

Abstract
Populus is an important model organism in forest biology, but levels of nucleotide polymorphisms and linkage disequilibrium have never been investigated in natural populations. Here I present a study on levels of nucleotide polymorphism, haplotype structure, and population subdivision in five nuclear genes in the European aspen Populus tremula. Results show substantial levels of genetic variation. Levels of silent site polymorphisms, πs, averaged 0.016 across the five genes. Linkage disequilibrium was generally low, extending only a few hundred base pairs, suggesting that rates of recombination are high in this obligate outcrossing species. Significant genetic differentiation was found at all five genes, with an average estimate of FST = 0.116. Levels of polymorphism in P. tremula are 2- to 10-fold higher than those in other woody, long-lived perennial plants, such as Pinus and Cryptomeria. The high levels of nucleotide polymorphism and low linkage disequilibrium suggest that it may be possible to map functional variation to very fine scales in P. tremula using association-mapping approaches.
QTL mapping is currently the key tool for identifying the genetic basis of quantitative traits. However, an approach that has gained in popularity recently is the use of natural populations to map traits by means of association analysis. Association analysis, or linkage disequilibrium (LD) mapping, has been used extensively to dissect traits in species where traditional QTL-mapping approaches are not feasible, such as in humans (Rafalski and Morgante 2004). Association methods have also been extended to plants and have helped increase the resolution considerably compared to standard mapping populations (Flint-Garcia et al. 2003). In principle, association studies can identify variation down to the single-nucleotide substitutions that are responsible for variation in phenotypes (quantitative trait nucleotides, QTNs). However, a move from a traditional QTL-mapping approach to association-based population surveys requires detailed knowledge about basic population genetic parameters, such as levels of genetic variation and the extent of linkage disequilibrium and population structure, and also how these parameters vary across the genome of the species in question.
The long generation times of most forest trees have led to slow progress in elucidating the genetic architecture of complex traits through traditional QTL-mapping approaches, and suggestions have been made that linkage-disequilibrium mapping approaches might be more fruitful (Brunner et al. 2004; Neale and Savolainen 2004). However, in plants, the majority of polymorphism data at the nucleotide level come from a few, well-studied model species such as Arabidopsis, maize, rice, and barley and from only a few studies that have estimated levels of nucleotide polymorphism and linkage disequilibrium in long-lived outcrossing plants, such as woody perennials (Dvornyk et al. 2002; Garcia-Gil et al. 2003; Kado et al. 2003; Semerikov and Lascoux 2003).
It is hard to know to what extent results from short-lived, selfing, or domesticated species generalize to plants with different life histories, mating systems, and/or domestication histories. For instance, even though levels of nucleotide polymorphism at the alcohol dehydrogenase (Adh) locus were comparable in selfing Arabidopsis thaliana and outcrossing A. lyrata, the distribution of segregating sites across the gene was very different in the two species (Savolainen et al. 2000). Furthermore, in predominantly selfing species like selfing A. thaliana and rice (Oryza sativa) linkage disequilibrium extends over large physical distances [>150 kb in Arabidopsis (Nordborg et al. 2000) and ∼100 kb in rice (Garris et al. 2003)] whereas in outcrossing maize, linkage disequilibrium declines to negligible levels in <1 kb (Remington et al. 2001).
The genus Populus has a long tradition as a model system for tree physiology and Populus has recently also been established as the de facto model species for tree genomics (Wullschleger et al. 2002; Brunner et al. 2004). The hope is that Populus will complement the “classical” plant model systems, such as Arabidopsis and rice, in elucidating areas such as wood formation, the transition from juvenility to maturity, and adaptation to seasonal and other environmental changes (Wullschleger et al. 2002; Brunner et al. 2004). Populus is dioecious, and hence outcrossing, and wind pollinated, and the expectation is that levels of linkage disequilibrium and population structure are generally low across the genome in Populus (Brunner et al. 2004). However, despite the long history of Populus as an experimental system for tree physiology and the recent public release of the complete genome sequence for a species of Populus (Populus trichocarpa), remarkably little is known about levels of naturally occurring genetic variation and this is especially true for polymorphisms at the nucleotide level. In this article I therefore provide the first characterization of levels of nucleotide polymorphism in a species of Populus, the European aspen (P. tremula), using data from five nuclear genes. I also characterize linkage disequilibrium and population structure by sampling from four natural populations across Europe. The results show substantial levels of nucleotide polymorphism in all five genes, comparable to levels in worldwide samples of Arabidopsis and maize. Linkage disequilibrium is generally low, extending only a few hundred base pairs. Estimates of population subdivision are low, although significant genetic differentiation among populations occurs at all five loci included in the study. Because of the high polymorphism levels and low linkage disequilibrium genome-wide linkage disequilibrium mapping approaches may not be feasible in Populus. However, very fine-scale mapping is possible if candidate gene approaches are used. The presence of significant, although modest, population structure and variation between loci in the decline of linkage disequilibrium within local populations highlights the care that has to be taken when sampling for association studies.
MATERIALS AND METHODS
Plant materials:
Leaf material was sampled from 24 different trees of P. tremula from four different sites throughout Europe (five to seven plants per site) in June/July 2002 and July 2003. Samples were taken within a few kilometers of Besancon in eastern France (FRA), Klagenfurt in southern Austria (AUT), Färjestaden in southeastern Sweden (SWE S), and Umeå in northern Sweden (SWE N). Three to four young, undamaged leaves were collected from each tree, dried in silica gel, and stored at room temperature until DNA extraction.
Loci studied:
Five loci were included in the study. The Alcohol dehydrogenase 1 locus (Adh1, EC 1.1.1.1) is a key enzyme for coping with low oxygen stress in higher plants. The Adh gene family has been extensively studied both at the functional and at the molecular level in a number of plant species and there are thus ample data to compare. Gibberellin 20-oxidase (GA20ox1, EC 1.14.11.-) is a multifunctional 2-oxoglutarate-dependent dioxygenase that is a key enzyme in controlling the synthesis of the plant hormone gibberellin (GAs). GAs are a group of powerful growth promoters in plants and have been implicated in such processes as seed germination, elongation growth, fruit development, and flowering (Eriksson and Moritz 2002). Glyceraldehyde-3-phosphate dehydrogenase (Gapdh, 1.2.1.12) is a tetrameric nicotinamide adenine dinucleotide-binding enzyme that plays an important role in glycolysis and glyconeogenesis. CI-1 is a cysteine protease inhibitor and TI-3 is a member of a small family of wound-inducible trypsin inhibitors that have recently been isolated in Populus (Haruta et al. 2001).
DNA extraction, PCR amplification, and sequencing:
Total genomic DNA was extracted from dried leaf tissue using the DNeasy plant mini-prep kit (QIAGEN, Valencia, CA). Primers to amplify P. tremula Adh1 and CI-1 genes were designed on the basis of BLAST searches in the publicly available Populus expressed sequence tag collection (http://poppel.fysbot.umu.se) using homologs from A. thaliana (Adh1, AF110456) and Malus x domestica (CI-1, AY173139). Primers to amplify GA20ox1 (Eriksson and Moritz 2002) and a TI-3 (Haruta et al. 2001) were designed from published sequences in GenBank (accession nos. AJ001326 and AF349443, respectively). Finally, part of the Gapdh gene was amplified in Populus using primers GPDX7F and GPDX9R from Strand et al. (1997). Primer sequences are available as supplementary material (supplementary Table 1).
TABLE 1.
Length of regions analyzed
| Coding
|
|||||
|---|---|---|---|---|---|
| Locus | Totala | Coding | Noncodinga | Nonsynonymous | Synonymous |
| Adh | 2192 | 909 | 1283 | 696.3 | 212.7 |
| CI-1 | 802 | 429 | 373 | 330.9 | 98.1 |
| G3PDH | 809 | 411 | 398 | 311.0 | 100.0 |
| GA20ox1 | 1746 | 1152 | 594 | 901.4 | 250.6 |
| TI-3 | 639 | 606 | 33 | 456.1 | 176.9 |
| Total | 6188 | 3507 | 2681 | 2695.7 | 838.3 |
Including indels.
PCR products were cloned into the pCR2.1 vector using a TA-cloning kit from Invitrogen (Carlsbad, CA). Clones were sequenced using BigDye chemistry (Applied Biosystems, Foster City, CA) on an ABI377 automated sequencer (Applied Biosystems) at the Umeå Plant Science Center sequencing facility. Four to 10 different clones of each fragment were sequenced to identify the presence of multiple haplotypes within individuals and to control for Taq polymerase errors. All sequences used in this article have been deposited in the EMBL/GenBank databases (accession nos. AJ842873, AJ842874, AJ842875, AJ842876, AJ842877, AJ842878, AJ842879, AJ842880, AJ842881, AJ842882, AJ842883, AJ842884, AJ842885, AJ842886, AJ842887, AJ842888, AJ842889, AJ842890, AJ842891, AJ842892, AJ842893, AJ842894, AJ842895, AJ842896, AJ842897, AJ842898, AJ842899, AJ842900, AJ842901, AJ842902, AJ842903, AJ842904, AJ842905, AJ842906, AJ842907, AJ842908, AJ842909, AJ842910, AJ842911, AJ842912, AJ842913, AJ842914, AJ842915, AJ842916, AJ842917, AJ842918, AJ842919, AJ842920, AJ842921, AJ842922, AJ842923, AJ842924, AJ842925, AJ842926, AJ842927, AJ842928, AJ842929, AJ842930, AJ842931, AJ842932, AJ842933, AJ842934, AJ842935, AJ842936, AJ842937, AJ842938, AJ842939, AJ842940, AJ842941, AJ842942, AJ842943, AJ842944, AJ842945, AJ842946, AJ842947, AJ842948, AJ842949, AJ842950, AJ842951, AJ842952 and AJ843576, AJ843713). Homologous sequences of Adh, CI-1, Gapdh, GA20-ox1, and TI-3 region were also obtained from P. trichocarpa by manually assembling contigs identified through BLAST searches in the Populus Genome Project database at Joint Genome Institute (JGI, http://genome.jgi-psf.org/).
Sequence analysis:
Sequences were verified manually and contigs were assembled using the computer program Sequencher v4.0 (Gene Codes, Ann Arbor, MI). Multiple sequence alignments were made using ClustalW (Thompson et al. 1994) and adjusted manually using BioEdit (http://www.mbio.ncsu.edu/BioEdit/bioedit.html). Estimates of nucleotide polymorphism (segregating sites, S, nucleotide diversity, π), the scaled mutation rate (θ = 4Nμ), and between-species divergences were obtained using the computer program DnaSP v4.00.5 (http://www.ub.es/dnasp/). DnaSP was also used to calculate a statistical test of neutrality, Tajima's D (Tajima 1989). The significance of D was evaluated using coalescent simulations of an island model with an average genetic differentiation equal to that estimated for each locus (see below). All simulations were run using a fixed number of segregating sites, equal to the observed number of segregating sites at each locus, and without recombination, making all tests conservative.
The decay of linkage disequilibrium with physical distance was estimated using nonlinear regression of linkage disequilibrium between polymorphic sites vs. the distance, in base pairs, between sites (Remington et al. 2001). This analysis was done both within populations and for the complete data set at each locus. Linkage disequilibrium was scored between pairs of polymorphic sites using the squared allele frequency correlations, r2 (Weir 1990). Under a simple drift-recombination model the expected value of r2 is E(r2) = 1/(1 + r), where r = 4Nc is the scaled recombination rate for the gene region. In the presence of mutations, the expectation changes to
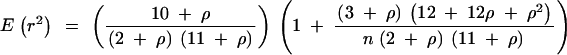 |
1 |
(Hill and Weir 1988), where n is number of haplotypes sampled. Equation 1 was fitted using the statistical package R (http://cran.us.r-project.org/). The nonlinear regression yields a least-squares estimate of r per base pair that may be biased because of the nonindependence between different sites (Remington et al. 2001). However, it may still be useful as a representation of the rate of decay of linkage disequilibrium with physical distance. Singletons were excluded in all linkage disequilibrium analyses.
Population subdivision was estimated as FST (Hudson et al. 1992; Charlesworth 1998) and significance of population subdivision was evaluated using the Snn-test statistic of Hudson (2000).
RESULTS
Nucleotide polymorphism and divergence:
Sequences were obtained from 17–24 individuals for the five genes studied. The length of the regions analyzed varied between ∼650 and ∼2200 bp [including insertions/deletions (indels), Table 1]. All regions contained both coding and noncoding sites (introns and/or flanking regions), although the amount of noncoding sites varied from as little as 30 bp in TI-3 to almost 1300 bp in Adh1 (Table 1).
Both indel polymorphisms and single-nucleotide polymorphisms (SNPs) were present in the sequenced regions. There were a total of nine indel polymorphisms, all in introns or flanking regions. In particular the 5′-UTR region of GA20ox1 was indel rich, with several short microsatellites that were highly polymorphic. There were no indels in the TI-3 region, which included only 30 noncoding sites. Indels were excluded in all further analyses.
SNP polymorphisms were substantially higher than indel polymorphisms; a total of 384 unique SNPs were found in the ∼6.2 kb sequenced from the five genes (Table 2) or ∼1 SNP every 60 bp. Total (πT), silent (synonymous sites and sites in introns, πsil), synonymous (πsyn), and nonsynonymous (πnonsyn) nucleotide diversities for the five genes are summarized in Table 2. Levels of polymorphism were generally high; a weighted average of silent site diversity across the five genes was 0.0160 and πsil varied between 9.4 × 10−3 at GA20ox1 and 22.9 × 10−3 at TI-3 (Table 2). Nonsynonymous diversities were generally lower than silent variation and the ratio of nonsynonymous to synonymous diversities ranged from 0.1 to 0.23 for Adh1, Gapdh, and GA20ox1, indicating strong purifying selection at most codons in these regions. CI-1 and TI-3 had about three- to fourfold higher levels of nonsynonymous diversity than the other three loci (Table 2), suggesting that the level of selective constraint varies between the genes.
TABLE 2.
Estimates of nucleotide diversity and divergence
| Polymorphism
|
Divergencea
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Locus | Population | N | S | θW | πT | πsil | πsyn | πnonsyn | πnonsyn/πsyn | DTajima | D | Ks | Ka | Ka/Ks |
| Adh | FRA | 6 | 15 | 0.0030 | 0.0027 | 0.0045 | 0.0063 | 0.0000 | 0.000 | 0.597 | ||||
| AUT | 10 | 58 | 0.0094 | 0.0080 | 0.0121 | 0.0167 | 0.0029 | 0.176 | −0.689 | |||||
| SWE S | 10 | 66 | 0.0107 | 0.0098 | 0.0132 | 0.0229 | 0.0037 | 0.162 | −0.398 | |||||
| SWE N | 8 | 49 | 0.0086 | 0.0089 | 0.0108 | 0.0233 | 0.0032 | 0.136 | 0.202 | |||||
| Total | 34 | 116 | 0.0131 | 0.0090 | 0.0119 | 0.0222 | 0.0028 | 0.128 | −1.816 | 0.034 | 0.045 | 0.012 | 0.259 | |
| SD | 0.0041 | 0.0080 | ||||||||||||
| CI-1 | FRA | 10 | 27 | 0.0121 | 0.0095 | 0.0097 | 0.0197 | 0.0092 | 0.466 | −1.018 | ||||
| AUT | 10 | 23 | 0.0107 | 0.0110 | 0.0128 | 0.0113 | 0.0085 | 0.753 | 0.378 | |||||
| SWE S | 12 | 24 | 0.0100 | 0.0079 | 0.0098 | 0.0204 | 0.0054 | 0.263 | −0.928 | |||||
| SWE N | 14 | 35 | 0.0139 | 0.0105 | 0.0118 | 0.0123 | 0.0085 | 0.694 | −1.074 | |||||
| Total | 46 | 62 | 0.0184 | 0.0101 | 0.0115 | 0.0157 | 0.0082 | 0.523 | −1.523 | 0.051 | 0.084 | 0.020 | 0.243 | |
| SD | 0.0054 | 0.0007 | ||||||||||||
| GAPDH | FRA | 10 | 37 | 0.0165 | 0.0145 | 0.0243 | 0.0198 | 0.0036 | 0.184 | −0.589 | ||||
| AUT | 12 | 31 | 0.0129 | 0.0132 | 0.0193 | 0.0246 | 0.0016 | 0.066 | 0.070 | |||||
| SWE S | 12 | 35 | 0.0146 | 0.0151 | 0.0200 | 0.0320 | 0.0047 | 0.146 | 0.148 | |||||
| SWE N | 14 | 33 | 0.0131 | 0.0131 | 0.0196 | 0.0339 | 0.0022 | 0.064 | 0.003 | |||||
| Total | 48 | 76 | 0.0176 | 0.0147 | 0.0223 | 0.0303 | 0.0031 | 0.102 | −1.128 | 0.024 | 0.040 | 0.002 | 0.042 | |
| SD | 0.0064 | 0.0006 | ||||||||||||
| GA20ox1 | FRA | 10 | 25 | 0.0053 | 0.0043 | 0.0062 | 0.0098 | 0.0027 | 0.270 | −0.887 | ||||
| AUT | 8 | 21 | 0.0048 | 0.0047 | 0.0074 | 0.0124 | 0.0027 | 0.220 | −0.110 | |||||
| SWE S | 12 | 34 | 0.0067 | 0.0061 | 0.0101 | 0.0120 | 0.0027 | 0.225 | −0.354 | |||||
| SWE N | 12 | 40 | 0.0079 | 0.0068 | 0.0119 | 0.0153 | 0.0037 | 0.242 | −0.616 | |||||
| Total | 42 | 81 | 0.0113 | 0.0059 | 0.0094 | 0.0130 | 0.0031 | 0.237 | −1.696 | 0.034 | 0.056 | 0.017 | 0.300 | |
| SD | 0.0034 | 0.0004 | ||||||||||||
| TI-3 | FRA | 10 | 24 | 0.0133 | 0.0124 | 0.0168 | 0.0202 | 0.0107 | 0.532 | −0.308 | ||||
| AUT | 12 | 32 | 0.0166 | 0.0161 | 0.0242 | 0.0291 | 0.0129 | 0.442 | −0.144 | |||||
| SWE S | 12 | 29 | 0.0150 | 0.0188 | 0.0288 | 0.0346 | 0.0149 | 0.430 | 1.121 | |||||
| SWE N | 14 | 25 | 0.0123 | 0.0087 | 0.0172 | 0.0206 | 0.0054 | 0.260 | −1.254 | |||||
| Total | 48 | 49 | 0.0216 | 0.0144 | 0.0229 | 0.0276 | 0.0112 | 0.406 | −0.586 | 0.055 | 0.074 | 0.046 | 0.623 | |
| SD | 0.0053 | 0.0172 | ||||||||||||
| Average | 0.0167 | 0.0111 | 0.0160 | 0.0220 | 0.0059 | 0.288 | 0.040 | 0.061 | 0.020 | 0.296 | ||||
Significant values are indicated in italic.
Jukes-Cantor-corrected divergence vs. P. trichocarpa.
There was also abundant nucleotide variation within populations at all five loci (Table 2), the only real exception being Adh1 samples from the FRA population, which had less than half the diversity of other populations, πsil = 0.0045. However, this is also the population from which the fewest samples were taken (N = 6), so this could be just a sampling artifact.
Silent site divergence, using P. trichocarpa as outgroup, was fairly constant across the five genes and averaged 0.049 (Table 2). However, the nonsynonymous substitution rate (Ka) varied >30-fold, from 0.0017 in G3PDH to 0.0463 in TI-3, suggesting that selective constraints and/or the history of adaptive evolution vary between loci. The low level of nonsynonymous divergence at Gapdh is consistent with strong selection for conserved amino acid sequence in this gene that plays a crucial role in glycolysis. There was a tendency across loci for an excess of singleton mutations, shown by negative values of Tajima's D, and this excess was significant in four out of five genes (Table 2).
Linkage disequilibrium:
The nonlinear regression model for analyzing the decay of linkage disequilibrium with distance showed that LD decays quite rapidly with distance when total samples were used; the expected value of r2 declined to <0.05 in around a few hundred bases (Figure 1). However, LD extends on average two to five times further within local populations, although there is substantial variation among populations. It should be noted that estimates of within-population rate of decay of LD are subject to much larger standard errors, due to the smaller number of sites that were polymorphic within populations. Despite the rapid decline of LD, several sites in Adh1 and GA20ox1 show extensive linkage disequilibrium over distances that approach the length of the sequenced region (Figure 1).
Figure 1.—





Plots showing the squared correlations of allele frequencies (r2) as a function of physical distance between sites for five genes in Populus tremula: (A) Adh1, (B) CI-1, (C) GA20ox1, (D) Gapdh, and (E) TI-3. The thick lines are fitted nonlinear regressions of the mutation-recombination-drift model given in Equation 1. Thin lines depict within-population decline in linkage disequilibrium (also fitted using Equation 1). It was not possible to fit Equation 1 for the FRA sample from TI-3 due to the low number of informative sites.
Population subdivision:
All five genes showed significant genetic differentiation among populations (Table 3) and using the combined data yielded an estimate of FST = 0.117, which is surprisingly high given that Populus is outbreeding, wind pollinated, and has seeds that are adapted to long-distance wind dispersal (Hamrick and Godt 1996). There was substantial variation across the five genes in levels of genetic differentiation, with the lowest estimate of FST = 0.04 for TI-3 and the highest estimate FST = 0.161 for Adh1. Most of the pairwise estimates of FST between populations were also significant, although in a few cases pairwise FST estimates were negative, probably reflecting low levels of genetic differentiation and/or stochastic variation associated with small sample sizes (supplementary Table 2 at http://www.genetics.org/supplemental/).
TABLE 3.
Estimates ofFST and tests of genetic differentiation
| All populations
|
||
|---|---|---|
| Locus | FSTa | Snnb |
| Adh | 0.161 | 0.647*** |
| CI-1 | 0.052 | 0.484*** |
| G3pdh | 0.071 | 0.461*** |
| GA20ox1 | 0.065 | 0.612*** |
| TI-3 | 0.040 | 0.361* |
| Total | 0.117 | 0.850*** |
Wright's fixation index (Hudson et al. 1992).
Statistical test of genetic differentiation (Hudson 2000).
To test whether there was evidence for any heterogeneity in the estimates of FST across loci, I ran a coalescent simulation (with 106 replicates). The simulations assumed an island model structure with a total of 20 populations and with a migration parameter, M = 4Nm, equal to 7.57. This produces an expected FST that matches the average FST observed from the five genes (FST = 0.117). A total of 34 chromosomes were simulated, distributed among 4 of the 20 populations using the same sample configuration as in the real samples (Table 2). The simulation was standardized to a locus 1 kb in length, with θ = 4Nμ matching the weighted average across loci. The approximate 95% confidence region for FST obtained from the simulation is 0.012–0.276 and all five loci have FST values that fall with this region, suggesting that the among-locus variation in FST across loci is not unusually high in this data set.
DISCUSSION
Nucleotide polymorphism and divergence:
There are substantial levels of nucleotide polymorphisms in all five genes included in this study, both species-wide and within populations (Table 2). Levels of silent site diversity within populations averaged 0.0145, although diversity varied almost threefold between loci, from 0.0089 in GA20ox1 to 0.022 in TI-3. These values are comparable to within-population diversity in outcrossing A. lyrata (pW = 0.014, Wright et al. 2003) and to the average within-population diversity seen in Drosophila melanogaster (πW = 0.014, Moriyama and Powell 1996). It is also about two to fivefold higher than nucleotide polymorphism in the BpMADS2 gene in Betula pendula, another long-lived woody perennial, which was sampled from two locations in Finland (πW = 0.0047, Järvinen et al. 2002).
Species-wide levels of silent polymorphism in P. tremula (πsil = 0.016) are also comparable to levels of polymorphism in several plant species. In selfing A. thaliana, πsil = 0.0114, whereas in the two closely related, outcrossing species A. lyrata ssp. petrea and A. halleri species-wide diversity at silent sites averaged 0.023 and 0.015, respectively (Aguade 2001; Wright et al. 2003; Ramos-Onsins et al. 2004). In two wild species of maize, Zea diploperennis and Z. perennis, silent site diversity in four genes averaged 0.012 and 0.013 (Tiffin and Gaut 2001). It is not surprising to see such similar levels of species-wide diversities, despite differences in breeding system, life histories, and/or demography since these factors are known to have far less effect on species-wide levels of polymorphism than on levels of polymorphism within populations (Ingvarsson 2002; Charlesworth 2003).
It thus appears that P. tremula harbors substantial genetic diversity, both within populations and across the species range. This is in agreement with earlier studies of allozyme variation that have shown that Populus has a substantially higher proportion of polymorphic loci and higher gene diversity than the average among long-lived, woody perennials (Jelinski and Cheliak 1992; Liu and Furnier 1993; Hamrick and Godt 1996).
This is in stark contrast to recent studies of several species of conifers, which appear to have substantially lower genome-wide levels of polymorphism. For instance, levels of silent polymorphism range from 0.0004 to 0.0049 in P. sylvestris (Dvornyk et al. 2002; Garcia-Gil et al. 2003) and from 0.00017 to 0.0081 in Cryptomeria japonica (Kado et al. 2003). Similarily, in Pinus taeda, silent site diversity averaged 0.0064 across 63 genes involved in wood formation and pathogen and drought resistance (Neale and Savolainen 2004).
Polymorphism in P. tremula is thus ∼2- to 10-fold higher than that in Pinus and Cryptomeria. P. tremula has, together with P. sylvestris, among the widest-known geographic distributions of any tree species and the samples analyzed in this article cover only a small fraction of the entire range of P. tremula, so species-wide diversities are, if anything, underestimated in P. tremula. This suggests that the low nucleotide diversities seen in Pinus and Cryptomeria are not a general phenomenon in long-lived tree species but rather appear to be unique for conifers.
Species-wide samples show an excess of low-frequency polymorphisms that is remarkably consistent across loci (Table 2). Selection seems like an unlikely cause of negative values of these statistics because the excess of low-frequency polymorphisms is consistent across loci. Also, sequences were obtained through cloning of PCR products and although care was taken to verify all singleton mutations in multiple clones, it is possible that some of the remaining singletons are the result of Taq polymerase errors. However, given the error rate of Taq of 1 × 10−4–2 × 10−5 (Cline et al. 1996), PCR errors would account for only between one and five singletons at each locus and thus do not appear to explain the significant excess of singleton mutations observed. Therefore, it is more likely that the departures of the frequency spectra from the standard neutral model are a product of demographic processes, such as postglacial expansion (Petit et al. 2003) or effects of population subdivision. Although population structure is expected to shift the frequency spectrum toward an excess of sites at intermediate frequencies (i.e., positive values of DTajima) under some scenarios, it is possible that population subdivision can also lead to negative Tajima's D, depending on strength of population subdivision and the actual sample configuration. Thus the reason for the overall excess of low-frequency mutation in Populus must be investigated in more detail.
Silent site divergence from P. trichocarpa averaged 6.1% and, using a silent substitution rate in the range 5.0–8.0 × 10−9 (Wolfe et al. 1987; Gaut 1998), yields an estimated time of divergence between P. tremula and P. trichocarpa between 3.8 and 6.2 million years. P. tremula and P. trichocarpa belong to different sections within the genus Populus (Leuce, aspens, and Tacamahaca poplars, respectively) and are morphologically quite distinct. It thus appears that the radiation that produced the ∼300 species contained in the genus Populus has been rapid and relatively recent. This view is also supported by other molecular data (Leskinen and Alström-Rapaport 1999) and by the prevalence of both intra- and intersectional hybridization in Populus (Brunner et al. 2004).
Linkage disequilibrium:
LD declines rapidly with distance in all five genes. In general, average linkage disequilibrium declined to negligible levels (r2 < 0.05) in <500 bp, although in some cases LD extends much further within local populations (up to 1 kb or more). Levels of LD observed in Populus are on par with those observed in maize, where LD also declines rapidly. Remington et al. (2001) showed that LD declined to negligible levels in ∼1 kb in 5 of 6 genes studied in a set of 102 inbred lines of maize. The sixth gene studied, su1, is part of the starch production pathway in maize and has been a target of selection during the domestication of maize (Whitt et al. 2002) and this artificial selection has presumably contributed to the extensive LD seen in su1. A study of a more limited set of elite maize lines showed that LD can extend over great distances also in maize, up to 100 kb, and that there was virtually no decline in r2 over distances of ∼500 bp in the collection of 18 genes they surveyed, consistent with a strong effect of breeding-induced bottlenecks and selection in producing the elite germplasm pool (Ching et al. 2002).
These results from Populus and maize, both outcrossers, are in stark contrast to the patterns of LD seen in primarily self-fertilizing species, such as A. thaliana and rice (Oryza spp.). Selfing dramatically reduced the effective recombination (Nordborg 2000), and in Arabidopsis LD generally extends over distances up to 250 kb (Nordborg et al. 2000) while in rice LD extends ∼100 kb (Garris et al. 2003). However, in wild barley (Hordeum vulgare ssp. spontaneum), another highly selfing species, some loci show high levels of LD, as expected in a selfing species, where as other loci show a surprisingly rapid decline of LD with distance (Lin et al. 2002). Although the reasons behind the different patterns of LD among loci in barley and also differences in LD patterns between Arabidopsis and barley are not fully understood, it is clear that mating system alone does not explain differences in LD and that other factors, such as the demographic history of a species, are important as well (Lin et al. 2002).
Population structure:
All five genes showed significant genetic differentiation among populations, although estimates of FST were moderate (Table 3). The estimates of FST are still surprisingly high given that Populus is outbreeding, wind pollinated, and has seeds that are adapted to long-distance wind dispersal (Hamrick and Godt 1996). There was a greater than fourfold variation among loci in estimates of FST, but a coalescent simulation suggested that this dispersion is no greater than expected. A recent study of genetic differentiation in maternally inherited cpDNA markers also showed moderate levels of genetic differentiation across Europe in P. tremula (FST = 0.11, Petit et al. 2003). Interestingly, the cpDNA estimate of FST is close to the average of the five nuclear genes studied here. This is somewhat surprising, since maternally inherited cpDNA markers are haploid and their lower effective population size in a dioecious species, like P. tremula, should result in an increased genetic differentiation, compared to nuclear genes [Ne(cpDNA)/Ne(nuclear) ≈ 0.25 with equal sex ratios, Laporte and Charlesworth 2002]. Sex ratios of Scandinavian P. tremula populations are also predominantly male biased (Blumenthal 1942), reinforcing the differences in effective population sizes between nuclear and organelle loci. More loci are needed to establish whether this discrepancy in genetic differentiation between organelle and nuclear markers is a real phenomenon or just a sampling artifact.
Conclusions:
This is the first study to quantify levels of nucleotide polymorphism and linkage disequilibrium from a member of the genus Populus. The results presented here show that P. tremula harbors significant amounts of genetic variation both within populations and across the species range. Since estimates of population subdivision are low (albeit significant), a substantial fraction of the species-wide diversity can be sampled from single populations. In the species-wide sample LD declines rapidly with physical distance, although it may extend much further within local populations.
The rate of decay of LD has implications for the potential utility of association approaches in mapping QTL as it effectively determines whether genome-wide scans are feasible or whether a candidate gene approach has to be taken (Gaut and Long 2003). The rapid decay of LD in P. tremula suggests that the number of markers needed to ensure adequate genome coverage may be simply too high for genome-wide scans to be feasible in this species and a more realistic approach might therefore be to search for functional variation in putative candidate loci. On the other hand, the rapid decay of LD suggests that it may be possible to map functional variation to very fine scales in P. tremula.
Population structure is low, but significant for all loci investigated. This structure creates additional linkage disequilibrium between loci that have different allelic frequencies in different subpopulations. Population structure may therefore interfere with association studies by producing spurious associations. However, methods to properly account for population structure have been developed (Pritchard et al. 2000) and applied successfully in other plant species (Thornsberry et al. 2001). Nevertheless, the presence of significant population structure and variation in rate of decay of LD in different populations emphasizes the care that has to be taken when sampling for association studies.
Acknowledgments
I thank Carin Olofsson for assistance in obtaining the DNA sequences presented in this article. Outi Savolainen and two anonymous reviewers provided comments that notably improved the article. Funding from the Swedish Research Council (Vetenskapsrådet) is also acknowledged.
References
- Aguade, M., 2001. Nucleotide sequence variation at two genes of the phenylpropanoid pathway, the FAH1 and F3H genes in Arabidopsis thaliana. Mol. Biol. Evol. 18: 1–9. [DOI] [PubMed] [Google Scholar]
- Blumenthal, B. E., 1942. Studier angående aspens förekomst i Finland. Silv. Fenn. 56: 1–63. [Google Scholar]
- Brunner, A. M., V. B. Busov and S. H. Strauss, 2004. Poplar genome sequence: functional genomics in an ecologically dominant plant species. Trends Plant Sci. 9: 49–56. [DOI] [PubMed] [Google Scholar]
- Charlesworth, B., 1998. Measures of divergence between populations and the effects of forces that reduce variability. Mol. Biol. Evol. 15: 538–543. [DOI] [PubMed] [Google Scholar]
- Charlesworth, D., 2003. Effects of inbreeding on the genetic diversity of populations. Philos. Trans. R. Soc. Lond. Biol. B Biol. Sci. 358: 1051–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching, A., K. S. Caldwell, M. Jung, M. Dolan, O. S. Smith et al., 2002. SNP frequency, haplotype structure and linkage disequilibrium in elite maize inbred lines. BMC Genet. 3: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline, J., J. C. Braman and H. H. Hogrefe, 1996. PCR fidelity of Pfu DNA polymerase and other thermostable DNA polymerases. Nucleic Acids Res. 24: 3546–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvornyk, V., A. Sirviö, M. Mikkonen and O. Savolainen, 2002. Low nucleotide diversity at the pal1 locus in the widely distributed Pinus sylvestris. Mol. Biol. Evol. 19: 179–188. [DOI] [PubMed] [Google Scholar]
- Eriksson, M. E., and T. Moritz, 2002. Daylength and spatial expression of a gibberellin 20-oxidase isolated from hybrid aspen (Populus tremula × P. tremuloides Michx.). Planta 214: 920–930. [DOI] [PubMed] [Google Scholar]
- Flint-Garcia, S. A., J. M. Thornsberry and E. S. Bucker, IV, 2003. Structure of linkage disequilibrium in plants. Annu. Rev. Plant Biol. 54: 357–374. [DOI] [PubMed] [Google Scholar]
- Garcia-Gil, M. R., M. Mikkonen and O. Savolainen, 2003. Nucleotide diversity at two phytochrome loci along a latitudinal cline in Pinus sylvestris. Mol. Ecol. 12: 1195–1206. [DOI] [PubMed] [Google Scholar]
- Garris, A. J., S. R. McCouch and S. Kresovich, 2003. Population structure and its effects on haplotype diversity and linkage disequilibrium surrounding the xa5 locus of rice (Oryza sativa L.). Genetics 165: 759–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaut, B. S., 1998. Molecular clocks and nucleotide substitution rates in higher plants. Evol. Biol. 30: 93–120. [Google Scholar]
- Gaut, B. S., and A. D. Long, 2003. The lowdown on linkage disequilibrium. Plant Cell 15: 1502–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamrick, J. L., and M. J. W. Godt, 1996. Effects of life history traits on genetic diversity in plant species. Philos. Trans. R. Soc. Lond. B Biol. Sci. 351: 1291–1298. [Google Scholar]
- Haruta, M., I. T. Major, M. E. Christopher, J. J. Patton and C. P. Constabel, 2001. A kunitz trypsin inhibitor gene family from trembling aspen (Populus tremuloides): cloning, functional expression and induction by wounding and herbivory. Plant Mol. Biol. 46: 247–259. [DOI] [PubMed] [Google Scholar]
- Hill, W. G., and B. S. Weir, 1988. Variances and covariances of squared linkage disequilibria in finite populations. Theor. Popul. Biol. 33: 54–78. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., 2000. A new test statistic for detecting genetic differentiation. Genetics 155: 2011–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., D. D. Boos and N. L. Kaplan, 1992. A statistical test for detecting geographic subdivision. Mol. Biol. Evol. 9: 138–151. [DOI] [PubMed] [Google Scholar]
- Ingvarsson, P. K., 2002. A metapopulation perspective of genetic diversity and differentiation in partially self-fertilizing plants. Evolution 56: 2368–2373. [DOI] [PubMed] [Google Scholar]
- Järvinen, P., J. Lemmetyinen, O. Savolainen and T. Sopanen, 2002. DNA sequence variation in BpMADS2 gene in two populations of Betula pendula. Mol. Ecol. 12: 369–384. [DOI] [PubMed] [Google Scholar]
- Jelinski, D. E., and W. M. Cheliak, 1992. Genetic diversity and spatial subdivision of Populus tremuloides (Salicaceae) in a heterogeneous landscape. Am. J. Bot. 79: 728–736. [Google Scholar]
- Kado, T., H. Yoshimaru, Y. Tsumura and H. Tachida, 2003. DNA variation in a conifer, Cryptomeria japonica (Cupressaceae sensu lato). Genetics 164: 1547–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laporte, V., and B. Charlesworth, 2002. Effective population size and population subdivision in demographically structured populations. Genetics 162: 501–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leskinen, E., and C. Alström-Rapaport, 1999. Molecular phylogeny of Salicaceae and closely related Flacourtiaceae: evidence from 5.8, ITS 1 and ITS 2 of the rDNA. Plant. Syst. Evol. 215: 209–227. [Google Scholar]
- Lin, J.-Z., P. L. Morrell and M. T. Clegg, 2002. The influence of linkage and inbreeding on patterns of nucleotide sequence diversity at duplicate alcohol dehydrogenase loci in wild barley (Hordeum vulgare ssp. spontaneum). Genetics 162: 2007–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Z., and G. R. Furnier, 1993. Comparison of allozyme, RFLP and RAPD markers for revealing genetic variation within and between trembling aspen and bigtooth aspen. Theor. Appl. Genet. 87: 97–105. [DOI] [PubMed] [Google Scholar]
- Moriyama, E. N., and J. R. Powell, 1996. Intraspecific nuclear DNA variation in Drosophila. Mol. Biol. Evol. 13: 261–277. [DOI] [PubMed] [Google Scholar]
- Neale, D. B., and O. Savolainen, 2004. Association genetics of complex traits in conifers. Trends Plant Sci. 9: 325–330. [DOI] [PubMed] [Google Scholar]
- Nordborg, M., 2000. Linkage disequilibrium, gene trees and selfing: an ancestral recombination graph with partial selfing. Genetics 154: 923–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordborg, M., J. O. Borevitz, J. Bergelson, C. C. Berry, J. Chory et al., 2000. The extent of linkage disequilibrium in Arabidopsis thaliana. Nat. Genet. 30: 190–193. [DOI] [PubMed] [Google Scholar]
- Petit, R. J., I. Aguinagalde, J. L. de Beaulieu, C. Bittkau, S. Brewer et al., 2003. Glacial refugia: hotspots but not melting pots of genetic diversity. Science 300: 1563–1565. [DOI] [PubMed] [Google Scholar]
- Pritchard, J. K., M. Stephens, N. A. Rosenberg and P. Donnelly, 2000. Association mapping in structured populations. Am. J. Hum. Genet. 67: 170–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafalski, A., and M. Morgante, 2004. Corn and humans: recombination and linkage disequilibrium in two genomes of similar size. Trends Genet. 20: 103–111. [DOI] [PubMed] [Google Scholar]
- Ramos-Onsins, S. E., B. E. Stranger, T. Mitchell-Olds and M. Agaude, 2004. Miltilocus analysis of variation and specialization in the closely related species Arabidopsis halleri and A. lyrata. Genetics 166: 373–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remington, D. L., J. M. Thornsberry, Y. Matsouka, L. M. Wilson, S. R. Whitt et al., 2001. Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc. Natl. Acad. Sci. USA 98: 11479–11484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savolainen, O., C. H. Langley, B. P. Lazzaro and H. Fréville, 2000. Contrasting patterns of nucleotide polymorphism at the alcohol dehydrogenase locus in the outcrossing Arabidopsis lyrata and the selfing Arabidopsis thaliana. Mol. Biol. Evol. 17: 645–655. [DOI] [PubMed] [Google Scholar]
- Semerikov, V. L., and M. Lascoux, 2003. Nuclear and cytoplasmic variation within and between Eurasian Larix (Pinaceae) species. Am. J. Bot. 90: 1113–1123. [DOI] [PubMed] [Google Scholar]
- Strand, A. E., J. Leebens-Mack and G. B. Milligan, 1997. Nuclear DNA-based markers for plant evolutionary biology. Mol. Ecol. 6: 113–118. [DOI] [PubMed] [Google Scholar]
- Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J. D., D. G. Higgins and T. J. Gibson, 1994. ClustalW: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22: 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornsberry, J. M., M. M. Goodman, J. Doebley, S. Kresovich, D. Nielsen et al., 2001. Dwarf8 polymorphisms associate with variation in flowering time. Nat. Genet. 28: 286–289. [DOI] [PubMed] [Google Scholar]
- Tiffin, P., and B. S. Gaut, 2001. Sequence diversity in the tetraploid Zea perennis and the closely related diploid Z. diploperennis: insights from four nuclear loci. Genetics 158: 401–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir, B. S., 1990 Genetic Data Analysis. Sinauer, Sunderland, MA.
- Whitt, S. R., L. M. Wilson, M. I. Tenallion, B. S. Gaut and E. S. Buckler, IV, 2002. Genetic diversity and selection in the maize starch pathway. Proc. Natl. Acad. Sci. USA 99: 12959–12962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe, K. H., W.-H. Li and P. M. Sharp, 1987. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast and nuclear DNAs. Proc. Natl. Acad. Sci. USA 84: 9054–9058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S., B. Lauga and D. Charlesworth, 2003. Subdivision and haplotype structure in natural populations of Arabidopsis lyrata. Mol. Ecol. 12: 1247–1263. [DOI] [PubMed] [Google Scholar]
- Wullschleger, S. D., S. Jansson and G. Taylor, 2002. Genomics and forest biology: Populus emerges as the perennial favorite. Plant Cell 14: 2651–2655. [DOI] [PMC free article] [PubMed] [Google Scholar]