

Abstract
We examined error-prone nonhomologous end joining (NHEJ) in Msh2-deficient and wild-type Chinese hamster ovary cell lines. A DNA substrate containing a thymidine kinase (tk) gene fused to a neomycin-resistance (neo) gene was stably integrated into cells. The fusion gene was rendered nonfunctional due to a 22-bp oligonucleotide insertion, which included the 18-bp I-SceI endonuclease recognition site, within the tk portion of the fusion gene. A double-strand break (DSB) was induced by transiently expressing the I-SceI endonuclease, and deletions or insertions that restored the tk-neo fusion gene's reading frame were recovered by selecting for G418-resistant colonies. Overall, neither the frequency of recovery of G418-resistant colonies nor the sizes of NHEJ-associated deletions were substantially different for the mutant vs. wild-type cell lines. However, we did observe greater usage of terminal microhomology among NHEJ events recovered from wild-type cells as compared to Msh2 mutants. Our results suggest that Msh2 influences error-prone NHEJ repair at the step of pairing of terminal DNA tails. We also report the recovery from both wild-type and Msh2-deficient cells of an unusual class of NHEJ events associated with multiple deletion intervals, and we discuss a possible mechanism for the generation of these “discontinuous deletions.”
MAMMALIAN cells contend with various forms of DNA damage on a daily basis. One type of DNA damage that is potentially quite deleterious is a double-strand break (DSB). DSBs can arise at stalled replication forks or following exposure to a variety of chemical or radiological agents. At least two general pathways for DSB repair in eukaryotes exist: homologous recombination, and nonhomologous end joining (NHEJ) (Chu 1997; Liang et al. 1998; Lin et al. 1999; Haber 2000; Karran 2000; Ferguson and Alt 2001; Johnson and Jasin 2001; Khanna and Jackson 2001; Norbury and Hickson 2001; Pastink et al. 2001; Pierce et al. 2001; van Gent et al. 2001; Bernstein et al. 2002; Jackson 2002; Helleday 2003; Valerie and Povirk 2003). Homologous recombination is an accurate repair pathway utilizing a homologous DNA template to correctly restore genetic information that may otherwise be lost at a DSB site. In contrast, NHEJ involves no template and is error prone because one or several nucleotides are usually deleted or inserted prior to DSB healing. NHEJ is considered to be a major DSB repair pathway in mammalian cells.
One approach toward gaining a better understanding of DSB repair pathways is to study the consequences of the loss of specific proteins that appear to be reasonable candidates for involvement in repair. It has been observed that NHEJ repair junctions often occur within short patches of homology, suggesting that the joining of DNA ends via NHEJ may be facilitated by terminal microhomologies (Chu 1997; Liang et al. 1998; Lin et al. 1999; Haber 2000; Karran 2000; Ferguson and Alt 2001; Johnson and Jasin 2001; Khanna and Jackson 2001; Norbury and Hickson 2001; Pastink et al. 2001; Pierce et al. 2001; van Gent et al. 2001; Bernstein et al. 2002; Jackson 2002; Helleday 2003; Valerie and Povirk 2003). One may envision that NHEJ involves interactions between single-stranded DNA tails with a concomitant “search” for homology as the strands align for subsequent joining. The homology search may be directed by specific proteins or may be driven by spontaneous base pairing. Interactions between mismatched DNA tails may produce short segments of heteroduplex DNA containing mispaired bases, and these mispairs may be substrates for the DNA mismatch repair (MMR) machinery. For this, and other reasons elaborated below, it seems reasonable to think that MMR proteins have a role in NHEJ.
In eukaryotes, Msh2 (MutS homologue 2) is a major player in MMR, functioning as a heterodimer in association with Msh3 or Msh6. Msh2 has been implicated in a variety of processes that serve to protect genomic integrity. In addition to its “spell-checking” function in postreplicative mismatch repair, Msh2 is involved in a generalized cellular response to DNA damage, with a role in triggering a signaling cascade that activates cell cycle checkpoints or apoptosis (Buermeyer et al. 1999; Harfe and Jinks-Robertson 2000; Aquilina and Bignami 2001; Bellacosa 2001; Bernstein et al. 2002; Wei et al. 2002; Brown et al. 2003; Li 2003; Schofield and Hsieh 2003; Fedier and Fink 2004). Msh2 is also a component of the BRCA-1-associated genome surveillance complex, a multiprotein complex involved in the recognition and response to abnormal DNA structures (Wang et al. 2000; De la Torre et al. 2003; Jhanwar-Uniyal 2003). With regard to DSB repair, Msh2 has been reported to localize to DSB sites in yeast (Evans et al. 2000) and has been directly implicated in DSB repair in yeast by playing a role in the removal of nonhomologous DNA tails and possibly assisting in a homology search (Saparbaev et al. 1996; Sugawara et al. 1997, 2004; Kijas et al. 2003). Msh2-deficient human and rodent cells display an increase in chromosomal damage and failure to form Mre11 and Rad51 foci in the G2 phase of the cell cycle following X irradiation (Franchitto et al. 2003). Recently, evidence suggesting that Msh2 may colocalize with Msh6, p53, BLM, and Rad51 at sites of DSBs at stalled replication forks in human cells and help regulate the processing of recombination intermediates and the repair of DSBs was reported (Yang et al. 2004). Deficiency in Msh2 results in increased mutation rate, global instability of microsatellite sequences, and increased rates of recombination between diverged sequences. Inherited defects in Msh2 are a cause of hereditary nonpolyposis colorectal cancer (HNPCC), a cancer predisposition syndrome (Watson and Lynch 2001; Peltomaki 2001; Heinen et al. 2002; Mitchell et al. 2002; Muller et al. 2003).
The multiple functions of Msh2 give this MMR protein the status of tumor suppressor and “caretaker” of the genome. The multiple roles of Msh2 in the maintenance of genomic integrity make Msh2 a likely player in DSB repair in mammalian cells, but little is known about how Msh2 may influence such repair in mammalian cells. In this report, we explore the role of Msh2 in error-prone NHEJ in mammalian cells using a stably integrated chromosomal substrate that enables the recovery of NHEJ events following DSB induction by endonuclease I-SceI. We compared NHEJ in Msh2-deficient Chinese hamster ovary (CHO) cell line clone B (Aquilina et al. 1988, 1989), with NHEJ in an isogenic wild-type cell line. Our results indicate that Msh2 deficiency has little effect on the efficiency of NHEJ or on the deletion size associated with NHEJ. However, relative to NHEJ events recovered from wild-type cells, significantly more NHEJ events recovered from Msh2 mutants involve the joining of DNA ends displaying no terminal microhomology. Our data suggest a role for Msh2 in responding to mispaired bases that are likely to arise at DNA termini interacting during NHEJ.
MATERIALS AND METHODS
Cell culture:
MT+ and clone B CHO cells were kindly provided by Margherita Bignami (Istituto Superiore di Sanità, Rome). Clone B was isolated from MT+ cells as an alkylating agent-resistant clone and was subsequently shown to be deficient in Msh2 (Aquilina et al. 1988, 1989, 1994). We confirmed that clone B cells are highly resistant to 6-thioguanine (data not shown), a phenotype associated with MMR-deficiency. MT+ and clone B CHO cells were cultured in minimum essential medium, α modification (Sigma, St. Louis) supplemented with 10% fetal bovine serum (growth medium). Cells were maintained at 37° in a humidified atmosphere of 5% CO2.
NHEJ substrate:
Plasmid pTNeo99-7 (Figure 1) was described previously (Bannister et al. 2004). pTNeo99-7 contains a herpes simplex virus type 1 (HSV-1) thymidine kinase (tk) gene fused to the coding region of the neo gene. The tk-neo gene in pTNeo99-7 is disrupted by a 22-bp oligonucleotide that contains the 18-bp recognition site for yeast endonuclease I-SceI. The 22-bp oligonucleotide was inserted into the unique SstI site at nucleotide position 964 of the HSV-1 tk sequence (tk gene nucleotide numbering according to Wagner et al. 1981).
Figure 1.—

DSB repair substrate pTNeo99-7. Shown is plasmid pTNeo99-7 linearized at the unique XhoI site (X) in the vector. The substrate contains a hygromycin-resistance gene (hyg) and a tk-neo fusion gene. The tk-neo fusion gene is disrupted by a 22-bp oligonucleotide containing the 18-bp recognition site for endonuclease I-SceI (underlined sequence); the sites of staggered cleavage by I-SceI are indicated by vertical arrows. Also shown are two BamHI sites (B) flanking the tk-neo fusion gene and the location of primers AW85 and AW91 (short horizontal arrows) used in PCR analysis. Primer AW85 maps within tk sequences and AW91 maps within neo sequences; the two primers are positioned 1.4 kb apart.
Establishing cell lines for studying DSB repair:
MT+ or clone B CHO cells (5 × 106) were resuspended in 800 μl of phosphate buffered saline (PBS) and electroporated with 2.5 μg of pTNeo99-7 using a Bio-Rad gene pulser (set at 1000 V, 25 μF). Cells were then plated into 150-cm2 flasks and allowed to grow for 2 days. At this point, 75-cm2 flasks containing growth medium supplemented with either 500 μg/ml of hygromycin (for clone B cell lines) or 400 μg/ml of hygromycin (for MT+ cell lines) were seeded with 1 × 106 cells each to initiate selection for clones stably transfected with pTNeo99-7. After 10 days of growth under selection, hygromycin-resistant colonies were picked, propagated, and screened by Southern blot analysis to isolate cell lines containing a single integrated copy of pTNeo99-7.
DSB induction and recovery of G418-resistant (G418R) clones:
pCMV3xnls-I-SceI (“pSce”) was generously provided by Maria Jasin (Sloan Kettering). This plasmid contains a gene encoding the I-SceI endonuclease under the control of the cytomegalovirus promoter and is expressible in mammalian cells. To induce a genomic DSB, cell lines stably transfected with pTNeo99-7 were electroporated with pSce. For each electroporation, 5 × 106 cells were resuspended in 800 μl of PBS containing 20 μg of supercoiled pSce (or PBS alone for controls) and electroporated as described above. Following electroporation, cells were propagated under no selection for 2 days, at which time cells were harvested by trypsinization and plated at a density of 5 ×105 cells/75-cm2 flask in growth medium supplemented with 1000 μg/ml of G418. G418 selection continued for approximately 10 days until G418R clones were counted and picked.
Southern blotting analysis:
Genomic DNA samples (8 μg each) were digested with appropriate restriction enzymes and resolved on 0.8% agarose gels. DNA was transferred to nitrocellulose membranes and hybridized with a 32P-labeled HSV-1 tk probe as described (Lukacsovich et al. 1994).
PCR amplification and DNA sequencing analysis:
Genomic DNA samples isolated from parent cell lines or DSB-induced G418R colonies were amplified using the primers AW85 (5′-TAATACGACTCACTATAGGGCCAGCGTCTTGTCATTGGCG-3′) and AW91 (5′-GATTTAGGTGACACTATAGCCAAGCGGCCGGAGAACCTG-3′) to produce a PCR product whose sequence spans the site of the I-SceI-induced DSB. AW85 is composed of nucleotides 308–327 from the coding strand of the HSV-1 tk gene with a T7 forward universal priming site appended to the 5′-end of the primer. AW91 is composed of 20 nucleotides from the noncoding strand of the neo gene mapping 25–44 bp downstream from the neo start codon, with an SP6 primer sequence appended to the 5′-end of the primer. The positions of AW85 and AW91 on pTNeo99-7 are indicated in Figure 1. The parental PCR product is 1432 bp in length, and products generated from G418R clones may be shorter due to deletions associated with NHEJ at the I-SceI-induced DSB. PCR reactions contained 0.5 μg of genomic template DNA in a final volume of 25 μl. PCR was carried out using Ready-To-Go PCR beads (Amersham Biosciences, Piscataway, NJ) and a “touchdown” PCR protocol. The annealing temperature was initially set to 72° and was progressively decreased in steps of 2° down to 62°, with two cycles at each temperature. An additional 20 cycles were run at an annealing temperature of 60°. Prior to sequencing, PCR products were treated with shrimp alkaline phosphatase and exonuclease I (USB, Cleveland). PCR products were then sequenced from a T7 primer or an SP6 primer using a Licor 4000L at the DNA Sequencing and Synthesis Core Facility in the Department of Biological Sciences at the University of South Carolina.
RESULTS
Recovery of NHEJ events from wild-type and Msh2-deficient CHO cells:
To study the potential role of Msh2 in NHEJ, MT+ and clone B (Msh2-deficient) CHO cells were stably transfected with pTNeo99-7 (Figure 1). The “tk-neo” fusion gene in pTNeo99-7 is disrupted by the insertion of a 22-bp oligonucleotide containing the 18-bp I-SceI recognition site. Three cell lines derived from MT+ cells (designated MT4, MT7, and MT19) and three cell lines derived from clone B (designated CB2, CB6, and CB9), each containing a single integrated copy of pTNeo99-7, were isolated. Cells containing pTNeo99-7 were electroporated with pSce to induce a genomic DSB at the I-SceI site within the integrated construct. NHEJ events in which the I-SceI-induced DSB was repaired in such a way to restore function to the tk-neo fusion gene were recovered by selecting for G418R clones. Restoration of function to the tk-neo gene required the deletion (or insertion) of an appropriate number of nucleotides to restore the correct reading frame to the fusion gene.
Frequencies of G418R colonies recovered following electroporation of cells with pSce are presented in Table 1. The frequency of G418R colonies recovered following mock electroporations of all cell lines with PBS alone was consistently <2 × 10−6 (data not shown). There was a notably greater line-to-line variability in the frequency of DSB-induced colonies for cell lines derived from clone B compared to cell lines derived from MT+ (Table 1). However, the mean frequency of DSB-induced G418R colonies recovered from MT+ cells was 3.15 ×10−4 while the mean colony frequency for clone B cell lines was 3.77 ×10−4, providing no evidence that the Msh2 deficiency of clone B-derived cells had an appreciable, consistent effect on the efficiency of NHEJ overall.
TABLE 1.
Recovery of G418R colonies following DSB induction
| Cell linea | Cells plated (millions)b |
No. of G418R colonies |
Colony frequency (×104)c |
|---|---|---|---|
| MT4 | 5 | 1931 | 3.86 |
| MT7 | 5 | 1152 | 2.30 |
| MT19 | 5 | 1643 | 3.28 |
| CB2 | 5 | 1530 | 3.06 |
| CB6 | 15 | 285 | 0.19 |
| CB9 | 5 | 4036 | 8.07 |
Cell lines designated “MT” were derived from MT+ cells that express functional Msh2; cell lines designated “CB” were derived from clone B cells that are Msh2 deficient.
Cells were electroporated with pSce and were plated into G418 selection two days post-transfection, as described in materials and methods.
Calculated as number of G418R colonies divided by number of cells plated into selection.
PCR and Southern blot analysis of DSB-induced G418R clones:
Genomic DNA samples isolated from 72 G418R clones recovered from MT+ cell lines and from 89 G418R clones recovered from clone B cell lines were PCR amplified using primers AW85 and AW91, which flank the original position of the I-SceI site in pTNeo99-7 (Figure 1). An illustrative representative analysis of PCR products generated from clones recovered from cell line CB2 is presented in Figure 2. Cell line CB2, like all parental cell lines, produced the expected 1.4-kb PCR product (Figure 2, lane 3). PCR products from most G418R clones from all cell lines appeared to be about 1.4 kb in length (see, for example, Figure 2, lanes 5–11, 14–17, 19, 23), suggesting that NHEJ was often accompanied by a relatively small deletion or insertion that did not alter the apparent mobility of the PCR product. Other clones generated PCR products that were visibly shorter than 1.4 kb (Figure 2, lanes 4, 12, 13, 18, 20, 21, 24–26), suggesting that these clones underwent more substantial deletions in association with NHEJ at the I-SceI site. Some clones produced no PCR product (Figure 2, lane 22) or multiple products (not shown), indicating either a large deletion or insertion or a more complex rearrangement. One clone recovered from cell line CB6 (not shown) displayed a PCR product notably larger than 1.4 kb.
Figure 2.—

Representative PCR products generated from DSB-induced G418R clones. Shown are PCR products generated from parent cell line CB2 (lane 3) and from DSB-induced G418R clones recovered from cell line CB2 (lanes 4–26). All PCR reactions were carried out using primers AW85 and AW91 (see Figure 1). Lanes 1 and 27 contain molecular weight markers; lane 2 displays a negative (no template) PCR control. See text for further discussion.
PCR products were further analyzed by digestion with I-SceI endonuclease to ascertain if the I-SceI site had indeed been lost by NHEJ. As expected, most PCR products were resistant to I-SceI cleavage and none were fully sensitive to I-SceI cleavage (data not shown). Somewhat surprisingly, however, PCR products from two G418R colonies from cell line MT4 and from 25 G418R colonies from cell line CB6 were partially sensitive to I-SceI, suggestive of a mixture of two PCR products in which one product could be cleaved with I-SceI and the other could not. These clones may have undergone a duplication of the tk-neo fusion gene in which one copy of the gene had a small deletion or insertion associated with NHEJ, while the other copy remained unaltered.
Genomic DNA samples from putative duplication clones were digested with either BamHI alone or BamHI plus I-SceI and analyzed on Southern blots along with additional clones that had apparently undergone simple NHEJ without duplication. In total, 28 MT+ and 39 clone B G418R clones were viewed on Southern blots using a tk-specific probe, and a representative analysis is presented in Figure 3. G418R clones that were produced by NHEJ associated with a small deletion or insertion were expected to display a 3.9-kb BamHI fragment (see Figure 1), which is evident for the clones presented in Figure 3, lanes 1, 3, 4, and 6–8. Clones that had undergone larger deletions produced smaller BamHI fragments (Figure 3, lanes 2 and 5). The clones whose BamHI digests are presented in lanes 3, 4, and 8 each generated a PCR product that was partially sensitive to I-SceI. An increased intensity of the 3.9-kb band is apparent for the clones in lanes 3, 4, and 8, relative to the BamHI fragments displayed by the clones in lanes 1, 2, and 5–7, consistent with a duplication of the tk-neo gene in the former clones. DNA samples from the clones shown in lanes 3, 4, and 8 were additionally subjected to a double digest with BamHI plus I-SceI. As shown in Figure 3, lanes 9–11, the double digest of each of these clones produced a 3.9-kb band as well as a 2.6-kb and a 1.3-kb band, indicating that the tk-neo gene had indeed undergone duplication in each of these clones and that one gene copy retained the I-SceI site while the other copy did not.
Figure 3.—

Representative Southern blot analysis of G418R clones. Genomic DNA samples (8 μg) isolated from eight G418R clones recovered from cell line CB6 were digested with BamHI and displayed on a blot using a tk-specific probe (lanes 1–8). DNA samples from the clones shown in lanes 3, 4, and 8 were additionally digested with BamHI plus I-SceI and displayed in lanes 9, 10, and 11, respectively. Lane 12 displays parental cell line DNA digested with BamHI plus I-SceI. As discussed in the text, the clones in lanes 2 and 5 produced a BamHI fragment notably shorter than 3.9 kb, indicative of relatively large deletions at the I-SceI site, and the clones displayed in lanes 3, 4, and 8 (and lanes 9–11) had undergone an apparent duplication of the integrated tk-neo gene. The origins of the fragments visualized are illustrated in Figure 1.
On the basis of PCR analysis and Southern blot analysis as described above, the DSB repair events responsible for the genesis of the recovered G418R clones were categorized as “ NHEJ,” “NHEJ with duplication,” or “complex” (Table 2). “Complex” events were those that produced no PCR products, multiple PCR products of different sizes, or unexpected bands upon Southern blotting. As presented in Table 2, there was no clear, consistent difference between MT+ cell lines vs. clone B cell lines regarding the types of repair events recovered, although we noted that one cell line, CB6, produced 25 NHEJ with duplication events of 44 events recovered from this line (Table 2, and data not shown).
TABLE 2.
Classification of DSB repair events
| No. of events recovered
|
||
|---|---|---|
| Type of event | MT+ lines | Clone B lines |
| NHEJ | 57 | 52 |
| NHEJ with duplication | 2 | 25a |
| Complex | 13 | 12 |
| Total analyzed | 72 | 89 |
All 25 duplication events were recovered from cell line CB6.
Analysis of nucleotide sequences across NHEJ repair junctions:
PCR products generated from 45 NHEJ events recovered from MT+ cell lines and from 45 NHEJ events recovered from clone B cell lines were sequenced. The clones that were sequenced were randomly selected from among the clones that had apparently undergone simple NHEJ on the basis of PCR and Southern blotting analysis. A summary of the sequence analysis is presented in Table 3. Detailed nucleotide sequence data for NHEJ junctions is available as supplemental material at http://www.genetics.org/supplemental/. As anticipated, most clones analyzed had undergone a single continuous deletion (or, more rarely, an insertion) of nucleotides that restored the correct reading frame to the tk-neo fusion gene. There was no striking difference between the deletion sizes for events recovered from MT+ cell lines vs. clone B cell lines. The median deletion size for MT+ lines and for clone B lines was 22 bp, with deletion sizes ranging from 1 to 1201 bp. The distributions of deletion sizes for MT+ and clone B cells were very similar, with several clones displaying deletions substantially larger than the median deletion size. Surprisingly, five NHEJ clones (MT4-7, MT19-10, MT7-11, CB6-191, and CB2-11, Tables 3 and 4) had two or three discrete deletions in the vicinity of the I-SceI site. A proposed mechanism for the generation of these “discontinuous deletions” is described in the discussion.
TABLE 3.
Analysis of sequences across NHEJ junctions
| NHEJ events from MT+ cell lines
|
NHEJ events from clone B cell lines
|
||||
|---|---|---|---|---|---|
| Clone namea | Deletion or insert size (bp)b |
Microhomologyc | Clone namea | Deletion or insert size (bp)b |
Microhomologyc |
| MT-13 | 2 (insert of AA) | A | CB6-71 | 638 (insertd) | 0, A |
| MT19-13 | 1 | A | CB6-212 | 2 (insert of AA) | A |
| MT19-16 | 1 | A | CB6-72 | 1 | 0 |
| MT19-15 | 7 | A | CB2-15 | 1 | A |
| MT19-14 | 7 | A | CB9-4 | 7 | 0 |
| MT19-12 | 7 | A | CB9-21 | 7 | 0 |
| MT4-7 | 8, 2 | TAA, CG | CB2-9 | 7 | G |
| MT4-9 | 10 | GG | CB2-13 | 7 | A |
| MT4-10 | 10 | GG | CB2-8 | 10 | 0 |
| MT19-3 | 10 | GG | CB2-1 | 10 | GG |
| MT7-4 | 10 | GG | CB9-13 | 10 | GG |
| MT4-14 | 16 | 0 | CB2-18 | 10 | 0 |
| MT4-16 | 16 | A | CB2-14 | 10 | 0 |
| MT7-6 | 16 | G | CB9-14 | 19 | GG |
| MT4-17 | 19 | A | CB9-16 | 19 | GG |
| MT4-2 | 22 | AGCT | CB6-21 | 22 | AGG |
| MT4-5 | 22 | AGCT | CB6-31 | 22 | 0 |
| MT4-8 | 22 | AGCT | CB6-41 | 22 | AGCT |
| MT7-1 | 22 | AGCT | CB6-42 | 22 | AGCT |
| MT7-7 | 22 | AGCT | CB6-152 | 22 | AGCT |
| MT7-12 | 22 | AGCT | CB6-202 | 22 | AGCT |
| MT7-15 | 22 | AGCT | CB9-18 | 22 | AGCT |
| MT7-17 | 22 | AGCT | CB9-20 | 22 | AGCT |
| MT19-7 | 22 | AGCT | CB2-5 | 22 | AGCT |
| MT19-8 | 22 | AGCT | CB2-6 | 22 | AGCT |
| MT19-23 | 22 | AGCT | CB2-16 | 22 | AGCT |
| MT7-24 | 22 | 0 | CB2-7 | 22 | AGG |
| MT7-13 | 22 | T | CB6-32 | 25 | 0 |
| MT4-15 | 25 | 0 | CB6-191 | 6, 22 | T, AGCT |
| MT4-21 | 25 | GGG | CB9-5 | 28 | GGG |
| MT19-11 | 34 | 0 | CB6-82 | 37 | 0 |
| MT4-24 | 34 | 0 | CB2-4 | 43 | GG |
| MT7-8 | 37 | C | CB9-8 | 76 | 0 |
| MT4-6 | 40 | T | CB2-2 | 100 | 0 |
| MT19-2 | 121 | GG | CB9-12 | 133 | CAGGGT |
| MT19-6 | 145 | AACA | CB9-6 | 136 | GCC |
| MT4-11 | 235 | ATA | CB9-3 | 190 | CACC |
| MT4-4 | 244 | GC | CB2-11 | 117, 60, 19 | 0, 0, 0 |
| MT7-5 | 385e | TAC, AGCe | CB6-92 | 202 | ATCG |
| MT19-10 | 348, 9, 427 | CT, AGGG, CC | CB9-10 | 244 | GGGT |
| MT7-11 | 183, 176, 650 | C, GA, GGCT | CB9-7 | 295 | GGT |
| MT7-18 | 1084 | G | CB6-92 | 310 | 0 |
| MT7-10 | 1111 | GCG | CB2-10 | 880 | TT |
| MT4-1 | 1144 | GC | CB6-121 | 1021 | C |
| MT7-14 | 1201 | T | CB9-17 | 1141 | CCG |
The name of the cell line from which each clone was recovered is indicated and precedes the hyphen in each clone name.
All sizes are deletion sizes, except where inserts are indicated. For clones displaying discontinuous deletions, the size of each discrete deletion interval is indicated.
For junctions displaying microhomology at the joined DNA termini, the actual sequence of microhomology shared between the joined termini is shown. Junctions in which no microhomology was found are indicated with a “0.” For clones displaying multiple deletions and, hence, multiple junctions, microhomologies for all junctions are shown.
The insert in clone CB6-71 contained the cytomegalovirus promoter and likely originated from transfected pSce DNA. Indicated are the microhomologies at the two junctions between the insert and the genomic DSB.
In addition to a 385-bp deletion, clone MT7-5 contained a 351-bp insertion of sequence from the hygromycin-resistance gene, likely copied from the integrated copy of pTNeo99-7. Indicated are the microhomologies at the two junctions between the insert and the genomic DSB.
TABLE 4.
Discontinuous deletions
| Clone name | Deletion structurea |
|---|---|
| MT4-7 | (−8)—81—(−2) |
| MT19-10 | (−48)—10—(−9)—114—(−427) |
| MT7-11 | (−183)—15—(−176)—34—(−650) |
| CB6-191 | (−6)—19—(−22) |
| CB2-11 | (−117)—7—(−60)—4—(−19) |
For each clone, the number of nucleotides deleted in each discrete deletion interval is indicated in parentheses. Also indicated is the length of sequence separating the deletion intervals.
Four clones had insertions of nucleotides at the site of the I-SceI-induced DSB. Clone MT4-13 had an insertion of 2 bp (AA) while clone MT7-5 had a 385-bp deletion in conjunction with a 351-bp insertion of sequence from the hygromycin-resistance gene, likely originating from the integrated copy of pTNeo99-7. Clone CB6-212 had an insertion of 2 bp (AA) and clone CB6-71 had an insertion of 638 bp containing a cytomegalovirus promoter sequence, likely originating from pSce, which had been electroporated into the cells to induce a DSB. Insertions of DNA sequences at genomic DSBs have been seen previously by us (Lin and Waldman 2001a,b) and others (Roth and Wilson 1986; Phillips and Morgan 1994; Liang et al. 1998; Allen et al. 2003). The relatively low frequency of recovery of clones that captured any significant length of DNA in the current work is likely a reflection of the fact that recovery of a clone required restoration of function to the tk-neo gene.
Microhomologies at the site of end joining were also examined for each repair junction (Table 3). The majority of NHEJ events for MT+ and clone B cells involved one or several bases of microhomology at the site where DNA termini were joined. However, we noted that when all NHEJ junctions are considered, 16 of 49 junctions recovered from clone B cells displayed no terminal bases of microhomology, while only 5 of 51 junctions recovered from MT+ cells involved no microhomology. This difference in the number of clones recovered that displayed no microhomology at NHEJ junctions is highly statistically significant (p = 0.0050 by a chi-square test). If we discount the data from cell line CB6, which yields a low frequency of DSB-induced G418R colonies (Table 1) and produces an unusually large number of duplications (Table 2), the difference in the number of NHEJ junctions that displayed no microhomology recovered from MT+ vs. clone B cells remains statistically significant (p = 0.0134 by a chi-square test). Our data demonstrate a more frequent use of microhomology in MT+ cells.
DISCUSSION
In this article, we investigated the role of Msh2 in error-prone DSB repair by studying NHEJ in wild-type and Msh2-deficient CHO cells. We used a system that allows us to induce a single well-defined DSB in the CHO cell genome and to recover error-prone events that result in deletion or insertion of nucleotides to restore function to a tk-neo fusion gene. Collectively, our results suggest that deficiency in Msh2 does not have a major impact on the overall efficiency of error-prone NHEJ. Neither the overall frequency nor the associated deletion size of recovered NHEJ events appears to be affected by Msh2 status in our experimental system. As in any study, our results may be influenced by the types of cells and DNA sequences used. We also recognize that since only events that restore function to the tk-neo fusion gene are recovered in our experimental system, a variety of DSB events may potentially occur and yet go undetected. Such events include any gene with the proper reading frame unrestored; introduction of a stop codon; large deletions by NHEJ; and large-scale gene conversions using a homolog that results in complete loss of the substrate. It is possible that experiments conducted using other experimental systems may reveal an influence of Msh2 on NHEJ efficiency and/or deletion size not uncovered in our investigation.
We noted that for both wild-type and Msh2-deficient cells, the deletion sizes associated with recovered NHEJ events displayed a somewhat unusual distribution in that several deletions were strikingly larger than the median deletion size of 22 bp (Table 3). The relatively common occurrence of deletions many-fold larger than the median-sized deletion suggests the possibility of two (or more) pathways leading to the removal of nucleotides during NHEJ, one producing relatively short deletions and one producing more substantial deletions.
Our data do reveal a significant difference between wild-type vs. Msh2-deficient cells in terms of the frequency of occurrence of NHEJ between DNA ends displaying no microhomology. The increased number of microhomology-independent events recovered from the Msh2-deficient (clone B) cells suggests that Msh2 may normally play a role in impeding the joining of mismatched DNA termini. We recently reported data supporting a fundamentally similar role for MMR protein Mlh1 in NHEJ in mouse fibroblasts (Bannister et al. 2004), suggesting, perhaps, a general role for the spell-checking function of MMR in modulating NHEJ. We envision that the MMR machinery may respond to mispaired bases produced as DNA termini interact during NHEJ. This engagement of MMR may disrupt the end-joining process and lead to an increased recovery of joining events occurring within patches of homology. Further work will be required to determine if the influence of MMR proteins on error-prone NHEJ is indeed mediated through spell checking or if other activities of the multifaceted MMR proteins come into play. Nonetheless, our current and previous work illustrate that the loss of a functional MMR machinery, such as is associated with HNPCC, can affect the manner in which DSBs are processed and possibly lead to more promiscuous end joining.
Somewhat unexpectedly, we recovered five NHEJ clones (MT4-7, MT19-10, MT7-11, CB6-191, and CB2-11, Tables 3 and 4) that each displayed more than one deleted interval of DNA sequence near the I-SceI site. An economical model for the generation of these discontinuous deletions is presented in Figure 4. In our model, which is a variation of “synthesis-dependent strand annealing” (SDSA) (reviewed in Prado et al. 2003), we propose that a broken chromatid first misaligns with an intact sister chromatid. The 3′ DNA terminus at the DSB then invades the sister chromatid, is extended via a short patch of DNA synthesis using the sister as a template, and is released from the template. The 3′-end of the nascent DNA strand is then rejoined via NHEJ to the DNA terminus at the other side of the DSB, in the case of clones with two deletion intervals. If, prior to end joining, there are additional cycles of strand invasion and short DNA synthesis, clones with three or more deletion intervals can be produced. Strand invasion and subsequent end joining occur at sites that may or may not display microhomology. Our model for discontinuous deletions suggests that, at least under certain circumstances, DNA synthesis during SDSA may not be highly processive and that SDSA may involve multiple rounds of strand invasion and synthesis. Evidence for multiple cycles of strand invasion during SDSA in Drosophila has also recently been reported (McVey et al. 2004). As presented in Table 4, the length of DNA synthesis tracts (the number of nucleotides between the deleted intervals) in our experiments ranges from a few nucleotides to ∼100 nucleotides.
Figure 4.—
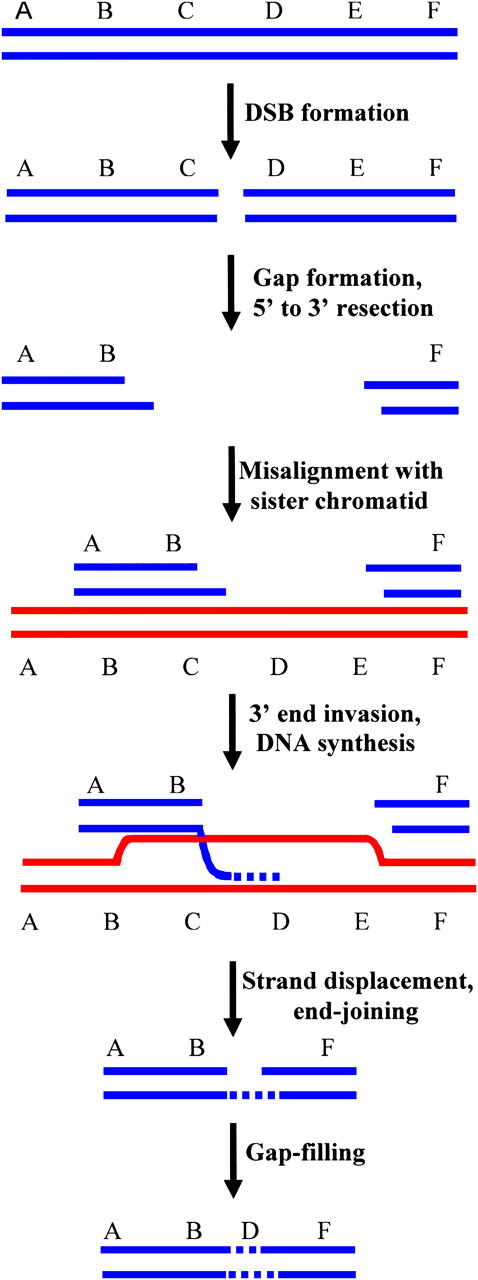
Model for the generation of discontinuous deletions. Following DSB formation, the DSB is enlarged into a gap, with 5′-end resection. The gapped molecule misaligns with a sister chromatid (or perhaps a homologous chromosome), and a 3′-end invades the sister chromatid and is extended by DNA synthesis. Following strand displacement, the nascent DNA strand is joined via NHEJ to a DNA terminus from the other side of the DSB. The single-strand gap is filled to complete the repair process. In this figure, DNA segments C and E are deleted from the broken chromosome while segment D is retained in the final repair product, thereby producing a discontinuous deletion. Repeated cycles of 3′-end invasion between misaligned chromatids followed by synthesis of short spans of DNA can produce clones with three or more discrete deletions.
We recognize the possibility that DNA end extension may be involved even in clones displaying a single, continuous deletion. In such cases, sister chromatids would be aligned in homologous register (as opposed to being misaligned) prior to 3′-end invasion. Along these lines, it is possible that the variation in deletion sizes noted above may be due to differences in the degree of DNA end extension rather than, or in addition to, a difference in the number of nucleotides initially removed from DNA ends.
We recovered clones that underwent duplications of the integrated tk-neo fusion gene as a consequence of a genomic DSB, and we have reported on DSB-induced sequence amplifications previously (Lin et al. 1999). Such amplifications could conceivably result from reiterated copying of sequences from one chromatid onto its broken sister, or from DSB-induced nondisjunctions. The possibility that certain genomic loci are particularly susceptible to such processes might explain the high frequency of duplications recovered from cell line CB6 (Table 2). Our current and previous studies (Bannister et al. 2004) do not provide evidence for a role for MMR in DSB-induced duplications. Analysis of the sequence of the human genome has revealed that the copy number of certain genes can vary significantly between normal individuals (Sebat et al. 2004). It is conceivable that some of the evolutionary events responsible for such copy-number polymorphisms were triggered by DSBs.
Acknowledgments
The authors are grateful to Yunfu Lin for constructing pTNeo99-7. This work was supported by Public Health Service grant GM47110 from the National Institute of General Medical Sciences to A.S.W.
References
- Allen, C., C. A. Miller and J. A. Nickoloff, 2003. The mutagenic potential of a single DNA double-strand break in a mammalian chromosome is not influenced by transcription. DNA Repair 2: 1147–1156. [DOI] [PubMed] [Google Scholar]
- Aquilina, G., and M. Bignami, 2001. Mismatch repair in correction of replication errors and processing of DNA damage. J. Cell. Physiol. 187: 145–154. [DOI] [PubMed] [Google Scholar]
- Aquilina, G., G. Frosina, A. Zijno, A. Di Muccio, E. Dogliotti et al., 1988. Isolation of clones displaying enhanced resistance to methylating agents in O6-methylguanine-DNA methyltransferase-proficient CHO cells. Carcinogenesis 9: 1217–1222. [DOI] [PubMed] [Google Scholar]
- Aquilina, G., A. Zijno, N. Moscufo, E. Dogliotti and M. Bignami, 1989. Tolerance to methylnitrosourea-induced DNA damage is associated with 6-thioguanine resistance in CHO cells. Carcinogenesis 10: 1219–1223. [DOI] [PubMed] [Google Scholar]
- Aquilina, G., P. Hess, P. Branch, C. Macgeoch, I. Casciano et al., 1994. A mismatch recognition defect in colon carcinoma confers DNA microsatellite instability and a mutator phenotype. Proc. Natl. Acad. Sci. USA 19: 8905–8909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister, L. A., B. C. Waldman and A. S. Waldman, 2004. Modulation of error-prone double-strand break repair in mammalian chromosomes by DNA mismatch repair protein Mlh1. DNA Repair 3: 465–474. [DOI] [PubMed] [Google Scholar]
- Bellacosa, A., 2001. Functional interactions and signaling properties of mammalian DNA mismatch repair proteins. Cell Death Differ. 8: 1076–1092. [DOI] [PubMed] [Google Scholar]
- Bernstein, C., H. Bernstein, C. M. Payne and H. Garewal, 2002. DNA repair/pro-apoptotic dual-role proteins in five major DNA repair pathways: fail-safe protection against carcinogenesis. Mutat. Res. 511: 145–178. [DOI] [PubMed] [Google Scholar]
- Brown, K. D., A. Rathi, R. Kamath, D. I. Beardsley, Q. Zhan et al., 2003. The mismatch repair system is required for S-phase checkpoint activation. Nat. Genet. 33: 80–84. [DOI] [PubMed] [Google Scholar]
- Buermeyer, A. B., S. M. Deschenes, S. M. Baker and R. M. Liskay, 1999. Mammalian DNA mismatch repair. Annu. Rev. Genet. 33: 533–564. [DOI] [PubMed] [Google Scholar]
- Chu, G., 1997. Double-strand break repair. J. Biol. Chem. 272: 24097–24100. [DOI] [PubMed] [Google Scholar]
- De la Torre, C., J. Pincheira and J. F. Lopez-Saez, 2003. Human syndromes with genomic instability and multiprotein machines that repair DNA double-strand breaks. Histol. Histopathol. 18: 225–243. [DOI] [PubMed] [Google Scholar]
- Evans, E., N. Sugawara, J. Haber and E. Alani, 2000. The Saccharomyces cerevisiae MSH2 mismatch repair protein localizes to recombination intermediates in vivo. Mol. Cell 5: 789–799. [DOI] [PubMed] [Google Scholar]
- Fedier, A., and D. Fink, 2004. Mutations in DNA mismatch repair genes: implications for DNA damage signaling and drug sensitivity. Int. J. Oncol. 24: 1039–1047. [PubMed] [Google Scholar]
- Ferguson, D. O., and F. W. Alt, 2001. DNA double strand break repair and chromosomal translocation: lessons from animal models. Oncogene 20: 5572–5579. [DOI] [PubMed] [Google Scholar]
- Franchitto, A., P. Pichierri, R. Piergentili, M. Crescenzi, M. Bignami et al., 2003. The mammalian mismatch repair protein MSH2 is required for correct MRE11 and RAD51 relocalization and for efficient cell cycle arrest induced by ionizing radiation in G2 phase. Oncogene 22: 2110–2120. [DOI] [PubMed] [Google Scholar]
- Haber, J., 2000. Partners and pathways repairing a double-strand break. Trends Genet. 16: 259–264. [DOI] [PubMed] [Google Scholar]
- Harfe, B. D., and S. Jinks-Robertson, 2000. DNA mismatch repair and genetic instability. Annu. Rev. Genet. 34: 359–399. [DOI] [PubMed] [Google Scholar]
- Heinen, C. D., C. Schmutte and R. Fishel, 2002. DNA repair and tumorigenesis: lessons from hereditary cancer syndromes. Cancer Biol. Ther. 1: 477–485. [DOI] [PubMed] [Google Scholar]
- Helleday, T., 2003. Pathways for mitotic homologous recombination in mammalian cells. Mutat. Res. 532: 103–115. [DOI] [PubMed] [Google Scholar]
- Jackson, S. P., 2002. Sensing and repairing DNA double-strand breaks. Carcinogenesis 23: 687–696. [DOI] [PubMed] [Google Scholar]
- Jhanwar-Uniyal, M., 2003. BRCA1 in cancer, cell cycle and genomic stability. Front. Biosci. 8: s1107–s1117. [DOI] [PubMed] [Google Scholar]
- Johnson, R. D., and M. Jasin, 2001. Double-strand-break-induced homologous recombination in mammalian cells. Biochem. Soc. Trans. 29: 196–201. [DOI] [PubMed] [Google Scholar]
- Karran, P., 2000. DNA double strand break repair in mammalian cells. Curr. Opin. Genet. Dev. 10: 144–150. [DOI] [PubMed] [Google Scholar]
- Khanna, K. K., and S. P. Jackson, 2001. DNA double-strand breaks: signaling, repair and the cancer connection. Nat. Genet. 27: 247–254. [DOI] [PubMed] [Google Scholar]
- Kijas, A. W., B. Studamire and E. Alani, 2003. Msh2 separation of function mutations confer defects in the initiation steps of mismatch repair. J. Mol. Biol. 331: 123–138. [DOI] [PubMed] [Google Scholar]
- Li, G. M., 2003. DNA mismatch repair and cancer. Front. Biosci. 8: d997–d1017. [DOI] [PubMed] [Google Scholar]
- Liang, F., M. Han, P. J. Romanienko and M. Jasin, 1998. Homology-directed repair is a major double-strand break repair pathway in mammalian cells. Proc. Natl. Acad. Sci. USA 95: 5172–5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y., and A. S. Waldman, 2001. a Capture of DNA sequences at double-strand breaks in mammalian chromosomes. Genetics 158: 1665–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y., and A. S. Waldman, 2001. b Promiscuous patching of broken chromosomes in mammalian cells with extrachromosomal DNA. Nucleic Acids Res. 29: 3975–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y., T. Lukacsovich and A. S. Waldman, 1999. Multiple pathways for repair of DNA double-strand breaks in mammalian chromosomes. Mol. Cell. Biol. 19: 8353–8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacsovich, T., D. Yang and A. S. Waldman, 1994. Repair of a specific double-strand break generated within a mammalian chromosome by yeast endonuclease I-SceI. Nucleic Acids Res. 22: 5649–5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVey, M., M. Adams, E. Staeva-Vieira and J. J. Sekelsky, 2004. Evidence for multiple cycles of strand invasion during repair of double-strand gaps in Drosophila. Genetics 167: 699–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell, R. J., S. M. Farrington, M. G. Dunlop and H. Campbell, 2002. Mismatch repair genes hMLH1 and hMSH2 and colorectal cancer: a HuGE review. Am. J. Epidemiol. 156: 885–902. [DOI] [PubMed] [Google Scholar]
- Muller, A., M. Korabiowska and U. Brinck, 2003. DNA-mismatch repair and hereditary nonpolyposis colorectal cancer syndrome. In Vivo 17: 55–59. [PubMed] [Google Scholar]
- Norbury, C. J., and I. D. Hickson, 2001. Cellular responses to DNA damage. Annu. Rev. Pharmacol. Toxicol. 41: 367–401. [DOI] [PubMed] [Google Scholar]
- Pastink, A., J. C. J. Eeken and P. H. M. Lohman, 2001. Genomic integrity and the repair of double-strand DNA breaks. Mutat. Res. 480/481: 37–50. [DOI] [PubMed] [Google Scholar]
- Peltomaki, P., 2001. Deficient DNA mismatch repair: a common etiologic factor for colon cancer. Hum. Mol. Genet. 10: 735–740. [DOI] [PubMed] [Google Scholar]
- Phillips, J. W., and W. F. Morgan, 1994. Illegitimate recombination induced by DNA double-strand breaks in a mammalian chromosome. Mol. Cell. Biol. 14: 5794–5803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce, A. J., J. M. Stark, F. D. Araujo, M. E. Moynahan, M. Berwick et al., 2001. Double-strand breaks and tumorigenesis. Trends Cell Biol. 11: S52–S59. [DOI] [PubMed] [Google Scholar]
- Prado, F., F. Cortes-Ledesma, P. Huertas and A. Aguilera, 2003. Mitotic recombination in Saccharomyces cerevisiae. Curr. Genet. 42: 185–198. [DOI] [PubMed] [Google Scholar]
- Roth, D. B., and J. H. Wilson, 1986. Nonhomologous recombination in mammalian cells: role for short sequence homologies in the joining reaction. Mol. Cell. Biol. 6: 4295–4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saparbaev, M., L. Prakash and S. Prakash, 1996. Requirement of mismatch repair genes MSH2 and MSH3 in the RAD1-RAD10 pathway of mitotic recombination in Saccharomyces cerevisiae. Genetics 142: 727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield, M. J., and P. Hsieh, 2003. DNA mismatch repair: molecular mechanisms and biological function. Annu. Rev. Microbiol. 57: 579–608. [DOI] [PubMed] [Google Scholar]
- Sebat, J., B. Lakshmi, J. Troge, J. Alexander, J. Young et al., 2004. Large-scale copy number polymorphism in the human genome. Science 305: 525–528. [DOI] [PubMed] [Google Scholar]
- Sugawara, N., F. Paques, M. P. Colaiacovo and J. E. Haber, 1997. Role of S. cerevisiae MSH2 and MSH3 repair proteins in double-strand break repair-induced recombination. Proc. Natl. Acad. Sci. USA 94: 9214–9219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara, N., T. Goldfarb, B. Studamire, E. Alani and J. E. Haber, 2004. Heteroduplex rejection during single-strand annealing requires Sgs1 helicase and mismatch repair proteins Msh2 and Msh6 but not Pms1. Proc. Natl. Acad. Sci. USA. 101: 9315–9320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valerie, K., and L. F. Povirk, 2003. Regulation and mechanisms of mammalian double-strand break repair. Oncogene 22: 5792–5812. [DOI] [PubMed] [Google Scholar]
- van Gent, D. C., J. H. J. Hoeijmakers and R. Kanaar, 2001. Chromosomal stability and the DNA double-stranded break connection. Nat. Rev. Genet. 2: 196–206. [DOI] [PubMed] [Google Scholar]
- Wagner, M. J., J. A. Sharp and W. C. Summers, 1981. Nucleotide sequence of the thymidine kinase of herpes simplex virus type 1. Proc. Natl. Acad. Sci. USA 78: 1441–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y., D. Cortez, P. Yazdi, N. Neff, S. J. Elledge et al., 2000. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 14: 927–939. [PMC free article] [PubMed] [Google Scholar]
- Watson, P., and H. T. Lynch, 2001. Cancer risk in mismatch repair gene mutation carriers. Fam. Cancer 1: 57–60. [DOI] [PubMed] [Google Scholar]
- Wei, K., R. Kucherlapati and W. Edelmann, 2002. Mouse models for human DNA mismatch-repair gene defects. Trends Mol. Med. 8: 346–353. [DOI] [PubMed] [Google Scholar]
- Yang, Q, R. Zhang, X. W. Wang, S. P. Linke, S. Sengupta et al., 2004. The mismatch DNA repair heterodimer, hMSH2/6, regulates BLM helicase. Oncogene 23: 3749–3756. [DOI] [PubMed] [Google Scholar]