

Abstract
In the budding yeast, Saccharomyces cerevisiae, control of cell proliferation is exerted primarily during G1 phase. The G1-specific transcription of several hundred genes, many with roles in early cell cycle events, requires the transcription factors SBF and MBF, each composed of Swi6 and a DNA-binding protein, Swi4 or Mbp1, respectively. Binding of these factors to promoters is essential but insufficient for robust transcription. Timely transcriptional activation requires Cln3/CDK activity. To identify potential targets for Cln3/CDK, we identified multicopy suppressors of the temperature sensitivity of new conditional alleles of SWI6. A bck2Δ background was used to render SWI6 essential. Seven multicopy suppressors of bck2Δ swi6-ts mutants were identified. Three genes, SWI4, RME1, and CLN2, were identified previously in related screens and shown to activate G1-specific expression of genes independent of CLN3 and SWI6. The other four genes, FBA1, RPL40a/UBI1, GIN4, and PAB1, act via apparently unrelated pathways downstream of SBF and MBF. Each depends upon CLN2, but not CLN1, for its suppressing activity. Together with additional characterization these findings indicate that multiple independent pathways are sufficient for proliferation in the absence of G1-specific transcriptional activators.
FAITHFUL execution of cell cycle events occurs as a consequence of coordinating the activity of key cell cycle regulators with the factors required for the execution of the events that they regulate. An important component of that regulation involves the coordinate expression of the genes encoding the relevant proteins during specific cell cycle phases. Although cell cycle-dependent transcriptional activation has been recognized for decades, the recent application of genome-wide transcriptional profiling has revealed the extent to which this form of regulation has been implemented (Cho et al. 1998; Spellman et al. 1998; Iyer et al. 2001). Those approaches have revealed a broad array of distinct patterns of cell cycle-dependent transcription and have contributed to the identification and characterization of the transcription factors responsible for those expression patterns.
G1-specific genes were among the first cell cycle-regulated genes to be recognized and are certainly among the best studied. Those genes are expressed specifically during late G1 phase preceding the events associated with cell cycle initiation. In the budding yeast, those events include formation of the bud, duplication of the spindle pole body, and DNA replication (Wittenberg and Flick 2003). Among the G1-specific genes are many encoding elements involved in the regulation and execution of those processes.
Our understanding of G1-specific transcription comes largely from the study of the expression of the HO gene, which encodes the initiating endonuclease for mating type switching (reviewed by Nasmyth 1993). Those studies led to the identification and characterization of the G1-specific transcription factor SBF that, together with a second factor MBF, is responsible for expression of this important gene family of genes (reviewed in Breeden 1996). SBF and MBF are both heterodimeric transcription factors composed of a unique DNA-binding component, Swi4 or Mbp1, respectively, and a common component, Swi6. The heterodimeric factors are sufficient for binding to promoter elements known as SCBs and MCBs, respectively. One or both of those elements are generally found in the promoters of G1-specific genes and the SBF and MBF transcription factors have been shown to bind to the majority of those promoters in vivo (Iyer et al. 2001).
Although binding of SBF or MBF to a promoter is necessary for transcriptional activation, it is not sufficient. In fact, both transcription factors are known to occupy their binding sites in the promoters of G1-specific genes during early G1 phase when those genes are transcriptionally inactive. Transcriptional activation occurs only during late G1 phase as a consequence of activation of the G1-specific cyclin-dependent protein kinase Cln3/Cdc28 (Tyers et al. 1993; Dirick et al. 1995; Stuart and Wittenberg 1995). Only then does the transcription factor activate expression of the genes, apparently by promoting recruitment of the RNA Pol II to join the mediator complex and complete the formation of the Pol II holoenzyme at the basal promoters (Cosma et al. 1999; Bhoite et al. 2001; Cosma et al. 2001).
Despite the importance of SBF and MBF for the concerted transcriptional activation of G1-specific promoters, neither transcription factor alone is essential for viability (although swi4Δ is lethal in some strains; Ogas et al. 1991). However, inactivation of both Swi4 and Mbp1, the DNA-binding components of these factors, results in the failure to proliferate (Koch et al. 1993; Nasmyth and Dirick 1991). Although mutations in Swi6, a common component of both factors, cause a severe morphological phenotype, it is not essential for viability unless Bck2 is also inactive (Epstein and Cross 1994; Di Como et al. 1995; Wijnen and Futcher 1999). Bck2 is required for the low level of cell cycle-dependent transcriptional activation of G1-specific genes observed in the absence of Swi6 via a mechanism that remains to be elucidated (Epstein and Cross 1994; Di Como et al. 1995).
The products of G1-specific genes are involved in diverse functions. Many of those are associated with events that occur as a consequence of start, including DNA replication, spindle pole body duplication, septin ring formation, and budding (Wittenberg and Flick 2003). The expression of those genes during G1 phase facilitates the coordinate execution of those events and reflects an economic use of cellular machinery. However, there is partial functional redundancy among the G1-specific regulators. For instance, among the SBF and MBF targets are CLN1, CLN2, CLB5, and CLB6, important regulators of late G1 events and DNA replication, which are not required individually for cell proliferation but lead to G1 arrest when coordinately inactivated (Schwob and Nasmyth 1993). Similarly, PCL1 and PCL2, two other SBF targets are essential in the absence of CLN1 and CLN2 (Espinoza et al. 1994; Measday et al. 1994). Other G1-specific genes encode essential products, such as DNA polymerases and other replication proteins. Yet, despite the importance of G1-specific gene products for the execution of a number of critical cell cycle events, inactivation of Swi6 does not lead to cell cycle arrest and, although inactivation along with Bck2 results in arrest, the terminal phenotype is not a characteristic G1 arrest phenotype (Nasmyth and Dirick 1991). Although poorly characterized, the phenotype of bck2Δ swi6Δ mutants arrests predominantly as budded cell or multibudded cells with a high degree of cell lysis consistent with defects in cytokinesis and cell wall synthesis (our unpublished observation). Thus, the consequences of a deficiency in SBF and MBF appear to reflect the inefficient execution of post-start events, including but not limited to septin ring formation and cell wall deposition, rather than the failure to progress out of G1 phase that is characteristic of the complete loss of G1- and S-phase cyclins.
In the interest of identifying genes that act upstream or in concert with Swi6 to activate G1-specific transcription, we performed a screen for dosage suppressors of a bck2Δ swi6-ts mutant. That screen resulted in the identification of a number of genes that when present in increased dosage in cells can relieve or reduce the temperature sensitivity of the screening strain. Each of the genes identified in this manner had been previously studied. On the basis of our current understanding they exhibit little apparent commonality of function. Our analysis of these suppressors has revealed that most act independently of SWI6 and BCK2. They are competent to bypass a null mutation of Swi6, suggesting that they either act downstream of Swi6 or facilitate a parallel pathway sufficient to bypass the deficiency. This study suggests that the roles of Swi6 targets are disparate in nature and that multiple independent defects contribute to the lethality resulting from the inactivation of G1-specific transcription.
MATERIALS AND METHODS
Yeast strains, cultures, and plasmids:
All yeast strains were isogenic to an arg4Δ derivative of BF264-15D (MATa ura3Δns ade1 his2 leu2-3, 112, trp1-1a; Richardson et al. 1989). The relevant genotypes of strains used in this study are provided in Table 1. All strains were grown in standard culture medium and standard yeast genetic methods were used.
TABLE 1.
Yeast strains used in this study
| Yeast strain | Genotype | Source |
|---|---|---|
| KFY193 | bck2::ARG4 swi6-4::KAN | This work |
| KFY194 | bck2::ARG4 swi6-12::KAN | This work |
| TAY477 | bck2::ARG4 swi6::TRP1 HIS2::GAL1:CLN2 | T. Kesti and C. Wittenberg (unpublished data) |
| TAY461 | bck2::ARG4 cln3::URA3 HIS2::GAL1:CLN2 | T. Kesti and C. Wittenberg (unpublished data) |
| KFY115 | mbp1::URA3 swi4::LEU2 HIS2::GAL:CLN2 | This work |
| KFY775 | bck2::ARG4 swi6-4::KAN cln1::URA3 | This work |
| KFY777 | bck2::ARG4 swi6-4::KAN cln2::URA3 | This work |
| KFY884 | bck2::ARG4 swi6-4::KAN whi3::HYG | This work |
| KFY430 | bck2::ARG4 swi6-4::KAN swe1::LEU2 | This work |
| KFY 707 | bck2::ARG4 swi6-4::KAN sic1::URA3 | This work |
To test temperature sensitivity and suppression cells were grown to log phase, counted, and diluted to ∼1000 cells per microliter. Tenfold serial dilutions were prepared and 5 μl of each dilution were spotted onto plates selective for the plasmid or on YEPD or YEPG plates. Plates were incubated 2–3 days at the temperatures indicated. Two or three independent transformants were used for each strain. The data presented are representative from at least two independent experiments.
Standard molecular biology techniques were used for all cloning and PCR procedures. Plasmids used in this study are listed in Table 2. Details on primers, plasmids, and cloning strategies are available on request.
TABLE 2.
Plasmids used in this study
| Plasmid | Parent and insert | Source |
|---|---|---|
| YEp24 | YEp24 | Struhl et al. (1979) |
| YEplac181 | YEplac181 | Gietz and Sugino (1988) |
| YEplac195 | YEplac195 | Gietz and Sugino (1988) |
| pRS416 | pRS416 | Sikorski and Hieter (1989) |
| SS6-78 | YEp24-genomic DNA | This study |
| SS6-395 | YEp24-genomic DNA | This study |
| SS6-396 | YEp24-genomic DNA | This study |
| SS6-406 | YEp24-genomic DNA | This study |
| pKF162 | YEplac195-FBA1 | This study |
| pKF142 | YEplac195-PAB1 | This study |
| pKF160 | YEplac195-GIN4 | This study |
| pKF158 | YEplac195-RPL40a | This study |
| pKF181 | YEplac181-PAB1-1 | This study |
| pKF182 | YEplac181-pab1ΔRRM1 | This study |
| pKF183 | YEplac181-pab1ΔRRM2 | This study |
| pKF184 | YEplac181-pab1ΔRRM3 | This study |
| pKF185 | YEplac181-pab1ΔRRM4 | This study |
| pKF186 | YEplac181-pab1ΔC | This study |
| pKF187 | YEplac181-pab1-180 | This study |
| pBD1378 | pRS316-SWI6 | Sidorova et al. (1995) |
| pKF163 | YEplac181-GIN4 | This study |
| YEp/gin4K48M | YEplac181-gin4K48M | Longtine et al. (1998) |
| pRS316-CDC3-GFP | pRS316-CDC3-GFP | Caviston et al. (2003) |
| pUB221 | pRS-CUP1-UBI-6xHis-Myc | Willems et al. (1996) |
Cell size analysis:
Cell size analysis was performed using a Coulter Z2 particle cell analyzer (Beckman-Coulter). Cell size distribution was analyzed using the Z2 AccuComp software (Beckman-Coulter).
Microscopy:
For visualization of Cdc3-GFP, cells were fixed in methanol at −70°. Differential interference microscopy (DIC) and fluorescence microscopy were performed using an Eclipse E800 microscope (Nikon, Melville, NY) with a ×100 objective. Cell images were captured with a Quantix CCD (Photometrics, Tucson, AZ) camera using IPLab Spectrum software (Signal Analytics, Vienna, VA). Phase-contrast microscopy was performed on an Axiostar plus (Zeiss) with a ×40 objective. Images were captured with a Coolpix 4500 (Fuji) camera.
RNA and protein analyses:
Cells were grown to log phase at permissive temperature. The culture was split and half of the cells remained at permissive temperature whereas the other half was shifted to 37° for 6 hr. Cell pellets collected for RNA and protein were frozen and stored at −80°.
Total RNA isolation and Northern blotting were performed as described previously (Stuart and Wittenberg 1994). The membrane was hybridized with radiolabeled probes (HiPrime) in buffer H (100 mm sodium phosphate buffer, pH 7.0, 400 mm NaCl, 5 mm EDTA, 1% SDS, 10% dextran sulfate, and 0.1 mg/ml denatured salmon sperm DNA). Northern blots were analyzed by Phospho-Imaging and ImageQuant software.
Protein extracts were prepared in RIPA buffer [1% doxycholic acid, 1% Triton-X100, 0.1% SDS, 250 mm NaCl, 50 mm Tris, pH 7.5, 10 mm sodium pyrophosphate, phosphatase inhibitors (5 mm EDTA, 5 mm EGTA, 50 mm NaF, and 0.1 mm orthovanadate), and protease inhibitors (1 mm PMSF, 2 μg/ml aproteine, leupeptin, and pepstatin)]. Cells were broken with glass beads three times for 40 sec in a FastPrep FP120 (Qbiogene, Carlsbad, CA). Proteins were separated on 10% SDS-PAGE, blotted onto a PVDF membrane, and detected by polyclonal rabbit anti-Swi6 (generous gift from Linda Breeden) or anti-Cdc28 antibody.
Construction of temperature-sensitive swi6 mutants:
Temperature-sensitive swi6 alleles were isolated by a modification of the technique described by Muhlrad et al. (1992). SWI6 was amplified from YIplac204-SWI6 with primers homologous to the MCS of pUC19 under mutagenic conditions (1 mm dCTP, dTTP, 0.2 mm dGTP, dATP, 30 pmol of each primer, 7 mm MgCl2, 0.5 mm MnCl2, 20 mm Tris-HCl, pH 8.4, 50 mm KCl, and 0.5 units of Taq DNA polymerase in a total volume of 100 μl). The PCR product was cotransformed with BamHI-digested YCplac22 into a bckΔ swi6Δ strain kept alive by GAL-CLN2. In vivo gap repair resulted in YCplac22 plasmids containing mutagenized SWI6. Transformants were grown on glucose-containing plates at 25° and then replica plated and colonies unable to grow on 37° were selected. Plasmids containing temperature-sensitive alleles of SWI6 were rescued and subcloned to replace wild-type SWI6 in a bck2Δ strain.
Multicopy suppressor screen:
Strains bck2Δ swi6-4 and bck2Δ swi6-12 were transformed with a genomic Yep24-based library (Carlson and Botstein 1982). We screened a total of 144,000 transformants for growth at 1° higher than maximal permissive temperature (34°–35°). A total of 529 colonies were isolated that were able to grow at the higher temperature. These clones were tested for loss of suppression on FOA plates. The remaining clones were classified according to the extent of suppression into three groups, 1°, 2°, or 3° above restrictive temperature. In total 258 clones were analyzed, 53 of the 1° suppressors, 222 of the 2° suppressors, and 18 of the 3° suppressors. Analysis was first done by restriction digest and clones appearing to be different as judged from three different digests (EcoRV, HindIII, and XbaI) were sequenced to identify the genomic inserts. The suppressing gene was then identified by subcloning and retesting of suppression. Once identified, genes were amplified by PCR using Vent polymerase (New England Biolabs, Beverly, MA) and cloned into YEplac195 or YEplac181. In the fully suppressing class (3°) all but 2 clones (containing SWI4) contained plasmids with the SWI6 gene. Most of the 2° suppressors contained SWI4 or SWI6 but RME1 (1 clone), CLN2 (4 clones), BCK2 (1 clone), PAB1 (5 clones), and GIN4 (10 clones) were also found in that class. RPL40a (3 clones) and FBA1 (1 clone) were found among the 1° suppressors. No other genes were confirmed as suppressors after rescreening.
RESULTS
Screen for high-copy suppressors of bck2Δ swi6-4 and bck2Δ swi6-12 mutants:
Temperature-sensitive swi6 mutants were constructed in vitro by PCR mutagenesis of SWI6 and screened for their ability to support growth of a bck2Δ swi6Δ mutant at 25° but not at 37°. The inactivation of BCK2 is required to render cells lacking Swi6 function inviable. Two such mutant alleles, swi6-4 and swi6-12 in a bck2Δ background, were chosen for the genetic screen on the basis of both their apparent lack of mutant phenotype at 25° and their inviability at 37°. These alleles were introduced at single genomic copy into a bck2Δ strain to yield bck2Δ swi6-4 and bck2Δ swi6-12. These strains have restrictive temperature of 34°–35° (Figure 1, A and B). As a measure of the functionality of these alleles, we analyzed cell size and morphology at both the permissive and restrictive temperature (Figure 1, D and E; data not shown). The swi6-4 allele has only a modest cell size phenotype and no morphological phenotype at the permissive temperature. This is in contrast to either the same allele at the restrictive temperature or the swi6Δ mutant, which exhibits a similar morphological defect (Figure 1E). Although inactivation of BCK2 results in an increase in cell size, it causes no morphological defect and combining bck2Δ with either allele results in no additional size or morphological phenotype. Nevertheless, it dramatically enhances the temperature-sensitive phenotype. The presence of SWI6 on a plasmid complements both the temperature sensitivity and morphological phenotypes of both the swi6-4 and swi6-12 mutant alone or in combination with bck2Δ. Finally, analysis of the Swi6 protein in the swi6-4 temperature-sensitive mutant demonstrates that the protein is thermolabile (Figure 1C). These observations suggest that these alleles of swi6 are largely wild type at permissive temperature but inactive at the restrictive temperature. Yet, strikingly, analysis of the accumulation of the transcripts of two Swi6-dependent genes, CLN1 and CLN2, revealed that are both significantly diminished at the permissive temperature and only slightly further affected at the restrictive temperature (Figure 5).
Figure 1.—
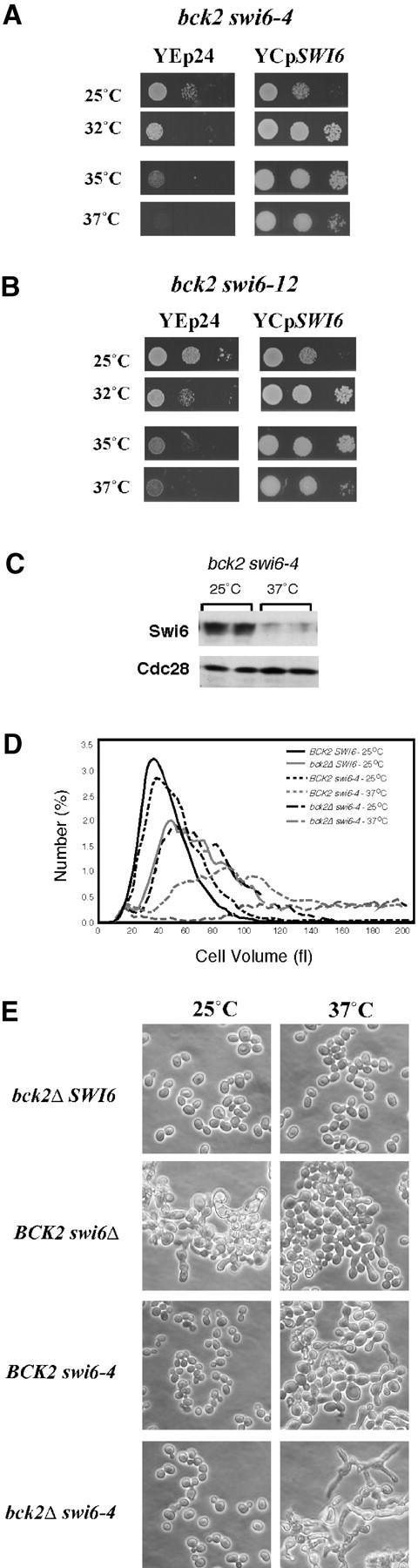
Characterization of bck2Δ swi6-ts mutants. (A and B) Thermosensitivity of bck2Δ swi6-4 and bck2Δ swi6-12 mutants. bck2 swi6-4 (A) or bck2 swi6-12 (B) with either control plasmid (Yep24) or YCp SWI6 plasmid were grown to log phase in −URA medium at 25°. Cells were counted and 10-fold serial dilutions with a starting dilution of ∼5000 cells were spotted onto −URA plates and incubated at the temperatures indicated for 2–3 days. (C) Swi6 protein is thermolabile in swi6-ts mutant. Cells were grown to log phase and then split and either grown at permissive temperature or shifted to 37° for 6 hr. After separation on a 10% SDS gel and Western blotting, Swi6 was detected with a polyclonal anti-Swi6 antibody. Cdc28 is shown as loading control. (D) Cell size of bck2 and swi6 mutants. Strains carrying the indicated SWI6 and BCK2 alleles were grown at 25° in YEPD and then either left untreated or shifted to 37° for 3 hr. Approximately 1.5 × 105 cells were sonicated and diluted into 20 ml of physiological saline solution and the mean cell volume in femtoliters was determined with a Coulter Z2 Channelyzer. Cell number in each size class is presented as a percentage of the total population. (E) Morphology of bck2 and swi6 mutants. Cells carrying the indicated BCK2 and SWI6 alleles were grown to log phase at 25° in YEPD and then either left untreated or shifted to 37° for 3 hr. Cells were imaged by phase-contrast microscopy on an Axiostar plus microscope (Zeiss) with a ×40 objective.
Figure 5.—

Analysis of G1 cyclin transcripts and protein in bck2Δ swi6-4 mutants containing multicopy PAB1. (A) Overexpression PAB1 leads to an increase in CLN1 and CLN2 transcript. Wild-type and bck2 swi6-4 cells were transformed with either control plasmid or plasmid overexpressing PAB1. Cells were grown to log phase in −Ura medium. The cells were then split and either grown at permissive temperature or shifted to 37° for 6 hr. CLN1, CLN2, and ACT1 transcript levels were determined by Northern blotting. CLN1 and CLN2 RNA levels were normalized to ACT1 RNA and are presented as the proportion of the RNA level in wild-type cells at the permissive temperature.
After transformation with a YEp24-based yeast genomic library, we screened ∼1.4 × 105 transformants for growth at 1° above the maximum permissive temperature. We have analyzed the majority of the suppressing plasmids from the 529 suppressed strains (see materials and methods for details of the screen). Most of those clones carried plasmids containing SWI4. The next most frequent isolated gene was SWI6. In addition, plasmids containing BCK2, CLN2, and RME1, genes previously shown to suppress mutations in swi6, were isolated (Epstein and Cross 1994; Di Como et al. 1995; Toone et al. 1995). Two genes, MSN1 or NHP6A, which had previously been shown to suppress the defect in ho::LacZ expression caused by the temperature-sensitive swi6-405 and swi6-406 alleles bearing mutations in the ankyrin repeat region of SWI6 (Sidorova and Breeden 1999), were not among those identified in this screen. In addition, we have isolated plasmids containing four genes not previously shown or predicted to suppress a bck2 swi6ts mutant (Figure 2A) each of which is discussed below.
Figure 2.—

Overview of bck2Δ swi6-ts suppressors. (A) Dosage suppressors of bck2Δ swi6-ts mutants. After screening ∼1 × 105 transformants at 1° higher than maximal restrictive temperature, plasmids SS6/78, SS6/395, SS6/396, and SS6/406 were isolated as dosage suppressors of bck2 swi6-ts. bck2 swi6-4 cells were retransformed with these plasmids, grown to log phase at 25° in −URA medium and spotted as described for Figure 1. The suppressing genes were identified by subcloning as PAB1, GIN4, FBA1, and RPL40a/UBI1, respectively. (B) Bypass of bck2Δ swi6Δ. FBA1, GIN4, and PAB1 not only are able to suppress the temperature sensitivity of bck2 swi6-ts, but also can bypass a swi6 deletion. A bck2Δ swi6Δ strain kept alive by GAL-CLN2 was transformed with the dosage suppressors shown in A. Cells were grown to log phase in −URA medium containing galactose, spotting was done as described above onto plates containing galactose or glucose, and plates were incubated at the indicated temperatures.
Enhanced expression of aldolase can bypass the requirement for Swi6:
Subcloning of plasmid SS6/396 revealed that the suppressing gene on that plasmid is FBA1, which encodes fructose-bisphosphate aldolase II, the glycolytic enzyme catalyzing the conversion of fructose 1,6-bisphosphate to dihydroxyacetone phosphate and glyceraldehyde 3-phoshophate. Glycolytic flux has been implicated in post-transcriptional regulation of MCM1 (Chen and Tye 1995). SWI4 transcription is dependent on MCM1 (McInerny et al. 1997). We reasoned that perhaps increasing FBA1 expression increased glycolytic flux, thereby increasing the expression of SWI4, which was the most abundant suppressor isolated in the same screen. To evaluate that possibility, we analyzed SWI4 transcripts in both bck2 swi6-ts and wild-type cells that were overexpressing FBA1. No significant difference in the level of SWI4 mRNA was observed in those strains compared to cells expressing a control plasmid (data not shown).
Surprisingly, FBA1 not only is able to suppress a bck2 swi6-ts strain, but also can bypass the requirement for Swi6 as shown by its ability to enable a bck2Δ swi6Δ GAL-CLN2 strain to grow on glucose (Figure 2B). Although the suppression by FBA1 appears to be dependent upon a functional CLN3 and/or BCK2 gene, it does not depend upon SBF or MBF (Table 3). This is surprising since the role of CLN3 is thought to be exerted through Swi6 (Wijnen et al. 2002). This remains to be investigated further.
TABLE 3.
Properties of multicopy suppressors ofswi6-ts
| Yeast strain | FBA1 | RPL40a/ UBI1 | GIN4 | PAB1 |
|---|---|---|---|---|
| bck2Δ swi6-4 | + | + | + | + |
| bck2Δ swi6Δ GAL1:CLN2a | + | − | + | (above 30°) |
| bck2Δ cln3Δ GAL:CLN2a | − | + | − | + |
| mbp1Δ swi4Δ GAL:CLN2a | + | − | − | − |
| bck2Δ swi6-4 cln1Δ | + | + | + | + |
| bck2Δ swi6-4 cln2Δ | − | − | − | − |
Suppression (+) of the double deletion strains by the designated multicopy plasmid indicates a capacity of those cells to form colonies on YEPD plates at 30° unless otherwise indicated. Suppression (+) of the strains carrying the temperature-sensitive swi6-4 allele indicates a capacity to form colonies on YEPD plates at 32°.
These strains carry GAL-CLN2 integrated at the HIS2 locus. CLN2 at its genomic locus is wild type.
Overexpression of ubiquitin suppresses bck2Δ swi6-ts:
The suppressing gene on clone SS6/406 (Figure 3A) was identified as RPL40a/UBI1, a naturally occurring fusion protein composed of ribosomal protein L40 (C-terminal half) and ubiquitin (N-terminal half). To determine whether the ribosomal protein L40 or ubiquitin is responsible for suppression, an open reading frame consisting solely of ubiquitin coding sequences was expressed from the CUP1 promoter on a 2μ plasmid. As shown in Figure 3A, ubiquitin is able to suppress bck2 swi6-ts to an extent similar to that of the full-length RPL40a/UBI1gene.
Figure 3.—
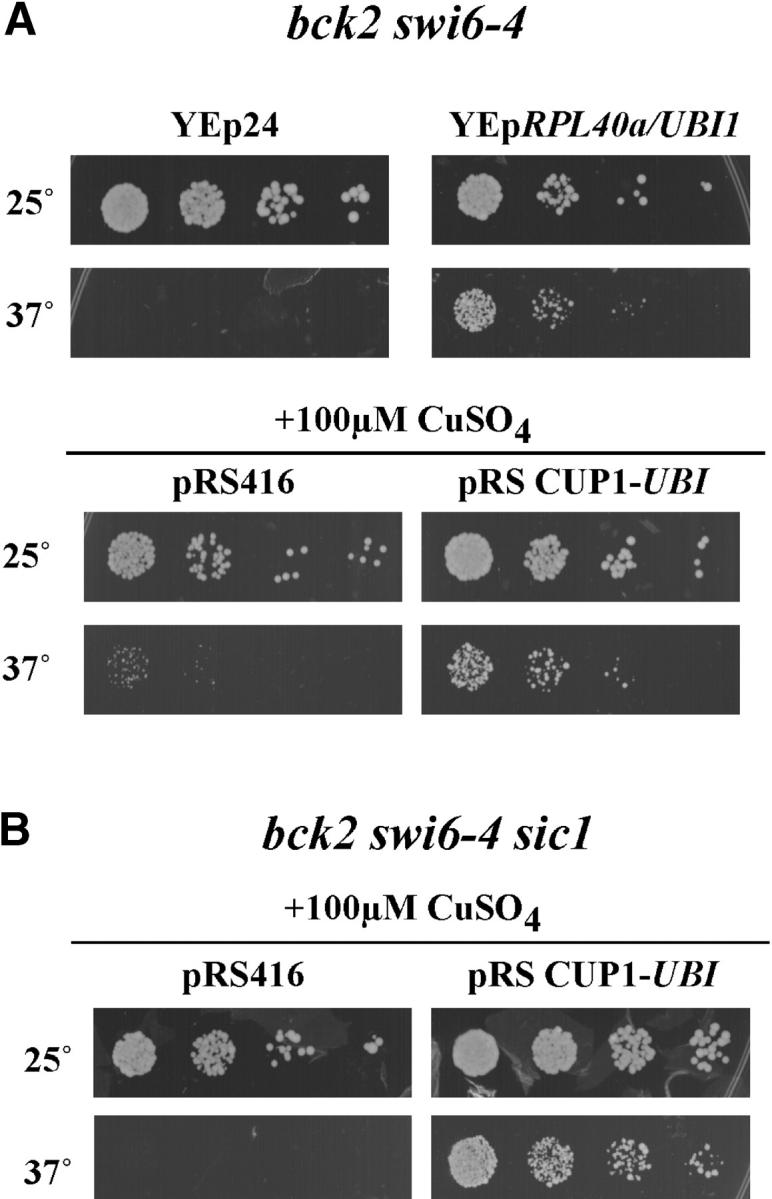
The ubiquitin moiety of RPL40a suppresses bck2Δ swi6-ts. (A) UBI1 is the suppressing moiety of RPL40a. bck2 swi6-4 cells containing either control plasmid (YEp24) or YEpRPL40a/UBI1 were grown and spotted as described in Figure 1. bck2 swi6-4 cells were transformed with control plasmid (pRS416) or a plasmid expressing UBI1 from the inducible CUP1 promoter. Cells were then grown to log phase at permissive temperature in −TRP medium, spotted onto −TRP plates containing 100 μm CuSO4, and incubated at the indicated temperatures for 3 days. (B) Suppression by UBI1 is not dependent upon SIC1. bck2 swi6-4 sic1 cells were transformed with control plasmid (pRS416) or a plasmid expressing UBI1 from the inducible CUP1 promoter. The spotting assay was performed as described in A.
Assessment of the ability of RPL40a/UBI1 to suppress various multiple gene mutants revealed that it is unable to bypass a complete deletion of SWI6 in the bck2Δ background and is dependent on both CLN3 and CLN2 for suppression of the temperature-sensitive allele (Table 3). This suggests that increased accumulation of ubiquitin suppresses by enhancing the effectiveness of Swi6-dependent transcriptional activation or of one or more Swi6 targets rather than by simply bypassing the requirement for Swi6.
It has been shown that overexpression of ubiquitin can suppress the temperature sensitivity of a mutant of CDC34, which encodes an E2 ubiquitin ligase required for cell cycle progression (Prendergast et al. 1995). Degradation of Sic1, a critical target of Cdc34, is required for induction of the G1/S transition. We considered the possibility that overexpression of ubiquitin would suppress the inviability of bck2 swi6 mutants by reducing Sic1 levels, thereby reducing the requirement for Cln1 and Cln2. To evaluate that possibility, SIC1 was deleted in a bck2 swi6-ts strain and growth in the presence and absence of overexpressed UBI1 was evaluated under nonpermissive conditions for the bck2 swi6-ts mutation (Figure 3B). Those experiments revealed that deletion of SIC1 fails to rescue a bck2 swi6-ts strain and, furthermore, that overexpression of ubiquitin is still able to suppress the bck2 swi6-ts mutant in the absence of SIC1. We conclude that suppression of Swi6 by ubiquitin is likely to be a consequence of perturbation of the ubiquitin-proteasome system caused by the increased abundance of ubiquitin.
The G1-specific gene GIN4 can bypass the requirement for SWI6:
The suppressing gene on clone SS6/395 (Figure 2A) was identified as GIN4, one of a family of Nim1-like protein kinases of Saccharomyces cerevisiae. GIN4 was isolated as a mutant that causes synthetic lethality with cln1Δ cln2Δ (Cvrckova et al. 1995; Benton et al. 1997) and later was shown to be a member of the G1-specific gene family (Cho et al. 1998; Spellman et al. 1998). GIN4 is able to not only suppress bck2Δ swi6-ts when expressed from a multicopy plasmid, but also bypass the requirement for SWI6 in a bck2Δ background (Figure 2B). That suppression is strictly dependent on CLN2 (Table 3). Furthermore, deletion of GIN4 in a bck2 swi6-ts severely enhances the temperature sensitivity (data not shown). Together, these findings underscore the importance of coordinate SWI6-dependent regulation of GIN4, CLN1, and CLN2.
The Wee1-related kinase Swe1 specifically inhibits the mitotic form of Cdc28 (Booher et al. 1993) and delays entry into mitosis in response to defects in septin ring formation (Barral et al. 1999) and bud emergence (Sia et al. 1996). SWE1, like GIN4 and the other genes encoding Nim1-like protein kinases, KCC4 and HSL1, are SWI6-dependent G1-specific genes (Spellman et al. 1998; Iyer et al. 2001). It has been suggested that GIN4 may act redundantly with HSL1 and KCC4 to negatively regulate SWE1 and thereby induce entry into mitosis (Barral et al. 1999). Because we found severe septin ring defects in bck2 swi6-ts mutants that are at least partially suppressed by GIN4 on a multicopy plasmid (Figure 4A), we evaluated whether the inactivation of SWE1 abolishes the suppressing effect of GIN4. As shown in Figure 4B deletion of SWE1 fails to rescue a bck2Δ swi6ts mutant. Furthermore, suppression by GIN4 is independent of SWE1 (Figure 4B).
Figure 4.—
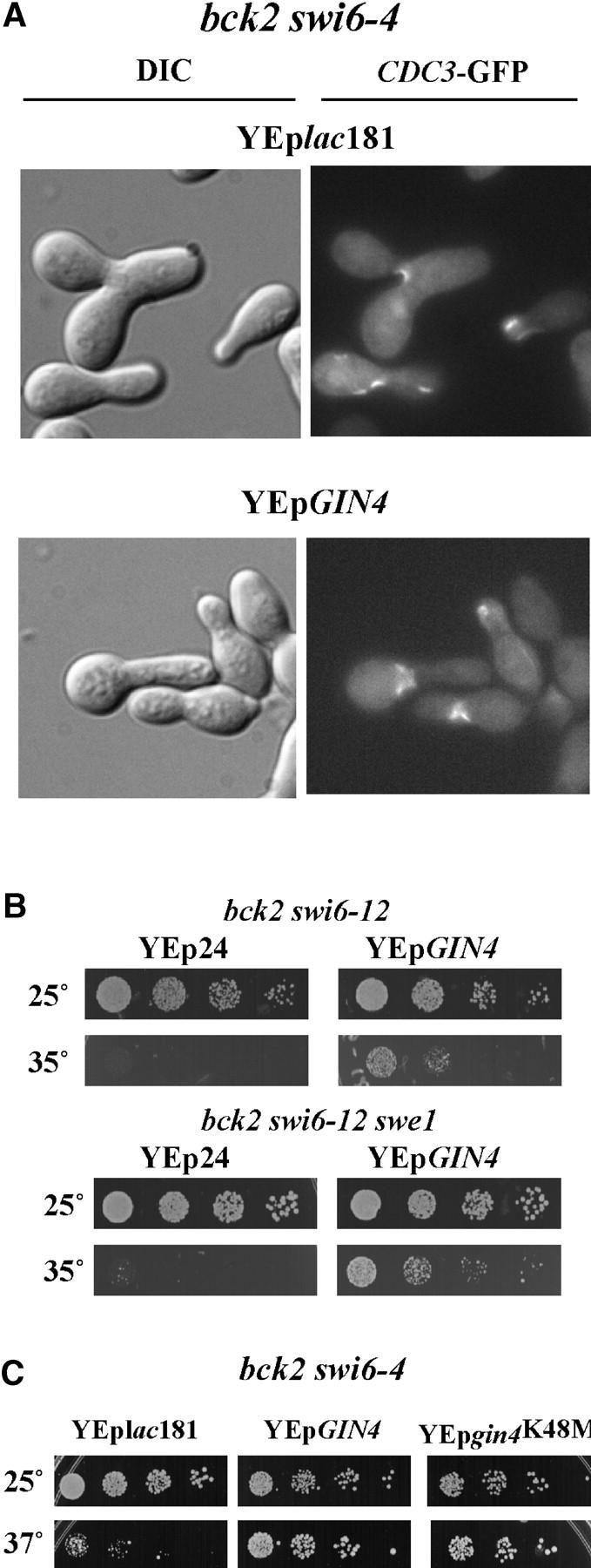
Suppression of bck2Δ swi6-ts by GIN4. (A) Morphogenetic and septin ring defects in bck2Δ swi6-ts mutants with and without multicopy GIN4. bck2 swi6-4 strains carrying either control plasmid (YEplac181) or YEpGIN4 were transformed with YCpCDC3-GFP. Cells were grown to log phase at permissive temperature and then shifted to 35° for a time course; the 10-hr point is shown here. For microscopic analysis, cells were fixed in methanol at −70°. DIC and fluorescence microscopy were performed using an Eclipse E800 microscope with a ×100 objective. (B) Suppression by GIN4 is not dependent upon SWE1. bck2 swi6-12 cells (top) and bck2 swi6-12 swe1 (bottom) were transformed with a control plasmid (Yep24) or YEpGIN4. Spotting assays were done as described in Figure 1. (C) Suppression by GIN4 is not dependent upon a functional protein kinase domain. bck2 swi6-4 cells were transformed with a control plasmid (YEplac181), YEpGIN4, or YEpgin4K48M. YEpgin4K48M harbors a mutation that renders GIN4 kinase inactive (Longtine et al. 1998). Cell growth and spotting assays were performed as described above.
To evaluate the mechanism by which GIN4 might suppress defects arising in the bck2Δ swi6-ts mutant, we asked whether rescue of the mutation was dependent upon Gin4 protein kinase activity. It has been previously established that gin4K48M, a gin4 mutant in which an invariant lysine in the kinase domain is altered to alanine (Longtine et al. 1998; Figure 4C), is able to localize to septin rings but unable to suppress either the temperature sensitivity of septin mutant cdc12-6 or the synthetic lethality arising from combined mutations in cdc12 and gin4. Surprisingly, gin4K48M, when present on a multicopy plasmid, retains the capacity to suppress the bck2 swi6-ts to an extent similar to that of the wild-type gene. Although it is unclear whether the capacity of Gin4 to suppress bck2Δ swi6Δ mutants and normalize their septin ring structure (Figure 4A) is dependent upon localization of Gin4 to septin rings, its protein kinase activity is dispensable for this function.
Poly(A)-binding protein PAB1 can bypass the requirement for SWI6 at high temperature:
The suppressing gene on clone SS6/78 (Figure 2A) was shown by subcloning to be PAB1, which encodes poly(A)-binding protein. PAB1 has several functions. First, it plays roles in polyadenylation, poly(A) length control, and mRNA turnover via the deadenylation/decapping pathway (Morrissey et al. 1999; Otero et al. 1999) and, second, it plays a role in poly(A)-dependent translation (Sachs et al. 1997). The latter function is mediated by the ability of PAB1 to bind to the 5′-cap-binding protein eIF4G and thereby stimulate the recruitment of the 40S ribosomal subunit (Tarun and Sachs 1995).
To evaluate whether overexpression of PAB1 affects CLN1 and CLN2 RNA we analyzed the abundance of those transcripts in cells carrying PAB1 on a multicopy plasmid (Figure 5). Whereas both CLN1 and CLN2 RNA levels are reduced in the bck2Δ swi6-4 mutant relative to wild-type cells at both permissive and restrictive temperatures, the levels of both transcripts are increased by multicopy PAB1 with CLN2 increasing to nearly the wild-type level. The increase in expression of those genes in cells carrying multicopy PAB1 was approximately twofold at 37° and approached wild-type levels. A similar effect of PAB1 is seen in wild-type cells (Figure 5). An increase in expression of SWI4 and GIN4 was also observed. These findings suggest that overexpression of PAB1 suppresses bck2 swi6-ts through a mechanism similar to that of G1 cyclins. However, in synchronized populations of the same strains, no increase of CLN2 transcripts could be demonstrated (data not shown). This suggests that the increased levels in CLN2 mRNA observed in asynchronous cells is due to an effect on cell cycle distribution (data not shown) and not a more direct effect of PAB1 on gene expression.
Suppression of bck2 swi6-ts by PAB1 is very efficient, enabling cells to grow well at 37° and enhancing the growth of bck2 swi6-ts cells even at permissive temperature when grown on solid medium. A similar effect on growth rate was not detectable when the same cells were grown at the permissive temperature on liquid medium (data not shown). Multicopy PAB1 is able to not only suppress the conditional lethality of the bck2 swi6-ts strain, but also bypass a bck2Δ swi6Δ mutant (Table 3). However, that suppression is effective only at elevated temperatures (32°), not at lower temperatures (18° and 25°; Figure 2B and data not shown). The relevance of this requirement for increased temperature is unclear.
The Pab1 protein contains four N-terminal RNA recognition motifs (RRMs; Figure 6). RRMs are found in a number of different RNA-binding proteins and have been associated with the capacity to interact with RNA. RRM2 is the most important RRM for Pab1 function and has been shown to participate in both poly(A) binding and the interaction of Pab1 with eIF4G (Deardorff and Sachs 1997; Kessler and Sachs 1998). A point mutation in RRM2, pab1-180 has been shown to compromise the ability of Pab1 to bind to eIF4G. In contrast, RRM4 plays a role in the interaction of Pab1 with nonpolyadenyalated RNAs. In vitro data suggest that RRM1, RRM4, and the C terminus of Pab1 mediate the translation function associated with the Pab1-poly(A) tail interaction. No specific function has been attributed to RRM3 (Otero et al. 1999).
Figure 6.—

Suppression of bck2Δ swi6-4 by RNA-binding motif mutants of PAB1. bck2 swi6-4 cells were transformed with control plasmid or with plasmids containing mutant forms of PAB1 depicted on the left side of the table. The relative translational efficiency observed in Pab1-depleted in vitro translation extracts supplemented with the indicated Pab1 mutant proteins is derived from Kessler and Sachs (1998). Cells were spotted as described and the minimum restrictive temperature of the strain is shown.
To test which domain of PAB1 is responsible for suppression of a bck2 swi6-ts we have cloned PAB1 mutants (Kessler and Sachs 1998; Otero et al. 1999) into multicopy plasmids and evaluated their capacity to suppress the bck2 swi6-ts mutation (Figure 6). Whereas YEp-pab1ΔRRM2, YEp-pab1ΔRRM4, and YEp-pab1ΔC have lost the ability to suppress bck2 swi6-ts, YEp-pab1ΔRRM1, YEp-pab1ΔRRM3, and YEp-pab1-180 suppress to an extent similar to that of wild type PAB1 (YEp-PAB1-1). On the basis of these results we conclude that the ability of PAB1 to suppress bck2 swi6-ts, while dependent upon specific domains, is not dependent upon domains associated with a specific Pab1-associated function. Instead, it appears to be dependent upon the overall efficiency of translation supported by each of the pab1 mutations, which, on the basis of their capacity to complement the defect in in vitro translation observed in Pab1-depleted extracts, is reduced in the pab1ΔRRM2, pab1ΔRRM4, and pab1ΔC mutants (Kessler and Sachs 1998; Figure 6).
Whi3, another RNA-binding protein with RRM motifs, has been shown to delay G1-specific transcription and progression out of G1 phase (Nash et al. 2001). Although it has been proposed to act by directly binding to the CLN3 mRNA, CLN2 mRNA also appears to be a target (Gari et al. 2001). Because PAB1 suppresses a defect in a G1-specific transcriptional activator, we reasoned that one of its roles might be to antagonize the G1-specific function of Whi3. To evaluate that possibility we first asked whether inactivation of WHI3, like overexpression of PAB1, suppressed the thermosensitivity of a bck2 swi6-4 mutant. That analysis revealed that a bck2Δ swi6-4 whi3Δ strain grew at substantially higher temperatures than the control strain without whi3Δ (Figure 7A). However, that suppression was not nearly as effective as that conferred by multicopy PAB1 (Figure 7B, left). Furthermore, it did not suppress the lethality associated with bck2Δ swi6Δ (data not shown). Because the strain carrying whi3Δ grew significantly less well than one carrying multicopy PAB1 at temperatures above 35°, we could ask whether the suppression by PAB1 was dependent upon WHI3. To do so, YEplac195-PAB was introduced into the bck2Δ swi6-4 whi3 strain and the thermolability of that strain was compared to the thermolability of the strain lacking the plasmid. Strikingly, PAB1 conferred no suppression of bck2Δ swi6-4 additional to that conferred by whi3Δ (Figure 7B, right), suggesting that it depends upon WHI3 to suppress swi6-ts mutants. Although it is difficult to establish without further analysis, this finding suggests that Pab1 might act to directly antagonize the effect of Whi3 on specific mRNA including, but not limited to, that encoding G1 cyclins.
Figure 7.—
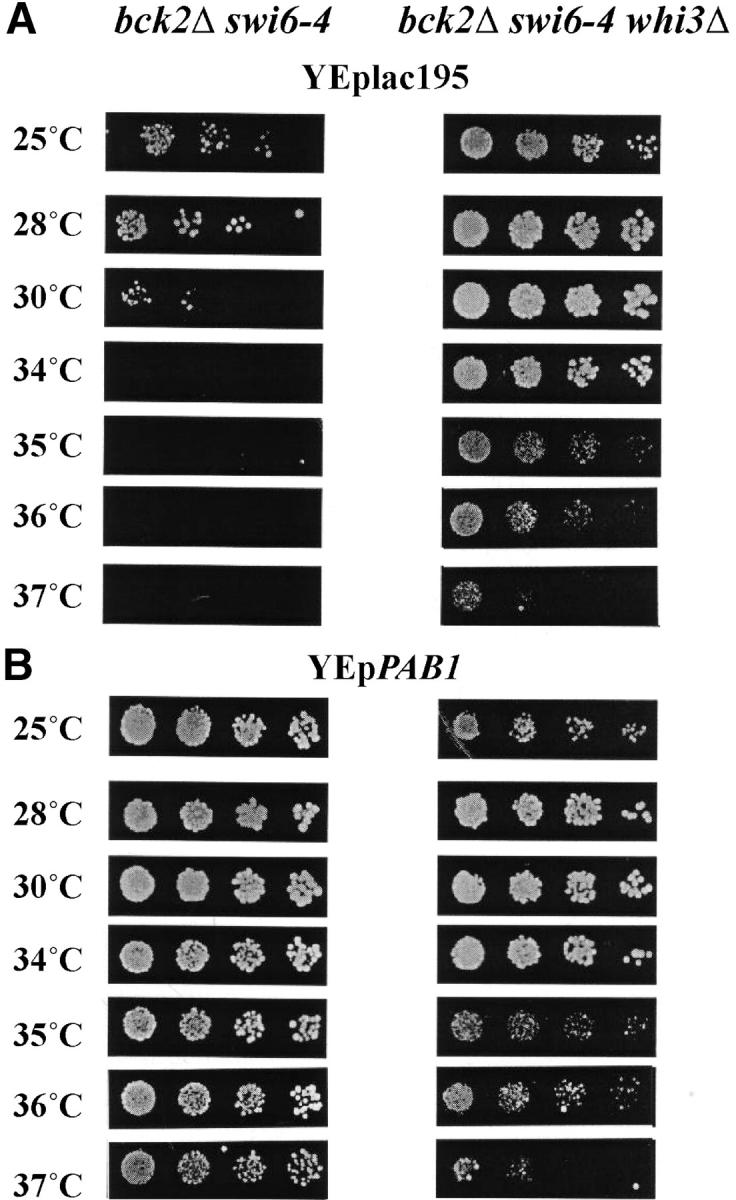
Role of WHI3 in suppression of bck2Δ swi6-4. (A) Inactivation of WHI3 suppresses bck2Δ swi6-4. bck2 swi6-4 (left) and bck2 swi6-4 whi3 (right) cells carrying a control plasmid were spotted onto −URA plates as described and incubated for 2 days at the temperature indicated. (B) Multicopy PAB1 depends upon WHI3 for suppression of bck2Δ swi6-4. bck2 swi6-4 (left) and bck2 swi6-4 whi3 (right) cells carrying YEpPAB1 were analyzed as described in A.
DISCUSSION
A screen for multicopy suppressors of temperature-sensitive alleles of SWI6 was performed under conditions in which those mutations were rendered conditionally lethal by the inactivation of BCK2. Although the intention of the screen was to identify elements of the G1-specific transcriptional machinery, from the outset several mechanisms were envisioned by which the inviability of the bck2Δ swi6-ts strain might be suppressed. First, overexpression of a gene might stabilize the defective Swi6 or otherwise enhance the activity of either MBF or SBF, or both. Second, genes that are dependent upon SWI6 for expression might be expressed sufficiently when present in multiple copies to bypass that dependence. Third, overexpression of genes that can activate G1-specific transcriptional targets independent of SBF and MBF might lead to suppression, as had been previously described for RME1 (Toone et al. 1995). Fourth, suppression might result from overexpression of genes that bypass the requirement for SWI6 by bypassing the specific requirement for an essential target of Swi6-dependent transcription. Consequently, in addition to identifying genes encoding proteins that become rate limiting when SWI6 is inactivated, this screen provides insight into processes that become essential in the absence of SWI6. Finally, because bck2Δ is required for the lethality of the swi6-ts mutants, suppressors of bck2Δ could also be isolated.
In addition to SWI6 and BCK2, suppressors that fall into each of these categories on the basis of prior analysis were identified in the course of the screen. As expected we isolated SWI4, an element of the SBF transcription factor that is able to act independent of SWI6 when sufficiently expressed (Andrews and Moore 1992; Primig et al. 1992; Sidorova and Breeden 1993) and, therefore, that falls into both the first and third classes. Unlike SWI4, another suppressor, RME1, falls into the third class because it activates CLN2 transcription independent of SBF, MBF, or their promoter binding sites. CLN2, a target of SBF that is known to be sufficient to bypass the requirement for G1-specific transcription, belongs to the second class. All four remaining genes isolated in the screen appear to suppress downstream of SWI6. However, with the exception of GIN4, they are not targets of SBF or MBF and, therefore, fall into one of the last two categories.
Whether any of these suppressors act specifically by suppressing the deficiency in bck2Δ is very difficult to address. None of the mutants efficiently suppresses the morphological phenotype that results from the swi6 mutation. However, neither does CLN2 or RME1, both of which clearly act by overcoming the defect in CLN2 expression. Next, GIN4, as a target of SWI6, is likely suppressing by overcoming the defect in GIN4 expression. Finally, because BCK2 affects the same targets as SWI6 (Wijnen and Futcher 1999), it will be suppressed downstream by the same genes that suppress swi6.
Although the isolation of SWI4 would appear to support the argument that interactors or upstream components of SBF can suppress a swi6-ts mutant by direct interaction, it has been established that overexpression of SWI4 alone is sufficient to induce SBF-dependent gene expression in the absence of BCK2 and SWI6 (Andrews and Moore 1992; Primig et al. 1992; Sidorova and Breeden 1993). BCK2 has been shown to be sufficient in the absence of SWI6 to promote low-level expression of G1-specific genes, although the mechanism by which it affects transcription of SBF-dependent genes is unknown (Epstein and Cross 1994; Di Como et al. 1995; Wijnen and Futcher 1999). Thus, these observations are not sufficient to confirm the efficacy of the screen. Finally, it has also been established that either increasing the copy number of CLN2, a target of SBF, or inducing its expression via increasing the copy number of RME1, a transcriptional activator that acts independently of SBF, is sufficient to suppress a bck2Δ swi6Δ. Nevertheless, our failure to isolate genes having a clearly identifiable role in transcriptional activation argues that such positively acting molecules might not exist or, perhaps more likely, may not be accessible via this screen. One likely explanation for the failure to identify novel targets of Cln3/CDK in this screen is that the relevant targets for activation of Swi6-dependent transcription act to negatively regulate that process. We have recently identified one such target as an SBF-associated transcriptional inhibitor (de Bruin et al. 2004).
More surprising than the finding that this screen did not identify genes encoding factors that behaved as transcriptional activators was the precise nature of the genes that were isolated. Perhaps aldolase, a highly abundant glycolytic enzyme, is the most surprising suppressor. Although it is possible that the suppression by FBA1 is a nonspecific effect on osmoregulation or cell integrity due solely to its abundance, other abundant cytosolic proteins were not isolated. It is more likely to be associated with the enzymatic function of FBA1. Overproduction of this enzyme might induce a signal of a nutrition-rich environment. CLN3 expression is regulated by nutrition availability (Gallego et al. 1997; Polymenis and Schmidt 1997; Wu et al. 1999; Newcomb et al. 2003) although it is unclear how CLN3 would act in the absence of the transcription factor. The potential for such overlap in function has been documented (Miller and Cross 2000; Edgington and Futcher 2001). Alternatively, aldolase could have a direct and heretofore unrecognized role in transcriptional activation, as has been recently reported for glyceraldehyde 3-phosphate dehydrogenase (encoded by the TDH1-TDH3 genes in yeast; Zheng et al. 2003).
PAB1, like FBA1, is involved in a general metabolic role in cells: namely, global regulation of translation. However, a number of observations led us to believe that suppression of bck2Δ swi6-ts by PAB1 occurs via a mechanism more directly related to G1-specific gene expression. First, human PAB1 can overcome the G1 arrest occurring in response to mating pheromone (Edwards et al. 1997). In that circumstance the effect of human PAB1 was shown to be dependent on CLN1. In contrast, we found the suppression of bck2 swi6-ts by multicopy PAB1 to be strictly dependent upon CLN2 (Table 1). However, it is likely that this distinction is not due to differences in the source of PAB1 but rather to the nature of the screen. Another observation that appears to link translation efficiency specifically to G1-specific transcription is the identification of SWI4 as synthetic lethal with eIF4G-DN300, a mutant of the translation initiation factor eIF4G unable to bind to PAB1 (A. Sachs, personal communication). Finally, we isolated EAP1, a gene recently described as a binding partner of eIF4E (Chial et al. 2000) in a screen for genes that, like CLN3 and SWI6, are synthetic lethal with BCK2 (T. Kesti and C. Wittenberg, unpublished observation). All of these observations are consistent with a specific effect of PAB1 on G1-phase progression or gene expression.
Despite these connections, we found little direct evidence for a specific effect on the cell cycle machinery. Nevertheless it remains possible that a more general effect of PAB1 on translation rate manifests itself as a specific effect on the cell cycle via an effect on a rate-limiting G1-specific transcriptional target. The finding that suppression of bck2 swi6-ts by multicopy PAB1 depends upon WHI3 suggests that Pab1 can act, at least in part, to antagonize Whi3 function, perhaps by abrogating its effect on CLN2 or CLN3 mRNA.
Ubiquitin is also involved in a process of general importance to the cell. Yet, suppression of the Swi6 deficiency by overexpression of ubiquitin appears consistent with the critical role for G1-specific transcription products in directing the degradation of proteins that restrict proliferation. We hypothesized that enhanced degradation of Sic1 might explain the suppressive effect of increased ubiquitin on bck2Δ swi6-ts but found that inactivation of Sic1 is not sufficient to suppress bck2Δ swi6-ts or to interfere with the capacity of ubiquitin to suppress that mutant. Thus, if increased ubiquitin leads to enhanced degradation of a natural target of the G1-specific cell cycle machinery, it does so via one or more proteins other than Sic1. Alternatively, overexpression of ubiquitin may interfere with other functions responsible for the lethality of these mutants.
Expression of CLN2 from a heterologous promoter is sufficient to bypass the lethality of bck2Δ cln3Δ, bck2Δ swi6Δ, or cln1Δ cln2Δ cln3Δ mutants. In contrast, inactivation of Sic1 bypasses the lethality caused by a CLN deficiency, but not that caused by either of the other mutants. Because CLN2 is sufficient to induce phosphorylation-dependent degradation of Sic1, we must assume that it plays additional essential roles in the context of bck2Δ cln3Δ and bck2Δ swi6Δ mutants. In the absence of Cln3, GAL-expressed CLN2 is an effective activator of G1-specific transcription (Cross and Tinkelenberg 1991; Dirick and Nasmyth 1991), a function that is likely to be important in the suppression of bck2Δ cln3Δ mutants, which are severely compromised in that regard. However, CLN2 must play a quite different role in the bck2Δ swi6Δ strain where neither SBF nor MBF is functional and, as a consequence, the targets of Cln/CDK required for transcriptional activation cannot act. In this context it is likely that Cln2 suppresses via its capacity to directly promote cell cycle progression through either normal or ectopic mechanisms. Thus, there are clearly multiple distinct pathways via which Cln2 can act to suppress a deficiency in G1-specific functions.
The nature of suppression by CLN2 must be taken into account when considering the mechanisms utilized by the multicopy suppressors because suppression by each depends upon CLN2 (Table 3). The bck2Δ swi6-ts strain must remain competent to express CLN2 and its expression or function may be facilitated by the overexpression of the suppressing gene. Although it is possible that PAB1 acts by facilitating Cln2 translation, its effect is not noticeable in terms of the level of Cln2 protein in an asynchronous culture. Alternatively, the suppressing gene might act by facilitating a pathway that is sufficient in the presence of CLN2 but not in its absence. GIN4, which functions in the pathway that is required for viability in the absence of CLN2, may suppress by such a mechanism. We have established that, as targets of the G1-specific transcriptional machinery, both GIN4 and CLN2 are poorly expressed in a bck2Δ swi6-ts mutant (Figure 5 and data not shown). Although increasing the copy number of GIN4 is sufficient to support viability, that suppression is dependent upon Cln2. We suggest that either the level of GIN4 expression remains sufficiently low in the swi6 mutant to require CLN2 or the poor expression of other Swi6-dependent gene products creates a situation in which both GIN4 and CLN2 become essential.
Together, these data suggest that defects in the G1-specific transcriptional apparatus can be suppressed via diverse pathways. Although this is consistent with the involvement of G1-specific genes in many functions, it appears that a relatively limited subset of those pathways is sufficient for viability. Whereas some of the suppressors (e.g., PAB1 and UBI1) are associated with rather general functions, others (e.g., CLN2 and GIN4) appear to be more specific. Although the specific mechanisms of suppression are unclear, it may be that CLN2 and GIN4 define the G1-specific pathways that are essential for viability whereas PAB1 and UBI1 act via more general mechanisms to facilitate deficiencies in those pathways.
Acknowledgments
We thank Cathy Yao, Dana Vukajlovich, and Marisela Guaderrama for excellent technical support and Tapio Kesti, Peter Kaiser, Robertus de Bruin, and the members of The Scripps Research Institute Cell Cycle Group for helpful discussion and comments on the manuscript. We also thank Alan Sachs for sharing unpublished results. This work was supported by U.S. Public Health Service grants GM-59441 and GM-43487 to C.W. K.F. acknowledges support from an Austrian Program for Advanced Research and Technology Fellowship of the Austrian Academy of Sciences.
References
- Andrews, B. J., and L. Moore, 1992. Mutational analysis of a DNA sequence involved in linking gene expression to the cell cycle. Biochem. Cell Biol. 70: 1073–1080. [DOI] [PubMed] [Google Scholar]
- Barral, Y., M. Parra, S. Bidlingmaier and M. Snyder, 1999. Nim1-related kinases coordinate cell cycle progression with the organization of the peripheral cytoskeleton in yeast. Genes Dev. 13: 176–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton, B. K., A. Tinkelenberg, I. Gonzalez and F. R. Cross, 1997. Cla4p, a Saccharomyces cerevisiae Cdc42p-activated kinase involved in cytokinesis, is activated at mitosis. Mol. Cell. Biol. 17: 5067–5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhoite, L. T., Y. Yu and D. J. Stillman, 2001. The Swi5 activator recruits the Mediator complex to the HO promoter without RNA polymerase II. Genes Dev. 15: 2457–2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booher, R. N., R. J. Deshaies and M. W. Kirschner, 1993. Properties of Saccharomyces cerevisiae wee1 and its differential regulation of p34CDC28 in response to G1 and G2 cyclins. EMBO J. 12: 3417–3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breeden, L., 1996. Start-specific transcription in yeast. Curr. Top. Microbiol. Immunol. 208: 95–127. [DOI] [PubMed] [Google Scholar]
- Carlson, M., and D. Botstein, 1982. Two differentially regulated mRNAs with different 5′ ends encode secreted with intracellular forms of yeast invertase. Cell 28: 145–154. [DOI] [PubMed] [Google Scholar]
- Caviston, J. P., M. Longtine, J. R. Pringle and E. Bi, 2003. The role of Cdc42p GTPase-activating proteins in assembly of the septin ring in yeast. Mol. Biol. Cell 14: 4051–4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y., and B. K. Tye, 1995. The yeast Mcm1 protein is regulated posttranscriptionally by the flux of glycolysis. Mol. Cell. Biol. 15: 4631–4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chial, H. J., A. J. Stemm-Wolf, S. McBratney and M. Winey, 2000. Yeast Eap1p, an eIF4E-associated protein, has a separate function involving genetic stability. Curr. Biol. 10: 1519–1522. [DOI] [PubMed] [Google Scholar]
- Cho, R. J., M. J. Campbell, E. A. Winzeler, L. Steinmetz, A. Conway et al., 1998. A genome-wide transcriptional analysis of the mitotic cell cycle. Mol. Cell 2: 65–73. [DOI] [PubMed] [Google Scholar]
- Cosma, M. P., T. Tanaka and K. Nasmyth, 1999. Ordered recruitment of transcription and chromatin remodeling factors to a cell cycle- and developmentally regulated promoter. Cell 97: 299–311. [DOI] [PubMed] [Google Scholar]
- Cosma, M. P., S. Panizza and K. Nasmyth, 2001. Cdk1 triggers association of RNA polymerase to cell cycle promoters only after recruitment of the mediator by SBF. Mol. Cell 7: 1213–1220. [DOI] [PubMed] [Google Scholar]
- Cross, F. R., and A. H. Tinkelenberg, 1991. A potential positive feedback loop controlling CLN1 and CLN2 gene expression at the start of the yeast cell cycle. Cell 65: 875–883. [DOI] [PubMed] [Google Scholar]
- Cvrckova, F., C. De Virgilio, E. Manser, J. R. Pringle and K. Nasmyth, 1995. Ste20-like protein kinases are required for normal localization of cell growth and for cytokinesis in budding yeast. Genes Dev. 9: 1817–1830. [DOI] [PubMed] [Google Scholar]
- de Bruin, R. A. M., W. H. MacDonald, T. I. Kalashnikova, J. R. R. Yates and C. Wittenberg, 2004. Cln3 Aativates G1-specific transcription via phosphorylation of the SBF-bound repressor, Whi5. Cell 117: 887–898. [DOI] [PubMed] [Google Scholar]
- Deardorff, J. A., and A. B. Sachs, 1997. Differential effects of aromatic and charged residue substitutions in the RNA binding domains of the yeast poly(A)-binding protein. J. Mol. Biol. 269: 67–81. [DOI] [PubMed] [Google Scholar]
- Di Como, C. J., H. Chang and K. T. Arndt, 1995. Activation of CLN1 and CLN2 G1 cyclin gene expression by BCK2. Mol. Cell. Biol. 15: 1835–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirick, L., and K. Nasmyth, 1991. Positive feedback in the activation of G1 cyclins in yeast. Nature 351: 754–757. [DOI] [PubMed] [Google Scholar]
- Dirick, L., T. Bohm and K. Nasmyth, 1995. Roles and regulation of Cln-Cdc28 kinases at the start of the cell cycle of Saccharomyces cerevisiae. EMBO J. 14: 4803–4813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgington, N. P., and B. Futcher, 2001. Relationship between the function and the location of G1 cyclins in S. cerevisiae. J. Cell Sci. 114: 4599–4611. [DOI] [PubMed] [Google Scholar]
- Edwards, M. C., N. Liegeois, J. Horecka, R. A. DePinho, G. F. Sprague, Jr. et al., 1997. Human CPR (cell cycle progression restoration) genes impart a Far- phenotype on yeast cells. Genetics 147: 1063–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein, C. B., and F. R. Cross, 1994. Genes that can bypass the CLN requirement for Saccharomyces cerevisiae cell cycle START. Mol. Cell. Biol. 14: 2041–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinoza, F. H., J. Ogas, I. Herskowitz and D. O. Morgan, 1994. Cell cycle control by a complex of the cyclin HCS26 (PCL1) and the kinase PHO85. Science 266: 1388–1391. [DOI] [PubMed] [Google Scholar]
- Gallego, C., E. Gari, N. Colomina, E. Herrero and M. Aldea, 1997. The Cln3 cyclin is down-regulated by translational repression and degradation during the G1 arrest caused by nitrogen deprivation in budding yeast. EMBO J. 16: 7196–7206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gari, E., T. Volpe, H. Wang, C. Gallego, B. Futcher et al., 2001. Whi3 binds the mRNA of the G1 cyclin CLN3 to modulate cell fate in budding yeast. Genes Dev. 15: 2803–2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz, R. D., and A. Sugino, 1988. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74: 527–534. [DOI] [PubMed] [Google Scholar]
- Iyer, V. R., C. E. Horak, C. S. Scafe, D. Botstein, M. Snyder et al., 2001. Genomic binding sites of the yeast cell-cycle transcription factors SBF and MBF. Nature 409: 533–538. [DOI] [PubMed] [Google Scholar]
- Kessler, S. H., and A. B. Sachs, 1998. RNA recognition motif 2 of yeast Pab1p is required for its functional interaction with eukaryotic translation initiation factor 4G. Mol. Cell. Biol. 18: 51–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch, C., T. Moll, M. Neuberg, H. Ahorn and K. Nasmyth, 1993. A role for the transcription factors Mbp1 and Swi4 in progression from G1 to S phase. Science 261: 1551–1557. [DOI] [PubMed] [Google Scholar]
- Longtine, M. S., H. Fares and J. R. Pringle, 1998. Role of the yeast Gin4p protein kinase in septin assembly and the relationship between septin assembly and septin function. J. Cell Biol. 143: 719–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInerny, C. J., J. F. Partridge, G. E. Mikesell, D. P. Creemer and L. L. Breeden, 1997. A novel Mcm1-dependent element in the SWI4, CLN3, CDC6, and CDC47 promoters activates M/G1-specific transcription. Genes Dev. 11: 1277–1288. [DOI] [PubMed] [Google Scholar]
- Measday, V., L. Moore, J. Ogas, M. Tyers and B. Andrews, 1994. The PCL2 (ORFD)-PHO85 cyclin-dependent kinase complex: a cell cycle regulator in yeast. Science 266: 1391–1395. [DOI] [PubMed] [Google Scholar]
- Miller, M. E., and F. R. Cross, 2000. Distinct subcellular localization patterns contribute to functional specificity of the Cln2 and Cln3 cyclins of Saccharomyces cerevisiae. Mol. Cell. Biol. 20: 542–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrissey, J. P., J. A. Deardorff, C. Hebron and A. B. Sachs, 1999. Decapping of stabilized, polyadenylated mRNA in yeast pab1 mutants. Yeast 15: 687–702. [DOI] [PubMed] [Google Scholar]
- Muhlrad, D., R. Hunter and R. Parker, 1992. A rapid method for localized mutagenesis of yeast genes. Yeast 8: 79–82. [DOI] [PubMed] [Google Scholar]
- Nash, R. S., T. Volpe and B. Futcher, 2001. Isolation and characterization of WHI3, a size-control gene of Saccharomyces cerevisiae. Genetics 157: 1469–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasmyth, K., 1993. Regulating the HO endonuclease in yeast. Curr. Opin. Genet. Dev. 3: 286–294. [DOI] [PubMed] [Google Scholar]
- Nasmyth, K., and L. Dirick, 1991. The role of SWI4 and SWI6 in the activity of G1 cyclins in yeast. Cell 66: 995–1013. [DOI] [PubMed] [Google Scholar]
- Newcomb, L. L., J. A. Diderich, M. G. Slattery and W. Heideman, 2003. Glucose regulation of Saccharomyces cerevisiae cell cycle genes. Eukaryotic Cell 2: 143–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogas, J., B. J. Andrews and I. Herskowitz, 1991. Transcriptional activation of CLN1, CLN2, and a putative new G1 cyclin (HCS26) by SWI4, a positive regulator of G1-specific transcription. Cell 66: 1015–1026. [DOI] [PubMed] [Google Scholar]
- Otero, L. J., M. P. Ashe and A. B. Sachs, 1999. The yeast poly(A)-binding protein Pab1p stimulates in vitro poly(A)-dependent and cap-dependent translation by distinct mechanisms. EMBO J. 18: 3153–3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymenis, M., and E. V. Schmidt, 1997. Coupling of cell division to cell growth by translational control of the G1 cyclin CLN3 in yeast. Genes Dev. 11: 2522–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prendergast, J. A., C. Ptak, T. G. Arnason and M. J. Ellison, 1995. Increased ubiquitin expression suppresses the cell cycle defect associated with the yeast ubiquitin conjugating enzyme, CDC34 (UBC3). Evidence for a noncovalent interaction between CDC34 and ubiquitin. J. Biol. Chem. 270: 9347–9352. [DOI] [PubMed] [Google Scholar]
- Primig, M., S. Sockanathan, H. Auer and K. Nasmyth, 1992. Anatomy of a transcription factor important for the start of the cell cycle in Saccharomyces cerevisiae. Nature 358: 593–597. [DOI] [PubMed] [Google Scholar]
- Richardson, H. E., C. Wittenberg, F. R. Cross and S. I. Reed, 1989. An essential G1 function for cyclin-like proteins in yeast. Cell 59: 1127–1133. [DOI] [PubMed] [Google Scholar]
- Sachs, A. B., P. Sarnow and M. W. Hentze, 1997. Starting at the beginning, middle, and end: translation initiation in eukaryotes. Cell 89: 831–838. [DOI] [PubMed] [Google Scholar]
- Schwob, E., and K. Nasmyth, 1993. CLB5 and CLB6, a new pair of B cyclins involved in DNA replication in Saccharomyces cerevisiae. Genes Dev. 7: 1160–1175. [DOI] [PubMed] [Google Scholar]
- Sia, R. A., H. A. Herald and D. J. Lew, 1996. Cdc28 tyrosine phosphorylation and the morphogenesis checkpoint in budding yeast. Mol. Biol. Cell 7: 1657–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidorova, J., and L. Breeden, 1993. Analysis of the SWI4/SWI6 protein complex, which directs G1/S-specific transcription in Saccharomyces cerevisiae. Mol. Cell. Biol. 13: 1069–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidorova, J., and L. Breeden, 1999. The MSN1 and NHP6A genes suppress SWI6 defects in Saccharomyces cerevisiae. Genetics 151: 45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidorova, J. M., G. E. Mikesell and L. L. Breeden, 1995. Cell cycle-regulated phosphorylation of Swi6 controls its nuclear localization. Mol. Biol. Cell 6: 1641–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski, R. S., and P. Hieter, 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122: 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellman, P. T., G. Sherlock, M. Q. Zhang, V. R. Iyer, K. Anders et al., 1998. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol. Biol. Cell 9: 3273–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl, K., D. T. Stinchcomb, S. Scherer and R. W. Davis, 1979. High-frequency transformation of yeast: autonomous replication of hybrid DNA molecules. Proc. Natl. Acad. Sci. USA 76: 1035–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart, D., and C. Wittenberg, 1994. Cell cycle-dependent transcription of CLN2 is conferred by multiple distinct cis-acting regulatory elements. Mol. Cell. Biol. 14: 4788–4801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart, D., and C. Wittenberg, 1995. CLN3, not positive feedback, determines the timing of CLN2 transcription in cycling cells. Genes Dev. 9: 2780–2794. [DOI] [PubMed] [Google Scholar]
- Tarun, S. Z., Jr., and A. B. Sachs, 1995. A common function for mRNA 5′ and 3′ ends in translation initiation in yeast. Genes Dev. 9: 2997–3007. [DOI] [PubMed] [Google Scholar]
- Toone, W. M., A. L. Johnson, G. R. Banks, J. H. Toyn, D. Stuart et al., 1995. Rme1, a negative regulator of meiosis, is also a positive activator of G1 cyclin gene expression. EMBO J. 14: 5824–5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyers, M., G. Tokiwa and B. Futcher, 1993. Comparison of the Saccharomyces cerevisiae G1 cyclins: Cln3 may be an upstream activator of Cln1, Cln2, and other cyclins. EMBO J. 12: 1955–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijnen, H., and B. Futcher, 1999. Genetic analysis of the shared role of CLN3 and BCK2 at the G1-S transition in Saccharomyces cerevisiae. Genetics 153: 1131–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijnen, H., A. Landman and B. Futcher, 2002. The G(1) cyclin Cln3 promotes cell cycle entry via the transcription factor Swi6. Mol. Cell. Biol. 22: 4402–4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems, A. R., S. Lanker, E. E. Patton, K. L. Craig, T. F. Nason et al., 1996. Cdc53 targets phosphorylated G1 cyclins for degradation by the ubiquitin proteolytic pathway. Cell 86: 453–463. [DOI] [PubMed] [Google Scholar]
- Wittenberg, C., and K. Flick, 2003 Cell cycle regulation during G1 phase in yeast: decisions, decisions, decisions, pp. 14–39 in G1 Phase Progression, edited by J. Boonstra. Landes Biosciences, Georgetown, TX.
- Wu, M., L. Newcomb and W. Heideman, 1999. Regulation of gene expression by glucose in Saccharomyces cerevisiae: a role for ADA2 and ADA3/NGG1. J. Bacteriol. 181: 4755–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, L., R. G. Roeder and Y. Luo, 2003. S phase activation of the histone H2B promoter by OCA-S, a coactivator complex that contains GAPDH as a key component. Cell 114: 255–266. [DOI] [PubMed] [Google Scholar]