

Abstract
The simultaneous analysis of multiple genomic loci is a powerful approach to studying the effects of population history and natural selection on patterns of genetic variation of a species. By surveying nucleotide sequence polymorphism at 334 randomly distributed genomic regions in 12 accessions of Arabidopsis thaliana, we examined whether a standard neutral model of nucleotide sequence polymorphism is consistent with observed data. The average nucleotide diversity was 0.0071 for total sites and 0.0083 for silent sites. Although levels of diversity are variable among loci, no correlation with local recombination rate was observed, but polymorphism levels were correlated for physically linked loci (<250 kb). We found that observed distributions of Tajima's D- and D/Dmin- and of Fu and Li's D-, D*- and F-, F*-statistics differed significantly from the expected distributions under a standard neutral model due to an excess of rare polymorphisms and high variances. Observed and expected distributions of Fay and Wu's H were not different, suggesting that demographic processes and not selection at multiple loci are responsible for the deviation from a neutral model. Maximum-likelihood comparisons of alternative demographic models like logistic population growth, glacial refugia, or past bottlenecks did not produce parameter estimates that were more consistent with observed patterns. However, exclusion of highly polymorphic “outlier loci” resulted in a fit to the logistic growth model. Various tests of neutrality revealed a set of candidate loci that may evolve under selection.
THE structure of genetic polymorphism in a genome is influenced by different evolutionary processes. They can be grouped into processes that affect the whole genome (historical population growth or geographic population structure) and into processes that act locally or are variable across the genome (mutation, recombination, and selection). A frequent goal of studies of DNA sequence variation is to elucidate whether a gene of interest has been the target of recent natural selection. This goal is achieved by comparing patterns of variation observed at a locus with the variation expected under a neutral model that assumes no significant fitness effect of the polymorphisms segregating at a locus. Such comparisons can be confounded by the demographic history of a population because demographic processes may lead to patterns of variation that are similar to those observed under selection. For example, if there is an excess of low-frequency polymorphisms at a locus, this could result from a recent population expansion, from purifying selection against deleterious polymorphisms, or from the recent selective fixation of an advantageous allele at this locus.
Therefore it is important to disentangle the effects of different evolutionary processes on sequence variation if one attempts to identify genes involved in adaptation. Theoretical analyses showed that the simultaneous analysis of genetic variation at multiple loci is a powerful approach to identifying genome-wide acting processes (e.g., Wall 1999). If common patterns of polymorphism are observed at different independent loci, they likely result from a genome-wide acting evolutionary process rather than from multiple independent processes acting locally on individual genes (Glinka et al. 2003; Hamblin et al. 2004; Akey et al. 2004; Tenaillon et al. 2004).
Over the past decade, Arabidopsis thaliana has become an important model organism for the analysis of genetic variation in plants (Mitchell-Olds 2001), and sequence variation at more than a dozen loci was surveyed to elucidate the role of selection. Two major patterns emerged from these studies. At some loci, sequence variation is characterized by a pattern of two or three distinct and sometimes highly divergent haplotypes (e.g., Hanfstingl et al. 1994; Kawabe et al. 1997; Caicedo et al. 1998; Stahl et al. 1999; Kawabe et al. 2000; Hauser et al. 2001; Olsen et al. 2002; Tian et al. 2002; Kroymann et al. 2003; Clauss and Mitchell-Olds 2004) and at other loci by an excess of rare polymorphisms (e.g., Kawabe and Miyashita 1999; Purugganan and Suddith 1999; Kuittinen and Aguadé 2000; Hagenblad and Nordborg 2002; Olsen et al. 2002). Tests of neutrality with both groups of loci frequently rejected the null hypothesis of neutral evolution. These results suggest that in A. thaliana numerous genes are subject to balancing selection (excess of intermediate-frequency polymorphisms or haplotypes) or selective sweeps (excess of rare polymorphisms) at or near the surveyed genes.
Although some of the analyzed genes were selected due to a putative role in adaptive evolution, the nonneutral patterns of genetic variation may also be influenced by the demographic history and other characteristics of this species and thus confound tests of neutrality. A. thaliana is a highly self-fertilizing species (99%; Abbott and Gomes 1989; Bergelson et al. 1998). This level of inbreeding reduces effective population size and the effective rate of recombination (reviewed by Charlesworth 2003). In comparison to outcrossing species, a lower nucleotide diversity is expected due to background selection (Charlesworth et al. 1993) or hitchhiking with selective sweeps (Maynard Smith and Haigh 1974). Average levels of linkage disequilibrium are expected to be increased due to a low effective recombination rate (Nordborg and Donnelly 1997), resulting in a strong haplotype structure in samples obtained from a single deme (Nordborg 2000). Such a haplotype structure may resemble patterns observed under balancing selection and confound neutrality tests such as Tajima's D (Tajima 1989). Furthermore, there is evidence of a large-scale genetic population structure, which reflects a history of glacial refugia and postglacial recolonization of the native species range (Sharbel et al. 2000), suggesting that recent changes in both population size and population structure have played an important role in shaping the current structure of genetic variation in A. thaliana.
The purpose of this study is to investigate the effect of demographic history and self-fertilization on genome-wide patterns of sequence variation in A. thaliana and to test the hypothesis that a standard neutral model is a suitable null model for the identification of genes involved in adaptation. We analyzed several hundred short genomic regions (sequence-tagged sites, STS) in up to 12 accessions and fitted the observed patterns of variation to various demographic models using coalescent simulations and maximum-likelihood analysis. Most of the sequence data used for this study were taken from a previous survey aimed at large-scale discovery of single-nucleotide polymorphism (SNP) markers in A. thaliana (Schmid et al. 2003). We reject a neutral model of sequence polymorphism, but did not obtain a different demographic model that performed significantly better. Our analysis indicates that both the selfing nature and the demographic history of A. thaliana have a significant effect on genome-wide sequence polymorphism and that the standard neutral model is not appropriate for tests of neutrality in this species. By using empirical distributions of descriptive statistics such as Tajima's D, we were able to identify new candidate loci that may have been targets of recent selection.
MATERIALS AND METHODS
Plant material:
Twelve accessions from A. thaliana were included in this survey, which consist of five accessions previously used in genetic mapping (Col-0, Cvi-0, Ler, Nd-0, and Ws-0) and an additional 7 accessions (Ei-2, CS22491, Gü-0, Lz-0, Wei-0, Ws-0, and Yo-0) with a high average genetic distance to other accessions (Sharbel et al. 2000). These lines are available from the stock centers. Single accessions each from the closely related species A. lyrata ssp. lyrata and from Boechera drummondii were used as outgroups. These two species have an evolutionary distance to A. thaliana of 5–8 and 10 million years, respectively (Koch et al. 2000).
Sequencing, quality trimming, and annotation:
A total of 595 STS loci were chosen essentially randomly from the A. thaliana genome sequence and constitute protein-coding and noncoding regions (Schmid et al. 2003). PCR, sequencing, and base calling were done as described in Schmid et al. (2003). Heterozygous sequences were identified by tagging ambiguous base calls with the polyphred program (Nickerson et al. 1997) and subsequent visual inspection of the corresponding trace files. STS loci with heterozygous sequences were excluded from further analysis (n = 27) because they may consist of paralogous sequences that were amplified with the same primer pair. To avoid the inclusion of additional nonallelic (i.e., paralogous) sequences every sequence was compared to the Col-0 genome sequence using the BLASTN program. If the sequences of a STS locus had best hits with different regions in the genome, the STS was excluded from further analysis (n = 26). After these filtering steps, the consensus sequences were aligned separately for every STS locus with ClustalW (Thompson et al. 1994) and the alignment was manually corrected with a sequence editor. Only STS loci with at least 100 alignment positions and a sequence from at least eight accessions were retained for further analysis. Coding regions of alignments were annotated using the Col-0 reference sequences and the MIPS annotation (version 11, July 2003; ftpmips.gsf.de/cress). Before the analysis, alignments were trimmed to exclude regions or sequences with missing or low-quality data. With the exception of the visual control of the alignment quality and problematic annotations, the whole assembly and annotation process was performed in an automated fashion and controlled by programs written in the Python programming language.
Population genetic analysis:
The standard population genetic analyses were carried out with the program DnaSP 3.99 (Rozas and Rozas 1999). Nucleotide diversity of a multiple alignment was calculated as π (or θT), the average pairwise nucleotide divergence (Tajima 1983; Nei 1987), and θW, the number of segregating sites (Watterson 1975). Nucleotide frequencies were analyzed by calculating the following test statistics: Tajima's D (Tajima 1989); Fu and Li's D*, F* (no outgroup) and D, F (with outgroup; Fu and Li 1993); Fay and Wu's H (Fay and Wu 2000); and a modification of Tajima's D-statistic, D/Dmin (Schaeffer 2002). The latter was calculated as D/Dmin = (π − S/an)/|πmin − S/an| and πmin = 2S/n, where S is the number of segregating sites and 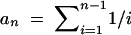 . In an analogous manner, we also calculated H/Hmin for Fay and Wu's H-statistic. In this case,
. In an analogous manner, we also calculated H/Hmin for Fay and Wu's H-statistic. In this case, 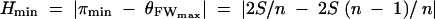 . Under a given demographic model the expected values of these statistics are homogeneous for different loci with different numbers of segregating sites, which thus facilitates comparisons among loci (Schaeffer 2002). The test of McDonald and Kreitman (1991) was calculated for STS with >80 aligned codons and with an outgroup sequence. Population genetic analyses based on haplotype structure were not performed, because this information was incomplete for many of the studied loci (i.e., internal columns of the alignment were masked as a consequence of the automated masking of low-quality sequences). Sequence divergence for silent, synonymous, and nonsynonymous variation was calculated using the correction of Jukes and Cantor (1969), as implemented in DnaSP 3.99 (Rozas and Rozas 1999).
. Under a given demographic model the expected values of these statistics are homogeneous for different loci with different numbers of segregating sites, which thus facilitates comparisons among loci (Schaeffer 2002). The test of McDonald and Kreitman (1991) was calculated for STS with >80 aligned codons and with an outgroup sequence. Population genetic analyses based on haplotype structure were not performed, because this information was incomplete for many of the studied loci (i.e., internal columns of the alignment were masked as a consequence of the automated masking of low-quality sequences). Sequence divergence for silent, synonymous, and nonsynonymous variation was calculated using the correction of Jukes and Cantor (1969), as implemented in DnaSP 3.99 (Rozas and Rozas 1999).
Effect of sequencing errors:
Tajima's D-, Fu and Li's D*-, and Fay and Wu's H-statistics are sensitive to rare polymorphisms and should be affected by sequencing errors. To investigate the effect of sequencing errors, we conservatively assumed that all errors result in singleton polymorphisms. The number of expected false-positive singleton polymorphisms (i.e., sequencing errors) was calculated by multiplying the total number of base pairs across all alignments with a given error rate of false base calls. From the set of observed singletons, the corresponding number was randomly selected and subsequently removed from the alignments. Then, Tajima's D, Fu and Li's D*, and Fay and Wu's H were calculated using these alignments. The last three steps were repeated 100 times per error rate, and the averages and standard deviations were calculated from individual simulations. Sequencing error rates investigated ranged from 10−7 to 10−2.
Although the observed mean of these statistics changes with sequencing errors, changes are minor over a wide range of error rates (Figure 1). We used the neighbor quality standard (NQS; Altshuler et al. 2000) for quality trimming. A phred quality score of 30 for the focal base call and a score of 20 for the five neighboring base calls on the left and right sides, respectively, were used as cutoff scores. With these parameters the expected sequencing error rate for single reads is 5 × 10−5 false base calls per base (Altshuler et al. 2000). However, since two-thirds of total bases were sequenced from both directions, the real error rate is likely to be much lower. On the basis of simulation results, we expect that sequencing errors do not significantly affect our analyses of polymorphism.
Figure 1.—

Effect of sequencing errors on estimates of genetic variation. Sequencing error rates correspond to the proportion of false positive singleton polymorphisms in the sample.
Multilocus analysis:
An estimate of genome-wide nucleotide polymorphism, θ, was obtained by maximum-likelihood (ML) analysis using an analytical result from coalescence theory that infers the probability of θ given the number of segregating sites, S, and sample size n at a locus (Tavaré 1984, Equation 9.5):
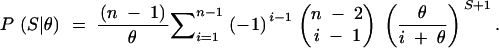 |
We first calculated the probabilities of individual loci for a range of θ per nucleotide ranging from a minimum of 0.0001 to a maximum of 0.10. To correct for differences in the alignment length, θ was multiplied by the sequence length and then used to calculate the probabilities. The multilocus likelihood for every value of θ was then calculated as the sum of the log probabilities across loci for a given value of θ. To test the hypothesis that the data are better described by more than one value of θ, likelihoods were calculated for two (M2), three (M3), and four (M4) different θ values for the whole sample and individual values for every locus (M no. loci). We first sorted loci numerically according to locus-specific ML estimates of θ. Then, for the M2 model, we used this sorted list to separate the loci into two different groups and searched for the best likelihood values in each group. The two exclusive groups with the best likelihood were then chosen. We also used this strategy to obtain θ values for the M3 and M4 models, although a heuristic search was performed because not all possible groups of loci could be investigated. In this search, all loci were first grouped randomly for 500 iterations, and then 500 additional iterations were performed with the best groups of loci to estimate the likelihood. Likelihood-ratio tests (LRT) for comparisons between different models were performed. The distribution of the LRT statistic was obtained by coalescent simulations and not from a χ2-distribution since it was difficult to ascertain the degrees of freedom. We simulated 1000 new samples using the simplest model (for example, comparing model M1 vs. M2, we simulated 1000 times the number of segregating sites for each locus given a θ value per nucleotide estimated by the model M1). We then recalculated the maximum likelihood for each simulated sample and stored the LRT value of each iteration. Finally, the LRT value for the observed data was contrasted with the simulated LRT distribution.
Levels of variation at synonymous and in noncoding sites were calculated by ML. We also calculated the likelihood under the assumption that both groups of sites have the same level of polymorphism (one θ is estimated) and that θ values differ between the two groups of genes. The likelihoods were then compared in a LRT.
To compare the observed genome-wide distribution of descriptive statistics with the expected distribution under a standard neutral model, we performed coalescent simulations conditioning on the genome-wide estimate of θ (using one or more θ values) obtained by maximum-likelihood estimation (Hudson 1990, 1993), using programs by S. Ramos-Onsins (unpublished data) and R. Hudson (Hudson 2000). For multilocus analyses, conditioning on θ is preferable to S (S. Ramos-Onsins, unpublished data). Simulations were carried out individually for each locus and repeated 10,000 times. We calculated two kinds of statistics:
The average and the population variance for a given statistic were calculated for all the loci combined in each iteration, as described by J. Hey in the HKA program (http://lifesci.rutgers.edu/heylab). For statistics that use an outgroup sequence (Fu and Li's D and F and Fay and Wu's H), simulations were carried out by taking into account recurrent mutations at the same position, and we included the ratio of transition vs. transversion (s/v) and the time of divergence from the outgroup species. When A. lyrata was taken as the outgroup, we used s/v = 1.2 (observed) and the time to the ancestral species was calculated as Tout ≃ divergenceobserved/2θ (Hudson et al. 1987), equal to eight measured N generations. In the case of B. drummondii, s/v = 1.0 and Tout ≃ 12. P-values were calculated as the probability that a neutral, simulated value of a given statistic was smaller or larger than the observed value.
Sign tests for a given statistic were performed by comparing the observed value with the median (i.e., 50% of the values) obtained by coalescent simulations and assigning a positive (negative) unit if the observed value was larger (smaller) than the median (Sokal and Rohlf 1995). Finally, the sum of all positive and negative values was compared to a binomial distribution having a probability of P = 0.5 to obtain the critical values of the test.
Multilocus HKA tests (Hudson et al. 1987) were calculated for silent positions using the silent segregating sites and the silent divergence value (corrected after Jukes and Cantor 1969). HKA tests were calculated separately for loci with A. lyrata and B. drummondii as outgroup sequences. The significance of the HKA tests was obtained using a χ2-distribution (Hudson et al. 1987).
Alternative demographic models:
The population history of A. thaliana appears to be determined by separated populations during the Pleistocene and subsequent expansion into Central and Eastern Europe during the past 18,000 years (Sharbel et al. 2000). For this reason, three alternative demographic models were evaluated that take the demographic history into account.
Logistic growth model:
This model estimates the ML parameters under logistic growth given the frequency spectrum of segregating sites (with or without outgroup). In the logistic growth model (e.g., Fu 1997), the population size changes back in time as follows:
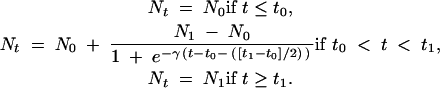 |
Here, Nt is the population size at time t in the past (expressed in N0 generations), N0 is the population size at the present time t0, and N1 is the population size at time t1. The population growth rate, γ, was fixed to γ = 10/(t1 − t0). Estimated model parameters include θ, the start and end time points of the logistic growth phase, and the growth rate N0/N1 (setting N0 = 1 as the current population size). Likelihood estimates were calculated by generating 100–200 coalescent trees for each set of parameters with no intralocus recombination. We then calculated the probability of the same number of mutations occurring with frequency i as observed in the data by using a Poisson distribution (J. Wakeley, personal communication). The log-likelihood value was calculated for each locus and the sum of all loci was stored. The best parameters were searched for by using a grid of 10,000 parameter values. Also, Metropolis-Hastings Markov chain Monte Carlo (Metropolis et al. 1953; Hastings 1970; Kuhner et al. 1995) sampling was performed. We used the simplest algorithm for sampling parameters and accepted the new parameters in the chain if
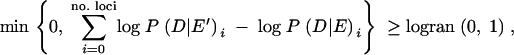 |
where logran(0, 1) is the logarithm of a random number between 0 and 1, and log P(D|E′)i is the logarithm of the probability that the observed frequency spectra will have the new sampled parameters in the locus i, and log P(D|E)i is the same but given the previous parameters in the Markov chain. Otherwise, we rejected the new parameters and accepted the previous parameters for the current step in the chain. The range of displacement for each parameter and for each iteration was adjusted empirically. Five chains of 106 iterations starting from distant parameter values were run first, and then a final chain was run to obtain the best parameters for this model. Using this approach, the likelihood values for the same parameter values varied depending on the number of coalescent simulations performed. Therefore, a LRT assuming a χ2-distribution might be too liberal. For this reason, we also calculated averages and variances across all loci for all summary statistics using the parameters estimated with the logistic growth model to evaluate whether the estimated parameters fit the observed data.
Refugia and bottleneck models:
The second demographic model assumes a single panmictic population in the distant past and then a subdivision into several refugia for a certain time period and a subsequent admixture into a single current population (Figure 2; Hudson 2000; Wall 2000). The time (in generations) from the present to the past split into several small populations (refugia), the number of refugia, the relative population size of each refugium (considering the present population size as N0 = 1), and the time until refugia merged into a single population were taken as general parameters for all loci. Values of θ and the relative contribution of each refugial population to the present population (freqr) were individual parameters for each locus. To be more conservative, the recombination parameter was set to zero, and, to simplify, the parameter freqr was considered equal for all loci. In the case of the bottleneck model, we used the same parameters as in the refugia model but the number of refugia was fixed to one. A ML analysis of the refugia model requires estimation of a large number of model parameters and is currently too expensive computationally, but we performed an exploratory analysis with a limited number of parameter combinations. In these analyses, we assumed a present effective population size of 2N = 106 and one generation per year. The time (from present to past) until refugia merged into a single population was fixed to cold periods at 104 generations ago (2 × 10−2 relative to N generations) and 2 × 104 (4 × 10−2), and the refugia were maintained for 103 or 5 × 103 generations (10−2) until a split into refugial populations. The relative population sizes in the refugia phase were set to 0.05, 0.01, 0.005, and 0.001 in relation to the present, and the number of refugia was set to 1 (bottleneck), 2, 5, and 10 refugia. The relative ancestral population sizes of these refugial populations (Nancestral/N0) were assumed to have been smaller than the current one and were fixed to 0.1, but 1 was also considered. To better adjust the model, we also tried additional parameter values in the vicinity of the best-fitting parameter combinations.
Figure 2.—

Schematic of the refugia model. From the present to the past, a single population with population size N0 was split into several small refugia before Ts generations. Each refugium has a population size Nri. After Tm generations, the refugial populations merge into a single population of size Na. Each refugium contributed freqri alleles to the present population.
Analysis of recombination rates and codon usage:
Local rates of recombination were obtained by comparing the physical and genetic distance of markers by polynomial regression as described by Zhang and Gaut (2003), who essentially applied the method of Kliman and Hey (1993). Centromeric regions were defined by taking the physical position of the genetically defined centromeres (Copenhaver et al. 1999) on the pseudochromosomes (obtained from MIPS, v270703) and then extension of these regions in both directions from the putative midpoint of the centromere by adding the estimated length of centromeres (Haupt et al. 2001).
Correlations of genetic diversity with GC content at silent sites (GC3) and estimated local recombination rate were analyzed by linear, quadratic, and quantile regression analysis using the R statistical package (http://www.r-project.org). The latter method is suitable for testing whether the edges of distributions with polygonal shapes reflect a random pattern or a significant relationship between two parameters (Scharf et al. 1998; Koenker and Hallock 2001). We used the quantreg package of R and a quadratic model to compare the quadratic regression coefficients for 50–95% quantiles and calculated the significance of observed regression coefficients by comparing 10,000 permutations of value pairs.
Data availability:
The sequences generated for this study were submitted to the STS section of GenBank under accession nos. CW672529, CW672721. Annotated alignments can be obtained from http://kiwi.ice.mpg.de/athapop.
RESULTS
Summary of polymorphism and divergence:
Of 595 STS loci sequenced by Schmid et al. (2003), 334 were retained for further analysis after quality trimming. For the present study, we obtained 118 STS (20%) from A. lyrata and 82 STS (14%) from B. drummondii. Of these sequences, we included 31 from A. lyrata, 26 from B. drummondii, and 14 from both outgroup species in the analysis. Outgroup sequences were then available for 71 STS loci. A summary table and a physical map of STS loci in A. thaliana is provided as supplementary information at http://www.genetics.org/supplemental/.
Average levels of polymorphism are summarized in Table 1. The majority of loci are distributed close to the mean of the silent variation (θW = 0.006), and few loci (39) are highly polymorphic (θW > 0.02; Figure 3). The relationship between synonymous and nonsynonymous polymorphism in coding regions is shown in Figure 4A. Among the set of 153 STS, which cover protein-coding regions of at least 80 codons, 90 (59%) have a ratio of πn/πs < 1, suggesting purifying selection against deleterious amino acid replacement polymorphisms. Among the most polymorphic genes, five have a πn/πs ratio of close to 1. They are annotated as “hypothetical” or “putative” and may constitute redundant, nonfunctional, or incorrectly annotated genes. Three of these genes contain at least one allele with a premature stop codon or an out-of-frame insertion/deletion, suggesting that they are pseudogenes.
TABLE 1.
Summary information on sequence alignments
| Parameter | Value |
|---|---|
| No. of alignments | 334 |
| Total no. of alignment positions | 139,038 |
| Mean no. of positions per alignment | 414 |
| Mean no. of alleles per alignmenta | 10 |
| Total no. of nucleotides includeda | 1,484,106 |
| Total no. of genes covered | 357 |
| Mean no. of codons per gene analyzed | 49 |
| Nucleotide variation, θW | |
| Total sites | 0.007 |
| Synonymous coding sites | 0.010 |
| Nonsynonymous coding sites | 0.001 |
| Synonymous coding and noncoding sites | 0.009 |
| Noncoding | 0.008 |
Only A. thaliana sequences are counted.
Figure 3.—

Distribution of levels of silent (synonymous and noncoding) variation in 334 STS loci. Levels of diversity are expressed as average pairwise differences, π (Tajima 1983), and as nucleotide diversity, θ (Watterson 1975).
Figure 4.—

Relationship between synonymous (πs) and nonsynonymous (πn) polymorphism (A) and synonymous (dS) and nonsynonymous (dN) divergence (B). Only alignments with >80 codons are included. The lines corresponds to πs = πn and dN = dS, respectively, and indicate expected levels of polymorphisms under complete neutrality.
Levels of sequence variation were not correlated with codon usage measured as the GC content at silent sites (GC3; R2 = 0.0036, P > 0.5). Diversity at noncoding sites is 1.2-fold lower than that at coding synonymous sites and 7.6-fold lower at nonsynonymous than at synonymous sites of coding regions (Table 1). When noncoding and synonymous variation were estimated by ML (θnoncoding = 0.0091, θsynonymous = 0.0088, ML = −414.33 − 398.41 = −812.74) and compared to a θsilent value estimated by combining all sites (θsilent = 0.0090, ML = −812.82), the diversity between the two types of sites was not significantly different (LRT = 0.148, P = 0.70). Therefore, we combined synonymous and noncoding sites in the following analyses.
Effect of recombination:
The levels of variation across the genome can be affected by different local recombination rates. Polymorphism levels do not differ between centromeric and noncentromeric regions (Table 2) and within noncentromeric regions there is no correlation between recombination rate and silent diversity (not shown). In regions of low and high recombination, polymorphism levels tend to be reduced (Figure 5), but linear (R2 = 0.04; P > 0.1), quadratic (R2 = 0.06; P > 0.1), and quantile regression coefficients (50–95% quantiles; P > 0.05) were not significant. In A. thaliana, pairwise linkage disequilibrium can extend up to 250 kb (Nordborg et al. 2002), suggesting that neighboring loci may have similar levels of polymorphism. This was confirmed by our observation of a higher proportion of loci with similar polymorphism levels among closely located (<250 kb) than among distant loci (using a small difference of 0.005 as cutoff; G-test, P = 0.039). Therefore, to be conservative, we used only loci that are separated by at least 250 kb for the analysis of demographic models (195 loci).
TABLE 2.
Polymorphism in centromeric (N = 38) and noncentromeric (N = 297) chromosome regions
| Mean π (SD)
|
||||
|---|---|---|---|---|
| Site type | Centromeric | Noncentromeric | t-test | P |
| Total sites | 0.0081 (0.0069) | 0.0059 (0.0073) | 1.827 | 0.07 |
| Silent sites | 0.0099 (1.1239) | 0.0079 (0.2619) | 1.124 | 0.26 |
Figure 5.—

Relationship between estimated recombination rate and levels of silent (synonymous and noncoding) nucleotide diversity.
Testing a neutral panmictic model of evolution:
We first asked whether the data are more consistent with a panmictic neutral model with 1, 2, 3, 4, or 195 different θsilent values. By comparing observed values with distributions obtained by coalescent simulations and testing for differences in an LRT (Table 3), we found that a model with two different estimates for silent θ (M2; θ1 = 0.0041, θ2 = 0.0213) best explained the distribution of silent variation under a neutral model. These estimates were used in the following simulations.
TABLE 3.
Maximum-likelihood estimates of θsilent per nucleotide
| Model | θ1 (no. loci) | θ2 (no. loci) | θ3 (no. loci) | θ4 (no. loci) | ML | LRT | Pb |
|---|---|---|---|---|---|---|---|
| M1 | 0.0092 (195) | −684.27 | |||||
| M2 | 0.0041 (128) | 0.0213 (67) | −485.77 | 397.00 | 0.000* (M1 vs. M2) | ||
| M3 | 0.0021 (63) | 0.0072 (82) | 0.0253 (50) | −441.11 | 89.32 | 0.104 (M2 vs. M3) | |
| M4 | 0.0011 (46) | 0.0051 (64) | 0.0122 (58) | 0. 0344 (27) | −422.86 | 36.49 | 0.671 (M3 vs. M4) |
| M195a | −388.69 | 68.33 | 0.959 (M4 vs. M195) |
Significant P-value (P < 0.05).
In the model with 195 different θ's, the value of each θ is not shown.
Probability calculated with a LRT distribution obtained by coalescent simulations.
Simulated distributions of Tajima's D, D/Dmin, and Fu and Li's D*- and F*-test statistics were compared with the observed distributions (Table 4 and Figure 6). Since the results obtained with D, D/Dmin, and Fu and Li's F*- and D*-statistics are very similar to those with Tajima's D, they are not mentioned further in the text but are shown in the tables. Simulated and observed distributions differed for most summary statistics, indicating that patterns of nucleotide polymorphism in our data are not consistent with a panmictic neutral model. There is an excess of low-frequency mutations as indicated by a negative average of Tajima's D and larger than expected variances of empirical distributions. It should be noted that these analyses are conservative because they assume no intralocus recombination (Tajima 1989).
TABLE 4.
Neutrality tests using silent polymorphisms
| No. loci
|
Sign testb
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Average | Pa | Variance | Pa | +2.5% | −2.5% | + | − | P | |
| n = 195 loci without outgroup | |||||||||
| Tajima's D | −0.4712 | <0.0001* | 1.0173 | 0.1158 | 3 | 17 | 50 | 127 | <0.001* |
| Fu and Li's D* | −0.4745 | <0.0001* | 1.2352 | <0.0001* | 7 | 15 | 64 | 111 | <0.001* |
| D/Dminc | −0.2944 | <0.0001* | 0.3578 | 0.9140 | 2 | 5 | 49 | 126 | <0.001* |
| n = 43 loci with A. lyrata outgroup | |||||||||
| Fu and Li's D | −0.4265 | 0.0050* | 1.4813 | 0.0012* | 2 | 6 | 10 | 21 | 0.071 |
| Fay and Wu's H | −0.3506 | 0.8610 | 2.5091 | 0.6028 | 3 | 2 | 22 | 16 | 0.418 |
| H/Hmind | −0.0277 | 0.8280 | 0.0475 | 0.6982 | 0 | 2 | 16 | 15 | 1.000 |
| n = 20 loci with B. drummondii outgroup | |||||||||
| Fu and Li's D | −0.6809 | 0.0004* | 1.1118 | 0.3920 | 0 | 2 | 4 | 13 | 0.049* |
| Fay and Wu's H | −0.7451 | 0.5748 | 6.7061 | 0.2118 | 1 | 0 | 12 | 7 | 0.359 |
| H/Hmind | 0.0328 | 0.1068 | 0.0242 | 0.4020 | 0 | 0 | 9 | 6 | 0.607 |
Significant P-values (P ≤ 0.05).
Two-tailed probability.
Number of loci with values larger (+) and smaller (−) than the median in coalescent simulations.
Tajima's D divided by its minimum (Schaeffer 2002).
Fay and Wu's H divided by its minimum (see materials and methods).
Figure 6.—

Comparison of empirical and simulated distributions of descriptive statistics analyzed in this study. Error bars indicate the 95% confidence interval of simulated means.
For the analyses of Fu and Li's D- and F- and Fay and Wu's H-test statistics, which require an outgroup sequence, we used 43 STS loci with A. lyrata and 20 STS loci with B. drummondii as outgroups. These loci are unlinked (physical distance >250 kb). Averages and variances of Fu and Li's D- and F-statistics differed significantly, but Fay and Wu's H-statistic was concordant with the neutral panmictic model (Table 4 and Figure 6). Thus, the deviation from the neutral panmictic model is not a consequence of an excess of a high frequency of derived mutations.
Since two groups of loci with low and high levels of variation were observed, we tested whether distributions of summary statistics differ between them (Table 6). In both cases, the average of Tajima's D is negative and significantly different from the expectations of the neutral model. When variances are considered, only the highly polymorphic group differs from the neutral model. The observation of an increased variance of test statistics in the group of highly polymorphic loci may be interpreted as a footprint of selection or of other evolutionary forces (see below).
TABLE 6.
Neutrality tests using silent polymorphisms based on groups of loci with low and high levels of variation
| No. loci
|
Sign testb
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Average | Pa | Variance | Pa | +2.5% | −2.5% | + | − | P | |
| n = 128 loci without outgroup (low θ) | |||||||||
| Tajima's D | −0.5634 | <0.0001* | 0.7813 | 0.0864 | 1 | 9 | 25 | 86 | <0.001* |
| Fu and Li's D* | −0.5405 | <0.0001* | 1.0330 | 0.4658 | 1 | 8 | 36 | 75 | <0.001* |
| D/Dminc | −0.3658 | <0.0001* | 0.3512 | 0.0616 | 0 | 0 | 25 | 86 | <0.001* |
| n = 67 loci without outgroup (high θ) | |||||||||
| Tajima's D | −0.3157 | 0.0316* | 1.4093 | <0.0001* | 2 | 8 | 25 | 41 | 0.064 |
| Fu and Li's D* | −0.3632 | 0.0096* | 1.5954 | <0.0001* | 6 | 7 | 28 | 36 | 0.382 |
| D/Dminc | −0.1739 | 0.0262* | 0.3563 | 0.0016 | 2 | 5 | 24 | 42 | 0.036* |
| n = 31 loci with A. lyrata outgroup (low θ) | |||||||||
| Fu and Li's D | −0.4576 | 0.0376* | 1.3092 | 0.1378 | 0 | 2 | 6 | 13 | 0.167 |
| Fay and Wu's H | 0.0219 | 0.7748 | 0.5658 | 0.7378 | 2 | 1 | 17 | 9 | 0.169 |
| n = 12 loci with A. lyrata outgroup (high θ) | |||||||||
| Fu and Li's D | −0.3615 | 0.0636 | 2.1195 | 0.0014* | 2 | 4 | 4 | 8 | 0.388 |
| Fay and Wu's H | −1.3131 | 0.725 | 6.8636 | 0.5724 | 1 | 1 | 5 | 7 | 0.774 |
Significant P-values (P ≤ 0.05).
Two-tailed probability.
Numbers of loci with a value above (+) and below (−) the median as calculated by coalescent simulations.
Tajima's D divided by its minimum (Schaeffer 2002).
Multilocus HKA tests based on silent polymorphisms were computed for all loci with outgroup sequences. The HKA test results were highly significant for 43 loci with an A. lyrata outgroup sequence (χ2 = 146.87 with 42 d.f., P < 0.0001; Figure 7) and for 20 loci with a B. drummondii outgroup sequence (χ2 = 54.38 with 19 d.f., P < 0.0001). Six of 43 loci (14%) were mainly responsible for the high significance of the HKA test in the comparison between A. thaliana and A. lyrata (AtV23, TIGR3437, GOLM25, At3est48, AtV11, and TIGR1736), and 4 of 20 loci (20%) between A. thaliana vs. B. drummondii (GOLM66, TIGR1744, TIGR1518, and GOLM80). Most of these loci have higher levels of polymorphism and lower levels of divergence than expected and are included in the group of highly polymorphic genes (Tables 3 and 6). Significant multilocus HKA tests are not consistent with the neutral panmictic model and may result from selection. It should be noted that a deviation from the assumption of panmixia may increase the variance of the observed data and contribute to the significant test result (Hudson et al. 1987; Ramos-Onsins et al. 2004).
Figure 7.—

Results of the multilocus HKA test for comparisons of A. thaliana with A. lyrata (A) and B. drummondii (B), respectively. The histogram describes the number of loci with a given χ2-value (left y-axis) and the line gives the deviation from the expected divergence given the polymorphism for the loci of each class (right y-axis).
As a final test of the neutral model, we compared the levels of polymorphism and divergence between synonymous and nonsynonymous sites for loci with >80 codons, using the McDonald-Kreitman (MK) test (McDonald and Kreitman 1991). Individual MK tests were significant only in 3 of 63 loci (AtIV20, AtV9, and GOLM29). This result is expected under the assumption of a neutral panmictic model with a 5% rejection probability for each locus (3 rejections/63 total loci = 4.8%).
Testing alternative demographic models:
Since a standard neutral model is not consistent with the observed data, we considered alternative demographic models. A. thaliana occurs frequently at disturbed sites, which have expanded over the last 6000 years with the spread of human agriculture; hence it seems plausible to consider a model of recent population expansion. Furthermore, we observed a significant negative average of Tajima's D, which may be interpreted as a footprint of recent population expansion (Tajima 1989). We first studied a logistic growth model with four parameters (see materials and methods) and found little statistical support for this model, because a higher likelihood was obtained with a model of constant population size (195 STS; ML = −1557.3) than with logistic growth (ML = −1562 for an estimated growth rate of 0.74). A higher likelihood for the constant population size model suggests that population expansion alone is not sufficient for explaining observed patterns of polymorphism.
We also compared our data against parameter combinations that take into account population expansion, such as those estimated by Innan and Stephan (2000) using a smaller number of loci (θ = 0.10, N1/N0 = 0.57, Tstart = 0, and Tend = 0.6, measured in N generations). Using these parameters, we always found a decrease in the variance of Tajima's D relative to the observed variance (see Figure 6 and Table 5). The high variance exhibited in the observed distribution of Tajima's D also suggests that our data are not consistent with a simple population expansion model.
TABLE 5.
Neutrality tests under alternative models
| Tajima's D
|
D/Dmin
|
Fu and Li's D
|
Fay and Wu's H
|
|||||
|---|---|---|---|---|---|---|---|---|
| Models | Pavg | Pvar | Pavg | Pvar | Pavg | Pvar | Pavg | Pvar |
| Expansion (IS)a | 0.0000*** | 0.9891* | 0.0001*** | 0.2163 | 0.0419 | 0.9977** | 0.1415 | 0.9160 |
| Expansion modelb | 0.4297 | 0.9992** | 0.6196 | 0.1170 | 0.5767 | 0.9952** | 0.0002*** | 0.9978** |
| Refugia modelc | 0.0427 | 0.1750 | 0.2151 | 0.0000*** | 0.5190 | 0.9920* | 0.0000*** | 1.0000*** |
| Bottleneck modeld | 0.8680 | 0.7834 | 0.9819 | 0.0329 | 0.7072 | 0.9825* | 0.0001*** | 1.0000*** |
P is the probability of having a smaller value than that observed. P ≤ 0.0250 and P ≥ 0.9750 are considered significant. For Tajima's D and D/Dmin, n = 195 loci and for Fu and Li's D and Fay and Wu's H, n = 43. Levels of variation were the same as used in Table 4.
P ≤ 0.025 or P ≥ 0.9750,
P ≤ 0.005 or P ≥ 0.995,
P ≤ 0.0005 or P ≥ 0.9995.
N1/N0 = 0.57, t0 = 0, and t1 = 0.6 in N generations.
N1/N0 = 0.21, t0 = 0.07, and t1 = 0.25 in N generations.
Two refugia, Nanc/N0 = 0.1, Nrefugia/N0 = 0.001; time of merging refugia was set to 5.6 × 10−2 and the duration of the refugia was 5 × 10−3 expressed in N generations.
Bottleneck, Nanc/N0 = 0.15, Nbottleneck/N0 = 0.003; time of bottleneck was set to 4 × 10−2 and the duration of the bottleneck was 10−2 expressed in N generations.
During the Pleistocene, A. thaliana may have been restricted to several isolated refugial populations before expanding and merging into the widespread distribution seen in the present (Sharbel et al. 2000). To account for such a population structure, we considered the refugia and bottleneck models (materials and methods). Again, we were unsuccessful in explaining the observed data, although only a few combinations of parameters were considered (Table 5). For some parameter combinations, we obtained negative averages for Tajima's D and the Fu and Li statistics, but did not observe the high variances seen in the simulations of the neutral model, although they also can be a consequence of population subdivision (Ramos-Onsins et al. 2004). In no case did the simulated distributions correspond with the negative averages and high variances of Tajima's D in the observed data. The same results were obtained with the bottleneck model, which is a refugia model with a single refugium (Table 5). This suggests that a more complex model incorporating additional parameters is necessary to explain the observed data.
Identification of nonneutrally evolving outlier loci:
The genome-wide distribution of nucleotide variation is a mixture of distributions resulting from demographic processes and selection. For this reason, the high variance we observe in the distribution of the test statistics may be caused by “outlier loci” that evolve under positive or balancing selection. Our data set contains 28 STS loci that reject a standard null model in Tajima's D, Fu and Li's D*, Fay and Wu's H, and the HKA and McDonald-Kreitman tests of neutrality and thus may have been targets of recent selection (supplementary information at http://www.genetics.org/supplemental/). These outlier loci may be responsible for the high variance observed in the empirical distributions and the rejection of a neutral model. A better fit to a neutral demographic model may be observed if they are excluded from the analysis (Luikart et al. 2003).
To evaluate this possibility, we identified all loci in the 2.5% tails on both sides of the empirical distributions of all statistics (see supplementary information). Thirteen loci (6.8%) were excluded from the set of 195 loci and 12 loci (27.9%) from the set with an outgroup sequence from A. lyrata. In the latter case, we also excluded loci with significant MK and HKA tests. The removal of outlier loci resulted in smaller variances, but they still differed significantly from expected variances (not shown). However, we obtained a better fit to the logistic growth model with the set of loci containing an outgroup sequence (n = 31, θ = 0.011, N1/N0 = 0.235, Tstart = 0.0158, and Tend = 0.0817, measured in N generations; ML = −140.85 when compared to the constant size neutral model, θ = 0.004, ML = −145.44). No better fit was observed with the larger data set.
We also evaluated the effect of outlier loci by removing loci with high estimates of θ identified by the M2 model described above (Table 3). Using the remaining 128 loci (66%), we obtained a better fit to the logistic growth model (θ = 0.0103, N1/N0 = 0.211, Tstart = 0.0180, and Tend = 0.0796, measured in N generations; ML = −585.01) than to the standard neutral model (θ = 0.004, ML = −603.15). The parameter estimates for the logistic growth model are similar to the ones in the previous analysis. In addition, observed averages and variances of all test statistics were compared with simulated distributions using the estimated parameters of the logistic growth model (not shown). No test showed a significant difference between observed and expected distributions, suggesting that a logistic growth model is more consistent with the patterns of variation at the 128 loci than is a neutral model of constant population size. Thus, some of the 67 high-diversity loci may have been either targets of selection or affected by demographic processes not included in our models. In the latter case, the exclusion of outlier loci may have resulted in an artificial reduction of the variance and thus an improved fit to the demographic model.
DISCUSSION
Genetic diversity in A. thaliana:
The main goal of our study was to conduct a multilocus analysis of nucleotide polymorphism in A. thaliana to identify patterns of variation that are visible at the genome-wide level. An understanding of such patterns is important for the interpretation of genetic variation at individual loci of interest. We analyzed patterns of genetic variation using a sequencing survey of 334 loci from 8–12 accessions at randomly chosen regions of the A. thaliana genome and in the outgroup species A. lyrata ssp. lyrata and B. drummondii. We used summary statistics to describe the observed patterns of variation and performed ML analyses to estimate population parameters from the data.
Sequence diversity in A. thaliana has been analyzed previously at more than a dozen loci, but the relative roles of demography and selection on patterns of genetic variation have not been rigorously studied, except by Innan and Stephan (2000). The average level of silent nucleotide diversity observed among all loci in our data (θW = 0.00896) is very similar to the mean diversity of 14 genes surveyed previously (0.009; Shepard and Purugganan 2003). Furthermore, we find an excess of low-frequency polymorphisms as indicated by an average negative estimate of Tajima's D, which has also been noted in earlier studies (Purugganan and Suddith 1999; Kuittinen and Aguadé 2000; Shepard and Purugganan 2003). Although previous studies focused on specific regions or protein-coding genes of particular interest, our data agree with levels and patterns of variation observed in these studies.
The large number of loci investigated in this study allowed us to search for possible causes of different levels of polymorphism among loci. A higher diversity at synonymous sites than at noncoding sites was also found in maize, Drosophila, and humans (Tenaillon et al. 2001), but the differences are more pronounced (1.5- to 2-fold) in these species than in our study (1.2-fold). Furthermore, synonymous diversity and GC3 were not correlated. The low level of codon usage bias of A. thaliana (Duret and Mouchiroud 1999) suggests that selection for optimal codon usage is not very strong and that synonymous polymorphisms are completely neutral, possibly because of a reduced effective population size due to the high selfing rate (Bustamante et al. 2002).
The local rate of recombination is correlated with nucleotide diversity in a number of plant (Dvorák et al. 1998; Kraft et al. 1998; Stephan and Langley 1998) and animal species (e.g., Begun and Aquadro 1992; Glinka et al. 2003). We did not find such a relationship in A. thaliana, suggesting that either differences in the effective rate of recombination are too small to have a measurable effect on nucleotide diversity or background or positive selection are not strong enough to cause the observed relationship between genetic diversity and recombination found in other species. Similarly, the local recombination rate is not correlated with the distribution of transposable elements (Wright et al. 2003) and only weakly correlated with the frequency of tandemly repeated genes (Zhang and Gaut 2003). Variation in recombination rates does not appear to be a strong force in structuring the genome of A. thaliana. On the other hand, the observation of similar levels of nucleotide diversity among neighboring genes (<250 kb) may result from a low effective recombination rate and extended regions of the genome then have similar evolutionary histories (Nordborg and Tavaré 2002). This finding is supported by the observation of correlated polymorphism levels within a 40-kb genomic region around the CLAVATA2 locus (Shepard and Purugganan 2003) and within a 170-kb region around the MAM locus of A. thaliana (Haubold et al. 2002). Correlated patterns of polymorphism among physically linked loci that result from background or positive selection occurring in a particular genomic region may interfere with the analysis of the demographic processes. Therefore, to minimize the effect of selection on our analysis of demographic models, we included only physically distant loci.
Rejection of a neutral panmictic model of evolution:
Our data show a significant, genome-wide deviation from a standard mutation-drift model of evolution (Table 6), which may result from the absence of panmixia, temporary changes in population size and structure, or selection at independent loci. We first want to consider possible effects of population structure and population growth on our analyses. The accessions included in this survey are genetically distantly related, as indicated by a genealogy with long terminal branches (Schmid et al. 2003). Every accession therefore is assumed to represent a single individual from different local demes, making it impossible to observe a deviation from the standard mutation-drift model at the level of the deme (e.g., Wakeley 2004). Using Wakeley's terminology (Wakeley 1999), our accessions represent the “collecting phase” of the coalescent of a metapopulation (i.e., the coalescing of lines from different demes) and not the “scattering phase” (coalescence events within individual demes). The collecting phase of a coalescent is equivalent to a single standard population (Wakeley and Aliacar 2001) and can be analyzed with the general coalescent as we have done in this study. This conclusion seems robust enough to be applicable to different metapopulation structures (Wakeley 2001; Wakeley and Aliacar 2001).
Under this assumption, deviations at a genomic level from the neutral panmictic model might be a consequence of those events that affect the entire metapopulation structure, like temporary changes in population size (expansion, bottlenecks), or also subdivision of the metapopulation (i.e., subdivision of this species in isolated refugia for a certain time period). The significant excess of low-frequency polymorphisms could be explained by an expansion process, as has been suggested (Purugganan and Suddith 1999; Innan and Stephan 2000; Kuittinen and Aguadé 2000), but ML estimates of an alternative logistic growth model were not different from the standard neutral model. Furthermore, the high variance observed in our empirical distributions is not expected under population expansion and a simple expansion model is not sufficient to explain the difference of observed data from a neutral model. We were able to obtain a better fit to a logistic growth than to a standard neutral model when highly polymorphic loci were excluded. However, this result needs to be interpreted with caution because we removed one-third of all loci from the analysis on the basis of a ML analysis of θ values under a neutral model without knowing whether these loci are highly polymorphic due to selection or due to a neutral process such as a locally increased mutation rate or admixture from previously separated subpopulations representing glacial refugia (Sharbel et al. 2000). To account for the latter possibility, a refugia model and bottleneck models were also considered. Although we investigated only a few biologically realistic parameter combinations, we could not detect a combination that was consistent with the observed data.
A second explanation for the observed deviation from a neutral model is selection. If selection occurred only at single loci, its effect on the distribution of summary statistics should be minor given the large number of loci analyzed. On the other hand, selective sweeps or balancing selection at many loci may contribute to the significant deviation of the mean and the variance of Tajima's D from the expectation of a neutral model. Such an explanation is not supported by the analysis of a subset of loci, for which we were able to obtain a sequence from one of outgroup species, because the observed distribution of Fay and Wu's H was not significantly different from the expectation of a standard neutral model. Thus, the negative averages of Tajima's D and Fu and Li's statistics do not seem to result from positive selection at or hitchhiking of multiple loci.
Alternatively, purifying selection against slightly deleterious mutations may have caused the excess of low-frequency polymorphisms. The efficacy of selection against deleterious mutations may be weak compared to that of other species (Bustamante et al. 2002), but it is nevertheless operating in A. thaliana, as indicated by the lower nonsynonymous than synonymous nucleotide diversity (Figure 4). The effect of purifying selection on the frequency spectrum is difficult to quantify, especially in noncoding regions, because little is known about their functional and evolutionary constraints. As discussed above, synonymous polymorphisms may not be exposed to purifying selection and thus contribute little to the deviation from a neutral model. Although it does not appear that positive or negative selection is solely responsible for the observed deviation from a neutral model, the observation of highly negative values of Fay and Wu's H at some loci and the high variance in the distribution of summary statistics may result from variable selection pressures at different loci.
It should be noted that we did not take the geographic origin of accessions used for this study into account. The excess of rare polymorphisms may also result from fixation of locally occurring polymorphisms by selection and thus represent geographic differentiation between demes of a self-fertilizing species (Hedrick and Holden 1979). Such an interpretation is consistent with a weak, but significant presence of a geographic population structure in the natural species range (Sharbel et al. 2000). However, our current sample of 12 accessions is not large enough to address this question.
Modified tests of neutrality:
A frequent goal of surveys of sequence variation is to evaluate whether a particular gene evolves under selection. Furthermore, genome-wide analyses of genetic variation aim at identifying novel “adaptive trait genes” that were subject to positive or balancing selection (reviewed by Luikart et al. 2003). In sequence surveys, the rejection of the null hypothesis of neutral evolution in one of the numerous available tests of neutrality is often taken as evidence that a gene evolves under selection (e.g., Purugganan and Suddith 1998; Olsen et al. 2002; Kroymann et al. 2003; Mauricio et al. 2003). According to our results, a standard neutral model should not be used as a null hypothesis for neutrality tests in A. thaliana because of the effects of demographic history on nucleotide diversity. Thus, if one attempts to test the hypothesis that a given gene has been the target of natural selection, the challenge consists of differentiating between the effects of demography and selection on genetic variation.
One approach to account for demography in tests of selection is to use a modified null model that incorporates the demographic history of a species and selection at independent loci. This allows the estimation of demographic parameters and of the likelihood that individual loci evolved under selection. Using our data, we could not formulate an alternative model for such a purpose, because we were not able to identify all the demographic and selective forces that have shaped observed variation. It will be a considerable challenge to develop such a model for a species with a complex demographic history given the large number of parameters involved. An alternative approach is to use empirical distributions of various descriptive statistics derived from randomly sequenced genomic loci and to identify the outlier loci in such statistics (Black et al. 2001; Luikart et al. 2003). Outlier loci are defined as falling into the extreme tails of the empirical distribution and thus exhibit unusual patterns of variation. We have calculated the critical values for various descriptive statistics using the empirical distributions obtained from our data (Table 7) and they may be useful for neutrality tests in future sequence surveys of novel genes of interest. In this case, however, one would have to consider the effect of using different accessions and different sequence lengths on descriptive statistics of nucleotide diversity before comparing them to such a distribution (Pluzhnikov and Donnelly 1996). A modification of this empirical approach is to use combinations of test statistics and to identify those genes that reject more than one neutrality test. In our data set, a small number of loci (e.g., AtV9, AtV11, AtV23, and GOLM80) fulfill this criterion. Candidate adaptive trait genes can be easily found by such an approach, but further investigations will be necessary, because the short genomic segments studied here are not sufficient to fully characterize patterns of polymorphism at a locus and thus to infer the role of selection. Despite these concerns, the use of empirical distributions appears to be a useful alternative to a model-based analysis to identify genes that may have been targets of selection because the demographic history is taken into account.
TABLE 7.
Averages and critical values for the outer 25% tails of empirical distributions of descriptive statistics
| −2.5% | Average | +2.5% | |
|---|---|---|---|
| n = 195 loci without outgroup | |||
| θW | ≤0.0000a | 0.0099 | ≥0.0624 |
| π | ≤0.0000a | 0.0092 | ≥0.0466 |
| Tajima's D | ≤−1.9437 | −0.4712 | ≥1.7121 |
| Fu and Li's D* | ≤−2.3888 | −0.4745 | ≥1.4525 |
| Fu and Li's F* | ≤−2.5799 | −0.5323 | ≥1.6234 |
| D/Dminb | ≤−1.0000c | −0.2944 | ≥1.0506 |
| n = 43 loci with A. lyrata outgroup | |||
| Divergenced | ≤0.006 | 0.141 | ≥0.342 |
| Fu and Li's D | ≤−2.2382 | −0.4265 | ≥1.8324 |
| Fu and Li's F | ≤−2.4493 | −0.5220 | ≥2.1240 |
| H/Hmine | ≤−0.8500 | −0.0277 | ≥0.1905 |
Distributions are based on silent sites.
The minimum value is 0.0. The percentage of 0.0 values is 7.7%.
Tajima's D divided by its minimum (Schaeffer 2002).
The minimum value is −1.0. The percentage of −1.0 values is 22%.
The interspecific divergence was corrected after Jukes and Cantor (1969).
Fay and Wu's H divided by its minimum (see materials and methods).
Acknowledgments
We thank the Gesellschaft für wissenschaftliche Datenverarbeitung (GWDG) in Göttingen for allowing us to use their computing facilities. N. Spies and T. Heinze helped with the programming, and I. Schumacher provided advice on implementing the HKA test. We thank L. Zhang and B. Gaut for providing us with the estimates of recombination rates and M. Clauss, D. de Lorenzo, M. Hamblin, A. Lawton-Rauh, S. Schaeffer, and E. Wheeler for discussion and comments on the manuscript. This work was funded by the German Ministry of Science project grants to T.M.-0.(0312275C/4) and to B.W. (0312275D/7), by the Emmy-Noether program of the Deutsche Forschungsgemeinschaft to K.J.S. (Schm 1354/2-2), and by the Max-Planck Society.
References
- Abbott, R. J., and M. F. Gomes, 1989. Population genetic structure and outcrossing rate of Arabidopsis thaliana. Heredity 42: 411–418. [Google Scholar]
- Akey, J., M. Eberle, M. Rieder, C. Carlson, M. Shriver et al., 2004. Population history and natural selection shape patterns of genetic variation in 132 genes. PLoS Biol. 2: e286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altshuler, D., V. Pollara, C. C. W. Van Etten, J. Baldwin, L. Linton et al., 2000. SNP map of the human genome generated by reduced representation sequencing. Nature 407: 513–516. [DOI] [PubMed] [Google Scholar]
- Begun, D., and C. Aquadro, 1992. Levels of naturally occurring DNA polymorphism correlate with recombination rates in D. melanogaster. Nature 356: 519–520. [DOI] [PubMed] [Google Scholar]
- Bergelson, J., C. Purrington and G. Wichmann, 1998. Promiscuity in transgenic plants. Nature 395: 25. [DOI] [PubMed] [Google Scholar]
- Black, W., C. Baer, M. Antolind and N. DuTeau, 2001. Population genomics: genome-wide sampling of insect populations. Annu. Rev. Entomol. 46: 441–469. [DOI] [PubMed] [Google Scholar]
- Bustamante, C., R. Nielsen, S. Sawyer, K. Olsen, M. Purugganan et al., 2002. The cost of inbreeding in Arabidopsis. Nature 416: 531–534. [DOI] [PubMed] [Google Scholar]
- Caicedo, A. L., B. A. Schaal and B. N. Kunkel, 1998. Diversity and molecular evolution of the RPS2 resistance gene in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 96: 302–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth, B., M. Morgan and D. Charlesworth, 1993. The effect of deleterious mutations on neutral molecular variation. Genetics 134: 1289–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth, D., 2003. Effects of inbreeding on the genetic diversity of populations. Philos. Trans. R. Soc. Lond. B 358: 1051–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clauss, M., and T. Mitchell-Olds, 2004. Functional divergence in tandemly duplicated Arabidopsis thaliana trypsin inhibitor genes. Genetics 166: 1419–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copenhaver, G., K. Nickel, T. Kuromori, M. Benito, S. Kaul et al., 1999. Genetic definition and sequence analysis of Arabidopsis centromeres. Science 286: 2468–2474. [DOI] [PubMed] [Google Scholar]
- Duret, L., and D. Mouchiroud, 1999. Expression pattern and, surprisingly, gene length shape codon usage in Caenorhabditis, Drosophila, and Arabidopsis. Proc. Natl. Acad. Sci. USA 96: 4482–4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorák, J., M. Luo and Z. Yang, 1998. Restriction fragment length polymorphism and divergence in the genomic regions of high and low recombination in self-fertilizing and cross-fertilizing Aegilops species. Genetics 148: 423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay, J. C., and C.-I Wu, 2000. Hitchhiking under positive Darwinian selection. Genetics 155: 1405–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y.-X., 1997. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147: 915–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y.-X., and W.-H. Li, 1993. Statistical tests of neutrality of mutations. Genetics 133: 693–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glinka, S., L. Ometto, S. Mousset, W. Stephan and D. De Lorenzo, 2003. Demography and natural selection have shaped genetic variation in Drosophila melanogaster: a multi-locus approach. Genetics 165: 1269–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagenblad, J., and M. Nordborg, 2002. Sequence variation and haplotype structure surrounding the flowering time locus FRI in Arabidopsis thaliana. Genetics 161: 289–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamblin, M., S. Mitchell, G. White, J. Allego, R. Kukatla et al., 2004. Comparative population genetics of the Panicoid grasses: sequence polymorphism, linkage disequilibrium and selection in a diverse sample of Sorghum bicolor. Genetics 167: 471–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanfstingl, U., A. Berry, E. E. Kellog, J. T. Costa, III, W. Rüdiger et al., 1994. Haplotypic divergence coupled with lack of diversity at the Arabidopsis thaliana alcohol dehydrogenase locus: roles for both balancing and directional selection. Genetics 138: 811–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings, W., 1970. Monte Carlo sampling methods using Markov chains and their applications. Biometrika 57: 57–109. [Google Scholar]
- Haubold, B., J. Kroymann, A. Ratzka, T. Mitchell-Olds and T. Wiehe, 2002. Recombination and gene conversion in a 170-kb genomic region of Arabidopsis thaliana. Genetics 161: 1269–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt, W., T. Fischer, S. Winderl, P. Fransz and R. Torres-Ruiz, 2001. The CENTROMERE1 (CEN1) region of Arabidopsis thaliana: architecture and functional impact of chromatin. Plant J. 27: 285–296. [DOI] [PubMed] [Google Scholar]
- Hauser, M.-T., B. Harr and C. Schlötterer, 2001. Trichome distribution in Arabidopsis thaliana and its close relative Arabidopsis lyrata: molecular analysis of the candidate gene GLABROUS1. Mol. Biol. Evol. 18: 1754–1763. [DOI] [PubMed] [Google Scholar]
- Hedrick, P., and L. Holden, 1979. Hitch-hiking: an alternative to coadaptation for the barley and slender wild oat examples. Heredity 43: 79–86. [Google Scholar]
- Hudson, R., 1993 The how and why of generating gene genealogies, pp. 23–36 in Mechanisms of Molecular Evolution, edited by N. Takahata and A. Clark. Sinauer Associates, Sunderland, MA.
- Hudson, R., 2000. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18: 337–338. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., 1990 Gene genealogies and the coalescent process, pp. 1–44 in Oxford Surveys in Evolutionary Biology, Vol. 7, edited by D. Futuyma and J. Antonovics. Oxford University Press, London/New York/Oxford.
- Hudson, R. R., M. Kreitman and M. Aguadé, 1987. A test of neutral molecular evolution based on nucleotide data. Genetics 116: 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innan, H., and W. Stephan, 2000. The coalescent in an exponentially growing metapopulation and its application to Arabidopsis thaliana. Genetics 155: 2015–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jukes, T., and C. Cantor, 1969 Evolution of protein molecules, pp. 21–132 in Mammalian Protein Metabolism, edited by H. Munro. Academic Press, New York.
- Kawabe, A., and N. Miyashita, 1999. DNA variation in the basic chitinase locus (ChiB) region of the wild plant Arabidopsis thaliana. Genetics 153: 1445–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabe, A., H. Innana, R. Terauchi and N. T. Miyashita, 1997. Nucleotide polymorphism in the acidic chitinase locus (ChiA) region of the wild plant Arabidopsis thaliana. Mol. Biol. Evol. 14: 1303–1315. [DOI] [PubMed] [Google Scholar]
- Kawabe, A., K. Yamane and N. Miyashita, 2000. DNA polymorphism at the cytosolic phosphoglucose isomerase (PgiC) locus of the wild plant Arabidopsis thaliana. Genetics 156: 1339–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliman, R., and J. Hey, 1993. DNA sequence variation at the period locus within and among species of the Drosophila melanogaster species complex. Genetics 133: 375–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch, M., B. Haubold and T. Mitchell-Olds, 2000. Comparative evolutionary analysis of chalcone synthase and alcohol dehydrogenase loci in Arabidopsis, Arabis and related genera (Brassicaeae). Mol. Biol. Evol. 17: 1483–1498. [DOI] [PubMed] [Google Scholar]
- Koenker, R., and K. Hallock, 2001. Quantile regression. J. Econ. Perspect. 15: 143–156. [Google Scholar]
- Kraft, T., T. Sall, I. Magnusson-Rading, N. Nilsson and C. Hallden, 1998. Positive correlation between recombination rates and levels of genetic variation in natural populations of sea beet (Beta vulgaris subsp. maritima). Genetics 150: 1239–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroymann, J., S. Donnerhacke, D. Schnabelrauch and T. Mitchell-Olds, 2003. Evolutionary dynamics of an Arabidopsis insect resistance quantitative trait locus. Proc. Natl. Acad. Sci. USA 100: 14587–14592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhner, M., J. Yamato and J. Felsenstein, 1995. Estimating effective population size and mutation rate from sequence data using Metropolis-Hastings sampling. Genetics 140: 1421–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuittinen, H., and M. Aguadé, 2000. Nucleotide variation at the CHALCONE ISOMERASE locus in Arabidopsis thaliana. Genetics 155: 863–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikart, G., P. England, D. Tallmon, S. Jordan and P. Taberlet, 2003. The power and promise of population genomics: from genotyping to genome typing. Nat. Rev. Genet. 4: 981–994. [DOI] [PubMed] [Google Scholar]
- Mauricio, R., E. Stahl, T. Korves, D. Tian, M. Kreitman et al., 2003. Natural selection for polymorphism in the disease resistance gene Rps2 of Arabidopsis thaliana. Genetics 163: 735–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard Smith, J., and J. Haigh, 1974. The hitch-hiking effect of a favorable gene. Genet. Res. 23: 23–35. [PubMed] [Google Scholar]
- McDonald, J., and M. Kreitman, 1991. Adaptive evolution at the Adh locus in Drosophila. Nature 351: 652–654. [DOI] [PubMed] [Google Scholar]
- Metropolis, N., A. Rosenbluth, M. Rosenbluth, A. Teller and E. Teller, 1953. Equations of state calculations by fast computing machines. J. Chem. Phys. 21: 1087–1091. [Google Scholar]
- Mitchell-Olds, T., 2001. Arabidopsis thaliana and its wild relatives: a model system for ecology and evolution. Trends Ecol. Evol. 16: 693–700. [Google Scholar]
- Nei, M., 1987 Molecular Evolutionary Genetics. Columbia University Press, New York.
- Nickerson, D., V. O. Tobe and S. L. Taylor, 1997. PolyPhred: automating the detection and genotyping of single nucleotide substitutions using fluorescence-based resequencing. Genome Res. 25: 2745–2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordborg, M., 2000. Linkage disequilibrium, gene trees and selfing: an ancestral recombination graph with partial self-fertilization. Genetics 154: 923–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordborg, M., and P. Donnelly, 1997. The coalescent process with selfing. Genetics 146: 1185–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordborg, M., and S. Tavaré, 2002. Linkage disequilibrium: what history has to tell us. Trends Genet. 18: 83–90. [DOI] [PubMed] [Google Scholar]
- Nordborg, M., J. Borevitz, J. Bergelson, C. Berry, J. Chory et al., 2002. The extent of linkage disequilibrium in Arabidopsis thaliana. Nat. Genet. 30: 190–193. [DOI] [PubMed] [Google Scholar]
- Olsen, K., A. Womack, A. Garrett, J. Suddith and M. Purugganan, 2002. Contrasting evolutionary forces in the Arabidopsis thaliana floral developmental pathway. Genetics 160: 1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluzhnikov, A., and P. Donnelly, 1996. Optimal sequencing strategies for surveying molecular genetic diversity. Genetics 144: 1247–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purugganan, M., and J. Suddith, 1999. Molecular population genetics of floral homeotic loci: departures from the equilibrium-neutral model at the APETALA3 and PISTILLATA genes of Arabidopsis thaliana. Genetics 151: 839–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purugganan, M. D., and J. I. Suddith, 1998. Molecular population genetics of the Arabidopsis CAULIFLOWER regulatory gene: nonneutral evolution and naturally occurring variation in floral homeotic function. Proc. Natl. Acad. Sci. USA 95: 8130–8134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Onsins, S., B. Stranger, T. Mitchell-Olds and M. Aguadé, 2004. Multilocus analysis of variation and speciation in the closely related species Arabidopsis halleri and A. lyrata. Genetics 166: 372–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas, J., and R. Rozas, 1999. DnaSP version 3: an integrated program for molecular population genetics and molecular evolution analysis. Bioinformatics 15: 174–175. [DOI] [PubMed] [Google Scholar]
- Schaeffer, S., 2002. Molecular population genetics of sequence length diversity in the Adh region of Drosophila pseudoobscura. Genet. Res. 80: 163–175. [DOI] [PubMed] [Google Scholar]
- Scharf, F., F. Juanes and M. Sutherland, 1998. Inferring ecological relationships from the edges of scatter diagrams: comparison of regression techniques. Ecology 79: 448–460. [Google Scholar]
- Schmid, K., T. Rosleff-Sörensen, R. Stracke, O. Törjek, T. Altmann et al., 2003. Large-scale identification and analysis of genome-wide single-nucleotide polymorphisms for mapping in Arabidopsis thaliana. Genome Res. 13: 1250–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharbel, T., B. Haubold and T. Mitchell-Olds, 2000. Genetic isolation by distance in Arabidopsis thaliana: biogeography and postglacial colonization of Europe. Mol. Ecol. 9: 2109–2118. [DOI] [PubMed] [Google Scholar]
- Shepard, K., and M. Purugganan, 2003. Molecular population genetics of the Arabidopsis CLAVATA2 region: the genomic scale of variation and selection in a selfing species. Genetics 163: 1083–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokal, R. R., and F. J. Rohlf, 1995 Biometry. Sinauer Associates, Sunderland, MA.
- Stahl, E., G. Dwyer, R. Mauricio, M. Kreitman and J. Bergelson, 1999. Dynamics of disease resistance polymorphism at the Rpm1 locus of Arabidopsis. Nature 400: 667–671. [DOI] [PubMed] [Google Scholar]
- Stephan, W., and C. Langley, 1998. DNA polymorphism in Lycopersicon and crossing-over per physical length. Genetics 150: 1585–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, F., 1983. Evolutionary relationship of DNA sequences in finite populations. Genetics 105: 437–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavaré, S., 1984. Line-of-descent and genealogical processes, and their applications in population genetics models. Theor. Popul. Biol. 26: 119–164. [DOI] [PubMed] [Google Scholar]
- Tenaillon, M., M. Sawkins, A. Long, R. Gaut, J. Doebley et al., 2001. Patterns of DNA sequence polymorphism along chromosome 1 of maize (Zea mays ssp. mays L). Proc. Natl. Acad. Sci. USA 98: 9161–9166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenaillon, M., J. U'Ren, O. Tenaillon and B. Gauth, 2004. Selection versus demography: a multilocus investigation of the domestication process in maize. Mol. Biol. Evol. 21: 1214–1225. [DOI] [PubMed] [Google Scholar]
- Thompson, J. D., D. G. Higgins and T. J. Gibson, 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22: 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, D., H. Araki, E. Stahl, J. Bergelson and M. Kreitman, 2002. Signature of balancing selection in Arabidopsis. Proc. Natl. Acad. Sci. USA 99: 11525–11530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeley, J., 1999. Non-equilibrium migration in human history. Genetics 153: 1863–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeley, J., 2001. The coalescent in an island model of population subdivision with variation among demes. Theor. Popul. Biol. 59: 133–144. [DOI] [PubMed] [Google Scholar]
- Wakeley, J., 2004. Metapopulation models for historical inference. Mol. Ecol. 13: 865–875. [DOI] [PubMed] [Google Scholar]
- Wakeley, J., and N. Aliacar, 2001. Gene genealogies in a metapopulation. Genetics 159: 893–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall, J., 1999. Recombination and the power of statistical tests of neutrality. Genet. Res. 74: 65–79. [Google Scholar]
- Wall, J., 2000. Detecting ancient admixture in humans using sequence polymorphism data. Genetics 154: 1271–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson, G. A., 1975. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7: 256–276. [DOI] [PubMed] [Google Scholar]
- Wright, S., N. Agrawal and T. Bureau, 2003. Effects of recombination rate and gene density on transposable element distributions in Arabidopsis thaliana. Genome Res. 13: 1897–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L., and B. Gaut, 2003. Does recombination shape the distribution and evolution of tandemly arrayed genes (TAGs) in the Arabidopsis thaliana genome? Mol. Biol. Evol. 13: 2533–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The sequences generated for this study were submitted to the STS section of GenBank under accession nos. CW672529, CW672721. Annotated alignments can be obtained from http://kiwi.ice.mpg.de/athapop.