) of this 260-kb sequence is single stranded. This value is of a similar order to the amount of ssDNA required to arrest cell division in cells with a single unrepaired DSB (between 4.6 and 25 kb; Vaze et al. 2002) or with stalled replication forks (Sogo et al. 2002). This comparison argues that exposed telomeric ssDNA is as efficient as ssDNA generated at DSBs elsewhere in the genome in activating checkpoint-dependent arrest.
Acknowledgments
We thank all members of our lab and Elspeth Stewart for comments on the manuscript. We thank Francis Fabre, Franz Klein, Wilfried Kramer, Alain Nicolas, David Shore, Lorraine Symington, and Hideo Tsubouchi for providing strains and/or plasmids and Marco Foiani and Jim Haber for communicating information prior to publication. We particularly thank Ed Louis for providing sequences and alignments for X and Y′ repeats. We thank Laura Maringele and Richard Blankley for providing type I and type II survivors. Finally we thank anonymous reviewers for helpful input and advice. This work was supported by the award of a Wellcome Senior Research Fellowship in Basic Biomedical Science to D.L.
References
- Adams, A., D. E. Gottshcling, C. A. Kaiser and T. Stearns, 1997 Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Aquilina, G., M. Crescenzi and M. Bignami, 1999. Mismatch repair, G(2)/M cell cycle arrest and lethality after DNA damage. Carcinogenesis 20: 2317–2326. [DOI] [PubMed] [Google Scholar]
- Bessho, T., and A. Sancar, 2000. Human DNA damage checkpoint protein hRAD9 is a 3′ to 5′ exonuclease. J. Biol. Chem. 275: 7451–7454. [DOI] [PubMed] [Google Scholar]
- Blackburn, E. H., 2001. Switching and signaling at the telomere. Cell 106: 661–673. [DOI] [PubMed] [Google Scholar]
- Booth, C., E. Griffith, G. Brady and D. Lydall, 2001. Quantitative amplification of single-stranded DNA (QAOS) demonstrates that cdc13-1 mutants generate ssDNA in a telomere to centromere direction. Nucleic Acids Res. 29: 4414–4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes, R. B., and V. Lundblad, 2002. Mechanisms of chromosome-end protection. Curr. Opin. Cell Biol. 14: 351–356. [DOI] [PubMed] [Google Scholar]
- Davis, T. W., C. Wilson-Van Patten, M. Meyers, K. A. Kunugi, S. Cuthill et al., 1998. Defective expression of the DNA mismatch repair protein, MLH1, alters G2-M cell cycle checkpoint arrest following ionizing radiation. Cancer Res. 58: 767–778. [PubMed] [Google Scholar]
- Diede, S. J., and D. E. Gottschling, 2001. Exonuclease activity is required for sequence addition and Cdc13p loading at a de novo telomere. Curr. Biol. 11: 1336–1340. [DOI] [PubMed] [Google Scholar]
- Ellison, V., and B. Stillman, 2003. Biochemical characterization of DNA damage checkpoint complexes: clamp loader and clamp complexes with specificity for 5′ recessed DNA. PLoS Biol. 1: E33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, H. Y., K. K. Cheng and H. L. Klein, 1996. Mutations in the RNA polymerase II transcription machinery suppress the hyperrecombination mutant hpr1 delta of Saccharomyces cerevisiae. Genetics 142: 749–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira, M. G., K. M. Miller and J. P. Cooper, 2004. Indecent exposure: when telomeres become uncapped. Mol. Cell 13: 7–18. [DOI] [PubMed] [Google Scholar]
- Fiorentini, P., K. N. Huang, D. X. Tishkoff, R. D. Kolodner and L. S. Symington, 1997. Exonuclease I of Saccharomyces cerevisiae functions in mitotic recombination in vivo and in vitro. Mol. Cell. Biol. 17: 2764–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freire, R., J. R. Murguia, M. Tarsounas, N. F. Lowndes, P. B. Moens et al., 1998. Human and mouse homologs of Schizosaccharomyces pombe rad1(+) and Saccharomyces cerevisiae RAD17: linkage to checkpoint control and mammalian meiosis. Genes Dev. 12: 2560–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garvik, B., M. Carson and L. Hartwell, 1995. Single-stranded DNA arising at telomeres in cdc13 mutants may constitute a specific signal for the RAD9 checkpoint. Mol. Cell. Biol. 15: 6128–6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert, C. S., C. M. Green and N. F. Lowndes, 2001. Budding yeast Rad9 is an ATP-dependent Rad53 activating machine. Mol. Cell 8: 129–136. [DOI] [PubMed] [Google Scholar]
- Grandin, N., C. Damon and M. Charbonneau, 2001. Cdc13 prevents telomere uncapping and Rad50-dependent homologous recombination. EMBO J. 20: 6127–6139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green, C. M., H. Erdjument-Bromage, P. Tempst and N. F. Lowndes, 2000. A novel Rad24 checkpoint protein complex closely related to replication factor C. Curr. Biol. 10: 39–42. [DOI] [PubMed] [Google Scholar]
- Harrington, L., 2004. Those dam-aged telomeres! Curr. Opin. Genet. Dev. 14: 22–28. [DOI] [PubMed] [Google Scholar]
- Hartwell, L. H., and T. A. Weinert, 1989. Checkpoints: controls that ensure the order of cell cycle events. Science 246: 629–634. [DOI] [PubMed] [Google Scholar]
- Horowitz, H., P. Thorburn and J. E. Haber, 1984. Rearrangements of highly polymorphic regions near telomeres of Saccharomyces cerevisiae. Mol. Cell. Biol. 4: 2509–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, L. C., K. C. Clarkin and G. M. Wahl, 1996. Sensitivity and selectivity of the DNA damage sensor responsible for activating p53-dependent G1 arrest. Proc. Natl. Acad. Sci. USA 93: 4827–4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia, X., T. Weinert and D. Lydall, 2004. Mec1 and Rad53 inhibit formation of single-stranded DNA at telomeres of Saccharomyces cerevisiae cdc13-1 mutants. Genetics 166: 753–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, R. E., G. K. Kovvali, L. Prakash and S. Prakash, 1998. Role of yeast Rth1 nuclease and its homologs in mutation avoidance, DNA repair, and DNA replication. Curr. Genet. 34: 21–29. [DOI] [PubMed] [Google Scholar]
- Khazanehdari, K. A., and R. H. Borts, 2000. EXO1 and MSH4 differentially affect crossing-over and segregation. Chromosoma 109: 94–102. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick, D. T., J. R. Ferguson, T. D. Petes and L. S. Symington, 2000. Decreased meiotic intergenic recombination and increased meiosis I nondisjunction in exo1 mutants of Saccharomyces cerevisiae. Genetics 156: 1549–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo, T., T. Wakayama, T. Naiki, K. Matsumoto and K. Sugimoto, 2001. Recruitment of Mec1 and Ddc1 checkpoint proteins to double-strand breaks through distinct mechanisms. Science 294: 867–870. [DOI] [PubMed] [Google Scholar]
- Lee, B. I., and D. M. Wilson III, 1999. The RAD2 domain of human exonuclease 1 exhibits 5′ to 3′ exonuclease and flap structure-specific endonuclease activities. J. Biol. Chem. 274: 37763–37769. [DOI] [PubMed] [Google Scholar]
- Lee, S. E., J. K. Moore, A. Holmes, K. Umezu, R. D. Kolodner et al., 1998. Saccharomyces Ku70, Mre11/Rad50, and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell 94: 399–409. [DOI] [PubMed] [Google Scholar]
- Lee, S. E., D. A. Bressan, J. H. J. Petrini and J. E. Haber, 2002. Complementation between N-terminal Saccharomyces cerevisiae mre11 alleles in DNA repair and telomere length maintenance. DNA Repair 1: 27–40. [DOI] [PubMed] [Google Scholar]
- Leroy, C., S. E. Lee, M. B. Vaze, F. Ochsenbien, R. Guerois et al., 2003. PP2C phosphatases Ptc2 and Ptc3 are required for DNA checkpoint inactivation after a double-strand break. Mol. Cell 11: 827–835. [DOI] [PubMed] [Google Scholar]
- Lewis, L. K., G. Karthikeyan, J. W. Westmoreland and M. A. Resnick, 2002. Differential suppression of DNA repair deficiencies of yeast rad50, mre11 and xrs2 mutants by EXO1 and TLC1 (the RNA component of telomerase). Genetics 160: 49–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey-Boltz, L. A., V. P. Bermudez, J. Hurwitz and A. Sancar, 2001. Purification and characterization of human DNA damage checkpoint Rad complexes. Proc. Natl. Acad. Sci. USA 98: 11236–11241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis, E. J., E. S. Naumova, A. Lee, G. Naumov and J. E. Haber, 1994. The chromosome end in yeast: its mosaic nature and influence on recombinational dynamics. Genetics 136: 789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowndes, N. F., and J. R. Murguia, 2000. Sensing and responding to DNA damage. Curr. Opin. Genet. Dev. 10: 17–25. [DOI] [PubMed] [Google Scholar]
- Luhr, B., J. Scheller, P. Meyer and W. Kramer, 1998. Analysis of in vivo correction of defined mismatches in the DNA mismatch repair mutants msh2, msh3 and msh6 of Saccharomyces cerevisiae. Mol. Gen. Genet. 257: 362–367. [DOI] [PubMed] [Google Scholar]
- Lydall, D., 2003. Hiding at the ends of yeast chromosomes: telomeres, nucleases and checkpoint pathways. J. Cell Sci. 116: 4057–4065. [DOI] [PubMed] [Google Scholar]
- Lydall, D., and T. Weinert, 1995. Yeast checkpoint genes in DNA damage processing: implications for repair and arrest. Science 270: 1488–1491. [DOI] [PubMed] [Google Scholar]
- Lydall, D., and T. Weinert, 1997. a G2/M checkpoint genes of Saccharomyces cerevisiae: further evidence for roles in DNA replication and/or repair. Mol. Gen. Genet. 256: 638–651. [DOI] [PubMed] [Google Scholar]
- Lydall, D., and T. Weinert, 1997. b Use of cdc13–1-induced DNA damage to study effects of checkpoint genes on DNA damage processing. Methods Enzymol. 283: 410–424. [DOI] [PubMed] [Google Scholar]
- Majka, J., and P. M. Burgers, 2003. Yeast Rad17/Mec3/Ddc1: a sliding clamp for the DNA damage checkpoint. Proc. Natl. Acad. Sci. USA 100: 2249–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maringele, L., and D. Lydall, 2002. EXO1-dependent single-stranded DNA at telomeres activates subsets of DNA damage and spindle checkpoint pathways in budding yeast yku70Δ mutants. Genes Dev. 16: 1919–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti, T. M., C. Kunz and O. Fleck, 2002. DNA mismatch repair and mutation avoidance pathways. J. Cell. Physiol. 191: 28–41. [DOI] [PubMed] [Google Scholar]
- Melo, J. A., J. Cohen and D. P. Toczyski, 2001. Two checkpoint complexes are independently recruited to sites of DNA damage in vivo. Genes Dev. 15: 2809–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau, S., E. A. Morgan and L. S. Symington, 2001. Overlapping functions of the Saccharomyces cerevisiae Mre11, Exo1 and Rad27 nucleases in DNA metabolism. Genetics 159: 1423–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naureckiene, S., and W. K. Holloman, 1999. DNA hydrolytic activity associated with the Ustilago maydis REC1 gene product analyzed on hairpin oligonucleotide substrates. Biochemistry 38: 14379–14386. [DOI] [PubMed] [Google Scholar]
- Nugent, C. I., G. Bosco, L. O. Ross, S. K. Evans, A. P. Salinger et al., 1998. Telomere maintenance is dependent on activities required for end repair of double-strand breaks. Curr. Biol. 8: 657–660. [DOI] [PubMed] [Google Scholar]
- Nugent, C. I., T. R. Hughes, N. F. Lue and V. Lundblad, 1996. Cdc13p: a single-strand telomeric DNA-binding protein with a dual role in yeast telomere maintenance. Science 274: 249–252. [DOI] [PubMed] [Google Scholar]
- Nyberg, K. A., R. J. Michelson, C. W. Putnam and T. A. Weinert, 2002. Toward maintaining the genome: DNA damage and replication checkpoints. Annu. Rev. Genet. 36: 617–656. [DOI] [PubMed] [Google Scholar]
- Paques, F., and J. E. Haber, 1999. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 63: 349–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polotnianka, R. M., J. Li and A. J. Lustig, 1998. The yeast Ku heterodimer is essential for protection of the telomere against nucleolytic and recombinational activities. Curr. Biol. 8: 831–834. [DOI] [PubMed] [Google Scholar]
- Pryde, F. E., and E. J. Louis, 1999. Limitations of silencing at native yeast telomeres. EMBO J. 18: 2538–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryde, F. E., H. C. Gorham and E. J. Louis, 1997. Chromosome ends: all the same under their caps. Curr. Opin. Genet. Dev. 7: 822–828. [DOI] [PubMed] [Google Scholar]
- Rizki, A., and V. Lundblad, 2001. Defects in mismatch repair promote telomerase-independent proliferation. Nature 411: 713–716. [DOI] [PubMed] [Google Scholar]
- Rouse, J., and S. P. Jackson, 2002. Lcd1p recruits Mec1p to DNA lesions in vitro and in vivo. Mol. Cell 9: 857–869. [DOI] [PubMed] [Google Scholar]
- Sogo, J. M., M. Lopes and M. Foiani, 2002. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 297: 599–602. [DOI] [PubMed] [Google Scholar]
- Strathdee, G., O. J. Sansom, A. Sim, A. R. Clarke and R. Brown, 2001. A role for mismatch repair in control of DNA ploidy following DNA damage. Oncogene 20: 1923–1927. [DOI] [PubMed] [Google Scholar]
- Szankasi, P., and G. R. Smith, 1995. A role for exonuclease I from S. pombe in mutation avoidance and mismatch correction. Science 267: 1166–1169. [DOI] [PubMed] [Google Scholar]
- Tishkoff, D. X., A. L. Boerger, P. Bertrand, N. Filosi, G. M. Gaida et al., 1997. Identification and characterization of Saccharomyces cerevisiae EXO1, a gene encoding an exonuclease that interacts with MSH2. Proc. Natl. Acad. Sci. USA 94: 7487–7492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toczyski, D. P., D. J. Galgoczy and L. H. Hartwell, 1997. CDC5 and CKII control adaptation to the yeast DNA damage checkpoint. Cell 90: 1097–1106. [DOI] [PubMed] [Google Scholar]
- Tomita, K., A. Matsuura, T. Caspari, A. M. Carr, Y. Akamatsu et al., 2003. Competition between the Rad50 complex and the Ku heterodimer reveals a role for Exo1 in processing double-strand breaks but not telomeres. Mol. Cell. Biol. 23: 5186–5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran, P. T., N. Erdenez, S. Dudley and R. M. Liskay, 2002. Characterization of nuclease-dependent functions of Exo1p in Saccharomyces cerevisiae. DNA Repair 1: 895–912. [DOI] [PubMed] [Google Scholar]
- Tsubouchi, H., and H. Ogawa, 2000. Exo1 roles for repair of DNA double-strand breaks and meiotic crossing over in Saccharomyces cerevisiae. Mol. Biol. Cell 11: 2221–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto, Y., A. K. Taggart and V. A. Zakian, 2001. The role of the Mre11-Rad50-Xrs2 complex in telomerase-mediated lengthening of Saccharomyces cerevisiae telomeres. Curr. Biol. 11: 1328–1335. [DOI] [PubMed] [Google Scholar]
- Usui, T., H. Ogawa and J. H. Petrini, 2001. A DNA damage response pathway controlled by Tel1 and the Mre11 complex. Mol. Cell 7: 1255–1266. [DOI] [PubMed] [Google Scholar]
- Vaze, M. B., A. Pellicioli, S. E. Lee, G. Ira, G. Liberi et al., 2002. Recovery from checkpoint-mediated arrest after repair of a double-strand break requires Srs2 helicase. Mol. Cell 10: 373–385. [DOI] [PubMed] [Google Scholar]
- Wang, Y., and J. Qin, 2003. MSH2 and ATR form a signaling module and regulate two branches of the damage response to DNA methylation. Proc. Natl. Acad. Sci. USA 100: 15387–15392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert, T. A., and L. H. Hartwell, 1993. Cell cycle arrest of cdc mutants and specificity of the RAD9 checkpoint. Genetics 134: 63–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzeler, E. A., C. I. Castillo-Davis, G. Oshiro, D. Liang, D. R. Richards et al., 2003. Genetic diversity in yeast assessed with whole-genome oligonucleotide arrays. Genetics 163: 79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, T., J. E. Schupp, H. S. Hwang, M. W. Wagner, S. E. Berry et al., 2001. Loss of DNA mismatch repair imparts defective cdc2 signaling and G(2) arrest responses without altering survival after ionizing radiation. Cancer Res. 61: 8290–8297. [PubMed] [Google Scholar]
- Zhou, B. B., and S. J. Elledge, 2000. The DNA damage response: putting checkpoints in perspective. Nature 408: 433–439. [DOI] [PubMed] [Google Scholar]
- Zou, L., and S. J. Elledge, 2003. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300: 1542–1548. [DOI] [PubMed] [Google Scholar]
- Zou, L., D. Cortez and S. J. Elledge, 2002. Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin. Genes Dev. 16: 198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]