

Abstract
We studied microsatellite frequency and distribution in 21.76-Mb random genomic sequences, 0.67-Mb BAC sequences from the Z chromosome, and 6.3-Mb EST sequences of Bombyx mori. We mined microsatellites of ≥15 bases of mononucleotide repeats and ≥5 repeat units of other classes of repeats. We estimated that microsatellites account for 0.31% of the genome of B. mori. Microsatellite tracts of A, AT, and ATT were the most abundant whereas their number drastically decreased as the length of the repeat motif increased. In general, tri- and hexanucleotide repeats were overrepresented in the transcribed sequences except TAA, GTA, and TGA, which were in excess in genomic sequences. The Z chromosome sequences contained shorter repeat types than the rest of the chromosomes in addition to a higher abundance of AT-rich repeats. Our results showed that base composition of the flanking sequence has an influence on the origin and evolution of microsatellites. Transitions/transversions were high in microsatellites of ESTs, whereas the genomic sequence had an equal number of substitutions and indels. The average heterozygosity value for 23 polymorphic microsatellite loci surveyed in 13 diverse silkmoth strains having 2–14 alleles was 0.54. Only 36 (18.2%) of 198 microsatellite loci were polymorphic between the two divergent silkworm populations and 10 (5%) loci revealed null alleles. The microsatellite map generated using these polymorphic markers resulted in 8 linkage groups. B. mori microsatellite loci were the most conserved in its immediate ancestor, B. mandarina, followed by the wild saturniid silkmoth, Antheraea assama.
MICROSATELLITES or simple sequence repeats (SSRs), tandemly repeated units of one to six nucleotides, are abundant in prokaryotic and eukaryotic genomes (Weber 1990; Field and Wills 1996). They are ubiquitously distributed in the genome, both in protein-coding and in noncoding regions (Toth et al. 2000). The advent of polymerase chain reaction (PCR) and the availability of high-throughput automated sequencers have increased the use, detectability, and popularity of microsatellite markers, which have become a highly informative and versatile class of genetic markers (Litt and Luty 1989; Tautz 1989; Weber and May 1989; Schlötterer 2004).
Information about the distribution and variability of microsatellite sequences in the genome of a species can elucidate its genetic history from the standpoint of evolution and artificial selection. Population and evolutionary studies in humans and Drosophila have shown that highly polymorphic microsatellite markers can provide information on the differentiation of populations and can detect recent selective sweeps and bottleneck events (Di Rienzo et al. 1994; Slatkin 1995; Schlötterer et al. 1997). On the other hand, more stable SSR markers with lower variability can be used to reconstruct more ancient evolutionary events (Meyer et al. 1995). Since mutation rates at microsatellite sequences vary substantially between species and between loci (reviewed in Schlötterer 2000), it is important to investigate and account for factors influencing microsatellite variability while using SSR markers. These include the structure and length of SSRs (Brinkmann et al. 1998; Schlötterer 1998; Streelman et al. 1998). In a variety of organisms, it has been demonstrated that microsatellite mutation rates are positively correlated with repeat number (Wierdl et al. 1997; Schlötterer et al. 1998). Initial studies of humans reported a higher mutation rate of tetranucleotide repeats (Weber and Wong 1993), whereas a later study that compared microsatellite variability in different human populations found strong evidence for an inverse correlation of microsatellite repeat unit length and mutation rate (Chakraborty et al. 1997). More work in different genomes is required to answer this open issue and to find out common patterns, if any. It has been shown that SSRs are less abundant in exons than in noncoding regions and that different taxa exhibit relative abundance of different SSR types (Toth et al. 2000; Morgante et al. 2002). The silkworm, Bombyx mori is an insect of agronomic importance and a lepidopteran molecular model (Nagaraju et al. 2000). Development of molecular markers is important in silkworm for construction of linkage maps, fingerprinting of strains for breeding purposes, and marker-assisted selection. In earlier studies, fingerprinting analyses of silkworm strains have been carried out using RAPDs (Nagaraja and Nagaraju 1995); a heterologous minisatellite probe, Bkm(2)8 (Nagaraju et al. 1995; Sharma et al. 1999); inter-(I)SSR-PCR (Reddy et al. 1999b); and microsatellite loci (Reddy et al. 1999a). Genetic linkage maps of silkworm have been constructed using RAPDs (Promboon et al. 1995; Yasukochi 1998), RFLPs (Shi et al. 1995; Kadono-Okuda et al. 2002), AFLPs (Tan et al. 2001), and SSRs and ISSRs (our unpublished data). Considering the advantages offered by SSR loci, we started an SSR discovery program in the silkworm genome. In the previous study, we analyzed 28 microsatellite loci cloned and characterized from a B. mori subgenomic library (Reddy et al. 1999a). Given the task of constructing a high-density linkage map for silkworm, the need for microsatellites for marker-assisted selection, and the high cost of developing and sequencing new genomic libraries for microsatellite discovery, we sought an alternative means of increasing our collection of these highly informative markers. Hence, in this study we used an in silico approach to characterize the distribution and frequency of different types of microsatellite motifs in the silkworm genome by analyzing the sequences derived from whole-genome shotgun (WGS) sequencing (Mita et al. 2004), BACs from the Z chromosome (Koike et al. 2003), and silkworm expressed sequence tags (ESTs; Mita et al. 2003). We also analyzed microsatellite flanking sequences for their sequence content and mutations in different classes of microsatellite repeats. In addition, we designed primers for 198 SSR flanking sequences and tested for their polymorphism in two divergent parental strains, Nistari and NB4D2, which show contrasting characters for various visible traits. Using a biphasic backcross strategy, we constructed a framework linkage map of microsatellite markers. We also estimated allele frequencies for 23 polymorphic loci using 13 strains of B. mori. Further, extending our study, we also tested the conservation of a selected number of loci in different lepidopteran insects, including B. mandarina, the nearest wild relative to silkworm, a group of saturniids, and a noctuid.
MATERIALS AND METHODS
Sequence analysis:
The sequences analyzed for microsatellites included 21.76-Mb random sequences from WGS sequence data downloaded from DDBJ (http://www.ddbj.nig.ac.jp/whatsnew/040423-e.html; Mita et al. 2004), 0.67-Mb BAC sequences from the Z chromosome (Koike et al. 2003), and ∼9300 nonredundant ESTs accounting for 6.3 Mb, downloaded from SilkBase (http://www.ab.a.u-tokyo.ac.jp/silkbase; Mita et al. 2003). To avoid the double counting, the Z chromosome BAC sequences were removed from the genomic sequences on the basis of similarity search using BLAST (Altschul et al. 1990). The three sets of sequences from WGS, Z chromosome BACs, and ESTs are referred to as genomic, Z chr-BAC, and EST, respectively in this article. The sequences were analyzed for microsatellites and their flanking sequences using SSR finder (SSRF, http://210.212.212.7/MIC/index.html) and Allflank (to be published elsewhere), respectively. In our analysis, only those microsatellites with ≥5 repeat units were counted except in the case of mononucleotide tracts, where ≥15 bases were considered.
Microsatellite validation:
Some microsatellite loci identified by the above analysis, composed of di-, tri-, tetra-, penta-, hexanucleotide compound and imperfect repeats with varying lengths, were chosen for the study of allele frequency, polymorphism, and conservation. Primers were designed for the flanking sequences of these loci using the Amplify program (http://bip.weizmann.ac.il/mb/bioguide/pcr/PCRsofAmplify.html). Primers were also designed for microsatellites identified in B. mori gene sequences archived in the GenBank database. The microsatellite primers developed in an earlier study (Reddy et al. 1999a) were also used. All the primers were standardized for optimum annealing temperature and MgCl2 concentration using genomic DNA from the Nistari strain of B. mori. The details of the primer sequences, size range of the alleles, and PCR amplification conditions for each of the 198 microsatellite loci developed and validated in this study are available at our website (http://210.212.212.7:9999/PHP/SILKSAT/index.php?f=design_primer).
Silkworm strains and genomic DNA:
The detection of polymorphism of the newly identified loci was carried out in two silkmoth strains, Nistari (male individuals) and NB4D2 (female individuals), which have nondiapausing and diapausing characters, respectively. These two strains are quite divergent for various qualitative and quantitative traits and produce highly heterotic hybrids when they are crossed and are being used as parental strains to construct a mapping population. Thirteen diverse silkworm strains, which included 6 diapausing (HU204, KA, NB1, NB7, NB18, and NB4D2) and 7 nondiapausing (C.nichi, Moria, Nistari, Pure Mysore, Daizo, Gungnong, and Sarupat) strains, were used for microsatellite analysis. In each of the strains, DNA was extracted from 10 individuals, except for NB4D2, in which DNA from only 5 individuals was extracted. The saturniid silkmoths, Antheraea mylitta, A. assama, A. roylei, A. proylei, A. pernyi, A. yamamai, and Samia cynthia ricini; the bombycid, B. mandarina; and the noctuid moth, Helicoverpa armigera, were used to study the conservation of B. mori microsatellite loci in heterologous lepidopterans. DNA was isolated from adult moths as described earlier (Prasad and Nagaraju 2003).
Choice of primers and locus detection:
For population analysis, 26 microsatellite loci involving di-, tri-, and tetranucleotide repeats with 11 loci identified from the SilkBase, 12 from GenBank, and 3 from subgenomic library screening were used. For polymorphism analysis, 198 loci (34 from ESTs, 91 from total genomic sequences, 19 from subgenomic library screening, and 54 from GenBank) involving di, tri-, tetra-, penta-, hexanucleotide, imperfect and compound repeats were used. For the evaluation of conservation of B. mori microsatellites in the heterologous lepidopteran species, 30 loci (12 loci identified from the subgenomic library, 16 from GenBank, and 2 from SilkBase) were tested. Out of these, 4 loci that amplified in most of the heterologous species were cloned, sequenced, and analyzed.
PCR amplification and electrophoresis:
PCR was typically performed in a 5-μl reaction volume containing 50 μm dNTPs, 0.2 μm fluorescent dUTP (Tamara or R110 or R6G; PE Biosystems), 0.4 μm of each primer, 0.25 units of AmpliTaq Gold polymerase (PE Biosystems), 1× PCR buffer (100 mm Tris-HCl pH 8.8, 500 mm KCl, 0.8% Nonidet P40), 1.5–4 mm MgCl2, and ∼5 ng genomic DNA. Thermal cycling was carried out for 30 cycles on a Thermal Cycler PTC 100 (MJ Research, Watertown, MA) with locus-specific annealing temperature. Samples containing 1 μl of PCR products, 0.3 μl of GeneScan 500 ROX size standard (PE Biosystems), and 2.0 μl of loading buffer (4 parts de-ionized formamide: 1 part 500 mg/ml blue dextran, 25 mm EDTA) were heated at 92° for 5 min and then placed on ice. Denatured samples (1 μl) were immediately loaded on 5% denaturing (6 m urea) Long ranger (FMC, Vallensbaek, Denmark) gels (36 cm well-to-read) in 1× TBE buffer (89 mm Tris, 89 mm borate, 2 mm EDTA pH 8.3) and electrophoresed at 3000 V for 3 hr on an automated DNA sequencer (PE Biosystems 377). Allelic sizes were determined using GeneScan v 2.1 software (PE Biosystems) and data were compiled using Genotyper v 3.0 (PE Biosystems). Polymorphic status for 198 microsatellite loci between two parental strains used to construct the mapping population was tested on 3% 3:1 Metaphor agarose (FMC) and agarose (United States Biochemicals, Cleveland; Amersham Pharmacia, Piscataway, NJ) gel electrophoresis. The primer sets that did not reveal polymorphism on agarose gels were subjected to GeneScan analysis and the polymorphic loci, if any, were detected.
Construction of genetic linkage map:
In B. mori, achiasmatic oogenesis results in absolute linkage in females. In males, linkage depends upon the crossover events occurring during spermatogenesis. This biphasic linkage behavior typical to lepidopterans aids mapping by sequential approach (Heckel et al. 1999; Nagaraju and Goldsmith 2002). F1 individuals from a cross NB4D2 × Nistari were backcrossed to Nistari (the recurrent parent) to generate the mapping population. In the first step, F1 females were backcrossed with Nistari males [backcross (BC)-I] to identify the linkage groups. In a second backcross, F1 males were mated with Nistari females, generating recombinant progeny (BC-II) to obtain the order of the markers within each linkage group. Forty individuals from BC-I and 60 individuals from BC-II were genotyped along with the parents and F1. The data matrix was input into MAPMAKER version 3.0 (Lander et al. 1987). Two-point linkage was determined at LOD = 3.0 and multipoint analysis based on Kosambi function (Kosambi 1944) yielded the order of the markers in each linkage group.
Statistical analyses:
Heterozygosity values (H) for alleles found in silkworm populations were calculated using
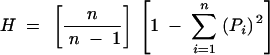 |
in which n is the number of alleles scored and Pi is the frequency of the ith allele. Significance of the differences in abundance and length of different repeat types within and between three sequence sources was measured using student's t-test. Nei's standard genetic distance (Nei 1972) between 13 silkworm populations based on 26 microsatellite loci was computed. Departure from mutation drift equilibrium was tested using the Bottleneck program (Cornuet and Luikart 1996) for both diapausing and nondiapausing populations.
Cloning, sequencing, and phylogeny:
Bmsat020, Bmsat110, Bmsat103, and Bmsat109 loci were used to study the conservation of B. mori loci in heterologous lepidopteran insects (Table 8). These loci were amplified by PCR as described earlier. The PCR products were purified with a column (QIAGEN, Valencia, CA) and cloned in pCR 2.1 vector (Invitrogen, San Diego). The recombinant plasmids were sequenced with the primers used for PCR for both strands with the ABI PRISM BigDye terminator cycle sequencing ready reaction kit on a 3100 genetic analyzer (Applied Biosystems, Foster City, CA) and the sequences were aligned using CLUSTAL W with manual corrections.
TABLE 8.
The species used for evaluation of conservation ofB. mori microsatellite loci
| Species name | Chromosome no. (n) | Family | Common name | Geographical distribution |
|---|---|---|---|---|
| Bombyx mori | 28 | Bombycidae | Domestic silkmoth | Karnataka, India |
| Bombyx mandarina | 27 | Bombycidae | Wild silkmoth | Tokyo, Japan |
| Antheraea mylitta | 31 | Saturniidae | Indian tropical tasar silkmoth | Bihar, India |
| A. roylei | 30, 31, 32a | Saturniidae | Indian temperate tasar silkmoth | Jammu and Kashmir, India |
| A. pernyi | 49 | Saturniidae | Chinese Oak silkmoth | China |
| A. proyleib | 49 | Saturniidae | Synthetic Oak silkmoth | Jammu and Kashmir, India |
| A. assama | 15 | Saturniidae | Indian muga silkmoth | Assam, India |
| A. yamamai | 31 | Saturniidae | Japanese Oak silkmoth | Japan |
| Samia cynthia ricini | 13 | Saturniidae | Indian eri silkmoth | Karnataka, India |
| Helicoverpa armigera | Noctuidae | Cotton bollworm | Karnataka, India |
A. roylei exhibits chromosome number polymorphism (Jolly et al. 1979).
F72 generation of an interspecific hybrid of A. pernyi × A. roylei (Nagaraju and Jolly 1986).
Phylogeny based on mitochondrial sequences:
For amplification of mitochondrial 12S ribosomal DNA (rDNA) from B. mori, wild silkmoths, and H. armigera, the forward primer SR-J-14199, tactatgttacgacttat, and reverse primer SR-N-14594, aaactaggattagataccc, were used. The amplicons were purified and cloned in the vector pCR 2.1. The recombinant plasmids were sequenced on a 3100ABI Genetic Analyzer (Applied Biosystems). rDNA sequences were submitted to GenBank with the following accession numbers: AF389421–22 and AY037827–35. The sequences were aligned using CLUSTAL W and a phylogenetic tree was constructed using the Neighbor-Joining program of PHYLIP software with Drosophila nasuta 12S rDNA as an outgroup.
RESULTS
Microsatellite distribution:
We examined the distribution of microsatellites in genomic, Z chr-BAC, and EST sequences. We found that microsatellites were widely distributed in the B. mori genome, with ∼3 kb of repeats per megabase of genomic and EST sequences (Table 1). We estimated that the silkworm genome of 530 Mb accounted for 1.63 Mb of microsatellite repeats, equivalent to 0.31% of the genome (Table 1). Under this consideration, mono-, di-, tri-, tetra-, penta-, and hexanucleotide repeats represented 0.110, 0.116, 0.053, 0.018, 0.006, and 0.003% of the genome, respectively.
TABLE 1.
Distribution of different types of microsatellites in the silkworm genomic, Z chr-BAC, and EST sequences
| No. of repeat units per Mb of sequence |
kb of repeats per Mb of sequence
|
Repeats in the whole genomea |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Repeat type (nucleotide) |
Repeat unit |
Genomic | EST | Z chr-BAC | Genomic | EST | Z chr-BAC | No.b | Mbc |
| Mono- | A | 1094 | 1645 | 133 | 1.094 | 1.645 | 0.133 | 579.820 | 0.580 |
| C | 11 | 5 | 0 | 0.011 | 0.005 | 0.000 | 5.830 | 0.006 | |
| Total | 1105 | 1650 | 133 | 1.105 | 1.650 | 0.133 | 585.650 | 0.586 | |
| Di- | CA | 242 | 128 | 160 | 0.484 | 0.255 | 0.320 | 128.260 | 0.257 |
| TA | 375 | 160 | 334 | 0.749 | 0.320 | 0.668 | 198.750 | 0.397 | |
| GA | 207 | 130 | 116 | 0.414 | 0.260 | 0.232 | 109.710 | 0.219 | |
| CG | 8 | 36 | 15 | 0.016 | 0.071 | 0.030 | 4.240 | 0.008 | |
| Total | 832 | 454 | 625 | 1.163 | 0.906 | 1.250 | 440.960 | 0.616 | |
| Tri- | TAA | 92.10 | 34.70 | 132.80 | 0.276 | 0.104 | 0.398 | 48.810 | 0.146 |
| CAA | 4.31 | 17.30 | 9.00 | 0.013 | 0.052 | 0.027 | 2.280 | 0.007 | |
| CCA | 2.73 | 16.70 | 16.40 | 0.008 | 0.050 | 0.049 | 1.450 | 0.004 | |
| CGA | 0.67 | 7.30 | 10.40 | 0.002 | 0.022 | 0.031 | 0.360 | 0.001 | |
| CGG | 4.34 | 13.70 | 10.40 | 0.013 | 0.041 | 0.031 | 2.300 | 0.007 | |
| GAA | 21.70 | 22.00 | 0.00 | 0.065 | 0.066 | 0.000 | 11.500 | 0.034 | |
| GCA | 0.67 | 8.00 | 10.40 | 0.002 | 0.024 | 0.031 | 0.360 | 0.001 | |
| GGA | 2.34 | 10.00 | 0.00 | 0.007 | 0.030 | 0.000 | 1.240 | 0.004 | |
| GTA | 24.20 | 10.00 | 25.40 | 0.072 | 0.030 | 0.076 | 12.830 | 0.038 | |
| TGA | 24.70 | 14.30 | 7.50 | 0.074 | 0.043 | 0.023 | 13.090 | 0.039 | |
| Total | 277.60 | 154.00 | 222.30 | 0.532 | 0.462 | 0.666 | 147.130 | 0.282 | |
| Tetra- | 0% AT | 0.00 | 0.00 | 0.00 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| 25% AT | 5.57 | 4.30 | 0.00 | 0.022 | 0.017 | 0.000 | 2.950 | 0.012 | |
| 50% AT | 5.84 | 4.50 | 0.00 | 0.023 | 0.018 | 0.000 | 3.100 | 0.012 | |
| 75% AT | 15.20 | 4.50 | 0.00 | 0.061 | 0.018 | 0.000 | 8.060 | 0.032 | |
| 100% AT | 20.02 | 3.50 | 0.03 | 0.080 | 0.014 | 0.000 | 10.610 | 0.042 | |
| Total | 46.63 | 16.80 | 0.03 | 0.186 | 0.067 | 0.000 | 24.720 | 0.098 | |
| Penta- | 0% AT | 0.21 | 0.00 | 0.00 | 0.001 | 0.000 | 0.000 | 0.110 | 0.001 |
| 20% AT | 1.23 | 0.00 | 0.00 | 0.006 | 0.000 | 0.000 | 0.650 | 0.003 | |
| 40% AT | 0.22 | 1.23 | 0.00 | 0.001 | 0.006 | 0.000 | 0.120 | 0.001 | |
| 60% AT | 2.61 | 0.84 | 0.00 | 0.013 | 0.004 | 0.000 | 1.380 | 0.007 | |
| 80% AT | 6.83 | 1.82 | 0.00 | 0.034 | 0.009 | 0.000 | 3.620 | 0.018 | |
| 100% AT | 0.22 | 1.61 | 0.00 | 0.001 | 0.008 | 0.000 | 0.120 | 0.001 | |
| Total | 11.32 | 5.50 | 0.00 | 0.056 | 0.027 | 0.000 | 6.000 | 0.030 | |
| Hexa- | 0% AT | 0.00 | 1.33 | 0.00 | 0.000 | 0.008 | 0.000 | 0.000 | 0.000 |
| 16.7% AT | 0.67 | 0.00 | 8.96 | 0.004 | 0.000 | 0.054 | 0.360 | 0.002 | |
| 33.3% AT | 0.33 | 2.67 | 0.00 | 0.002 | 0.016 | 0.000 | 0.170 | 0.001 | |
| 50% AT | 0.34 | 0.83 | 0.00 | 0.002 | 0.005 | 0.000 | 0.180 | 0.001 | |
| 66.7% AT | 2.67 | 5.17 | 0.00 | 0.016 | 0.031 | 0.000 | 1.420 | 0.008 | |
| 83.3% AT | 0.17 | 8.02 | 0.00 | 0.001 | 0.048 | 0.000 | 0.090 | 0.001 | |
| 100% AT | 0.52 | 5.17 | 14.93 | 0.003 | 0.031 | 0.090 | 0.280 | 0.002 | |
| Total | 4.70 | 23.19 | 23.89 | 0.028 | 0.139 | 0.144 | 2.490 | 0.015 | |
| Grand total | 2277.25 | 2304.00 | 1004.00 | 3.070 | 3.251 | 2.193 | 1206.950 | 1.626 | |
The sequences analyzed included 21.76 Mb of genomic sequences, 0.67 Mb of Z chr-BAC sequences, and 6.3 Mb of EST sequences.
The silkworm haploid genome size was considered to be 530 Mb and the values were obtained by extrapolating data from genomic sequences.
Expressed in thousands.
The microsatellite content is expressed in megabases.
A/T mononucleotide stretches were much more abundant than C/G stretches in genomic, Z chr-BAC, and EST sequences (Figures 1 and 2). Greater than 20 repeat unit tracts of A/T were common in genomic and EST sequences (Figure 2). Among dinucleotides, CG repeats were found to be the least abundant. Among dinucleotides in ESTs CA and GA were as abundant as TA repeats, and CG repeats were relatively more compared to genomic and Z chr-BAC sequences (Figure 2). Among trinucleotides, TAA repeats were the most abundant repeats in the genomic sequences, composing almost 50% of trinucleotide repeats followed by GTA and TGA (Table 1). These three trinucleotide repeat types were significantly less abundant in ESTs (P < 0.01). All the remaining repeats were significantly overrepresented in ESTs (Figure 1). CGA was the least represented among the trinucleotide repeats.
Figure 1.—
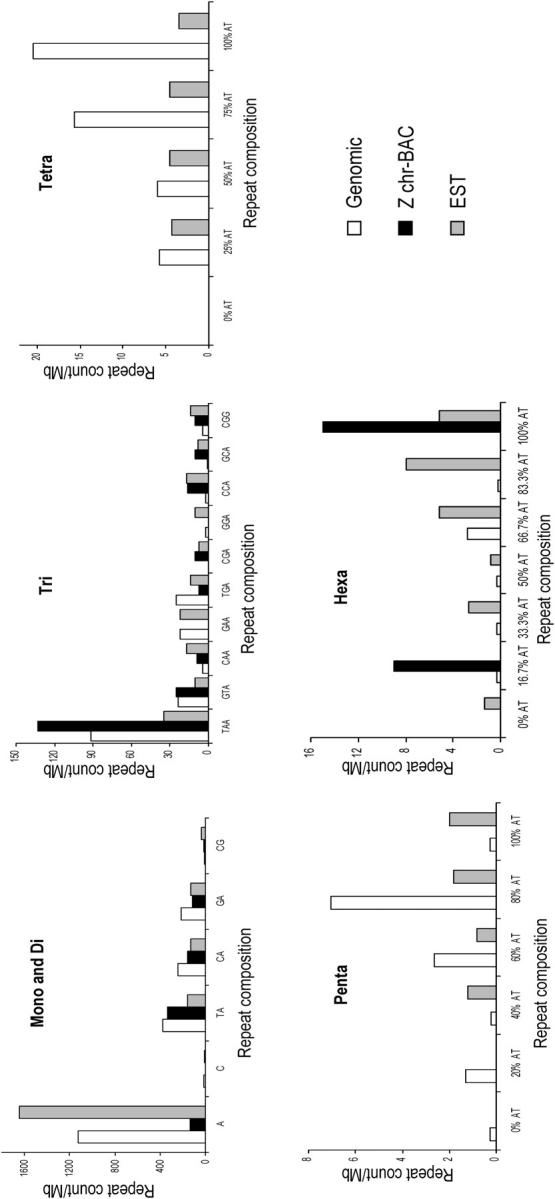
Number of repeat units per megabase of genomic, Z chr-BAC, and EST sequences. The x-axis represents the repeat type. In tetra-, penta-, and hexanucleotide repeat types, AT content of the repeat motif is expressed in percentage. The y-axis represents the repeat counts per megabase of sequence.
Figure 2.—


Distribution of microsatellites in genomic, Z chr-BAC, and EST sequences. The x-axis represents length of the repeat motifs (in base pairs) and the y-axis represents repeat counts extracted from 21.76 Mb of genomic, 0.67 Mb of Z chr-BAC, and 6.3 Mb of EST sequences.
Since tetra-, penta-, and hexanucleotide repeats have large groups of repeat types, we classified them on the basis of AT percentage from 0 to 100. For example, ATTT was considered to be a 100% AT-rich tetranucleotide repeat, CAAA was a 75% AT-rich tetranucleotide repeat, ACGT was a 50% AT-rich tetranucleotide repeat, and so on. The maximum number of tetranucleotide repeats was observed in the 100% AT-rich repeats followed by the 75% AT-rich category (with one C/G in the repeat) in genomic sequences whereas in EST sequences the 50 and 75% AT-rich categories contributed the most (Figure 2). Eighty percent (with one C/G) and 100% AT-rich repeat types together constituted >80 and >60% of the total number of pentanucleotide repeats in the genomic and EST sequences, respectively. Three groups of hexanucleotide repeats, 66.7% (with two C/G), 83.3% (with one C/G), and 100% AT rich, were found to be significantly bigger in ESTs compared to the genomic sequences. Overall, we observed a common phenomenon whereby repeat number was inversely proportional to repeat length; this was more pronounced in trinucleotide repeats, especially because of their abundance in both the genome and ESTs (Table 1). The average length of mononucleotides was more as compared to other repeat classes. There was no significant difference in the lengths of repeats among genomic, ESTs, and Z chr-BAC sequences (Table 2).
TABLE 2.
Average repeat numbers of microsatellites observed in genomic, Zchr-BAC, and EST sequences
| Repeat type (nucleotide) |
Repeat unit |
Genomic | Zchr-BAC | EST |
|---|---|---|---|---|
| Mono- | A | 19.6 ± 0.5 | 18.1 ± 3.0 | 20.9 ± 0.7 |
| C | 16.1 ± 0.4 | — | 15.5 ± 0.6 | |
| Average | 17.9 ± 0.5 | 18.1 ± 3.0 | 18.2 ± 0.7 | |
| Di- | CA | 6.5 ± 0.3 | 7.7 ± 0.7 | 7.0 ± 0.3 |
| TA | 6.0 ± 0.2 | 6.1 ± 0.6 | 7.3 ± 0.3 | |
| GA | 6.8 ± 0.3 | 7.8 ± 0.7 | 7.1 ± 0.3 | |
| CG | 5.2 ± 0.2 | 5.0 ± 0.5 | 5.8 ± 0.2 | |
| Average | 6.1 ± 0.2 | 6.6 ± 0.6 | 6.8 ± 0.3 | |
| Tri- | TAA | 5.7 ± 0.5 | 5.2 ± 0.5 | 5.9 ± 0.7 |
| CAA | 6.2 ± 0.6 | 6.0 ± 0.1 | 6.1 ± 0.8 | |
| CCA | 5.6 ± 0.5 | 5.5 ± 0.1 | 5.5 ± 0.7 | |
| CGA | 6.0 ± 0.5 | 7.0 ± 0.2 | 5.2 ± 0.7 | |
| CGG | 6.6 ± 0.6 | 7.0 ± 0.1 | 5.8 ± 0.7 | |
| GAA | 5.5 ± 0.5 | — | 5.8 ± 0.7 | |
| GCA | 5.5 ± 0.5 | 7.0 ± 0.2 | 5.7 ± 0.7 | |
| GGA | 5.2 ± 0.5 | — | 5.7 ± 0.7 | |
| GTA | 6.4 ± 0.6 | 8.5 ± 0.3 | 5.6 ± 0.7 | |
| TGA | 6.1 ± 0.6 | 5.0 ± 0.1 | 6.5 ± 0.8 | |
| Average | 5.9 ± 0.5 | 6.4 ± 0.2 | 5.8 ± 0.7 | |
| Tetra- | 0% AT | — | — | — |
| 25% AT | 7.9 ± 0.7 | — | 5.2 ± 1.7 | |
| 50% AT | 5.2 ± 0.5 | — | 5.8 ± 1.9 | |
| 75% AT | 5.5 ± 0.5 | — | 5.6 ± 1.9 | |
| 100% AT | 5.6 ± 0.5 | 5.0 ± 0.0 | 5.5 ± 1.8 | |
| Average | 6.1 ± 0.6 | 5.0 ± 0.0 | 5.5 ± 1.8 | |
| Penta- | 0% AT | 6.0 ± 0.8 | — | — |
| 20% AT | 9.4 ± 1.2 | — | — | |
| 40% AT | 5.0 ± 0.6 | — | 6.1 ± 2.0 | |
| 60% AT | 8.2 ± 1.0 | — | 5.1 ± 1.7 | |
| 80% AT | 5.9 ± 0.7 | — | 11.2 ± 3.7 | |
| 100% AT | 6.0 ± 0.8 | — | 10.0 ± 1.7 | |
| Average | 6.7 ± 0.9 | — | 8.1 ± 2.3 | |
| Hexa- | 0% AT | — | — | 8.0 ± 0.9 |
| 16.7% AT | 7.0 ± 1.2 | 6.0 ± 3.1 | — | |
| 33.3% AT | 7.0 ± 1.2 | — | 17 ± 1.9 | |
| 50% AT | 7.0 ± 1.2 | — | 5.0 ± 0.6 | |
| 66.7% AT | 8.6 ± 1.4 | — | 10.7 ± 1.2 | |
| 83.3% AT | 5.0 ± 0.9 | — | 10.0 ± 1.1 | |
| 100% AT | — | 5.0 ± 2.5 | 8.0 ± 0.9 | |
| Average | 6.9 ± 1.2 | 5.5 ± 2.8 | 9.8 ± 1.1 | |
| Grand average | 9.2 ± 0.5 | 13.9 ± 0.7 | 7.8 ± 0.7 |
Z chromosome vs. other chromosomes and ESTs:
Our analysis of mononucleotide repeat tracts in Z chr-BAC sequences yielded only A/T motifs (Table 1). Among trinucleotide repeat tracts, TAA repeats were significantly overrepresented in Z chr-BAC compared to genomic and EST sequences. GCA, CGA, and CGG were significantly more represented than those in genomic sequences whereas GAA and GGA were completely absent. Tetra-, penta-, and hexanucleotide repeat motifs were very scarce in the Z chr-BACs (Table 1).
Flanking sequence analysis:
To test the base composition surrounding the microsatellite repeats, we calculated GC content for 100 bases upstream and 100 bases downstream flanking different types of repeat motifs. Overall, the GC content of the sequences flanking different repeat types was more or less similar in both genomic and EST sequences. Also, there was no significant difference in AT/GC composition between upstream and downstream flanking sequences except in the case of 100% AT-rich pentanucleotide repeats, where downstream flanking sequences showed 5% lower GC content compared to that in upstream sequences in both genomic and EST sequences (data not shown). Hence, the upstream and downstream flanking sequences of both genomic and EST sequences were combined and analyzed. GC content was between 30 and 40% in sequences flanking A, T, AT, and AAT repeat tracks and increased up to 60% with increase in G/C content of the repeat unit (Figure 3). In other words, the GC contents of the repeat unit and the flanking sequences were positively correlated.
Figure 3.—

Sequence composition of microsatellite motifs and their flanking regions. Repeat types are mapped on the x-axis and each bar represents a class of repeat type based on percentage of GC content of the repeat motif indicated inside the bar. The y-axis is percentage of GC content of the flanking sequence (combined average of 100 bases upstream and downstream sequences flanking the microsatellite repeats derived from total-genomic and EST sequence).
Mutations:
We analyzed silkworm genomic and EST sequences for imperfections in di-, tri-, tetra-, and pentanucleotide repeats. Incorporation of imperfections in perfect repeat tract by means of mutations was predominantly seen in the center of the repeat tract (398 out of 491). Mutations toward the 3′ end of the repeat motif were less abundant (152/491), whereas mutations toward the 5′ end of the repeat motif were rarely found although they do exist (41/491; supplementary Table S1 at http://www.genetics.org/supplemental/). This distribution was irrespective of the repeat length and we did not observe any length-dependent distribution. The shortest imperfect repeats were identified in di-, tri-, and tetranucleotide repeats of 15 bases in length. The longest was a tetranucleotide repeat of 611 bases (supplementary Table S1).
We determined if the mutations were transitions, transversions, insertions, or deletions. An insertional mutation within the repeat unit, e.g., (ATC)3G(ATC)3, (TGA)3TCGA(TGA)2, and a deletion of a base or more in the repeat tract, e.g., (ATT)3_TT(ATT)2, disturb the continuity of the perfect repeat units.
Transitions (29/74) were most abundant in EST sequences compared to other types of mutations, whereas insertions were the least abundant (7/74; Table 3). Changes from A to G and T to A were the most common mutations disrupting repeat continuity (Table 4). Transition/transversions accounted for 74% of the interruptions of perfect repeats, whereas insertion/deletions were observed in only 26% of the interruptions in ESTs (Table 3). In genomic sequences, repeats were interrupted most often by deletions (153/417) and least by insertions (19/417; Table 3). A to G transitions were the most common substitution disrupting the repeat continuity (Table 4). The ratio of transition/transversion to insertion/deletion was found to be ∼3:2 (Table 3). Double mutations, in which two types of mutations coexisted, were observed in 53 repeats (4 in ESTs and 49 in genomic). Among these, double transitions and double transversions occurred together in 10 and 5 repeat motifs, respectively; transitions and transversions were together in 11/53; insertions/deletions (indels) were together in 4/53; and indels and substitutions were associated in 23/53 imperfect repeats. Triple mutations occurred together in 9 repeat motifs while one (AC)n motif showed all four possible mutations.
TABLE 3.
Number and type of mutations disrupting perfect microsatellite motifs in EST and total-genomic sequences
| Transition | Transversion | Deletion | Insertion | |
|---|---|---|---|---|
| EST | 29 (39%)a | 26 (35%) | 12 (16%) | 7 (10%) |
| Total-genomic | 124 (30%) | 121 (29%) | 153 (37%) | 19 (4%) |
Total-genomic sequences include 21.76 Mb random genomic sequences and 0.67 Mb BAC sequences from the Z chromosome.
Percentages of total mutations are given in parentheses.
TABLE 4.
Number of transitions and transversions found in microsatellite motifs of EST and total-genomic sequences
| T to C | A to T | A to G | A to C | T to A | G to T | C to T | C to A | G to C | C to G | G to A | T to G | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EST | 6 | 6 | 7 | 4 | 6 | 2 | 7 | 5 | 2 | 1 | 8 | 1 |
| Total-genomic | 36 | 26 | 37 | 9 | 37 | 11 | 28 | 13 | 8 | 4 | 23 | 13 |
Total-genomic sequences include 21.76 Mb random genomic sequences and 0.67 Mb BAC sequences from the Z chromosome.
Allelic distribution:
The allelic distribution of 26 loci was tested in 13 different strains of diapausing and nondiapausing silkworm strains. Polymorphisms were detected for 23 of the 26 loci. Two microsatellite loci (Bmsat012 and Bmsat015), which were present in the coding region, and one locus (Bmsat109) with an unknown location were found to be monomorphic. The number of alleles scored at each locus in the 13 strains varied from as few as 2 to as many as 14 (Table 5). The heterozygosity values varied from 0.20 to 0.87. We did not observe any association of allele number to any particular repeat type or repeat length.
TABLE 5.
Repeat motif, number of alleles, and heterozygosity for different microsatellite loci in 13 strains ofB. mori
| Locus | Repeat motif | Total no. of alleles |
Allele size range (bp)a |
Heterozygosity value |
|---|---|---|---|---|
| Bmsat007 | (TCA)9 | 6 | 177–201 | 0.76 |
| Bmsat008 | (TAA)13 | 3 | 275–350 | 0.67 |
| Bmsat010 | (CAGC)18 | 4 | 137–220 and null | 0.52 |
| Bmsat011 | (GT)2 AGC (GTT)4 GT (GC)2 GT | 2 | 150–225 | 0.43 |
| Bmsat013 | (CT)2 T (CT)9N12 (CA)3 | 2 | 360 and null | 0.00 |
| Bmsat014 | (AT)9 | 2 | 210–220 | 0.49 |
| Bmsat016 | (GT)7 N13 (GT)4 | 3 | 350, 375 and null | 0.50 |
| Bmsat018 | (CTA)2 TTA (CTA)5 | 3 | 217–230 and null | 0.20 |
| Bmsat020 | (TTA)4 A (TA)2 N39 (TTA)2 | 2 | 175–200 | 0.31 |
| Bmsat057 | (ATT)6 | 2 | 197–230 | 0.21 |
| Bmsat060 | (ATG)5 | 2 | 180–200 | 0.50 |
| Bmsat061 | (CAT)12 | 3 | 130–140 | 0.21 |
| Bmsat062 | (TAC)3 TAG (TAC)2 N12 (TAC)3 N9 (TAC)7 | 4 | 150–250 | 0.69 |
| Bmsat063 | (TA)10 | 2 | 140 and null | 0.00 |
| Bmsat066 | (TA)7 | 2 | 148–215 | 0.40 |
| Bmsat067 | (GT)16 N12 (GT)5 | 2 | 175–200 | 0.65 |
| Bmsat068 | (TG)19 N10(TG)5 | 5 | 130–159 | 0.48 |
| Bmsat069 | (CTA)7 | 4 | 170–185 | 0.71 |
| Bmsat070 | (ATTT)10 | 10 | 107–148 | 0.87 |
| Bmsat071 | (TCA)13 | 3 | 105–115 | 0.58 |
| Bmsat103 | (GT)22N12(GT)7 | 14 | 130–190 | 0.87 |
| Bmsat110 | (TA)4 C (TA)4 (GATA) GAA (GATA)3 GAA (GATA)3 GAA (GATA)3 GAA (GATA)10 |
3 | 90–185 | 0.54 |
| Bmsat132 | (GT)10 | 6 | 96–262 | 0.82 |
Absence of amplification for a locus was scored as a null allele.
The average heterozygosity value for 23 polymorphic microsatellite loci was 0.54. Bmsat103, an imperfect dinucleotide repeat locus, was found to be the most polymorphic with 14 alleles and a heterozygosity value of 0.87. The lowest heterozygosity value was found for the trinucleotide locus Bmsat018 with two alleles. A few loci, like Bmsat010, Bmsat013, Bmsat016, Bmsat018, and Bmsat063, showed null alleles. Three loci, Bmsat132, Bmsat066, and Bmsat110, revealed alleles specific to diapausing and nondiapausing strains (Table 6). The 134-bp allele in locus Bmsat132 was specific to all diapausing strains, whereas the 146-bp allele was specific to all nondiapausing strains, except for C.nichi, which did not share either of these alleles. The 215-bp allele in locus Bmsat066 was specific only to diapausing strains except for HU204, which did not share this allele. Likewise, the 90-bp allele of Bmsat110 was unique to diapausing strains.
TABLE 6.
Diapausing and nondiapausing strain-specific alleles of the loci Bmsat110, Bmsat132, and Bmsat66
| Diapausing strains
|
Nondiapausing strains
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Locus | Allele size | HU204 | KA | NB1 | NB7 | NB18 | NB4D2 | C.nichi | GNM | Moria | Nistari | PM | Diazo | Sarupat |
| Bmsat066 | 148 | + | + | + | + | + | + | + | + | + | + | + | ||
| 215 | +a | +a | +a | +a | +a | |||||||||
| Bmsat110 | 90 | +a | +a | +a | +a | +a | +a | |||||||
| 105 | + | + | + | |||||||||||
| 185 | + | + | + | + | + | + | + | + | + | |||||
| Bmsat132 | 96 | + | + | + | + | + | ||||||||
| 97 | + | + | ||||||||||||
| 99 | + | + | + | + | + | |||||||||
| 134 | +a | +a | +a | +a | +a | +a | ||||||||
| 138 | + | + | + | + | + | + | + | |||||||
| 146 | +b | +b | +b | +b | +b | +b | ||||||||
Presence of allele is represented by +.
Diapausing strain-specific alleles.
Nondiapausing strain-specific alleles.
We tested the influence of domestication of diapause and nondiapause silkworm strains on the level of heterozygosity using bottleneck analysis. The observed heterozygosity exceeded the average of the corresponding distribution of heterozygosities expected at equilibrium in both diapausing and nondiapausing strains. Under the infinite-allele model (IAM) 17 of the 18 polymorphic loci (P = 0.000) and in the stepwise mutation model (SMM) 15 of the 18 polymorphic loci (P = 0.01243) showed an excess of heterozygotes in the case of diapausing populations. Similarly, for nondiapausing strains, all 17 polymorphic loci in IAM (P = 0.000) and 16 of 17 polymorphic loci in SMM (P = 0.000) showed a significant heterozygosity excess. Both the bottleneck tests reject the hypothesis of mutation drift equilibrium and show the occurrence of a recent bottleneck in both diapausing and nondiapausing populations, i.e., a recent decline in effective population size (Ne). These results are not surprising since the populations are highly domesticated and are being maintained by inbreeding. Nei's standard genetic distance based on 23 polymorphic loci in 13 silkworm populations yielded population trees that reflected their geographic origin, voltinism (diapausing/nondiapausing), and pedigree (supplementary Figure 1 at http://www.genetics.org/supplemental/).
Polymorphism:
The detection of polymorphism of the newly identified loci was carried out in two silkmoth strains, Nistari (male individuals) and NB4D2 (female individuals), which have nondiapausing and diapausing characters, respectively. These two strains are quite divergent for various qualitative and quantitative traits and produce highly heterotic hybrids when they are crossed and are being used as parental strains to construct the mapping population. Out of 198 loci tested, which included 34 from the EST database, 19 from genomic library screening, 54 from GenBank, and 91 from genomic sequences, 36 loci showed polymorphism (18.2%) and 10 loci revealed null alleles (5.1%; Table 7). Perfect tetra- (36.7%), imperfect di- (33.3%), and trinucleotide repeats (23.5%) showed higher levels of polymorphism, respectively, whereas perfect tri- (11.4%) and imperfect tetra- (0%) and pentanucleotide (0%) repeats showed the lowest levels of polymorphism (Table 7).
TABLE 7.
Polymorphism of microsatellite loci between NB4D2 female and Nistari male strains
| Perfect repeat motifs (nucleotides)
|
Imperfect repeat motifs (nucleotides)
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Source | Mono- | Di- | Tri- | Tetra- | Penta- | Di- | Tri- | Tetra- | Penta- | Compound | Total |
| Subgenomic librarya | NDb | 3 + 1 (11)c | 0 (1) | ND | 1 (2) | 0 (2) | 0 (1) | ND | ND | 1d (2) | 3 + 2d (19) |
| GenBanka | 1 (4) | 0 (9) | 1 (11) | 1 + 1d (5) | 0 (3) | 1 (3) | 2 + 1d (7) | 0 (1) | 0 (1) | 0 (10) | 6 + 2d (54) |
| EST | ND | 1d (4) | 5 (19) | 1 (1) | ND | 3 (3) | 1 (3) | ND | ND | 0 (4) | 10 + 1d (34) |
| Total genomic | 0 (1) | 1 + 2d (13) | 0 (13) | 6 + 1d (23) | 1 (15) | 2 (7) | 1 + 1d (8) | 3 (5) | 0 (2) | 1 + 1d (4) | 14 + 5d (91) |
| Total | 1 (5) | 5 + 4d (37) | 6 (44) | 8 + 2d (29) | 2 (20) | 6 (15) | 4 + 2d (19) | 3 (6) | 0 (3) | 1 + 2d (20) | 36 + 10d (198) |
| % polymorphic | 20 | 13.5 | 13.6 | 27.5 | 10.0 | 40.0 | 21.0 | 50.0 | 0 | 5.0 | 18.2 |
| % null allele | 0 | 10.8 | 0 | 6.8 | 0 | 0 | 10.5 | 0 | 0 | 10 | 5.1 |
The microsatellite loci derived from our earlier study (Reddy et al. 1999a) and extracted from GenBank sequences were used only for the polymorphism analysis.
ND represents not determined.
Numbers in parentheses indicate total number of loci used for analysis of polymorphism.
Number of null alleles.
Genetic linkage map:
A total of 46 polymorphic SSR markers were detected in the study. Among them, 10 markers exhibited null alleles in the male parent, Nistari, and segregated as dominant markers. Four markers showed Z-specific inheritance, which was confirmed by homology search in Z chr-BACs. Four markers exhibited monomorphism among BC progeny and were thus excluded. We carried out χ2 analyses to carry forward only those markers fitting the expected 1:1 ratio in the BC-II population. Five of the markers showed segregation distortion at P = 0.05. Finally, 37 markers were employed for the construction of the SSR linkage map. Eight linkage groups including one Z-group were separated (Figure 4). They ranged from 58.7 cM (group 2) to 6.6 cM (group 5). Eight markers remained unlinked.
Figure 4.—

Microsatellite genetic map of B. mori linkage groups 1–8. Distances between locus positions are in centimorgans. Group 8 carries four Z-specific markers.
Conservation in heterologous species:
To test the conservation of B. mori microsatellite loci in heterologous species (Table 8), we used 30 B. mori microsatellite loci. All species except H. armigera were from the Bombycoidea superfamily. Twenty-two (73.3%) B. mori microsatellite loci were found to be conserved in B. mandarina, the ancestral species of B. mori, and 16 (53.3%) were conserved in A. assama, an Indian muga silkmoth, endemic to northeastern India. The species of S. c. ricini, A. mylitta, A. yamamai, and H. armigera shared four (13.3%); A. pernyi shared three (10%); whereas A. roylei and A. proylei shared only two (6.6%) conserved microsatellite loci with B. mori (supplementary Table S2 at http://www.genetics.org/supplemental/).
The alleles of four loci (Bmsat020, Bmsat110, Bmsat103, and Bmsat109) that showed amplification in most of the heterologous species were cloned and sequenced. We identified both repeat variations and mutations in the flanking sequences in most of the alleles. For example, out of two alleles of Bmsat020 we sequenced, one allele of 200 bp harbored one TTA repeat and the other of 171 bp contained both TTA and TA motifs in H. armigera (Figure 5). The locus Bmsat103 harboring an imperfect (GT) motif showed complex mutations characterized by variation in the repeat motif length and point mutations in the repeat motif as well as in the flanking sequences. The point mutations either created or destroyed the repeat perfection, in addition to creating a new class of repeat motif. In three species, A. roylei, A. yamamai, and H. armigera, a new pentanucleotide microsatellite repeat GGTTA was created in one of the alleles due to multiple point mutations (Figure 5). The locus Bmsat109 revealed an allele in A. pernyi, in which there was a duplication of 59 bp containing a GT repeat motif (Figure 5). The locus Bamsat110, which harbored both GATA and TA repeats, did not show much variation in repeat motif except in B. mandarina, which had only two repeat units for each of these motifs.
Figure 5.—

Repeat regions of the four microsatellite loci are mapped onto phylogeny of the silkmoths. The number of repeat units is indicated as subscripts. The allele sizes (in base pairs) are given in parentheses. Note that the number of alleles sequenced differs between species. NA indicates no PCR amplification and ND represents alleles for which sequence could not be determined. Subscript .. represents imperfections introduced in the repeat motif by mutations, the number and sequence of which can vary between different alleles. Superscript $ denotes duplication of 59-bp region containing (GT) repeat motif in one of the alleles of A. pernyi, and * represents creation of a new repeat motif by point mutations. Phylogeny of domestic silkmoth, wild silkmoths, and bollworm is based on 12S rRNA sequence of 430 bp. D. nasuta is used as an outgroup; bootstrap values are mentioned above the node (our unpublished data).
DISCUSSION
In this article we report in silico analysis of microsatellites and experimentally validate a few representative loci for allelic distribution, polymorphism, and conservation in heterologous species. Also, we mapped the polymorphic loci onto a backcross mapping population. Earlier reports on the distribution of microsatellites in insect species showed that, unlike higher animals, they are rich in short repeat tracts (Kruglyak et al. 1998; Chambers and Macavoy 2000; Li et al. 2002). In this study we mined microsatellites of ≥15 bases of mononucleotide repeats and ≥5 repeat units of other classes of repeats.
The B. mori genome is AT rich:
With the sequence data we used for analysis, poly(A/T) was far more abundant than poly(C/G) in both genomic and transcribed sequences, similar to earlier reports (Katti et al. 2001; Morgante et al. 2002). Among dinucleotide repeats TA was in excess in genomic sequences, whereas CG was the rarest (Table 1 and Figure 2). The excess of TA repeats found here deviated from the overall distribution found in arthropods, where CA was found to be the most frequent (Toth et al. 2000). Dinucleotide repeats have been found to be in excess, contributing nearly 50% in many species, and mononucleotide repeats are abundant in primates (Li et al. 2002). In B. mori, mono- and dinucleotide repeats accounted for 36 and 37.9% of the total repeats, respectively. Among trinucleotides, TAA was the most common trinucleotide repeat, whereas CGA and GCA were rare in both genomic and transcribed sequences. In Drosophila, where microsatellite distribution has been studied in detail, AGC is the highest, followed by AAC (Katti et al. 2001). The preponderance of AT-rich trinucleotide repeats in silkworm is consistent with findings in the genomes of Arabidopsis and the yeast Sacccharomyces cervisiae (Cardle et al. 2000; Young et al. 2000). On the contrary, the genomes of maize, human, and rice were rich in GC-rich microsatellites (Jurka and Pethiyagoda 1995; Chin et al. 1996; Temnykh et al. 2001). In accordance with the literature, most of the trinucleotide repeats were excess in ESTs compared to genomic sequences whereas other repeat types were fewer in ESTs (Li et al. 2002; Toth et al. 2000; Morgante et al. 2002). This increase was contributed by trinucleotide repeats other than TAA, GTA, and TGA (Table 1). Tetra-, penta-, and hexanucleotide repeats with one G or C in the repeat unit were found to be abundant in addition to 100% AT-rich repeat units. This kind of distribution might indicate that the presence of a single G/C in the repeat is probably stabilizing the repeat tract.
The most abundant tetra-, penta-, and hexanucleotide repeats were TAAA, CAAA, TTAA, CATA, TAAAA, TTAAA, TATAA, CAAAA, TAAAAA, TTAAAA, and CAAAAA. Long tetra- and pentanucleotide repeats were not found; instead, surprisingly, hexanucleotide repeat arrays were found to have longer repeat tracts (Figure 2). The occurrence of very high amounts of AT-rich repeats, irrespective of repeat type, can be explained by (a) the AT-rich genome of B. mori (∼60%); (b) low Tm, thus easy strand separation; and (c) mutations in poly(A/T) tracts at different time points leading to a combination of AT-rich repeats. It remains to be studied whether the AT-rich portion of the silkworm genome has any relationship with high-order chromatin structure similar to that documented for plant species (Pedersen et al. 1996; Schmidt and Heslop-Harrison 1996). In particular, this knowledge will be useful in light of the fact that silkworm chromosomes are holocentric in nature.
We estimated that microsatellites account for 0.31% of the genome of B. mori (Table 1), which is similar to that of yeast, less than that of plants and vertebrates, and greater than that of Caenorhabditis elegans. (Crollius et al. 2000; Toth et al. 2000; Morgante et al. 2002). The shorter repeat tracts were abundant in the Bombyx genome, similar to those in Drosophila (Table 2, Figure 2); repeat tracts >15 repeat units were sparse in contrast to those in mammals (Kruglyak et al. 1998). This shows that probably there is a limiting mechanism for the length of the repeat tract, as observed in most of the organisms studied (Schlötterer 2000). The lack of longer microsatellites with many repeats could be due to their short persistence time due to mutations and probably they are underrepresented (Harr and Schlötterer 2000).
Sex chromosomes have been found to differ in microsatellite density from autosomes in many eukaryotes. Human, rat (Beckman and Weber 1992), and mouse (Jarne et al. 1998) X chromosomes were found to have a lower abundance of microsatellites compared to autosomes, whereas the reverse is the case for dinucleotide repeats in the Drosophila X chromosome (Bachtrog et al. 1999). The B. mori Z chromosome, equivalent to the X chromosome of mammals and Drosophila, had a higher density of ATT repeats compared to both the autosomes and transcribed sequences (Table 1). These repeats accounted for 59.7% of the total trinucleotides in the Z chromosome. The relative overall trinucleotide microsatellite density was higher in the Z chromosome than in autosomes and ESTs. The Z chromosome sequences contained very few tetra- and hexanucleotide repeats and were devoid of pentanucleotides (Figure 2).
Flanking sequences and microsatellite origin:
Zhu et al. (2000) gave experimental evidence of microsatellite genesis from random sequences. It was shown that the birth of proto-microsatellites happens by short duplications of neighboring sequences without disturbing the stability of the surrounding region. The short repeat then expands by a replication slippage mechanism leading to variability. The generation and perpetuation of proto-microsatellites may be influenced by the nature and composition of the flanking sequences. Our results showed a positive correlation of the AT/GC distribution with flanking sequence, where the GC content of flanking sequence increased from a minimum of 30% to a maximum of 60% as the repeat composition increased from 0% GC to 100% GC (Figure 3). In a sequence that is AT rich, there is a higher possibility of an AT-rich repeat evolving rather than a GC-rich repeat or vice versa. This observation of positive correlation was similar irrespective of the length and type of the repeat motif analyzed. Although the origin and evolution of microsatellites is influenced by the flanking sequences, we did not observe any variation in the flanking sequence content with varying length of repeat unit. A similar observation of positive correlation was made by Bachtrog et al. (1999) in D. melanogaster with respect to TA repeat density, where length was independent of flanking AT content. Considering these results together, we propose that base composition of the flanking sequence may influence the origin and evolution of microsatellites but is independent of repeat unit length (Bachtrog et al. 1999).
Mutations in the repeat motifs:
Microsatellites grow until they reach a threshold length, after which they are abrogated by point mutations or indels disrupting the perfect repeats and thus preventing their infinite growth (Kruglyak et al. 1998; Ellegren 2000). The study of imperfections might indicate previous presence of perfect repeats, which can be reverted back to perfection by reverse mutations (Harr et al. 2000) or evolve into a different repeat motif by further mutations. Mutations in the center of the repeat motif accounted for 60.7% of the imperfect repeats as compared to 30.9% toward the 3′ end of the repeat and 8.4% toward the 5′ end (occurring rarely). This result is in sharp contrast with previous findings (Brohede and Ellegren 1999), which inferred from substitution rate in (CA)n microsatellites that the rate of nucleotide substitution is higher in the ends of the repeat regions than in the middle of the repeat regions. The discrepancy between our observations and that inferred by Brohede and Ellegren (1999) may be that their observations were solely based on 22 (CA)n repeats derived from noncoding regions whereas our observations are based on a large number of di- (138), tri- (120), tetra- (148), and pentanucleotide (85) repeats from both coding and noncoding regions (supplementary Table S1). Imperfect repeats <15 bases were not found (supplementary Table S1). Transitions and transversions constituted 74% of repeat disruptions in silkworm ESTs (Table 3) because they did not disturb the reading frame, which is not the case with indels. By contrast, in genomic sequences transitions/transversions and indels shared equal numbers of repeat disruptions without any bias toward any particular repeat type (Table 3).
Marker potential:
In this study, we went beyond in silico discovery to test the loci experimentally for allelic distribution and polymorphism. Eighty-nine alleles for 23 loci were identified in 13 diverse silkworm varieties (Table 5). Most of the loci showed 2–4 alleles. An imperfect dinucleotide repeat locus revealed as many 14 alleles, followed by a tetranucleotide locus with 10 alleles. Imperfect dinucleotide repeat motifs also showed higher polymorphism between Nistari and NB4D2 strains (Table 7). It has been observed that dinucleotide repeats have more slippage events per unit length of DNA than do other repeats (Kruglyak et al. 1998). Generally it is observed that, the longer the repeat stretch is, the higher the mutation rate and allele number are (Brinkmann et al. 1998), but in our results we did not observe any association of the repeat type/length with allele numbers. Our results may be explained by the small number of loci chosen for study combined with locus-specific mutation rates (Schlötterer et al. 1998).
The heterozygosity values ranged from 0.20 to 0.87 (Table 5), showing good variation in the 13 strains for the loci chosen. The three alleles, 215, 134, and 90 bp of Bmsat066, Bmsat132, and Bmsat110 loci, respectively, were present only in diapausing strains, and one allele of 146 bp of Bmsat132 was specific to nondiapausing strains (Table 6). However, further study of these loci may be useful to ascertain their demographic association. Bmsat012 and Bmsat015 loci derived from coding regions showed no polymorphism or allelic variation in the 13 silkmoth populations. This reinforced the observation that coding regions have lower variance than noncoding regions.
About 5% of 198 microsatellite loci revealed null alleles in the parental strains, Nistari and NB4D2, used for assaying for polymorphism. Most of these alleles inherited and segregated in Mendelian fashion (data not shown). Thus, the presence of segregating null alleles will not corrupt the linkage data. If undetected, a null allele will merely result in that individual being scored as a homozygote, resulting in loss of informativeness. However, in population studies, such null alleles, at an appreciable frequency, may result in depression of observed heterozygosity, compared with that expected on the basis of Hardy-Weinberg equilibrium. In almost all the articles reporting microsatellite markers in Lepidoptera, a significant departure from Hardy-Weinberg equilibrium was observed at many of the loci due to deficit of heterozygotes, suggesting the presence of null alleles (Bogdanovicz et al. 1997; Meglecz and Solignac 1998; Harper et al. 2000; Caldas et al. 2002; Cassel 2002; Flanagan et al. 2002; Keyghobadi et al. 2002; Amsellem et al. 2003).
Microsatellite mapping initiative:
Lepidoptera has received little attention in terms of genetics and genetic maps despite immense consequences in terms of economy and ecology. In B. mori, all the linkage maps except the RFLP map are based on dominant markers (Promboon et al. 1995; Shi et al. 1995; Yasukochi 1998; Tan et al. 2001; Kadono-Okuda et al. 2002). Here, we have presented a preliminary linkage map based on 37 microsatellite markers. This effort is a part of our ongoing program to develop a dense genetic linkage map of silkworm using fluorescent inter-simple sequence repeat (FISSR) markers (Nagaraju et al. 2002) and microsatellite markers. These mapped codominant markers will be useful in anchoring a genetic map on a physical map.
Cross-species amplification of microsatellite loci:
The cross-species amplification of silkworm microsatellite loci shows that mutation patterns of microsatellites are often complex: some loci tend to gain repeats (Weber and Wong 1993; Eichler et al. 1994), while others tend to lose repeats (Zhang et al. 1994), and size variations at microsatellite loci are caused by indels in the flanking sequences as well (Angers and Bernatchez 1997; Grimaldi and Crouau-Roy 1997; Colson and Goldstein 1999). In Drosophila, ∼60% of examined microsatellite loci divergence is in the lengths of flanking regions between species (Hutter et al. 1998; Colson and Goldstein 1999). In this study, 71 alleles sequenced from four loci revealed that complexity in interallelic variation is affected by both contraction and expansion of repeat motifs and indels in the flanking regions. At least 20 out of 71 alleles (28.2%) are affected by indels. Many alleles that are identical in length revealed sequence differences (identical by state) as against identical-sized alleles that have similar sequences (identical by descent). Such studies in lepidopteran microsatellites can be particularly useful for population studies and for increasing understanding of the origin, mutational processes, and structure of microsatellites, in addition to allowing more informed interpretation of genotyping data.
Of the 30 loci, 26 showed amplification in at least one of the heterologous species. Twenty-two loci showed amplification in B. mandarina and 15 showed amplification in A. assama. The number of alleles per locus observed was more in B. mori, the focal species, than in B. mandarina at 11 loci and the remaining 11 loci showed no difference in the number of alleles. Similarly 7 loci showed a greater number of alleles in B. mori than in A. assama while the remaining 8 loci showed no difference. The allelic length variation did not show any particular trend in the focal and nonfocal species. These preliminary results could be attributed to ascertainment bias (Ellegren et al. 1995; Forbes et al. 1995; Amos et al. 2003) rather than to directional evolution (Rubinsztein et al. 1995; Amos et al. 1996, 2003). Further studies on microsatellite variation by performing a reciprocal study surveying microsatellite allelic variation in number and length using microsatellite loci characterized from each of these species should shed additional light on directional evolution and ascertainment bias hypothesis.
The successful amplification of silkworm SSR markers across related wild species particularly in B. mandarina and A. assama provides “connectivity” for future consolidation of genetic and physical maps and provides the foundation for association of these maps with particular traits of interest. Also, it provides an opportunity to use SSR markers for investigating the wide range of genetic diversity that exists in wild species outside the gene pool of domesticated silkworm, B. mori.
It is observed that isolation of microsatellites from lepidopterans is very difficult (Meglecz et al. 2004; Zhang 2004) and thus we find as few as 20 reports describing microsatellites from lepidopterans in the literature in contrast with high numbers found in the hymenoptera. Hence, the present report on abundance of microsatellite motifs along with identification and validation of 198 microsatellite loci from B. mori and their utility in population studies, and conservation in heterologous moth species, will be of great use for researchers working on microsatellites in general and lepidopteran species in particular. In addition, the availability of the full sequence of the silkworm genome in the near future will greatly expand the repertoire of SSRs; the more polymorphic ones can be effectively exploited for mapping, annotation, and comparative genomics leading to a better utilization of silkworm, both as a model lepidopteran insect and as an economically important insect on which millions of rural people in the developing countries like China, India, and Thailand depend for their livelihood.
Acknowledgments
We particularly thank M. R. Goldsmith for useful suggestions on the manuscript. We thank K. P. Arun Kumar for assistance in in silico analysis of the sequence data and B. Srilatha and P. Chandrashekar for technical assistance. This work was supported by the Department of Biotechnology, Government of India by way of grants to J. Nagaraju.
References
- Altschul, S. F., W. Gish, W. Miller, E. W. Myers and D. J. Lipman, 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403–410. [DOI] [PubMed] [Google Scholar]
- Amos, W., S. J. Sawcer, R. W. Feakes and D. C. Rubinsztein, 1996. Microsatellites show mutational bias and heterozygote instability. Nat. Genet. 13: 390–391. [DOI] [PubMed] [Google Scholar]
- Amos, W., C. M. Hutter, M. D. Schug and C. F. Aquadro, 2003. Directional evolution of size coupled with ascertainment bias for variation in Drosophila microsatellites. Mol. Biol. Evol. 20: 660–662. [DOI] [PubMed] [Google Scholar]
- Amsellem, L., A. M. Risterucci and V. Benrey, 2003. Isolation and characterization of polymorphic microsatellite loci in Lobesia botrana Den. & Schiff. (Lepidoptera: Tortricidae). Mol. Ecol. Notes 3: 117–119. [Google Scholar]
- Angers, B., and L. Bernatchez, 1997. Complex evolution of a salmonid microsatellite locus and its consequences in inferring allelic divergence from size information. Mol. Biol. Evol. 14: 230–238. [DOI] [PubMed] [Google Scholar]
- Bachtrog, D., S. Weiss, B. Zangerl, G. Brem and C. Schlötterer, 1999. Distribution of dinucleotide microsatellites in the Drosophila melanogaster genome. Mol. Biol. Evol. 16: 602–610. [DOI] [PubMed] [Google Scholar]
- Beckman, J. S., and J. L. Weber, 1992. Survey of human and rat microsatellites. Genomics 12: 627–631. [DOI] [PubMed] [Google Scholar]
- Bogdanovicz, S. M., V. S. Mastro, D. C. Prasher and R. G. Harrison, 1997. Microsatellite variation among Asian and North American gypsy moths (Lepidoptera: Lymantriidae). Ann. Entomol. Soc. Am. 90: 768–775. [Google Scholar]
- Brinkmann, B., M. Klintschar, F. Neuhuber, J. Huhne and B. Rolf, 1998. Mutation rate in human microsatellites: influence of the structure and length of the tandem repeat. Am. J. Hum. Genet. 62: 1408–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brohede, J., and H. Ellegren, 1999. Microsatellite evolution: polarity of substitutions within repeats and neutrality of flanking sequences. Proc. R. Soc. Lond. Ser. B Biol. Sci. 266: 825–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldas, A., D. J. Hawthorne and P. Barbosa, 2002. Isolation and characterization of microsatellite markers from Zale galbanata (Lepidoptera: Noctuidae) and amplification in other members of the genus. Mol. Ecol. Notes 2: 296–297. [Google Scholar]
- Cardle, L., L. Ramsay, D. Milbourne, M. Macaulay, D. Marshall et al., 2000. Computational and experimental characterization of physically clustered simple sequence repeats in plants. Genetics 156: 847–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassel, A., 2002. Characterization of microsatellite loci in Coenonympha hero (Lepidoptera:Nymphalidae). Mol. Ecol. Notes 2: 566–568. [Google Scholar]
- Chakraborty, R., M. Kimmel, D. N. Stivers, L. J. Davison and R. Deka, 1997. Relative mutation rates at di-, tri-, and tetranucleotide microsatellite loci. Proc. Natl. Acad. Sci. USA 94: 1041–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers, G. K., and E. S. Macavoy, 2000. Microsatellites: consensus and controversy. Comp. Biochem. Physiol. B 126: 455–476. [DOI] [PubMed] [Google Scholar]
- Chin, E. C., M. L. Senior, H. Shu and J. S. Smith, 1996. Maize simple repetitive DNA sequences: abundance and allelic variation. Genome 39: 866–873. [DOI] [PubMed] [Google Scholar]
- Colson, I., and D. B. Goldstein, 1999. Evidence for complex mutations at microsatellite loci in Drosophila. Genetics 152: 617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornuet, J. M., and G. Luikart, 1996. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144: 2001–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crollius, H., O. Jaillon, C. Dasilva, C. Ozouf-Costaz, C. Fizames et al., 2000. Characterization and repeat analysis of the compact genome of the freshwater pufferfish Tetraodon nigroviridis. Genome Res. 10: 939–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Rienzo, A., A. C. Peterson, J. C. Garza, A. M. Valdes, M. Slatkin et al., 1994. Mutational processes of simple-sequence repeat loci in human populations. Proc. Natl. Acad. Sci. USA 91: 3166–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler, E. E., J. J. A. Holden, B. W. Popovich, A. L. Reiss, K. Snow et al., 1994. Length of uninterrupted CGG repeats determines instability in the FMR1 gene. Nat. Genet. 8: 88–94. [DOI] [PubMed] [Google Scholar]
- Ellegren, H., 2000. Heterogeneous mutation processes in human microsatellite DNA sequences. Nat. Genet. 24: 400–402. [DOI] [PubMed] [Google Scholar]
- Ellegren, H. C., R. Primmer and B. C. Sheldon, 1995. Microsatellite “evolution”: Directionality or bias? Nat. Genet. 11: 360–362. [DOI] [PubMed] [Google Scholar]
- Field, D., and C. Wills, 1996. Long, polymorphic microsatellites in simple microorganisms. Proc. R. Soc. Lond. Ser. B Biol. Sci. 263: 209–215. [DOI] [PubMed] [Google Scholar]
- Flanagan, N. S., M. J. Blum, A. Davison, M. Alamo, R. Albarrán et al., 2002. Characterization of microsatellite loci in neotropical Heliconius butterflies. Mol. Ecol. Notes 2: 398–401. [Google Scholar]
- Forbes, S. H., J. T. Hogg, F. C. Buchanan, A. M. Crawford and F. W. Allendorf, 1995. Microsatellite evolution in congeneric mammals: domestic and bighorn sheep. Mol. Biol. Evol. 12: 1106–1113. [DOI] [PubMed] [Google Scholar]
- Grimaldi, M. C., and B. Crouau-Roy, 1997. Microsatellite allelic homoplasy due to variable flanking sequences. J. Mol. Evol. 44: 336–340. [DOI] [PubMed] [Google Scholar]
- Harper, G. L., S. Piyapattanakorn, D. Goulson and N. Maclean, 2000. Isolation of microsatellite markers from the Adonis blue butterfly (Lysandra bellargus). Mol. Ecol. 9: 1948–1949. [DOI] [PubMed] [Google Scholar]
- Harr, B., and C. Schlötterer, 2000. Long microsatellite alleles in Drosophila melanogaster have a downward mutation bias and short persistence times, which cause their genome-wide under representation. Genetics 155: 1213–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harr, B., B. Zangerl and C. Schlötterer, 2000. Removal of microsatellite interruptions by DNA replication slippage: phylogenetic evidence from Drosophila. Mol. Biol. Evol. 17: 1001–1009. [DOI] [PubMed] [Google Scholar]
- Heckel, D. G., L. J. Gahan, Y. B. Liu and B. E. Tabashnik, 1999. Genetic mapping of resistance to Bacillus thuringiensis toxins in diamondback moth using biphasic linkage analysis. Proc. Natl. Acad. Sci. USA 20: 8373–8377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutter, C. M., M. D. Schug and C. F. Aquadro, 1998. Microsatellite variation in Drosophila melanogaster and Drosophila simulans: a reciprocal test of the ascertainment bias hypothesis. Mol. Biol. Evol. 15: 1620–1636. [DOI] [PubMed] [Google Scholar]
- Jarne, P., P. David and F. Viard, 1998. Microsatellites, transposable elements and the X chromosome. Mol. Biol. Evol. 15: 28–34. [DOI] [PubMed] [Google Scholar]
- Jolly, M. S., S. K. Sen, G. K. Prasad and A. K. Sengupta, 1979. Some cytological observations on interspecific hybrids of Antheraea pernyi and A. roylei. Cytologia 44: 259–263. [Google Scholar]
- Jurka, J., and C. Pethiyagoda, 1995. Simple repetitive DNA sequences from primates: compilation and analysis. J. Mol. Evol. 40: 120–126. [DOI] [PubMed] [Google Scholar]
- Katti, M. V., P. K. Ranjekar and V. S. Gupta, 2001. Differential distribution of simple sequence repeats in eukaryotic genome sequences. Mol. Biol. Evol. 18: 1161–1167. [DOI] [PubMed] [Google Scholar]
- Keyghobadi, N., J. Roland and C. Strobeck, 2002. Isolation of novel microsatellite loci in the Rocky Mountain apollo butterfly, Parnassius smintheus. Hereditas 136: 247–250. [DOI] [PubMed] [Google Scholar]
- Kadono-Okuda, K., E. Kosegawa, K. Mase and W. Hara, 2002. Linkage analysis of maternal EST cDNA clones covering all twenty-eight chromosomes in the silkworm, Bombyx mori. Insect Mol. Biol. 11: 443–451. [DOI] [PubMed] [Google Scholar]
- Koike, Y., K. Mita, M. G. Suzuki, S. Maeda, H. Abe et al., 2003. Genomic sequence of a 320-kb segment of the Z chromosome of Bombyx mori containing a kettin ortholog. Mol. Genet. Genomics 269: 137–149. [DOI] [PubMed] [Google Scholar]
- Kosambi, D. D., 1944. The estimation of map distances from recombination values. Ann. Eugen. 12: 172–175. [Google Scholar]
- Kruglyak, S., R. T. Durrett, M. D. Schug and C. F. Aquadro, 1998. Equilibrium distributions of microsatellite repeat length resulting from a balance between slippage events and point mutations. Proc. Natl. Acad. Sci. USA 95: 10774–10778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander, E. S., P. Green, J. Abrahamson, A. Barlow, M. J. Daly et al., 1987. MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1: 174–181. [DOI] [PubMed] [Google Scholar]
- Li, Y. C., A. B. Korol, T. Fahima, A. Beiles and E. Nevo, 2002. Microsatellites: genomic distribution, putative functions and mutational mechanisms: a review. Mol. Ecol. 11: 2453–2465. [DOI] [PubMed] [Google Scholar]
- Litt, M., and J. A. Luty, 1989. A hypervariable microsatellite revealed by in vitro amplification of a dinucleotide repeat within the cardiac muscle actin gene. Am. J. Hum. Genet. 44: 397–401. [PMC free article] [PubMed] [Google Scholar]
- Meglecz, E., and M. Solignac, 1998. Microsatellite loci for Parnassius mnemosyne (Lepidoptera). Hereditas 128: 179–180. [Google Scholar]
- Meglecz, E., F. Petenian, E. Danchin, A. Couur D'Acier, J. Y. Rasplus et al., 2004. High similarity between flanking regions of different microsatellites detected within each of two species of Lepidoptera: Parnassius apollo and Euphydryas aurinia. Mol. Ecol. 13: 1693–700. [DOI] [PubMed] [Google Scholar]
- Meyer, E., P. Wiegand, S. P. Rand, D. Kuhlmann, M. Brack et al., 1995. Microsatellite polymorphisms reveal phylogenetic relationships in primates. J. Mol. Evol. 41: 10–14. [DOI] [PubMed] [Google Scholar]
- Mita, K., M. Morimyo, K. Okano, Y. Koike, J. Nohata et al., 2003. The construction of an EST database for Bombyx mori and its application. Proc. Natl. Acad. Sci. USA 100: 14121–14126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mita, K., M. Kasahara, S. Sasaki, Y. Nagayasu, T. Yamada et al., 2004. The genome sequence of silkworm, Bombyx mori. DNA Res. 11: 27–35. [DOI] [PubMed] [Google Scholar]
- Morgante, M., M. Hanafey and W. Powell, 2002. Microsatellites are preferentially associated with non-repetitive DNA in plant genomes. Nat. Genet. 30: 194–200. [DOI] [PubMed] [Google Scholar]
- Nagaraja, G. M., and J. Nagaraju, 1995. Genomic fingerprinting of silkworm, Bombyx mori using random arbitrary primers. Electrophoresis 16: 1633–1638. [DOI] [PubMed] [Google Scholar]
- Nagaraju, J., and M. R. Goldsmith, 2002. Silkworm genomics—progress and prospects. Curr. Sci. 83: 415–425. [Google Scholar]
- Nagaraju, J., and M. S. Jolly, 1986. Interspecific hybrid of Antheraea roylei and A. pernyi—a cytogenetic reassessment. Theor. Appl. Genet. 72: 269–273. [DOI] [PubMed] [Google Scholar]
- Nagaraju, J., A. Sharma, B. N. Sethuraman, G. V. Rao and L. Singh, 1995. Molecular characterization of silkworm races using Bkm probe. Electrophoresis 16: 1639–1642. [DOI] [PubMed] [Google Scholar]
- Nagaraju, J., V. Klimenko and P. Couble, 2000 The silkworm Bombyx mori, a model genetic system, pp. 219–239 in Encyclopedia of Genetics, edited by E. Reeves. Fitzroy Dearborn, London.
- Nagaraju, J., M. Kathirvel, M. Muthulakshmi, E. V. Subbiah and L. D. Kumar, 2002. FISSR-PCR: a simple and sensitive assay for high throughput genotyping and genetic mapping. Mol. Cell. Probes 16: 67–72. [DOI] [PubMed] [Google Scholar]
- Nei, M., 1972. Genetic distance between populations. Am. Nat. 106: 283–291. [Google Scholar]
- Pedersen, C., S. K. Rasmussen and I. Linde-Laursen, 1996. Genome and chromosome identification in cultivated barley and related species of the Triticeae (Poaceae) by in situ hybridization with the GAA-satellite sequence. Genome 39: 93–104. [DOI] [PubMed] [Google Scholar]
- Prasad, M. D., and J. Nagaraju, 2003. A comparative phylogenetic analysis of full-length mariner elements isolated from the Indian tasar silkmoth, Antheraea mylitta (Lepidoptera: Saturniidae). J. Biosci. 28: 443–453. [DOI] [PubMed] [Google Scholar]
- Promboon, A., T. Shimada, H. Fujiwara and M. Kobayashi, 1995. Linkage map of random amplified polymorphic DNAs (RAPDs) in the silkworm Bombyx mori. Genet. Res. 66: 1–7. [Google Scholar]
- Reddy, K. D., E. G. Abraham and J. Nagaraju, 1999. a Microsatellites of the silkworm, Bombyx mori: abundance, polymorphism and strain characterization. Genome 42: 1057–1065. [PubMed] [Google Scholar]
- Reddy, K. D., J. Nagaraju and E. G. Abraham, 1999. b Genetic characterization of the silkworm, Bombyx mori by inter-simple sequence repeat (ISSR)-anchored PCR. Heredity 83: 681–687. [DOI] [PubMed] [Google Scholar]
- Rubinsztein, D. C., W. Amos, J. Leggo, S. Goodburn, S. Jain et al., 1995. Microsatellite evolution—evidence for directionality and variation in rate between species. Nat. Genet. 10: 337–343. [DOI] [PubMed] [Google Scholar]
- Schlötterer, C., 1998. Genome evolution: Are microsatellites really simple sequences? Curr. Biol. 8: 132–134. [DOI] [PubMed] [Google Scholar]
- Schlötterer, C., 2000. Evolutionary dynamics of microsatellite DNA. Chromosoma 109: 365–371. [DOI] [PubMed] [Google Scholar]
- Schlötterer, C., 2004. The evolution of molecular markers—just a matter of fashion? Nat. Rev. Genet. 5: 63–69. [DOI] [PubMed] [Google Scholar]
- Schlötterer, C., C. Vogl and D. Tautz, 1997. Polymorphism and locus-specific effects on polymorphism at microsatellite loci in natural Drosophila melanogaster populations. Genetics 146: 309–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlötterer, C., R. Ritter, B. Harr and G. Brem, 1998. High mutation rate of a long microsatellite allele in Drosophila melanogaster provides evidence for allele-specific mutation rates. Mol. Biol. Evol. 15: 1269–1274. [DOI] [PubMed] [Google Scholar]
- Schmidt, T., and J. S. Heslop-Harrison, 1996. The physical and genomic organization of microsatellites in sugar beet. Proc. Natl. Acad. Sci. USA 93: 8761–8765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, A., M. P. Niphadkar, P. Kathirvelu, J. Nagaraju and L. Singh, 1999. DNA fingerprinting variability within and among the silkworm Bombyx mori genotypes and estimation of their genetic relatedness using Bkm-derived probe. J. Hered. 90: 315–319. [Google Scholar]
- Shi, J., D. G. Heckel and M. R. Goldsmith, 1995. A genetic linkage map for the domesticated silkworm, Bombyx mori, based on restriction fragment length polymorphisms. Genet. Res. 66: 109–126. [Google Scholar]
- Slatkin, M., 1995. Hitchhiking and associative overdominance at a microsatellite locus. Mol. Biol. Evol. 12: 473–480. [DOI] [PubMed] [Google Scholar]
- Streelman, J. T., R. Zardoya, A. Meyer and S. A. Karl, 1998. Multilocus phylogeny of cichlid fishes (Pisces: Perciformes): evolutionary comparison of microsatellite and single-copy nuclear loci. Mol. Biol. Evol. 15: 798–808. [DOI] [PubMed] [Google Scholar]
- Tan, Y. D., C. Wan, Y. Zhu, C. Lu, Z. Xiang et al., 2001. An amplified fragment length polymorphism map of the silkworm. Genetics 157: 1277–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tautz, D., 1989. Hypervariability of simple sequences as a general source for polymorphic DNA markers. Nucleic Acids Res. 17: 6463–6471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temnykh, S., G. Declerck, A. Lukashova, L. Lipovich, S. Cartinhour et al., 2001. Computational and experimental analysis of microsatellites in rice (Oryza sativa L): frequency, length variation, transposon associations, and genetic marker potential. Genome Res. 11: 1441–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth, G., Z. Gaspari and J. Jurka, 2000. Microsatellites in different eukaryotic genomes: survey and analysis. Genome Res. 10: 967–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber, J. L., 1990. Informativeness of human (dC-dA)n. (dG-dT)n polymorphisms. Genomics 7: 524–530. [DOI] [PubMed] [Google Scholar]
- Weber, J. L., and P. E. May, 1989. Abundant class of human DNA polymorphisms which can be typed using the polymerase chain reaction. Am. J. Hum. Genet. 44: 388–396. [PMC free article] [PubMed] [Google Scholar]
- Weber, J. L., and C. Wong, 1993. Mutation of human short tandem repeats. Hum. Mol. Genet. 2: 1123–1128. [DOI] [PubMed] [Google Scholar]
- Wierdl, M., M. Dominska and T. D. Petes, 1997. Microsatellite instability in yeast: dependence on the length of the microsatellite. Genetics 146: 769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasukochi, Y., 1998. A dense genetic map of the silkworm, Bombyx mori, covering all chromosomes based on 1018 molecular markers. Genetics 150: 1513–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young, E. T., J. S. Sloan and K. Van Riper, 2000. Trinucleotide repeats are clustered in regulatory regions in Saccharomyces cerevisiae. Genetics 154: 1053–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, D., 2004. Lepidopteran microsatellite DNA: redundant but promising. Trends Ecol. Evol. 19: 507–509. [DOI] [PubMed] [Google Scholar]
- Zhang, L., E. P. Leeflang, J. Yu and N. Arnheim, 1994. Studying human mutations by sperm typing: instability of CAG trinucleotide repeats in the human androgen receptor gene. Nat. Genet. 7: 531–535. [DOI] [PubMed] [Google Scholar]
- Zhu, Y., J. E. Strassmann and D. C. Queller, 2000. Insertions, substitutions, and the origin of microsatellites. Genet. Res. 76: 227–236. [DOI] [PubMed] [Google Scholar]