

Abstract
We have investigated patterns of within-species polymorphism and between-species divergence for synonymous and nonsynonymous variants at a set of autosomal and X-linked loci of Drosophila miranda. D. pseudoobscura and D. affinis were used for the between-species comparisons. The results suggest the action of purifying selection on nonsynonymous, polymorphic variants. Among synonymous polymorphisms, there is a significant excess of synonymous mutations from preferred to unpreferred codons and of GC to AT mutations. There was no excess of GC to AT mutations among polymorphisms at noncoding sites. This suggests that selection is acting to maintain the use of preferred codons. Indirect evidence suggests that biased gene conversion in favor of GC base pairs may also be operating. The joint intensity of selection and biased gene conversion, in terms of the product of effective population size and the sum of the selection and conversion coefficients, was estimated to be ∼0.65.
DROSOPHILA miranda (a close relative of D. pseudoobscura) provides a model system for studying the evolutionary effects of reduced recombination. In this species, an autosome (Muller's element C) has become fused to the Y chromosome and does not recombine (the neo-Y), while its homolog (the neo-X) cosegregates with the X chromosome and recombines in the homogametic females (MacKnight 1939; Steinemann and Steinemann 1998). The neo-Y chromosome shows clear signs of incipient loss of gene function, including absence of genes, reduction in gene expression, and major changes (such as deletions) to some coding sequences (MacKnight 1939; Steinemann and Steinemann 1998; Bachtrog 2003a,b). The absence of genetic recombination on the neo-Y chromosome is expected to result in reduced levels of genetic variability and adaptation, reflecting a reduction in effective population size caused by various types of Hill-Robertson effects associated with selection acting on a nonrecombining block of genes (reviewed by Charlesworth and Charlesworth 2000). The degeneration of the neo-Y chromosome probably reflects the cumulative effects of this reduction in the efficacy of selection.
In accordance with theoretical expectation, silent-site diversities at neo-Y loci are reduced compared with their neo-X linked homologs (Bachtrog and Charlesworth 2002). In addition, data on protein evolution on the neo-sex chromosomes of this species (Yi and Charlesworth 2000; Bachtrog 2003a,b) suggest that there has been an accumulation of amino acid substitutions on the nonrecombining neo-Y. This probably reflects a weakening of the effectiveness of selection against deleterious amino acid substitutions. In addition, there is an apparent excess of fixations of synonymous mutations, creating unpreferred codons on both the neo-X and neo-Y chromosomes (Bachtrog 2003b). While a reduction in the effectiveness of selection for codon usage on the neo-Y chromosome of D. miranda is in accord with expectation, the reduction for neo-X genes is surprising, in view of the evidence for selection on codon usage in D. pseudoobscura (Akashi and Schaeffer 1997). But the effective population size (Ne) of D. miranda appears to be much smaller than that of D. pseudoobscura (Yi et al. 2003). Unless there is extreme mutational bias in favor of unpreferred codons (McVean and Charlesworth 1999; Takano-Shimizu 1999), this could have resulted in evolution toward reduced codon usage bias (Bachtrog 2003b), since lower Ne means that genetic drift is more likely to overcome the effect of selection and cause the fixation of weakly deleterious mutations (Kimura 1983). Examples of evolution toward reduced codon usage bias in species with small Ne, such as D. guanche (Perez et al. 2003), are consistent with this process.
The primary objective of this analysis was to examine the patterns of protein evolution and codon usage bias on autosomal and X-linked genes of D. miranda, to determine whether the patterns observed for genes located on the neo-sex chromosomes are a general feature of this species. We used the publicly available genome sequence of D. pseudoobscura (http://hgsc.bcm.tmc.edu/projects/drosophila/), and a set of DNA sequences that we determined from D. affinis, for the between-species divergence estimates. D. affinis is the only readily available relative of D. pseudoobscura and its sibling species (Powell 1997), yet has been little studied at the level of DNA sequences. The use of D. affinis together with D. pseudoobscura allows assignment of mutations to the two branches of the phylogeny connecting D. miranda and D. pseudoobscura to their common ancestor, which is extremely useful for inferring patterns of evolution and variation at synonymous and noncoding sites (Akashi 1996; Maside et al. 2004). In addition, its relatively high level of divergence from the other two species makes it useful for estimates of net between-species divergence.
The results indicate that efficient purifying selection is acting on amino acid replacement polymorphisms, and that selection is still maintaining codon usage at the loci that we studied. The apparent ineffectiveness of selection on codon usage on the neo-X chromosome (Bachtrog 2003b) is probably a consequence of polymorphic variants having been classed as fixed differences.
MATERIALS AND METHODS
Strains used:
We studied 12 D. miranda lines derived from single wild-caught females: 0101.3, 0101.4, 0101.5, 0101.7 (Port Coquitlam, British Columbia, Canada), 0101.9, MA28, MA32 (Mather, CA), SP138, SP235, SP295 (Spray, OR), MSH22, and MSH38 (Mount Saint Helena, CA). The flies were originally obtained from the National Drosophila Species Resource Center (Bowling Green, OH) and from M. Noor and W.W. Anderson. Two other lines from different species were used as outgroups: a strain of D. affinis from Nebraska (no. 0141.2; Drosophila Species Resource Center) and a strain of D. pseudoobscura from Mather, California (provided by J. Coyne). Stocks of all three species were reared on banana medium at 18°.
DNA extraction and PCR amplification:
The genes studied here were initially selected from the sequences of D. pseudoobscura available in GenBank until the release of its complete genomic sequence (http://hgsc.bcm.tmc.edu/projects/drosophila/), which subsequently allowed us to broaden the choice of loci. Some of these genes were included in a previous study of chromosomal and DNA sequence variation in D. miranda (Yi et al. 2003; see Table 1). We used a longer sequence for Gapdh2 than did Yi et al. (2003), so that the data for this locus are new. Twenty loci were used for the population survey results reported here: 6 of them are located on chromosome 2 of D. miranda (bcd, Bruce, Gld, hyd, sry-alpha, and rosy), 7 on chromosome 4 (ade3, Adh, amd, Ddc, Eno, Lam, and Uro), and 7 on the left arm of the X chromosome (AnnX, Cyp1, Gapdh2, scute, sesB, sisA, and swallow; Yi et al. 2003; our unpublished data). These chromosomes correspond to Muller's elements E, B, and A, respectively (Ashburner 1989). For the other genes, a single allele from each species was investigated. Descriptions of the sequences analyzed are given in Table 1.
TABLE 1.
Details of the genes studied
| Segment sequenced
|
||||||
|---|---|---|---|---|---|---|
| Gene | 5′ | 3′ | Codingb | Noncodingb | Totalb | Chromosome |
| bcd bicoid | Exon 2 | Exon 3 | 1068 | 72 | 1140 | 2 |
| Bruce Bruce | Exon 19 | Exon 23 | 627 | 306 | 933 | 2 |
| ftz fushi tarazu | Exon 1 | Exon 2 | 1017 | 111 | 1128 | 2 |
| Gld glucose dehydrogenase | Exon 3 | Exon 3 | 1350 | 0 | 1350 | 2 |
| hb hunchback | Exon 1 | Exon 1 | 2001 | 0 | 2001 | 2 |
| hyd hyperplastic discs | Exon 15 | Exon 19 | 903 | 270 | 1173 | 2 |
|
ninaE (rh1) neither inactivation nor after potential E |
Exon 2 | Exon 5 | 936 | 762 | 1698 | 2 |
| Nop56 Nop56 | Exon 2 | Exon 3 | 1275 | 63 | 1338 | 2 |
| RpL32 (rp49) ribosomal protein L32 | 5′-FID | 3′-FID | 402 | 814 | 1216 | 2 |
| rosy (Xdh) rosy | Exon 2 | Exon 3 | 2295 | 63 | 2358 | 2 |
| sry-alphaa serendipity α | Exon 1 | Exon 1 | 477 | 0 | 477 | 2 |
| Tl Toll | Exon 1 | Exon 2 | 2175 | 90 | 2265 | 2 |
| ade3 (Gart) adenosine 3 | Exon 2 | Exon 5 | 1377 | 801 | 2178 | 4 |
| Adha alcohol dehydrogenase | 5′-FID | 3′-FID | 762 | 1496 | 2258 | 4 |
| Adhr Adh-related | Exon 2 | Exon 3 | 678 | 63 | 741 | 4 |
| Amd α-methyl dopa-resistant | Exon 1 | Exon 2 | 912 | 465 | 1377 | 4 |
| Ddc dopa decarboxylase | Exon 3 | Exon 3 | 912 | 0 | 912 | 4 |
| dpp decapentaplegic | Exon 1 | Exon 2 | 1002 | 2295 | 3297 | 4 |
| Eno Enolase | Exon 2 | Exon 3c | 1050 | 69 | 1119 | 4 |
|
Gpdh glycerol-3-phosphate dehydrogenase |
Exon 1 | Exon 3 | 411 | 3544 | 3955 | 4 |
| Lam Lamin | Exon 1 | Exon 2d | 1500 | 90 | 1590 | 4 |
| smo smoothened | Exon 3 | Exon 6 | 1284 | 229 | 1513 | 4 |
| Uro urate oxidase | Exon 1 | Exon 2 | 864 | 69 | 933 | 4 |
| AnnX Annexin X | Exon 2 | Intron 3 | 612 | 264 | 876 | XL |
| Cyp1a cyclophylin 1 | Intron 1 | Intron 1 | 0 | 634 | 634 | XL |
|
Gapdh2 glyceraldehyde-3-phosphate dehydrogenase 2 |
Exon 1 | Exon 1 | 768 | 0 | 768 | XL |
| scutea scute | Exon 1 | 3′-FID | 684 | 407 | 1091 | XL |
| sesBa stress-sensitive B | Exon 2 | Exon 4 | 711 | 174 | 885 | XL |
| sisAa sisterless A | 5′-FID | 3′-FID | 636 | 1292 | 1928 | XL |
| swallowa swallow | Exon 1 | Exon 3 | 1026 | 144 | 1170 | XL |
| Est-5B esterase-5B | 5′-FID | Exon 2 | 1569 | 186 | 1755 | XR |
| Hsp83 (Hsp82) heat-shock protein 83 | Exon 1 | Exon 1 | 789 | 0 | 789 | XR |
| Sod superoxide dismutase | Exon 1 | Exon 2 | 318 | 399 | 717 | XR |
Names in parentheses represent the names used in previous studies. FID, flanking intergenic DNA.
Polymorphism data are from Yi et al. (2003).
Length in base pairs (including alignment gaps).
Exon 2 of D. melanogaster.
Exon 3 of D. melanogaster.
Primers were designed for regions conserved between D. melanogaster and D. pseudoobscura after identifying their orthologous sequences by means of a BLAST search from http://hgsc.bcm.tmc.edu/blast/?organism=Dpseudoobscura and subsequent alignment. Genomic DNA samples were extracted from a single male of each line using Puregene (Gentra Systems, Minneapolis). We employed standard PCR procedures using the Expand high-fidelity PCR system (Roche Diagnostics, Lewes, East Sussex, UK), gel purifying the products with Qiaquick (QIAGEN, Crawley, West Sussex, UK).
Cloning and sequencing:
Sequences were cloned from purified PCR products using TOPO-TA (Invitrogen, San Diego), except for the X-linked loci for which the use of a single male fly should ensure the hemizygosity of the templates. DNA sequencing was performed on an ABI3730 automatic sequencing machine using Dyenamic (Amersham Biosciences, Little Chalfont, Buckinghamshire, UK). To minimize errors in the sequencing procedure for autosomal loci, at least three plasmids from each cloning reaction were sequenced. Both strands were sequenced. All read-outs were checked for accurate base calling and assembled using Sequencher (Gene Codes, Ann Arbor, MI). Sequences have been deposited in GenBank (accession nos. AY754390, AY754609).
Sequence analyses:
Sequences were edited and manually aligned using Se-Al (A. Rambaut, http://evolve.zoo.ox.ac.uk/software.html?name=Se-Al). Noncoding DNA (introns and 5′- and 3′-flanking sequences) alignments were performed using McAlign (Keightley and Johnson 2004), a program that implements a statistical method based on an evolutionary model of the frequency distribution of gaps and substitutions observed in Drosophila. Slight differences from the alignment used by Yi et al. (2003) for the genes used in that study mean that there are some numerical differences from their estimates of divergence and polymorphism. Population genetic analyses were conducted with DnaSP (β v. 3.99; Rozas 1999).
Fop, the frequency of “optimal” codons in a gene (Marais and Duret 2001), was calculated for each gene of D. miranda using a C program, kindly provided by L. Duret, applying the table of optimal codons for D. pseudoobscura (Akashi and Schaeffer 1997). Amino acid mutations and synonymous substitutions were assigned to either the D. miranda or D. pseudoobscura branches of the phylogeny connecting these two close relatives to the outgroup species D. affinis, assuming parsimony (Akashi 1996). Only changes assigned to the D. miranda lineage were used in the analysis of patterns of polymorphism.
RESULTS
Sequence polymorphism data:
Nucleotide diversity within D. miranda at each locus is shown in Table 2. The most variable gene was rosy, in agreement with previous reports showing that it is highly polymorphic at the coding sequence level in several Drosophila species (Riley et al. 1992; Begun and Whitley 2002). The unweighted average silent pairwise nucleotide diversity for all the genes studied was 0.41% although a slight overall difference between autosomal and X-linked loci was detected (means of 0.48% vs. 0.28%, respectively). The results given in Table 3 show that there is no significant difference in mean Ks for silent or synonymous sites between autosomal and X-linked genes (the means and standard errors of the silent Ks-values between D. pseudoobscura and D. affinis are 23.4 ± 1.4% and 20.2 ± 2.3%, for autosomal and X-linked genes, respectively), so there is no evidence for an overall difference in mutation rate between autosomal and X-linked loci, as seems usually to be the case in Drosophila (Bauer and Aquadro 1997).
TABLE 2.
Nucleotide diversity withinD. miranda (values expressed as percentages)
| πb
|
θwc
|
||||||
|---|---|---|---|---|---|---|---|
| Gene | Replacement | Synonymous | Silent | Replacement | Synonymous | Silent | Tajima's D: All sites |
| bcd | 0.02 | 0.74 | 0.87 | 0.04 | 0.95 | 0.97 | −0.63 |
| Bruce | 0.04 | 0.12 | 0.19 | 0.07 | 0.23 | 0.30 | −1.53 |
| Gld | 0.02 | 0.75 | 0.75 | 0.03 | 0.60 | 0.60 | 0.51 |
| hyd | 0.05 | 0.00 | 0.13 | 0.10 | 0.00 | 0.23 | −1.79 |
| rosy | 0.24 | 1.75 | 1.69 | 0.25 | 1.69 | 1.63 | 0.02 |
| sry-alphaa | 0.31 | 0.30 | 0.30 | 0.36 | 0.28 | 0.28 | −0.45 |
| ade3 | 0.00 | 0.09 | 0.26 | 0.00 | 0.10 | 0.29 | −0.45 |
| Adha | 0.00 | 0.37 | 0.28 | 0.00 | 0.34 | 0.23 | 0.99 |
| amd | 0.02 | 0.61 | 0.27 | 0.05 | 0.61 | 0.35 | −1.00 |
| Ddc | 0.21 | 0.32 | 0.32 | 0.19 | 0.31 | 0.31 | 0.32 |
| Eno | 0.02 | 0.43 | 0.34 | 0.04 | 0.66 | 0.52 | −1.43 |
| Lam | 0.05 | 0.42 | 0.33 | 0.06 | 0.39 | 0.31 | 0.05 |
| Uro | 0.00 | 0.58 | 0.52 | 0.00 | 0.51 | 0.52 | −0.06 |
| AnnX | 0.04 | 0.00 | 0.00 | 0.07 | 0.00 | 0.00 | −1.14 |
| Cyp1a | NA | NA | 0.49 | NA | NA | 0.63 | −0.92 |
| Gapdh2 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | NA |
| scutea | 0.11 | 0.36 | 0.25 | 0.13 | 0.42 | 0.34 | −0.89 |
| sesBa | 0.00 | 0.10 | 0.32 | 0.00 | 0.20 | 0.41 | −0.74 |
| sisAa | 0.41 | 0.84 | 0.75 | 0.43 | 0.65 | 0.84 | −0.42 |
| swallowa | 0.05 | 0.18 | 0.11 | 0.09 | 0.15 | 0.09 | −0.83 |
| Average | 0.08 | 0.42 | 0.41 | 0.10 | 0.42 | 0.44 | −1.11 |
NA, not available.
Sequence data are from Yi et al. (2003) after realignment with McAlign (Keightley and Johnson 2004).
Pairwise nucleotide diversity (Nei 1987).
Nucleotide site variability is based on the number of segregating sites (Watterson 1975).
TABLE 3.
Synonymous (Ks), silent (Ksilent), and nonsynonymous (Ka) divergence betweenD. miranda, D. pseudoobscura, andD. affinis, expressed as percentages
|
D. miranda vs. D. pseudoobscura
|
D. miranda vs. D. affinis
|
D. pseudoobscura vs. D. affinis
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Ks | Ksilent | Ka | Ka/Ks | Ks | Ksilent | Ka | Ka/Ks | Ks | Ksilent | Ka | Ka/Ks |
| bcd | 4.13 | 3.58 | 0.14 | 0.03 | 22.32 | 21.32 | 0.91 | 0.04 | 20.89 | 19.78 | 1.02 | 0.05 |
| Bruce | 6.55 | 6.35 | 0.23 | 0.03 | 39.01 | 29.57 | 0.86 | 0.02 | 41.59 | 31.68 | 0.63 | 0.02 |
| ftz | 6.05 | 5.39 | 0.96 | 0.16 | 20.83 | 19.52 | 3.51 | 0.17 | 19.75 | 18.02 | 3.36 | 0.17 |
| Gld | 4.16 | 4.16 | 0.01 | 0.00 | 20.96 | 20.96 | 0.40 | 0.02 | 20.07 | 20.07 | 0.39 | 0.02 |
| hb | 3.77 | 3.75 | 0.32 | 0.09 | 23.87 | 23.74 | 0.93 | 0.04 | 22.96 | 22.83 | 1.06 | 0.05 |
| hyd | 2.50 | 2.37 | 0.60 | 0.24 | 20.42 | 18.35 | 0.87 | 0.04 | 21.78 | 18.85 | 0.72 | 0.03 |
| ninaE | 3.73 | 3.81 | 0.00 | 0.00 | 23.23 | 32.53 | 0.84 | 0.04 | 23.78 | 33.81 | 0.84 | 0.04 |
| nop56 | 4.11 | 3.97 | 0.31 | 0.08 | 17.15 | 19.14 | 0.52 | 0.03 | 18.82 | 20.20 | 0.62 | 0.03 |
| RpL32 | 3.21 | 2.15 | 0.00 | 0.00 | 13.68 | 13.16 | 0.00 | 0.00 | 17.59 | 13.71 | 0.00 | 0.00 |
| rosy | 6.03 | 5.81 | 0.52 | 0.09 | 28.40 | 27.00 | 2.19 | 0.08 | 30.66 | 28.93 | 2.22 | 0.07 |
| sry-alpha | 2.75 | 2.75 | 0.62 | 0.23 | 35.60 | 35.60 | 11.23 | 0.32 | 34.46 | 34.46 | 10.80 | 0.31 |
| Tl | 6.92 | 6.65 | 0.42 | 0.06 | 26.53 | 25.22 | 5.94 | 0.22 | 24.89 | 24.29 | 5.74 | 0.23 |
| ade3 | 5.27 | 5.91 | 0.83 | 0.16 | 22.34 | 27.16 | 1.18 | 0.05 | 25.49 | 29.10 | 0.68 | 0.03 |
| Adh | 4.49 | 3.21 | 1.06 | 0.24 | 20.34 | 26.41 | 1.78 | 0.09 | 20.07 | 25.94 | 2.14 | 0.11 |
| Adh-dup | 3.87 | 4.76 | 0.97 | 0.25 | 33.31 | 33.80 | 2.64 | 0.08 | 35.24 | 35.22 | 2.84 | 0.08 |
| amd | 2.30 | 2.83 | 0.30 | 0.13 | 22.92 | 16.54 | 1.47 | 0.06 | 23.03 | 17.54 | 1.45 | 0.06 |
| Ddc | 6.01 | 6.01 | 0.29 | 0.05 | 26.99 | 26.99 | 1.28 | 0.05 | 32.30 | 32.30 | 1.01 | 0.03 |
| dpp | 3.26 | 4.45 | 1.07 | 0.33 | 12.86 | 18.91 | 2.14 | 0.17 | 11.81 | 18.92 | 1.99 | 0.17 |
| Eno | 2.67 | 2.41 | 0.01 | 0.00 | 12.12 | 11.20 | 1.08 | 0.09 | 12.77 | 12.08 | 1.07 | 0.08 |
| Gpdh | 3.25 | 3.32 | 0.00 | 0.00 | 7.81 | 16.09 | 0.00 | 0.00 | 11.43 | 14.98 | 0.00 | 0.00 |
| Lam | 3.03 | 2.40 | 0.64 | 0.21 | 25.10 | 22.77 | 3.97 | 0.16 | 26.25 | 23.70 | 4.23 | 0.16 |
| smo | 3.74 | 2.80 | 0.10 | 0.03 | 14.41 | 17.06 | 0.56 | 0.04 | 13.41 | 16.30 | 0.41 | 0.03 |
| Uro | 6.96 | 5.82 | 0.31 | 0.04 | 25.50 | 23.84 | 1.23 | 0.05 | 26.41 | 25.04 | 1.23 | 0.05 |
| AnnX | 8.96 | 9.55 | 0.02 | 0.00 | 22.87 | 25.47 | 0.66 | 0.03 | 25.73 | 30.98 | 0.64 | 0.02 |
| Cyp1 | — | 2.11 | — | — | — | 14.48 | — | — | — | 14.04 | — | — |
| Est-5B | 5.78 | 4.79 | 0.96 | 0.17 | 31.82 | 24.20 | 4.47 | 0.14 | 31.35 | 24.17 | 4.47 | 0.14 |
| Gapdh2 | 3.22 | 3.22 | 0.00 | 0.00 | 15.07 | 15.07 | 0.35 | 0.02 | 17.02 | 17.02 | 0.35 | 0.02 |
| Hsp83 | 4.18 | 4.18 | 0.00 | 0.00 | 17.60 | 17.60 | 0.16 | 0.01 | 16.77 | 16.77 | 0.16 | 0.01 |
| scute | 2.13 | 3.98 | 0.26 | 0.12 | 23.18 | 13.84 | 2.08 | 0.09 | 24.63 | 16.55 | 2.22 | 0.09 |
| sesB | 2.44 | 3.83 | 0.37 | 0.15 | 7.15 | 16.42 | 0.65 | 0.09 | 7.11 | 15.63 | 1.02 | 0.14 |
| sisA | 3.91 | 3.50 | 1.16 | 0.30 | 29.73 | 25.09 | 10.56 | 0.36 | 31.97 | 27.40 | 9.33 | 0.29 |
| Sod | 5.60 | 1.79 | 0.41 | 0.07 | 16.55 | 9.55 | 1.66 | 0.10 | 16.57 | 9.81 | 1.24 | 0.07 |
| swallow | 4.64 | 3.70 | 1.11 | 0.24 | 34.64 | 29.59 | 8.07 | 0.23 | 37.21 | 30.18 | 7.71 | 0.21 |
| Average | 4.36 | 4.10 | 0.44 | 0.11 | 22.32 | 21.76 | 2.32 | 0.09 | 23.24 | 22.43 | 2.24 | 0.09 |
| SE | 0.284 | 0.287 | 0.068 | 0.017 | 1.359 | 1.146 | 0.498 | 0.015 | 1.422 | 1.241 | 0.469 | 0.015 |
Silent (Ksilent), synonymous (Ks), and nonsynonymous (Ka) divergence was estimated by the Jukes-Cantor correction for multiple hits. The data shown in this table are not corrected for within-species diversity.
To increase the power of the comparison of X-linked and autosomal variation, we combined our data with those reported by Yi et al. (2003), increasing the number of sex-linked loci to 12. To correct for differences in information among different loci, we weighted each locus by its estimated net variance of nucleotide diversity for the case of free recombination, which should approximate the true variance given the relatively low levels of linkage disequilibrium in D. miranda (Yi et al. 2003). This yielded an overall mean value of 0.47% for X-linked loci and 0.51% for autosomal loci, suggesting that sexual selection may be inflating the value of Ne for X-linked genes, as previously proposed by Yi et al. (2003). If the X-linked values are adjusted by a factor of 4/3, to take account of the fact that the mean diversity for X-linked genes is expected to be three-quarters of the autosomal value in the absence of sexual selection, the difference in mean becomes 0.12%, with a lower bootstrap 95% confidence limit of −0.13%. This reflects the very high among-locus variability in estimates of diversity. To reduce this variability, we removed runt (X-linked) and rosy (autosomal), which are outside the range of variability observed for other loci, as well as loci that showed evidence for significant departures from neutrality (per, swallow, and AnnX; see below and Yi et al. 2003). This increases the difference between the weighted means for adjusted X-linked and autosomal values (0.65 and 0.35%, respectively); the lower bootstrap 95% confidence limit for the difference is 0.00% and the difference in observed adjusted mean has P < 0.05 on a t-test (t = 2.26, 16 d.f.). This suggests that sexual selection may be acting to reduce autosomal variability in D. miranda, in agreement with the conclusion of Yi et al. (2003), but more data are clearly needed to resolve this point. A possible problem with this conclusion is that there is evidence for weak selection on synonymous variants (see below). However, the theoretical results of McVean and Charlesworth (1999) show that such selection reduces the ratio of X-linked to autosomal variability, if the deleterious effects of mutations are recessive or additive, as usually seems to be the case.
The frequency spectrum of variants at each locus was studied using Tajima's D statistic (Tajima 1989), for which a significantly negative value indicates that there are more low-frequency variants than expected under the neutral model. Fourteen out of the 20 genes exhibited negative D-values when silent and nonsynonymous sites were combined, although only hyd was individually significant. The most negatively skewed values corresponded to hyd and Bruce, with 5 of 5 and 4 of 5 variants being singletons, respectively. The mean values of π and θw for silent variants are very close to each other (mean paired difference of −0.038%, with standard error of 0.020%), with 7 of 18 comparisons giving positive values, so there is no significant evidence of an overall departure of silent variants from neutral expectation, in agreement with the conclusions of Yi et al. (2003). In contrast, only 1 of 14 loci with replacement polymorphism data have larger π than θw for nonsynonymous variants (the mean difference is −0.023%, SE 0.005), P = 0.001 on a sign test. This suggests the action of purifying selection on replacement polymorphisms (see below). Although the pooled frequency distribution of nonsynonymous variants was more skewed toward low-frequency variants than the distribution of synonymous variants, the distributions did not differ significantly on a Mann-Whitney U-test (data not shown).
Another way of testing whether the patterns of nucleotide variation and divergence are compatible with the standard neutral model is to apply the HKA test, which asks if polymorphism levels for each locus are proportional to divergence between species (Hudson et al. 1987). To do this, we used a maximum-likelihood version of this test (Wright and Charlesworth 2004), available at www.yorku.ca/stephenw. The application of this program to our data on silent sites, using D. affinis for measuring divergence, showed that only three loci departed significantly from neutral expectation (conservatively adjusting the expected diversity values for X-linked loci to three-quarters of those for autosomes): AnnX, swallow, and sry-alpha (P < 0.001, 0.003, and 0.02, respectively). All of them showed less variability than expected from their divergence levels, suggesting possible effects of selection. The result for sry-alpha is not significant if allowance is made for multiple tests. rosy, despite being unusually polymorphic, did not deviate significantly from the null hypothesis of neutrality, in agreement with Begun and Whitley (2002) and Riley et al. (1992). No evidence for departure from neutrality at rosy was obtained from other tests, such as haplotype tests. After removing AnnX and swallow, we compared the log-likelihood obtained when the expected diversities for X-linked loci were set to three-quarters of the autosomal values with that for the case of equal expected values for X-linked loci and autosomes (strong sexual selection). The resulting χ2 was 5.24, P = 0.023, supporting the above conclusion that D. miranda is subject to sexual selection.
Selective constraints on protein sequences:
When a gene is evolving neutrally, the ratio of nonsynonymous to synonymous or silent-site divergence (Ka/Ks) should be equal to one, but selective constraints on the protein sequence cause the ratio be lower (Ka/Ks < 1), because selection removes deleterious nonsynonymous mutations (Kimura 1983). To assess the levels of selective constraints on protein sequence in our sample, we estimated the proportions of replacement (Ka), silent (Ksilent), and synonymous substitutions (Ks) per site among D. miranda, D. pseudoobscura, and D. affinis (Table 3), using the Jukes-Cantor correction for multiple hits (Jukes and Cantor 1969).
On average, pairwise comparisons among the three species under analysis show very similar Ka/Ks ratios. Interestingly, the mean Ka-, Ks-, and Ka/Ks-values between D. miranda and D. pseudoobscura are extremely close to those for loci on the neo-X chromosome of D. miranda after excluding two loci that appear to be under positive selection (Bachtrog 2003b, Table 2). However, given the close relationship between D. miranda and its sibling species D. pseudoobscura (Yi et al. 2003), it is desirable to apply a correction for within-species genetic variation when comparing them. The silent pairwise nucleotide diversity for D. pseudoobscura (πsilent = 1.48%) was estimated by taking the mean of individual locus values from previous studies (Hamblin and Aquadro 1999; Kovacevic and Schaeffer 2000; Machado et al. 2002). The mean of this and the mean for D. miranda in the present analysis (πsilent = 0.34%, excluding the unusually highly variable rosy locus) were subtracted from the mean silent divergence between the two species (Ksilent = 4.10%), providing a slightly lower estimate of net divergence (Ksilent = 3.19%). An analysis of published value for DNA sequence polymorphism in D. pseudoobscura suggests that the replacement-site nucleotide diversity is fairly similar to that for D. miranda (V. Noël, C. Bartolomé and B. Charlesworth, unpublished data), so that the adjusted mean value of Ka is ∼0.36%. This yields a ratio of adjusted mean Ka to mean Ksilent of 0.11, which is the same as the mean of Ka/Ks. Given the much larger divergence from D. affinis, the lack of correction for within-species polymorphism will have only a small effect on the comparisons with D. affinis. As shown in Table 3, all genes are subject to purifying selection (Ka/Ks < 1).
However, it should be pointed out that there is some heterogeneity in selective constraints among loci: sry-alpha, sisA, and swallow seem to exhibit unusually fast rates of amino acid evolution, although there was no evidence for positive selection even when these three loci were pooled (see below). There is a slightly but not significantly higher mean Ka for X-linked genes (3.0 ± 1.1%, compared with 1.9 ± 0.50% for autosomes). This is consistent with the higher rate of protein sequence evolution observed for the right arm of the X in comparisons of D. pseudobscura and its relatives, relative to the same genes (which are autosomal) in comparisons of D. melanogaster and its relatives (Counterman et al. 2004).
To examine further the nature of selection on protein sequences, we identified polymorphic replacement and synonymous mutations within coding sequences of D. miranda and apparent fixed differences between D. miranda and D. affinis (Table 4). We then applied the McDonald-Kreitman test (McDonald and Kreitman 1991), which compares the ratios of polymorphism to divergence among different types of sites that are interspersed along the same sequence. Under the neutral model, the ratio of silent to replacement variants should be the same for polymorphisms as for fixed differences. Most genes did not show significant values of this ratio (except for Ddc and hyd). The existence of an excess of polymorphisms relative to fixations for replacement variants, compared with the ratio of synonymous polymorphisms to fixations, in the overall data set was evaluated by the Mantel-Haenszel statistic, z. This involves the sum over all the tables of the deviations of the observed numbers of replacement polymorphisms from the expected numbers when the cell frequencies for a table are the products of the row and column frequencies, divided by its sampling standard deviation (Snedecor and Cochran 1980). For the number of independent 2 × 2 tables used here, z should be close to a standard normal variate. This was checked by comparing the normal probability values to those from 10,000 resamplings of the 2 × 2 tables, keeping row and column numbers fixed; there was excellent agreement. Including all 18 relevant loci, z = 3.00, P < 0.001; if rosy is removed (which contributes a large fraction of the polymorphisms), z = 2.73, P < 0.01. If singletons are removed from the tables, the corresponding z-statistics become 1.28 and 1.01, respectively, which are nonsignificant. This suggests strongly that the low-frequency replacement polymorphisms are slightly deleterious. We estimated the value of Nes (where Ne is the effective population size, and s is the selection coefficient on a homozygous deleterious replacement variant), using a modification of the method of Maside et al. (2004) for estimating the intensity of selection on codon usage. This involves using the frequency spectrum for segregating mutations under selection with no dominance (Equation 9 of McVean and Charlesworth 1999) to calculate the expected proportion of singletons in a sample, yielding a maximum-likelihood estimate of Nes on the assumption of independence among sites with the same selection coefficient for each site. Pooling across loci, we obtained a value of 1.2, with 2-unit support limits (0.2, 2.7). Variation among sites in the selection coefficient is likely to cause this estimate to be downwardly biased (see below).
TABLE 4.
McDonald-Kreitman tests (coding regions)
| Fixed
|
Polymorphic
|
||||
|---|---|---|---|---|---|
| Gene | Synonymous | Nonsynonymous | Synonymous | Nonsynonymous | P |
| bcd | 46 | 7 | 7 | 1 | 1.00 |
| Bruce | 44 | 4 | 1 | 1 | 0.19 |
| Gld | 58 | 4 | 6 | 1 | 0.42 |
| hyd | 36 | 5 | 0 | 2 | 0.02* |
| rosy | 124 | 33 | 29 | 13 | 0.22 |
| sry-alpha | 34 | 34 | 1 | 4 | 0.36 |
| ade3 | 68 | 11 | 1 | 0 | 1.00 |
| Adh | 34 | 10 | 2 | 0 | 1.00 |
| amd | 42 | 10 | 4 | 1 | 1.00 |
| Ddc | 49 | 8 | 2 | 4 | 0.01** |
| Eno | 28 | 8 | 5 | 1 | 1.00 |
| Lam | 74 | 42 | 4 | 2 | 1.00 |
| Uro | 41 | 8 | 3 | 0 | 1.00 |
| AnnX | 28 | 3 | 0 | 1 | 0.13 |
| Gapdh2 | 26 | 2 | 0 | 0 | — |
| scute | 31 | 10 | 2 | 2 | 0.56 |
| sesB | 12 | 3 | 1 | 0 | 1.00 |
| sisA | 38 | 42 | 2 | 6 | 0.28 |
| swallow | 62 | 54 | 1 | 2 | 0.60 |
| Pooled | 875 | 298 | 71 | 41 | |
Synonymous and Nonsynonymous are the number of synonymous and nonsynonymous changes, respectively. D. affinis was used as an outgroup. P was calculated using the two-tailed Fisher's exact test, comparing numbers of synonymous vs. replacement changes in the fixed and polymorphic categories, respectively.
P < 0.05,
P ≤ 0.01.
Codon usage bias:
As described in materials and methods, we estimated codon usage bias from the frequency of optimal codons (Fop) for each gene, i.e., the fraction of optimal codons among all codons in the gene (Ikemura 1981; Duret and Mouchiroud 1999). The major codon preferences of D. pseudoobscura are very similar to those of D. melanogaster (Akashi and Schaeffer 1997), so that preferences in either species can be used to define optimal codons for D. miranda. To check this, we compared the values of Fop using the tables of preferences from both species and the results did not differ significantly (Table 5), except for those from sry-alpha, whose Fop-values were 0.42 and 0.57 using the D. pseudoobscura and D. melanogaster preferences, respectively. Given that D. miranda is much closer to D. pseudoobscura than to D. melanogaster, we used the D. pseudoobscura preferences in all the subsequent analyses.
TABLE 5.
Estimates of codon usage bias (Fop) inD. miranda
| Gene |
Fop (D. pseudoobscura) |
Fop (D. melanogaster) |
|---|---|---|
| bcd | 0.50 | 0.52 |
| Bruce | 0.55 | 0.56 |
| ftz | 0.60 | 0.56 |
| Gld | 0.60 | 0.57 |
| hb | 0.55 | 0.50 |
| hyd | 0.21 | 0.25 |
| ninaE | 0.57 | 0.62 |
| nop56 | 0.59 | 0.64 |
| RpL32 | 0.67 | 0.75 |
| rosy | 0.64 | 0.61 |
| sry-alpha | 0.42 | 0.57 |
| Tl | 0.66 | 0.63 |
| ade3 | 0.48 | 0.48 |
| Adh | 0.66 | 0.69 |
| Adh-dup | 0.56 | 0.57 |
| amd | 0.53 | 0.55 |
| Ddc | 0.60 | 0.62 |
| dpp | 0.44 | 0.41 |
| Eno | 0.76 | 0.77 |
| Gpdh | 0.48 | 0.46 |
| Lam | 0.64 | 0.64 |
| smo | 0.50 | 0.52 |
| Uro | 0.64 | 0.64 |
| AnnX | 0.69 | 0.65 |
| Cyp1 | 0.66 | 0.67 |
| Est-5B | 0.42 | 0.40 |
| Gapdh2 | 0.33 | 0.40 |
| Hsp83 | 0.67 | 0.70 |
| scute | 0.63 | 0.61 |
| sesB | 0.66 | 0.72 |
| sisA | 0.63 | 0.56 |
| Sod | 0.69 | 0.73 |
| swallow | 0.58 | 0.55 |
| Average | 0.57 | 0.58 |
Fop-values for D. miranda were calculated using the preferences table of D. pseudoobscura and D. melanogaster.
The major codon preference model assumes that selective forces on synonymous codons are weak (Bulmer 1991; Akashi 1995). Comparisons of sequence data within and between species thus provide a means of detecting these forces, which otherwise would be difficult to detect (Akashi 1995). Given that selection is expected to be less efficient at removing slightly deleterious mutations than preventing their fixation (Kimura 1983; Akashi 1995), one way of detecting selection at synonymous sites is to compare the ratio of polymorphism to divergence (rpd) between the two different classes of synonymous changes that change codon usage between preferred (P) and unpreferred (U) codons. If there is no selection, the rpd ratio for P → U changes should be equal to that for U → P changes. In contrast, higher ratios of polymorphism to divergence for P → U than for U → P changes are expected if there is selection against unpreferred (nonoptimal) codons (Akashi 1995).
To assess this, we classified synonymous changes as either polymorphic variants within D. miranda or fixed differences between D. miranda and D. pseudoobscura (Table 6). The ancestral state was inferred by parsimony using D. affinis as a distant outgroup (Akashi 1995), and mutational changes were assigned to the branches of the phylogeny leading to D. miranda and D. pseudoobscura. To avoid confounding effects of polymorphism within D. pseudoobscura, for which data are lacking in our study, we consider only fixed mutations assigned to the D. miranda branch. We found that rpd was much higher for P → U mutations than for U → P changes (1.9 vs. 0.5, P < 0.01, one-tailed contingency test), consistent with the action of weak selection against P → U changes. In addition, the numbers of P → U and U → P fixations do not differ significantly from equality (19 and 12, respectively), consistent with codon usage being in equilibrium in these two species (Bulmer 1991). If there had been a genome-wide relaxation of selection on codon bias (consistent with a recent decline in the effective population size, Ne), as seems to have happened in D. melanogaster (Akashi 1996), we would observe an excess of P → U fixations.
TABLE 6.
Synonymous changes (usingD. pseudoobscura preferences table)
| Fixed
|
Polymorphic
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | P-U | U-P | P-P | U-U | Total | NS | P-U | U-P | P-P | U-U | Total | NS |
| bcd | 3 | 0 | 0 | 0 | 3 | 0 | 3 | 0 | 0 | 2 | 5 | 1 |
| Bruce | 1 | 1 | 0 | 1 | 3 | 1 | 0 | 0 | 0 | 1 | 1 | 1 |
| Gld | 1 | 3 | 0 | 1 | 5 | 0 | 2 | 1 | 0 | 3 | 6 | 1 |
| hyd | 0 | 0 | 0 | 1 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 1 |
| rosy | 2 | 1 | 0 | 1 | 4 | 0 | 14 | 2 | 0 | 5 | 21 | 11 |
| sry-alpha | 0 | 0 | 0 | 2 | 2 | 0 | 0 | 0 | 0 | 1 | 1 | 4 |
| ade3 | 2 | 0 | 0 | 1 | 3 | 5 | 1 | 0 | 0 | 0 | 1 | 0 |
| Adh | 2 | 0 | 0 | 1 | 3 | 2 | 2 | 0 | 0 | 0 | 2 | 0 |
| amd | 1 | 1 | 0 | 0 | 2 | 0 | 3 | 0 | 0 | 0 | 3 | 1 |
| Ddc | 0 | 1 | 0 | 1 | 2 | 1 | 1 | 0 | 0 | 1 | 2 | 2 |
| Eno | 0 | 0 | 0 | 2 | 2 | 0 | 5 | 0 | 0 | 0 | 5 | 1 |
| Lam | 1 | 0 | 0 | 1 | 2 | 1 | 1 | 1 | 0 | 2 | 4 | 2 |
| Uro | 2 | 1 | 0 | 2 | 5 | 1 | 1 | 0 | 0 | 0 | 1 | 0 |
| AnnX | 2 | 2 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| Gapdh2 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| scute | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 2 | 2 |
| sesB | 0 | 2 | 0 | 0 | 2 | 0 | 0 | 1 | 0 | 0 | 1 | 0 |
| sisA | 0 | 0 | 0 | 1 | 1 | 4 | 1 | 1 | 0 | 1 | 3 | 6 |
| swallow | 1 | 0 | 0 | 2 | 3 | 3 | 1 | 0 | 0 | 0 | 1 | 2 |
| Total | 19 | 12 | 0 | 17 | 48 | 20 | 37 | 6 | 0 | 16 | 59 | 36 |
Conversely, a recent population expansion would produce an excess number of singletons compared to neutral expectation. To check for this, we performed a Fu and Li (1993) test. As shown in Table 7, there was no overall significant departure from neutral expectations for both coding and noncoding sequences. This is in agreement with the results of Yi et al. (2003), who found no convincing evidence for a recent population expansion in D. miranda from polymorphism data on a set of 12 autosomal, X, and neo-X linked genes, in contrast to its close relative D. pseudoobscura (Machado et al. 2002).
TABLE 7.
Fu and Li'sD-test statistics
| Gene | Coding | Noncoding |
|---|---|---|
| bcd | −0.23 | 0.97 |
| Bruce | −1.42 | −1.12 |
| Gld | −0.01 | NA |
| hyd | −0.45 | −1.07 |
| rosy | 0.29 | 0.95 |
| sry-alpha | −0.27 | NA |
| ade3 | 0.70 | 0.70 |
| Adh | 0.95 | 0.83 |
| amd | −0.27 | −1.42 |
| Ddc | 0.30 | NA |
| Eno | −2.12 | NA |
| Lam | 0.52 | NA |
| Uro | 1.11 | −1.42 |
| AnnX | −1.42 | NA |
| Cyp1 | NA | −1.34 |
| scute | −0.63 | −1.12 |
| sesB | −1.42 | −0.01 |
| sisA | −0.01 | −1.07 |
| swallow | −1.12 | NA |
| Combined sample | −0.40 | −1.55 |
NA, not available.
DISCUSSION
Nature of selection on protein sequences in D. miranda:
Our analysis of polymorphism and divergence data on 20 autosomal and X-linked loci of D. miranda suggests that there is a predominance of purifying selection on polymorphic amino acid replacement variants, as indicated by an excess of low-frequency nonsynonymous polymorphisms over neutral expectation and a significantly larger ratio of nonsynonymous polymorphism to divergence relative to the ratio for synonymous mutations, contributed by low-frequency variants (Tables 2 and 4). This contrasts with the results of the survey by Weinreich and Rand (2000) of data on 39 nuclear genes from various Drosophila species, which showed little evidence for purifying selection, although selection against low-frequency nonsynonymous variants has been inferred for D. melanogaster on somewhat different grounds (Fay et al. 2002). A high frequency of adaptive amino acid substitutions among nonsynonymous fixed differences has been suggested by recent applications by Smith and Eyre-Walker (2002) and Fay et al. (2002) of modifications of the McDonald-Kreitman test to comparisons between D. simulans and D. yakuba and between D. simulans and D. melanogaster, respectively. A likelihood-based extension of this approach by Bierne and Eyre-Walker (2004) estimated that ∼20% of amino acid substitutions between D. simulans and D. yakuba are driven by positive selection.
In contrast, application of the method of Smith and Eyre-Walker (2002) to the seven loci in our data set with more than five polymorphisms in their coding sequence yields an estimate of −0.32 for this proportion, with an upper 95% bootstrap confidence limit of 0.07. For this small set of genes, there is therefore no strong evidence for anything other than purifying selection on amino acid substitutions. The results of Bachtrog (2003a)(b) and Bachtrog and Charlesworth (2002) suggest that 2 of 10 neo-X-linked genes of D. miranda have been subject to positive selection for amino acid replacements since the divergence of the neo-X and neo-Y chromosomes. It is not clear whether this difference between the neo-X genes and the genes surveyed here is meaningful.
Maintenance of codon usage in D. miranda by selection:
Our finding that codon usage in D. miranda seems to be approximately in equilibrium ostensibly differs from the results for genes on the neo-sex chromosomes of D. miranda (Bachtrog 2003b), which suggested that selection was not maintaining codon bias. However, it seems likely that the excess of fixations of unpreferred mutations on the neo-X chromosome lineage observed by Bachtrog (2003b) is probably due to the use of only one sequence per locus, which causes some polymorphisms to be incorrectly classified as fixations. Given that selection in favor of preferred codons generates an excess of P → U over U → P polymorphisms (Akashi 1995), inclusion of polymorphisms among fixations will inflate the number of inferred P → U fixations.
To test this possibility, we reestimated the number of changes between D. miranda and D. pseudoobscura using a single, randomly chosen sequence from each gene. The number of substitutions to unpreferred codons was greatly overestimated when we employed a single sequence, with 37 P → U and 14 U → P fixations (P < 0.005, χ2-test against 1:1 expectation). When we compared our results with those shown in Table 4 of Bachtrog (2003b), we found no significant differences in the proportions of changes for either the neo-X or the neo-Y chromosomes using χ2-contingency tests. This strongly suggests that the use of a single allele inflates the estimates of numbers of P → U fixations for the highly polymorphic neo-X chromosome. We also examined the pattern of ostensible fixations for the loci sequenced in D. affinis for which polymorphism data are not available for D. miranda (Tables 1 and 2). We found 29 P → U vs. 5 U → P “fixations” on the D. miranda branch. This does not differ significantly from the value for the set with polymorphism data, when analyzed by using single sequences from D. miranda.
These results imply that codon usage in the recombining portion of the D. miranda genome is still being maintained by selection, contrary to the conclusion of Bachtrog (2003b) for the neo-X. More polymorphism and divergence data for the neo-X are clearly desirable to check this conclusion, and these are currently being collected. Given the low level of polymorphism on the neo-Y chromosome, the bias in this case is negligible, so that the results of Bachtrog (2003b) imply that P → U mutations are accumulating on the neo-Y chromosome, as would be expected from its exposure to Hill-Robertson effects due to its lack of recombination (Charlesworth and Charlesworth 2000).
However, other factors could have similar effects to selection on the ratio of polymorphism to divergence for synonymous mutations. Almost all preferred codons in D. pseudoobscura end in G or C (Akashi and Schaeffer 1997), so that GC-biased gene conversion (Galtier et al. 2001; Birdsell 2002), or recent changes in the intensity of mutational bias (Francino and Ochman 1999), could be confounded with the effects of selection on codon usage. Given that the former are nonselective mechanisms, they should have similar effects on coding and neighboring noncoding regions, so that the analysis of nucleotide substitutions in these two fractions of the genome should reveal which forces are involved.
We compared rpd (GC → AT) to rpd (AT → GC) in coding (rc) vs. noncoding DNA (rnc) and found that rc was much greater than rnc (Table 8). This is due to a substantial excess of GC → AT polymorphisms at synonymous sites compared with noncoding sites (P < 0.01, χ2-contingency test with Yates' correction). However, there is also a significant excess of GC → AT fixations among the coding sequences (P < 0.01), apparently conflicting with the above inference that base composition is at equilibrium. No such difference is found for the noncoding sequences, and the difference between the two types of sequence is significant (P < 0.01). The probable reason for the excess of GC → AT fixations at synonymous sites is that the expectation of equality of GC → AT and AT → GC fixations holds only for those mutations that arose in the D. miranda lineage from sites that were fixed at the time of divergence from the common ancestor with D. pseudoobscura. Given the low divergence between the two species compared with the within-species diversity in D. pseudoobscura (see results), it is likely that a significant fraction of fixations involve polymorphisms that were present in the common ancestor. It is easily shown that the ratio of the probabilities of fixation of deleterious and favorable variants is higher for polymorphic variants than for new mutations, since a relatively frequent deleterious variant has already avoided loss from the population. We would therefore expect an enrichment of deleterious variants among fixed differences that have arisen from ancestral polymorphisms; this hypothesis can be tested by determining the status of variants within D. pseudoobscura, by comparison with D. miranda and D. affinis, to see if there is evidence that they are often ancestral (V. Noël, C. Bartolomé and B. Charlesworth, unpublished data).
TABLE 8.
Polymorphic and fixed synonymous changes at coding and noncoding sites inD. miranda
| Sites | GC → AT | AT → GC | Fisher's exact test |
|
|---|---|---|---|---|
| Coding | ||||
| Fixed | 30 | 12 | P = 0.012 | |
| Polymorphic | 48 | 4 | ||
| rpd | 1.60 | 0.33 | rc = 4.80 | |
| Noncoding | ||||
| Fixed | 16 | 22 | P = 0.285 | |
| Polymorphic | 13 | 9 | ||
| rpd | 0.81 | 0.41 | rnc = 1.99 |
Since our results imply a significant difference in the substitution patterns between coding sequences and introns, we can conclude that biased gene conversion toward GC (BGCGC) and/or changes in mutational bias are not the major forces driving codon usage evolution. This does not, of course, completely exclude a role for these forces. BGCGC is expected to generate a correlation between the base compositions of adjacent coding and noncoding sequences (Galtier et al. 2001; Marais 2003). For the genes in Table 1, we found a nonsignificant correlation between the GC content of introns (GCi) and the GC content at the third codon position (GC3) of the genes in which they reside (Kendall's τ = 0.11, P = 0.43, two-tailed test; Figure 1). The pattern is the same when we use the corresponding D. pseudoobscura sequences. This is consistent with the weak correlations reported for D. melanogaster (Kliman and Hey 1994; Marais and Piganeau 2002), which could not be detected in the small sample of genes used here.
Figure 1.—

Correlation between GC content at the third codon position and GC content in introns. Solid line, including all loci; dashed line, discarding the outlier; arrow, indicates the outlier.
Estimates of the intensity of selection on codon usage:
To estimate the selection intensities at synonymous sites, we applied a maximum-likelihood method based on the frequencies of P → U mutations among P → U and U → P polymorphic sites (Maside et al. 2004). The scaled selection parameter 4Nes is denoted by γ, where 2s is the selection coefficient against a homozygous U variant (diploidy, no dominance, and equal selection coefficients at each site are assumed). For the pooled data set, the maximum likelihood of γ was 2.5 (2-unit support limits 1.5–3.8); this value did not differ significantly from those obtained after dividing the data set into two groups of genes with low bias (Fop < 0.60, γ = 2.6) and high bias (Fop > 0.63, γ = 2.2). This lack of an apparent difference between classes may reflect the limited range of Fop-values in our sample of genes: the average Fop-values for the low- and high-bias groups were 0.50 ± 0.024 and 0.66 ± 0.009, respectively.
With γ = 2.5, we have an Nes-value of 0.63 (with ∼95% confidence interval of 0.38–0.95). This implies the action of very weak natural selection on synonymous changes, given that Ne for D. miranda is of the order of 1 million (Yi et al. 2003). This value of Nes is lower than previous estimates obtained by different methods in other species of Drosophila (Akashi and Schaeffer 1997), but is very similar to that for D. americana, obtained by the present method (Maside et al. 2004). These differences probably reflect the sensitivity to demographic perturbations of allele frequency spectra of the methods used previously (Maside et al. 2004).
We also estimated the selection intensities from the proportion of U singletons among P → U and U → P) polymorphisms (Maside et al. 2004); the results were not significantly different from the above estimate (γ = 1.2, upper 2-unit support limit 3.1). This agrees with the absence of evidence for a recent population expansion in D. miranda, described above.
Effects of BGC:
The absence of evidence for BGCGC in our data on noncoding sequences may simply reflect the relatively small amount of polymorphism data. Following the approach of Maside et al. (2004), we have indirect evidence for effects of BGCGC. We can compare the expected value of GC3 with that expected from the estimated mutational bias in favor of GC → AT mutations and the intensity of selection on preferred codons. The former can be estimated from the GC content of introns (GCi); assuming equilibrium under neutrality, the mutational bias k for a gene (the ratio of the mutation rates for GC → AT and AT → GC mutations) can be estimated from the standard formula for statistical equilibrium under mutation pressure alone (Bulmer 1991) as (1/GCi) − 1. Taking the mean of 1/GCi over all 27 D. miranda genes for which data are available, the estimated mean value of k is 1.77, with a standard deviation of 0.49. Assuming as a rough approximation that the selection intensity for GC3 (γ′) is 80% of that estimated for preferred codon usage (i.e., γ′ = 2.0; Maside et al. 2004), the predicted value of GC3 from the equation for equilibrium under selection, drift, and mutation (Bulmer 1991) is 0.81, much larger than the observed value of 0.69.
This value might, in fact, be somewhat underestimated if there is variance in k among genes, as indicated by the substantial variance in GCi. A second-order Taylor series correction for the effect of variance in k on the equilibrium value of GC3 (p) yields the following prediction for mean GC3,
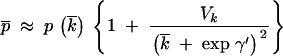 |
1a |
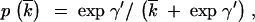 |
1b |
where overbars indicate mean values, and Vk is the variance in k. Substituting the estimated standard deviation of k into this expression increases the predicted mean GC3 by a factor of only 1.025, so the effect is negligible.
The larger predicted value of mean GC3 compared with the observed value thus suggests that BGCGC may have some effect on the base composition of both coding and noncoding sequences, since it causes an underestimation of the mutational bias parameter (Maside et al. 2004). A GC3 content of 0.69 with a γ′-value of 2.0 implies a mean k-value of 3.3; this in turn suggests a γ′-value of 0.75 for noncoding sequences, to account for the observed value of GCi. This is well within the 2-unit support limits for the maximum-likelihood estimate of γ′ from the noncoding polymorphism data in Table 8 (∼−1.0–1.8); further data on polymorphisms at noncoding sites are needed to examine this question further. This value of k requires a γ of 1.48 to yield the observed mean value of Fop (Table 5); this falls within the 2-unit support limits for the maximum-likelihood estimate of γ. This analysis does not, of course, distinguish between the effects of selection and BGC on noncoding sequences, but comparative studies of base composition across the genome tend to support a role for BGC (Marais 2003).
Effects of variation in the selection parameter:
Another question is the extent to which estimates of γ may be biased by variation in γ-values among different sites. This is relatively hard to examine in the context of maximum likelihood without using simulations, but is simple to model for the method of moments estimator obtained by equating the theoretical and observed values of the proportion of P → U polymorphisms among P → U and U → P polymorphisms. Unless the existence of variation in γ among sites has a large effect on the sampling distribution, this should yield some insight into the effects of variation in γ, since the method of moments estimator and the maximum-likelihood estimator must converge asymptotically.
Table 9 shows examples of three different methods of calculating the effect of a distribution of γ-values on estimates of mean γ. Each codon is assumed to be sampled independently from the relevant distribution. The left-hand part of the table shows the results of assuming a gamma distribution; i.e., the probability density of a given value of x is proportional to β−αxα−1 exp(−x/β). The shape parameter, α, was assigned arbitrarily (first column), and the expected proportion of P → U polymorphisms was calculated by numerically integrating the expression given by Equation 1 of Maside et al. (2004) over a gamma distribution with a given value of the β parameter (note that the sign of γ in the expression that follows their Equation 1 should be reversed). The value of β that equalizes observed and expected proportions of P → U polymorphisms for the assigned α-value was then determined iteratively (second column). The corresponding means and standard deviations were calculated from the standard formulas for a gamma distribution (third and fourth columns).
TABLE 9.
 |
| α | β |
|
σγ |
|
|
|||
|---|---|---|---|---|---|---|---|---|
| ∞ | 0 | 2.50 | 0.00 | 2.50 | 2.50 | |||
| 20 | 0.13 | 2.57 | 0.56 | 2.59 | 2.60 | |||
| 10 | 0.26 | 2.64 | 0.84 | 2.72 | 2.65 | |||
| 5 | 0.56 | 2.82 | 1.26 | 3.02 | 2.87 | |||
| 2 | 1.85 | 3.71 | 2.62 | 4.19 | — | |||
| 1 | 4.79 | 4.79 | 4.79 | 5.55 | — | |||
| 0.5 | 23.00 | 11.30 | 16.30 | 8.38 | — |
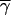
The first four columns relate to a gamma distribution of the scaled selection intensity γ; the fifth gives the estimate of mean γ obtained for a general distribution from the second-order Taylor series approximation, with the same variance as the gamma distribution; the sixth is the estimate for a normal distribution with this variance. See text for further details.
The corresponding mean values from the second-order Taylor series approximation for the expected proportion of P → U polymorphisms are shown in Table 9, column 5, and the value for a normal distribution with the same variance as the gamma distribution is shown in column 6, for that part of the parameter space where a normal distribution of γ produces only a negligible fraction of negative values of γ.
For the gamma distribution, it is evident that variation in γ causes the mean value of γ to be underestimated if the variance is ignored, as was done above (where γ was estimated as 2.5 by both maximum likelihood and method of moments). The underestimation is very large for α-values <1.0, but these generate coefficients of variation in γ that exceed 1 and hence represent very high levels of variability. The same result is seen for the approximation, which agrees quite well for relatively small variances, but increasingly underestimates mean γ as α decreases. In the regions where they are valid, the normal distribution values agree well with the other two.
However, even if we assume that the mutational bias is as high as 3.3, the predicted mean Fop-values, calculated by integrating Equation 1b over the gamma distribution, are all >80%, far higher than what is observed. This suggests that the estimates of mean γ from the polymorphism data are too high. Using the binomial distribution, the lower 95% confidence interval on the proportion of P → U polymorphisms is 0.757. Examination of the variance of the distribution of the proportion of P → U polymorphisms generated with variation in γ shows only a very small deviation from the binomial value, so this is likely to be a good approximation. Use of 0.757 instead of the observed value in the estimation equations yields lower predicted values of Fop, much closer to the observed. For example, for a gamma distribution with α-values of 5, 2, and 1, we obtain estimates of 1.6, 1.9, and 2.1 for mean γ, with predicted mean Fop-values of 0.61, 0.66, and 0.73, respectively, compared with a value of 0.61 without any variance. Thus, it would seem that a mean γ somewhat lower than that estimated from the polymorphism data and a high mutational bias are required to explain all features of the data. Moderate to high variability in γ requires higher mean γ-values than if variability is absent, and high variability is difficult to reconcile with the overall level of codon usage bias.
A similar analysis was also carried out for data on D. americana, previously analyzed by Maside et al. (2004) on the assumption of no variation in γ. Their data set was reduced to five alleles per gene for this purpose. Very similar results to the above were obtained; with a gamma distribution, the estimated value of mean γ increases from 2.58 to 10.3 as α changes from 20 to 0.5. However, these mostly predict too high a mean Fop, especially for the set of low codon usage bias genes, as was found for the case of no variation in γ by Maside et al. (2004). Using their estimate of k = 3.6 for low-bias genes, together with the lower 95% confidence interval for the proportion of P → U polymorphisms for low-bias genes, a gamma distribution with α-values of 5, 2, and 1 yields estimates of mean γ of 1.62, 1.92, and 2.12 and mean Fop-values of 0.59, 0.65, and 0.70, respectively, compared with an observed mean Fop of 0.59. Again, it seems that a relatively low variance in γ is most compatible with the data.
Acknowledgments
This work was funded by a grant from the Biotechnology and Biological Sciences Research Council UK to B.C., and a National Science Foundation doctoral dissertation improvement grant to SY. B.C. is supported by the Royal Society. We thank Peter Keightley and Laurent Duret for providing their computer programs, and two anonymous reviewers for their constructive comments on the manuscript.
References
- Akashi, H., 1995. Inferring weak selection from patterns of polymorphism and divergence at “silent” sites in Drosophila DNA. Genetics 139: 1067–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi, H., 1996. Molecular evolution between Drosophila melanogaster and D. simulans: reduced codon bias, faster rates of amino acid substitution, and larger proteins in D. melanogaster. Genetics 144: 1297–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi, H., and S. W. Schaeffer, 1997. Natural selection and the frequency distributions of “silent” DNA polymorphism in Drosophila. Genetics 146: 295–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner, M., 1989 Drosophila: A Laboratory Handbook. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Bachtrog, D., 2003. a Adaptation shapes patterns of genome evolution on sexual and asexual chromosomes in Drosophila. Nat. Genet. 34: 215–219. [DOI] [PubMed] [Google Scholar]
- Bachtrog, D., 2003. b Protein evolution and codon usage bias on the neo-sex chromosomes of Drosophila miranda. Genetics 165: 1221–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachtrog, D., and B. Charlesworth, 2002. Reduced adaptation of a non-recombining neo-Y chromosome. Nature 416: 323–326. [DOI] [PubMed] [Google Scholar]
- Bauer, V. L., and C. F. Aquadro, 1997. Rates of DNA sequence evolution are not sex-biased in Drosophila melanogaster and D. simulans. Mol. Biol. Evol. 14: 1252–1257. [DOI] [PubMed] [Google Scholar]
- Begun, D. J., and P. Whitley, 2002. Molecular population genetics of Xdh and the evolution of base composition in Drosophila. Genetics 162: 1725–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierne, N., and A. Eyre-Walker, 2004. The genomic rate of adaptive amino acid substitution in Drosophila. Mol. Biol. Evol. 21: 1350–1360. [DOI] [PubMed] [Google Scholar]
- Birdsell, J. A., 2002. Integrating genomics, bioinformatics, and classical genetics to study the effects of recombination on genome evolution. Mol. Biol. Evol. 19: 1181–1197. [DOI] [PubMed] [Google Scholar]
- Bulmer, M., 1991. The selection-mutation-drift theory of synonymous codon usage. Genetics 129: 897–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth, B., and D. Charlesworth, 2000. The degeneration of Y chromosomes. Philos. Trans. R. Soc. Lond. B 355: 1563–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counterman, B. A., D. Ortiz-Barrientos and M. A. Noor, 2004. Using comparative genomic data to test for fast-X evolution. Evolution 58: 656–660. [PubMed] [Google Scholar]
- Duret, L., and D. Mouchiroud, 1999. Expression pattern and, surprisingly, gene length shape codon usage in Caenorhabditis, Drosophila, and Arabidopsis. Proc. Natl. Acad. Sci. USA 96: 4482–4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay, J. C., G. J. Wyckoff and C. I. Wu, 2002. Testing the neutral theory of molecular evolution with genomic data from Drosophila. Nature 415: 1024–1026. [DOI] [PubMed] [Google Scholar]
- Francino, M. P., and H. Ochman, 1999. Isochores result from mutation not selection. Nature 400: 30–31. [DOI] [PubMed] [Google Scholar]
- Fu, Y. X., and W. H. Li, 1993. Statistical tests of neutrality of mutations. Genetics 133: 693–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galtier, N., G. Piganeau, D. Mouchiroud and L. Duret, 2001. GC-content evolution in mammalian genomes: the biased gene conversion hypothesis. Genetics 159: 907–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamblin, M. T., and C. F. Aquadro, 1999. DNA sequence variation and the recombinational landscape in Drosophila pseudoobscura: a study of the second chromosome. Genetics 153: 859–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., M. Kreitman and M. Aguade, 1987. A test of neutral molecular evolution based on nucleotide data. Genetics 116: 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemura, T., 1981. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes. J. Mol. Biol. 146: 1–21. [DOI] [PubMed] [Google Scholar]
- Jukes, T. H., and C. R. Cantor, 1969 Evolution of protein molecules, pp. 21–132 in Mammalian Protein Metabolism, edited by H. N. Munro. Academic Press, New York.
- Keightley, P. D., and T. Johnson, 2004. MCALIGN: stochastic alignment of noncoding DNA sequences based on an evolutionary model of sequence evolution. Genome Res. 14: 442–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, M., 1983 The Neutral Theory of Molecular Evolution. Cambridge University Press, Cambridge, UK.
- Kliman, R. M., and J. Hey, 1994. The effects of mutation and natural selection on codon bias in the genes of Drosophila. Genetics 137: 1049–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacevic, M., and S. W. Schaeffer, 2000. Molecular population genetics of X-linked genes in Drosophila pseudoobscura. Genetics 156: 155–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado, C. A., R. M. Kliman, J. A. Markert and J. Hey, 2002. Inferring the history of speciation from multilocus DNA sequence data: the case of Drosophila pseudoobscura and close relatives. Mol. Biol. Evol. 19: 472–488. [DOI] [PubMed] [Google Scholar]
- MacKnight, R. H., 1939. The sex-determining mechanism of Drosophila miranda. Genetics 24: 180–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marais, G., 2003. Biased gene conversion: implications for genome and sex evolution. Trends Genet. 19: 330–338. [DOI] [PubMed] [Google Scholar]
- Marais, G., and L. Duret, 2001. Synonymous codon usage, accuracy of translation, and gene length in Caenorhabditis elegans. J. Mol. Evol. 52: 275–280. [DOI] [PubMed] [Google Scholar]
- Marais, G., and G. Piganeau, 2002. Hill-Robertson interference is a minor determinant of variations in codon bias across Drosophila melanogaster and Caenorhabditis elegans genomes. Mol. Biol. Evol. 19: 1399–1406. [DOI] [PubMed] [Google Scholar]
- Maside, X., A. W. Lee and B. Charlesworth, 2004. Selection on codon usage in Drosophila americana. Curr. Biol. 14: 150–154. [DOI] [PubMed] [Google Scholar]
- McDonald, J. H., and M. Kreitman, 1991. Adaptive protein evolution at the Adh locus in Drosophila. Nature 351: 652–654. [DOI] [PubMed] [Google Scholar]
- McVean, G., and B. Charlesworth, 1999. A population genetic model for the evolution of synonymnous codon usage: patterns and predictions. Genet. Res. 74: 145–158. [Google Scholar]
- Nei, M., 1987 Molecular Evolutionary Genetics. Columbia University Press, New York.
- Perez, J. A., A. Munte, J. Rozas, C. Segarra and M. Aguade, 2003. Nucleotide polymorphism in the RpII215 gene region of the insular species Drosophila guanche: reduced efficacy of weak selection on synonymous variation. Mol. Biol. Evol. 20: 1867–1875. [DOI] [PubMed] [Google Scholar]
- Powell, J. R., 1997 Progress and Prospects in Evolutionary Biology: The Drosophila Model. Oxford Universtiy Press, Oxford.
- Riley, M. A., S. R. Kaplan and M. Veuille, 1992. Nucleotide polymorphism at the xanthine dehydrogenase locus in Drosophila pseudoobscura. Mol. Biol. Evol. 9: 56–69. [DOI] [PubMed] [Google Scholar]
- Rozas, J. R. R., 1999. DnaSP version 3: an integrated program for molecular population genetics and molecular evolution analysis. Bioinformatics 15: 174–175. [DOI] [PubMed] [Google Scholar]
- Smith, N. G., and A. Eyre-Walker, 2002. Adaptive protein evolution in Drosophila. Nature 415: 1022–1024. [DOI] [PubMed] [Google Scholar]
- Snedecor, G. W., and W. G. Cochran, 1980 Statistical Methods. Iowa State University Press, Ames, IA.
- Steinemann, M., and S. Steinemann, 1998. Enigma of Y chromosome degeneration: neo-Y and neo-X chromosomes of Drosophila miranda a model for sex chromosome evolution. Genetica 102/103: 409–420. [PubMed] [Google Scholar]
- Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano-Shimizu, T., 1999. Local recombination and mutation effects on molecular evolution in Drosophila. Genetics 153: 1285–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson, G. A., 1975. On the number of segregating sites in genetical models without recombination. Theor. Pop. Biol. 7: 256–276. [DOI] [PubMed] [Google Scholar]
- Weinreich, D. M., and D. M. Rand, 2000. Contrasting patterns of nonneutral evolution in proteins encoded in nuclear and mitochondrial genomes. Genetics 156: 385–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S. I., and B. Charlesworth, 2004. A maximum likelihood ratio test of the standard neutral model. Genetics 168: 1071–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, S., and B. Charlesworth, 2000. Contrasting patterns of molecular evolution of the genes on the new and old sex chromosomes of Drosophila miranda. Mol. Biol. Evol. 17: 703–717. [DOI] [PubMed] [Google Scholar]
- Yi, S., D. Bachtrog and B. Charlesworth, 2003. A survey of chromosomal and nucleotide sequence variation in Drosophila miranda. Genetics 164: 1369–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]