

Abstract
Plant transcription factors often contain domains that evolve very rapidly. Although it has been suggested that this rapid evolution may contribute substantially to phenotypic differentiation among species, this suggestion has seldom been tested explicitly. We tested the validity of this hypothesis by examining the rapidly evolving non-DNA-binding region of an R2R3-myb transcription factor that regulates anthocyanin expression in flowers of the genus Ipomoea. We first provide evidence that the W locus in Ipomoea purpurea, which determines whether flowers will be pigmented or white, corresponds to a myb gene segregating in southeastern U.S. populations for one functional allele and one nonfunctional allele. While the binding domain exhibits substantial selective constraint, the nonbinding region evolves at an average Ka/Ks ratio of 0.74. This elevated rate of evolution is due to relaxed constraint rather than to increased levels of positive selection. Despite this relaxed constraint, however, ∼20–25% of the codons, randomly distributed throughout the nonbinding region, are highly constrained, with the remainder evolving neutrally, indicating that the entire region performs important function(s). Our results provide little indication that rapid evolution in this regulatory gene is driven by natural selection or that it is responsible for floral-color differences among Ipomoea species.
THE uncoupling of rates of morphological evolution and molecular evolution, noted 25 years ago by Wilson and collaborators (King and Wilson 1975; Cherry et al. 1978), has continued to be an unsolved issue in evolutionary biology. One popular hypothesis for this uncoupling is that morphological evolution occurs primarily through evolution of regulatory sequences, rather than sequences coding for structural genes (Doebley 1993; Doebley and Lukens 1998; Purugganan 1998). In this context, regulatory sequences may be either the coding regions of transcription factors or promoter/enhancer regions of almost any gene. Substantial evidence indicates that sequence changes in either of these types of regulatory regions can alter morphology (Wray et al. 2003). Nevertheless, the relative importance of regulatory vs. structural genes in contributing to morphological evolution in nature remains unclear.
One observation suggesting the importance of regulatory genes to phenotypic diversification in plants is that transcription factors often exhibit increased ratios of nonsynonymous to synonymous substitution rates (ω), compared to structural genes (Purugganan and Wessler 1994; Purugganan et al. 1995; Purugganan 1998; Rausher et al. 1999, Barrier et al. 2001; Remington and Purugganan 2002). Moreover, in many cases, the elevated ω for the coding region as a whole is due to ω-values that approach 1 in some domains, while other domains are highly conserved (Purugganan and Wessler 1994; Purugganan et al. 1995; Rausher et al. 1999; Remington and Purugganan 2002). One explanation for these domain-specific elevated ω's is that repeated positive selection has occurred in these domains. If this explanation is correct, this positive selection is likely to reflect substitutions that contribute to the evolution of phenotypic diversity.
To date, however, information on whether domain-specific elevated ω's are caused by selection or, alternatively, simply by relaxed selective constraint is meager and inconclusive, in part because specific tests that distinguish positive selection from relaxed constraint in these regions have rarely been performed. Here, we apply such tests to a transcription factor that regulates anthocyanin structural gene expression and that has been shown previously to exhibit rapid evolution in its non-DNA-binding domain (Rausher et al. 1999).
The biosynthetic pathway for anthocyanins and other flavonoids has been a model system for the study of gene regulation in plants (Irani et al. 2003). The pathway, as well as its regulatory elements, is evolutionarily conserved across the angiosperms (Koes et al. 1994; Mol et al. 1998). Three regulatory factors have been identified: (1) an R2, R3 myb protein, (2) a helix-loop-helix protein of the myc family, and (3) a WD40 protein. According to the most recent model of how these proteins interact (Irani et al. 2003; Kroon 2004), the myb and myc factors form a complex and bind the cis-regulatory region of the target anthocyanin structural gene, while the WD40 protein is believed to be involved in post-translational control of the myb protein. Together, these factors coordinately regulate the expression of anthocyanin structural genes.
Here we focus on the myb protein. A previous comparison (Rausher et al. 1999) of homologs of this protein from maize, petunia, and snapdragon revealed two major regions evolving at different rates (Figure 1). The DNA-binding domain, which also is involved with binding to the myc protein (Kroon 2004), is ∼350 bp in length and is highly conserved, exhibiting 66% similarity at the amino acid level between maize and petunia (Quattrocchio et al. 1999). By contrast, the region outside the binding domain, consisting of ∼500–570 bp, is much less conserved, exhibiting no identifiable similarity between the same two species. We first describe cloning and functional characterization of a myb homolog from the morning glory Ipomoea purpurea that regulates floral anthocyanins. We then characterize the molecular evolution of this gene, using standard population-genetic approaches. We specifically ask whether rapid evolution of the variable region is due to repeated positive selection. The species chosen for analysis exhibit a wide diversity of floral hues and pigmentation patterns. Our analysis asks whether rapid evolution in this gene can account in part for this phenotypic diversification.
Figure 1.—

Domains of Ipmyb1 from Ipomoea purpurea. The shaded region is the R2R3 binding domain. Solid vertical lines are intron positions (i1 and i2). The open region is the variable domain. Arrows (labels) indicate positions (identity) of PCR primers. Triangles indicate deletions in ipmyb1. The asterisk indicates the position of the premature stop codon in ipmyb1.
MATERIALS AND METHODS
Species:
The genus Ipomoea (Convolvulacea) contains ∼1000 species distributed throughout the tropics and subtropics. For this study, we chose 13 species representative of the variation in floral color in the genus (Table 1). The phylogenetic relationships among these species are well supported (Miller et al. 1999; Manos et al. 2001) and portrayed in Figure 2. Plant material for RNA and DNA extraction was obtained either from naturally growing plants in the field or from specimens grown in our greenhouse. For I. purpurea, multiple accessions were collected from Orange and Durham counties, North Carolina, and from Oconee County, Georgia.
TABLE 1.
Floral color characteristics and sequence accession numbers for 13 Ipomoea species
| Species | Primary floral hue | Color patterna | Sequence accession nos. |
|---|---|---|---|
| I. purpurea | Purple | Variable intensity | AY986828 |
| I. lindheimeri | Pale lavender | Uniform | AY986826 |
| I. nil | Intense blue | Uniform | AY986825 |
| I. hederacea | Pale blue | Uniform | AY986827 |
| I. tricolor | Blue | Variable hue | AY986824 |
| I. alba | White | Uniform | AY986823 |
| I. coccinea | Red | Uniform | AY986829 |
| I. quamoclit | Red | Uniform | AY986830 |
| I. hederifolia | Red | Uniform | AY986831 |
| I. neei | Red | Uniform | AY986832 |
| I. lacunosa | White | Uniform | AY986835 |
| I. triloba | Pale lavender | Uniform | AY986833 |
| I. trifida | Pale lavender | Variable intensity | AY986834 |
Variable intensity, pigmentation varies in intensity among regions of the corolla; uniform, pigmentation of uniform intensity and hue throughout the corolla; variable hue, different hues on different parts of the corolla.
Figure 2.—

Phylogenetic relationship of the 13 species examined. Branch lengths convey no information. Numbers associated with each branch represent estimated dS (outside parentheses) and dN (inside parentheses) from model M1 (neutral model) for the variable region of Ipmyb1.
For detailed analysis of patterns of intraspecific variation, we focused on I. trifida, a species that grows throughout Central and northern South America. Because this species is closely related to cultivated sweet potato, I. batatas, numerous accessions are available from these regions. We sampled two greenhouse-grown plants from each of 16 accessions (appendix).
Cloning and characterizing Ipmyb1 from I. purpurea:
We employed a previously described cDNA library that was constructed from the distal half of flower buds of I. purpurea using the ZAP cDNA synthesis kit (Stratagene, La Jolla, CA) (Tiffin et al. 1998). A fragment of a putative myb gene corresponding to the W locus was isolated from this library using two rounds of PCR and degenerate primers designed from sequences of anthocyanin myb genes in maize (C gene) and Petunia (An2). The first round of amplification (35 cycles of 95° for 30 sec, 50° for 30 sec, and 72° for 1 min) used the vector-specific M13F primer and degenerate primer C1 (GGNGA[AG]GGNAA[AG]TGG), which is located in the binding domain 96 bp downstream of the start codon. This reaction produced a faint but well-defined band of ∼1100 bp, which was isolated and used as the template for a second round of amplification. This round used primer C1 along with a second degenerate primer C3 (CCNGGNAG[AG]CTNCCNGC[AGT]AT), which is also located in the binding domain, 291 bp downstream of the start codon. This reaction amplified a 195-bp fragment that exhibited 86% amino acid identity to the corresponding sequences of both maize C and petunia An2.
This fragment was used to probe the cDNA library for full-length clones. Approximately 300,000 plaques were screened and 10 potential positive clones isolated and sequenced. Of the 10 isolates, 3 contained partial genes and 7 contained the entire coding region of the same gene plus ∼100 additional bp upstream of the start codon. The locations and sequences of introns were determined by cloning most of the gene from genomic DNA using primers C27 (CAACGTAAGTACCCACTACG) from the 5′-untranslated region and C28 (CGGAAAGTCATCATCAGTTG) located ∼40 bp from the stop codon. In this and all other analyses, genomic DNA was isolated using the DNeasy DNA extraction kit (QIAGEN, Valencia, CA).
The corresponding gene was isolated from white-flowered I. purpurea plants using primers designed from the 5′- and 3′-untranslated ends of Ipmyb1: C27 (CAACGTAAGTACCCACTACG) and C38 (CGTATGATTTAAAGAC). Genomic DNA used as template for this analysis was extracted from a white-flowered inbred line (line X in our collection). Alleles sampled from natural populations were obtained through PCR of genomic DNA using the same set of primers.
Cosegregation of flower color and variation at the Ipmyb1 locus was examined in two groups of plants. The first group consisted of 24 F2 offspring that were homozygous for flower color and that were produced by selfing the F1 offspring of of crosses between homozygous purple (WW) and white (ww) parents. Homozygosity was determined by intensity of floral pigmentation: heterozygous (Ww) plants have pigmentation of distinctly lower intensity than do homozygous (WW) plants. The second group consisted of 74 homozygous offspring obtained by selfing lightly pigmented (Ww) parents collected from a natural population in Georgia. For these analyses, genotype at the Ipmyb1 locus was characterized for each plant by the size of a gene fragment amplified from genomic DNA. Our preliminary analyses indicated the segregation of a length polymorphism (either 356 or 381 bp) in this fragment in natural populations. Primers used for this analysis were C25 (GTAGTTAGCATGCATATGGC) and C28 (CGGAAAGTCATCATCAGTTG). Amplified fragments were scored visually for size on 1% agarose gels.
Cloning and sequencing binding and variable domains from other species:
On the basis of the sequence of Ipmyb1 from I. purpurea, we designed two PCR primer pairs to amplify the variable domain from cDNA of the other 12 species and, for comparison, the binding domain from 6 of these species. The pair for the binding domain was C31 (CCTGCCATGGTTAATTCTTC) and C34 (CCATATGCATGCTAACTACC). The pair for the variable domain was C42F (GTCGCTTATTGCTGGCAGAA) and C42R (AGGTCACATCAATCGGAAA). The locations of these primers are shown in Figure 1. cDNA was prepared for PCR using M-MLV reverse transcriptase (Invitrogen, San Diego) according to the manufacturer's instructions. PCR reactions were performed using AmpliTaq DNA polymerase (Applied Biosystems, Foster City, CA). PCR fragments were cloned into the TOPO 2.1 vector (Invitrogen) and sequenced off the vector using the Big Dye protocol (Applied Biosystems). Sequence data were collected by an ABI 3700 automated sequencer (Applied Biosystems). Both strands of at least two clones from each species were sequenced. The variable domain was cloned and sequenced from genomic DNA from I. trifida accessions using primers C42F and C42R and high-fidelity Pfx polymerase (Invitrogen).
Cloning and sequencing of chalcone synthase:
To assess whether the level of variation is reduced in the variable region of Ipmyb1, as would be expected if there had been a recent selective sweep, a second sequence is needed for comparison. For this second sequence we chose CHS-D, which codes for chalcone synthase, an anthocyanin structural gene (Fukada-Tanaka et al. 1997). This gene is one member of a five-member family of chalcone-synthase-like genes in Ipomoea. CHS-D is readily distinguishable in sequence from the other four members of this family and is the copy that is most highly expressed in most tissues (Fukada-Tanaka et al. 1997; Durbin et al. 2003).
An 870-bp fragment from the second exon of this gene was cloned from genomic DNA from two individuals of each of the I. trifida accessions that were sampled for the Ipmyb1 gene. Two degenerate PCR primers were used: CHSD1F (GCAYTTAACCGAGGAAATATTGAAGG) and CHSD2R (ATKGTAAGCCCRGGCCCAAAYCCAAACA).
Codon-based analysis of sequences:
Sequences analyzed for the binding domain consisted of the 285 bp between primers C31 and C34. Those for the variable domain consisted of the 510 bp between the locations of primers C42F and C42R. Sequences were initially aligned by translating nucleotide sequences into amino acid sequences, aligning the amino acid sequences manually, and adjusting the nucleotide sequence on the basis of the amino acid alignment. Manual adjustments were then made to the nucleotide alignment. Separate alignments were performed on the binding domain and variable domain.
As a first approach to characterizing the selective processes that have acted on the myb gene, we employed the likelihood approach developed by Yang and coworkers (Nielsen and Yang 1998; Yang and Nielsen 2000; Yang et al. 2000) and implemented in version 3.12 of the PAML software package (Yang 1997). Because the phylogenetic relationships among the species are well characterized, we assumed the tree topology in Figure 2. Preliminary maximum-likelihood reconstruction of the sequence tree recovered the same tree topology as in Figure 2, confirming that this topology is appropriate and that the genes examined are orthologous. We first determined whether the average dN/dS ratio, or ω, differed among branches by comparing constant-site models, in which all codons within the sequence are assumed to have the same value of ω. A model in which ω was constrained to be the same on all branches was compared to a free model, in which ω was estimated independently for each branch. We then determined the pattern of variation among amino acid sites, and whether some sites were subject to positive selection, by comparing constant-branch models, in which the mean value of ω is assumed to be the same for all branches. In this analysis, a series of nested models (Table 2) were compared using the standard likelihood-ratio criterion for significance (Hocking 1985). Model M0 represents the null hypothesis that all amino acid positions exhibit the same ω. Model M1 (neutral model) represents the hypothesis that all amino acid positions are either completely constrained (ω = 0) or neutral (ω = 1). Model M2 (selection model) is similar to M1, except that it allows for an additional category of amino acid sites that may be positively selected (ω > 1). Model M3 (discrete model) assigns each amino acid position to one of three categories, the ω for each category being unconstrained and estimated. If models M2 and/or M3 describe the data significantly better than models M0 and M1, and if at least one of the estimated values of ω is >1, positive selection is indicated. Models M7 (β-model) and M8 (β + ω-model) are similar in that they model the distribution of ω among amino acid positions as a β-distribution and estimate the parameters of that distribution. M8 differs from M7 in including an additional category of sites that are not part of the β-distribution. For these sites, a common ω is estimated. A test of whether M8 fits the data significantly better than M7 thus constitutes another test of whether positive selection has acted.
TABLE 2.
Results of codon-based models for detecting selection for binding domain
| Model | la | ϖb | Parametersc |
|---|---|---|---|
| M0 (single ω) | −442.31 | 0.235 | ω = 0.235 |
| M1 (neutral) | −442.53 | 0.273 | ω0 = 0 (p0 = 0.73) |
| ω1 = 1 (p1 = 0.27) | |||
| M2 (selection) | −442.31 | 0.235 | ω0 = 0 (p0 = 0.00) |
| ω1 = 1 (p1 = 0.00) | |||
| ω2 = 0.235 (p2 = 1.00) | |||
| M3 (discrete) | −442.31 | 0.235 | ω0 = 0 (p0 = 0.00) |
| ω1 = 0.008 (p1 = 0.00) | |||
| ω2 = 0.235 (p2 = 1.00) | |||
| M7 (β) | −442.32 | 0.236 | α = 30.54; β = 99 |
| M8 (β + ω) | −442.32 | 0.236 |
p0 = 1.00; α = 30.54; β = 99 |
| p1 = 0.00; ω = 30.55 |
Log-likelihood of data.
Mean dN/dS ratio for entire domain.
pi, the proportion of codons that falls in each category.
The locations of highly conserved amino acid sites in the variable region of Ipmyb1 were determined on the basis of Bayesian posterior probabilities that a given site should be assigned to particular discretized categories that approximate the β-distribution in model M7. In particular, because our analyses indicated that ∼20% of sites have ω = 0, we designated the 20% of sites with the highest posterior probabilities associated with the category ω = 0 as highly conserved sites. To test whether the spatial distribution of these sites was nonrandom, we employed a runs test (Siegel 1956). To test whether negatively charged, acidic amino acids are more likely to be in the set of highly conserved sites than in sites that are not highly conserved, we employed a G-test (Sokal and Rohlf 1969). To categorize each site as either acidic or nonacidic, we reconstructed the site's ancestral amino acid state using parsimony. These reconstructions were unambiguous with respect to categorization of the ancestral state as either acidic or nonacidic, although 19 sites representing insertions were omitted from the analysis.
Because most insertions and deletions in Ipmyb1 are confined to one or a very few closely related sequences (Figure 3), it was straightforward to distinguish insertions from deletions and to unambiguously assign 17 of 18 deletions to a particular lineage. To determine whether deletions tended to avoid conserved sites we simulated sets of random deletions. Specifically, each replicate of the simulation randomly placed 17 deletions on the presumed ancestral state of the gene (all deletions and insertions removed). The size distribution of the deletions was the same as that for the 17 observed deletions. The number of conserved sites, as defined above, included by the deletions was then tabulated. A total of 10,000 replicate simulations were run to provide a frequency distribution of the number of included conserved sites. The criterion for significant avoidance of conserved sites was whether the proportion of replicates with a number of included conserved sites less than or equal to the observed number was <0.05.
Figure 3.—
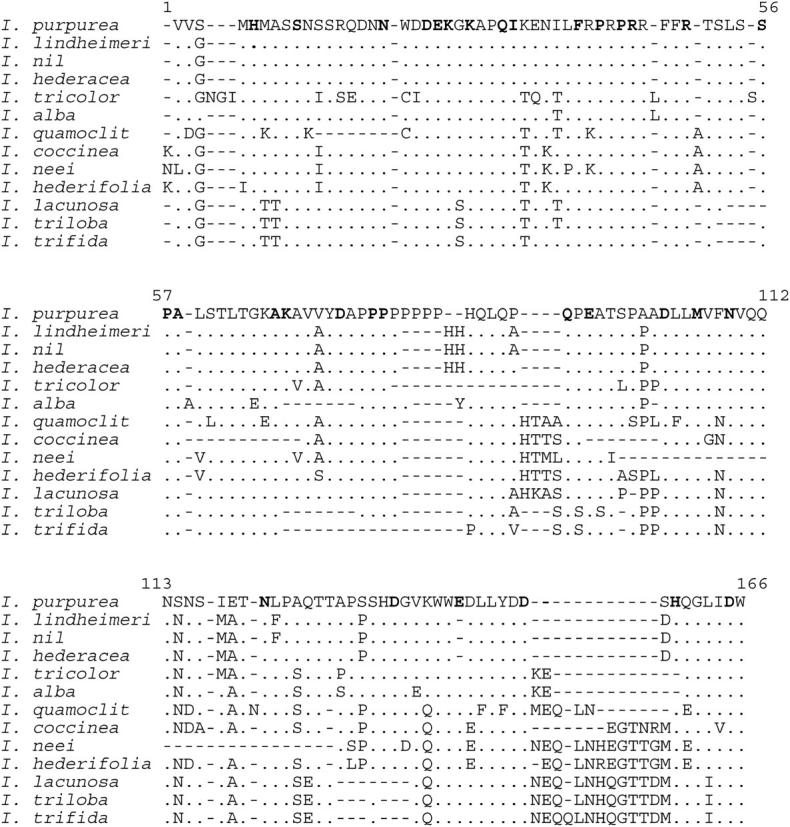
Amino acid alignment of variable-region sequences. Amino acid abbreviations in boldface type in the I. purpurea reference sequence indicate highly conserved sites. Dots indicate amino acids that do not differ from the I. purpurea reference sequence. Dashes indicate insertions/deletions.
Analysis of amino acid substitutions:
To determine whether the nature of amino acid substitutions among the species examined differed from that expected under neutrality, we compared the magnitude of observed changes in physicochemical properties with those expected if amino acid substitutions were not constrained to be substitutions involving similar amino acids. Because most of the amino acid substitutions observed were confined to one species or a small group of related species, it was possible to determine, using parsimony criteria, the ancestral and derived codons for 83 of 87 of the observed amino acid substitutions. Using the ancestral codons, we then simulated substitution under neutrality by allowing each codon to mutate randomly to a new codon specifying a different amino acid. One complete simulation chose a new amino acid corresponding to each of the 87 observed amino acid substitutions. For the four substitutions for which the ancestral codon could not be determined unambiguously, we randomly chose among the possible ancestral codons as the initial codon. For each simulation, we then calculated the mean absolute value of the change in each of three physicochemical properties (composition, polarity, and volume of Grantham 1974) and of the Grantham distance (a combination of change in these three properties; Grantham 1974) associated with each amino acid substitution. One thousand simulations were conducted to determine the expected frequency distribution for mean change for each of the four measures. To ascertain whether the degree of transition/transversion bias influenced the outcomes of the simulations, we conducted 1000 simulations for several values of the transition/transversion ratio: 2.0, 3.0, 3.56 (the empirically estimated value), and 4.0. We asked whether the observed mean of each of these measures differed significantly from the mean expected under neutrality by determining whether the observed mean lay outside the 99% confidence region of the frequency histogram. (We use a 99% confidence region to correct for multiple comparisons and maintain an overall type I error of 5%). A significantly lower mean would indicate that amino acid substitutions are constrained to those involving physicochemically similar amino acids. Alternatively, a significantly higher mean would suggest the repeated operation of positive selection in favoring nonconservative amino acid substitutions.
A nonsignificant deviation in this analysis may reflect either true neutrality of amino acid substitutions or a combination of constraint favoring conservative substitutions at some sites and positive selection favoring more radical substitutions at other sites. We attempted to distinguish between these two possibilities by comparing the variance of the change in each property to the distribution of variances from the simulations. If substitutions are truly neutral, the observed variance should be similar to the mean of the simulated variance distribution. By contrast, under a combination of constraint and positive selection, the variance is expected to be larger than that predicted by the simulations. To determine the significance of the observed variance from the simulation mean, we determined whether the observed mean lay in the upper 1% tail (to maintain an overall probability of type I error of 5%).
Patterns of intraspecific variation:
Patterns of intraspecific variation in the Ipmyb1 gene from I. trifida were compared to expectations under neutrality using standard population-genetic approaches. Deviations of allele frequency spectra from neutrality were assessed using Tajima's D- and Fu and Li's D- and F-statistics (Tajima 1989; Fu and Li 1993) as implemented by the software DnaSP (Rozas and Rozas 1999). Coalescent simulations involving both extremes of free recombination and no recombination were performed to evaluate the significance of these statistics. A McDonald-Kreitman test of nonneutral divergence was performed, also with DnaSP, using a single sequence from I. purpurea for calculating fixed differences between species. This species was used instead of the more closely related I. triloba or I. lacunosa because these two exhibited very little sequence divergence from I. trifida. Comparable analyses with other species used as an outgroup produced very similar results. Finally, to determine whether there is any evidence for positive selection in the form of a recent selective sweep, we compared silent-site variation between Ipmyb1 and CHS-D with an HKA test (Hudson et al. 1987) as implemented in DnaSP. In this test, I. purpurea was again used to assess divergence.
RESULTS
Cloning and characterization of an I. purpurea myb transcription factor:
To clone and characterize the I. purpurea floral myb gene, we utilized a floral color polymorphism common in populations of this species throughout the southeast United States (the W locus; Epperson and Clegg 1988). Homozygotes of one allele at this locus produce pigmented flowers, while homozygotes of the other allele produce white flowers with pigmented rays. In white flowers, six core structural genes of the anthocyanin pathway are greatly downregulated (Tiffin et al. 1998), indicating that this locus likely corresponds to a transcription factor.
Using PCR with degenerate primers, followed by cDNA library screening, we obtained from purple (WW) plants a 1072-bp clone, designated Ipmyb1, including the entire coding region of a gene with similarities to plant R2, R3 myb transcription factors. The deduced amino acid sequence corresponding to this gene has an N-terminal R2, R3 myb domain that is 86% identical at the amino acid level with the An2 anthocyanin regulator of Petunia (Quattrocchio et al. 1999). Moreover, a BLAST search revealed that this region exhibits higher similarity to An2 than to any other gene in the GenBank database, suggesting Ipmyb1 and An2 may be orthologs. Cloning and sequencing from genomic DNA indicated that this gene possesses two introns, the first ∼100 bp in length and the second ∼320 bp (Figure 1). These two introns correspond in position to introns 1 and 2 commonly found in plant R2R3-myb genes (Romero et al. 1998).
Using primers designed from the 5′- and 3′-untranslated portions of the Ipmyb1 sequence, we isolated a second clone, ipmyb1, from white (ww) plants. The sequence of this clone is identical to that of Ipmyb1, except for the presence of two deletions (Figure 1). The larger deletion produces a frameshift and a premature stop codon, such that 95 bp at the 3′ end of the Ipmyb1 gene are radically altered.
All of seven alleles sampled from homozygous purple-flowered plants from North Carolina and Georgia were identical to Ipmyb1, whereas all six alleles sampled from white-flowered plants from these two states were identical to ipmyb1. In addition, segregation analysis of 12 homozygous purple- and 12 white-flowered F2 plants from two crosses between homozygous purple and white parents demonstrated that these alleles cosegregated perfectly with flower color. Similarly, alleles from 36 homozygous purple- and 38 white-flowered progeny derived from self-fertilization of heterozygous parents collected in Georgia also cosegregated perfectly with flower color. These observations indicate that Ipmyb1 corresponds to the functional (pigment-producing) allele at the W locus and that Ipmyb1 is thus the myb transcription factor controlling expression of anthocyanin genes in the flowers of I. purpurea. Although the segregation analysis is consistent with the alternate hypothesis that Ipmyb1 is a separate locus in linkage disequilibrium with W, we believe the geographic sampling argues against this hypothesis, since in such samples past recombination would likely have broken up an association between the two genes.
Interspecific variation in Ipmyb1:
To characterize the molecular evolution of the variable region of Ipmyb1, we cloned and sequenced a 510-bp portion of this region from 13 Ipomoea species (Figure 3). In this sample, a total of 40 sites were variable (28 nonsynonymous, 12 synonymous). For comparison, we also cloned and sequenced the binding domain from 6 of these species (Figure 4). In this sample, there were a total of 10 variable sites (4 nonsynonymous, 6 synonymous). With these sequences, we first asked whether there was evidence for heterogeneity among lineages in ω. For the binding domain, the log-likelihoods for models with global and local ω's were −442.3 and −437.6, respectively, and were not significantly different (d.f. = 8, P > 0.2), providing no reason to reject the null hypothesis that ω was constant across all lineages. Analysis of variable region sequences also revealed no evidence for differences in ω among branches. For the entire set of 13 sequences, the log-likelihoods for the global and local ω-models were −1625.9 and −1618.5, respectively (d.f. = 23, P > 0.3). A similar analysis using subsets consisting of 6 of the 13 sequences, including the subset corresponding to that of the binding-domain analysis, also revealed no indication of heterogeneity among lineages (data not shown). It thus appears that both the binding domain and the variable domain of Ipmyb1 have evolved at an approximately constant rate.
Figure 4.—

Amino acid alignment of R2R3 binding-domain sequences. Dots indicate amino acids that do not differ from the I. purpurea reference sequence.
We next examined whether there was detectable selection on codons within either domain. For the binding domain, models M0–M3, M7, and M8 of PAML all gave virtually identical results. In particular, the log-likelihoods for all models were indistinguishable, and each estimated the average ω to be ∼0.235 (Table 2). Both the selection model (M2) and the discrete-category model (M3) indicated that all codons had a common ω of 0.235, with no codons that were absolutely constrained (ω = 0) or evolving neutrally (ω = 1). Similarly, the estimated parameters of the β-models (M7 and M8) yield a narrow distribution of ω-values centered on 0.235 (Figure 5A), and the discretized approximation to the β-distribution indicates that, for all codons, ω is between 0.177 and 0.299. This analysis indicates that the binding domain is subject to substantial selective constraint, but reveals no evidence for positive selection.
Figure 5.—

Estimated probability density distribution of ω from model M7. The distribution function is a β-distribution with parameters corresponding to the maximum-likelihood fit to the data. (A) Binding domain. (B) Variable domain.
A similar analysis reveals that the distribution of ω-values among codons in the variable region is markedly bimodal, with ∼20–25% of the codons subject to very strong purifying selection (ω ∼ 0) and the remainder evolving essentially neutrally (ω = 1) (Table 3). The neutral model (M1) fits substantially better than the single-ω-model (M0) (Λ = 8.18), despite no difference in degrees of freedom, and estimates that 26% of sites are highly conserved, while 74% are evolving neutrally. Despite additional parameters, neither the selection model (M2) nor the discrete model (M3) shows any improvement of likelihood over the neutral model, providing no evidence for positive selection on any codons. Moreover, in the selection model, the freely estimated ω for the third category of codons, 1.41, is barely larger than the value of 1.0 expected under neutrality and is easily explained by stochastic variation in the estimate of ω. Similarly, in the discrete model, one estimated ω is 0, while the other two are again close to 1.0 and consistent with neutrality. These three models thus are consistent with all codons being either rigidly constrained or neutral.
TABLE 3.
Results of codon-based models for detecting selection for variable domain
| Model | la | ϖb | Parametersc |
|---|---|---|---|
| M0 (single ω) | −1553.47 | 0.803 | ω = 0.803 |
| M1 (neutral) | −1549.38 | 0.737 | ω0 = 0 (p0 = 0.26) |
| ω1 = 1 (p1 = 0.74) | |||
| M2 (selection) | −1549.01 | 0.948 | ω0 = 0 (p0 = 0.28) |
| ω1 = 1 (p1 = 0.39) | |||
| ω2 = 1.40 (p2 = 0.33) | |||
| M3 (discrete) | −1548.91 | 0.852 | ω0 = 0 (p0 = 0.42) |
| ω1 = 1.36 (p1 = 0.37) | |||
| ω2 = 1.36 (p2 = 0.21) | |||
| M7 (β) | −1549.47 | 0.710 | α = 0.04; β = 0.014 |
| M8 (β + ω) | −1549.02 | 0.844 |
p0 = 0.30; α = 0.001; β = 1.83 |
| p1 = 0.70; ω = 1.20 |
Log-likelihood of data.
Mean dN/dS ratio for entire domain.
pi, the proportion of codons that falls in each category.
Models M7 and M8 lead to a very similar conclusion. In M7, the fit β-distribution shows sharp peaks at ω = 0 and ω = 1, with very little probability density at intermediate values (Figure 5B). The estimated discrete categories of this distribution indicate that 30% of codons have ω < 0.10, and the remainder have ω > 0.99. The model M8, which allows for an additional category of sites possibly under selection, shows no improvement in fit over M7 (Λ = 0.90, d.f. = 2, P > 0.5). Moreover, the ω-value associated with this additional category is only 1.20, again very little different from what is expected under neutrality. Thus, models M1–M3 and M7 and M8 are all consistent in indicating absence of positive selection on codons in the variable region and a markedly bimodal distribution of severity of constraint.
One possible explanation for this pattern of constraint is that the variable region contains previously unidentified domains that are highly conserved, are interspersed with sequences that have no function, and are evolving neutrally. When the locations of the conserved codons are plotted, however, this explanation becomes untenable. Thirty-four amino acid sites were identified by their Bayesian posterior probabilities as the most likely conserved sites (Figure 3). These sites are dispersed across the entire variable region and their positions do not differ from random expectation based on a runs test (observed number of runs is 51, expected number of runs is 55, z = −0.98, P > 0.3). Apparently the entire variable region performs important functions, although the identity of every amino acid within that region is not critical.
A second possible explanation is that the variable domain functions primarily in transcription activation. In general, activation domains of eukaryotic transcription factors can often be exchanged without loss of function, indicating that the overall configuration of that domain is not highly functionally constrained (Ptashne 1988). The basic requirement for an activation domain appears to be the presence of negatively charged, acidic amino acids, which are usually dispersed along one face of a secondary structure such as an α-helix (Ptashne 1988). This requirement predicts that there should be a number of highly conserved sites, representing acidic amino acids and sites crucial for their proper orientation, while most other sites should be subject to little constraint. The distribution of acidic amino acids among conserved and nonconserved sites in Ipmyb1 is consistent with this prediction: acidic amino acids are found at 27% of the highly conserved sites (9 of 33), but only at 5.4% of the nonconserved sites (6 of 112; G = 10.5, d.f. = 1, P = 0.0012). Additionally, 87.7% (13 of 15) of sites occupied by acidic amino acids are absolutely conserved, while only 55.1% (65 of 118) of sites occupied by nonacidic amino acids are absolutely conserved (Fisher's exact test, P = 0.016).
Insertions and deletions:
Within the binding domain, there are no insertions or deletions among the sequences examined. By contrast, within the variable domain, indels are numerous (Figure 3), although none cause frameshifts or premature stop codons. Because ∼20% of the amino acids in the variable region are highly conserved, and thus presumably perform a crucial function, we would expect that any deletions that occur in this region would tend to avoid these conserved codons. We tested this hypothesis using simulations of random deletions from the presumed ancestral state of the variable region of Ipmyb1 to ask how frequently the observed number or fewer conserved codons would be deleted by chance. In the genealogy of variable regions from the species examined in this study, 17 deletions include 14 conserved amino acid sites. In 10,000 simulations, the average number of conserved sites deleted was 21.4, and the proportion of runs in which 14 or fewer conserved sites were deleted was 0.047. It thus appears that although deletions do eliminate conserved sites, they do so significantly less often than if deletions were completely random.
Patterns of amino acid substitutions:
The codon-based analysis described above suggests that at sites within the variable region of Ipmyb1 that are not rigidly constrained amino acid substitutions occurred at the same rate as silent substitutions, implying that there are no constraints on nonsynonymous substitutions at these sites. If this inference is correct, then substitutions should be random with respect to physicochemical properties of amino acids. We tested this hypothesis by simulating the distribution of the mean value of changes in amino acid composition, polarity, volume, and a combined measure incorporating all three properties (Grantham distance), for the sites that actually exhibited amino acid substitutions in our data set. Because results from simulations incorporating different transition/transversion ratios are very similar, we report only those simulations in which the ratio was set to the observed value (3.56).
For composition and volume, the observed mean change is slightly greater than the simulation mean (Figure 6). By contrast, the mean changes in polarity and in Grantham distance are slightly lower than the simulation means. None of these differences, however, are statistically significant, since all four observed means fall within the 95% confidence region for the simulated mean (Figure 6). These results are consistent with the hypothesis that the observed amino acid substitutions were effectively neutral and random with respect to physicochemical properties. However, a similar pattern could also be produced if some sites were constrained to permit only conservative substitutions, while other sites were positively selected for more radical substitutions. In this situation, the observed variance of change in a property is expected to be greater than the simulated variance. For composition, volume, and Grantham distance, the observed variance was greater than the mean simulated variance, but none of these differences were statistically significant (P > 0.05 in all three cases). There is thus little reason to reject the null hypothesis that the observed amino acid substitutions were random with respect to physicochemical properties.
Figure 6.—

Frequency histograms of mean change in amino acid properties derived from simulations under the assumption that nonsynonymous mutations are neutral. For each property, 1000 simulations were performed. Arrows indicate mean of observed amino acid substitutions. Bars above histograms indicate the 1st, 5th, 95th, and 99th percentiles of the cumulative frequency distribution.
Intraspecific variation in Ipmyb1:
The interspecific, codon-based analysis of selection described above can be very conservative in its ability to detect positive selection. Specifically, it will not detect positive selection, even if it occurs, if there are no sites at which ω is actually >1, i.e., if advantageous substitutions are scattered across sites rather than occurring repeatedly at a subset of sites. In this situation, patterns of intraspecific variation may be more informative of the operation of selection.
There is no evidence that the distribution of allele frequencies among the I. trifida sample differed from neutral expectations. Considering both synonymous and nonsynonymous polymorphisms, Tajima's D-statistic was estimated to be 0.32, with a confidence interval under neutrality as determined by coalescent simulations of (−1.07, 1.08). Fu and Li's D- and F-statistics were 0.71 and 0.59, with confidence intervals of (−1.45, 1.24) and (−1.60, 1.42), respectively (these confidence intervals were determined assuming free recombination and are smaller than those determined assuming no recombination). Analyses using only segregating nonsynonymous sites or only synonymous sites produced similar nonsignificant results.
To determine the extent to which nonsynonymous substitutions between species could be ascribed to positive selection, we compared the ratio of nonsynonymous to synonymous divergence with the corresponding ratio of polymorphic sites using a standard McDonald-Kreitman test. The outgroup for estimating divergence was a single sequence from I. purpurea. Within I. trifida, there were 15 polymorphic sites, 9 of them segregating for nonsynonymous substitutions and 6 for synonymous substitutions. Between I. trifida and I. purpurea, there were 19 nonsynonymous and 9 synonymous fixed differences. While there appears to be a slight excess of fixed nonsynonymous differences, the difference between the ratios for polymorphism and divergence is not significant (Fisher's exact test, P > 0.73). This analysis thus provides little evidence indicating that nonsynonymous substitutions in the variable region are not neutral.
A final way in which positive selection could be detected is if a recent selective sweep had left a signature of reduced variation in the variable region of Ipmyb1. For this gene, synonymous nucleotide diversity in the I. trifida sample was π = 0.021, while for CHSD, the comparable value was π = 0.067. There thus appears to be less diversity in Ipmyb1, consistent with the recent occurrence of a selective sweep. However, this difference in diversity is not statistically significant, as assessed by a standard HKA test using I. purpurea to assess divergence (χ2 = 0.39, P > 0.50). The result of this analysis is thus consistent with those of the previous analyses, in that they provide no evidence for positive selection acting on the variable region of Ipmyb1.
DISCUSSION
Evolution of the variable domain:
It has frequently been speculated that evolutionary changes in transcription factors contribute substantially to morphological and physiological differences among species because of their control over developmental processes (Doebley and Lukens 1998; Purugganan 1998; Stern 1998; Cubas et al. 1999; Arthur 2002). Consistent with this suggestion, a number of investigations have noted that both plant and animal transcription factors often evolve rapidly compared to structural genes and that this rapid evolution is frequently due to high nonsynonymous substitution rates in certain gene domains (Tucker and Lundrigan 1993; Whitfield et al. 1993; Purugganan and Wessler 1994; Purugganan et al. 1995; de Bono and Hodgkin 1996; Purugganan 1998; Rausher et al. 1999; Barrier et al. 2001; Remington and Purugganan 2002). However, because even in these domains the dN/dS ratio is typically <1, it has been unclear whether these elevated rates of substitution are due primarily to relaxed selective constraint or to an increased frequency of adaptive substitutions. Our results indicate that for one transcription factor, the anthocyanin structural gene activator Ipmyb1, elevated substitution rates are due almost exclusively to relaxed constraint in the variable domain. This relaxed constraint appears to take the form of almost complete neutrality at ∼75–80% of the amino acid sites in this domain. Consistent with the notion that most amino acid substitution in this domain is due to genetic drift, the average ω does not differ detectably across lineages.
Results from five different analyses are consistent in supporting the conclusion that nonsynonymous substitutions observed in the variable region of Ipmyb1 do not differ from neutral expectations and in particular are seldom caused by positive selection: (1) the between-species, codon-based analysis failed to detect selection at any site; (2) analysis of changes in amino acid properties indicated that both the mean and the variance of changes in amino acid composition, polarity, and volume, as well as in Grantham distance, were consistent with expectations under the assumption of random substitution; (3) analysis of the allele-frequency spectrum within I. trifida revealed no indication that polymorphisms within this species were nonneutral; (4) the McDonald-Kreitman analysis detected no positive selection, despite 19 fixed nonsynonymous substitutions between I. trifida and I. purpurea; and (5) the HKA analysis failed to provide evidence that a recent selective sweep had occurred in I. trifida. While each of these analyses separately has its limitations, their collective consistency indicates that very few, if any, of the numerous nonsynonymous substitutions that have occurred during the evolution of the variable region of Ipmyb1 were advantageous. Most appear to have been fixed by genetic drift.
These results argue against the hypothesis that rapid evolution of Ipmyb1 contributes to differentiation of floral hue and color patterning among the Ipomoea species examined. More generally, they argue against the hypothesis that rapid evolution of transcription factors contributes substantially to phenotypic divergence among species (Purugganan 1998, 2000; Remington and Purugganan 2002). This hypothesis also failed to be upheld by Remington and Purugganan (2002), who detected no positive selection on the coding regions of growth-regulating transcription factors in Hawaiian Silverswords. By contrast, analyses of HOX gene evolution in vertebrates have revealed that the non-DNA-binding region of these genes shows both high rates of nonsynonymous substitution and evidence of repeated positive selection (van de Peer et al. 2001; Fares et al. 2003). However, positive selection observed in HOX genes tends to be associated with gene duplication and probably reflects adaptive divergence of function of duplicate copies. By contrast, the rapid evolution examined here and in studies of the evolution of many other transcription factors (Tucker and Lundrigan 1993; Whitfield et al. 1993; Purugganan and Wessler 1994; Purugganan et al. 1995; Barrier et al. 2001; Remington and Purugganan 2002) is within a single ortholog that is believed to have a conserved function in activating anthocyanin genes across angiosperms (Mol et al. 1998). These observations suggest that variable domains of transcription factors may often evolve rapidly primarily because of relaxed constraint in these domains, but that selection may contribute to rapid evolution in the period immediately following gene duplication.
This conclusion naturally leads to the question of whether the amino acid composition of large, contiguous portions of transcription factor variable domains is irrelevant to function, i.e., whether large portions of the domain are not involved in specific interactions between components of the transcription complex. While the binding domain of R2R3-myb anthocyanin transcription factors is believed to function in both DNA sequence recognition and binding to partner myc transcription factors (Goff et al. 1992; Sainz et al. 1997; Williams and Grotewold 1997; Grotewold et al. 2000), little is known about the function of the variable domain, other than that it contains a transcription-activation region (Goff et al. 1991). It is thus possible that large portions of this domain serve only as spacers to link the activation region to the binding domain. In such a situation, one would expect to see blocks of unconserved amino acid sites, corresponding to spacer regions, in the variable domain. Two types of evidence argue against this possibility. First, highly conserved amino acid sites are not clumped, but are scattered throughout the variable region (Figure 3). Second, deletions, when they occur, tend to avoid conserved sites. These patterns suggest that the entire variable region is made up of one or more functional units in close proximity. Although the locations of the conserved sites on the three-dimensional structure of an anthocyanin myb gene might suggest possible functions for this region, such a structure has unfortunately not been determined.
One possibility is that transcriptional activation is the only function of the variable domain. In anthocyanin myb transcription factors, this domain is known to facilitate transcription activation (Goff et al. 1991). In eukaryotes, transcription-activation domains tend not to have specific three-dimensional configurations, but tend to have acidic, negatively charged amino acids dispersed along one edge of α-helices or other secondary structures (Ptashne 1988). Mutational loss of acidic amino acids in these regions tends to reduce activation, and the level of activation seems to be determined primarily by the number of acidic amino acids present (Ptashne 1988). These observations suggest that in activation domains acidic amino acids will be subject to strong purifying selection, while a large proportion of the nonacidic amino acids may be subject to little constraint. Our observations that acidic amino acids are substantially more common at conserved sites than at variable sites, and that a large proportion of sites are evolving neutrally, are consistent with these expectations and thus suggest that much of the variable domain of Ipmyb1 may function primarily in transcriptional activation.
Evolution of the binding domain:
In an examination of the R2R3-myb gene families of rice and of Arabidopsis, Jia et al. (2003)(2004) found evidence of substantial positive selection on sites in the binding domains. By contrast, we detected no evidence of positive selection having occurred in the binding domain of Ipmyb1 in morning glories. Although this difference could be ascribed to a small sample size in our study, and thus low power to detect selection, there is little indication that any sites exhibited a value of ω > ∼0.5. Rather, we suspect the difference between Jia et al.'s and our study is again connected with gene duplication. Jia et al. examined divergence of paralogs and ascribe the positive selection they observed to the evolution of divergent function in different paralogs. This divergence presumably occurred early in the evolutionary history of the divergent lineages. By contrast, we have examined evolutionary change in orthologous genes. During the period of divergence of these orthologs, a common function for the binding domain (activation of anthocyanin structural genes) has been maintained. Consequently, we would not expect to see major functional divergence, and thus substantial positive selection, in this domain, and our observations are in accord with this expectation.
Acknowledgments
We thank Dave Des Marais for useful suggestions. Robert Jarret provided the I. trifida accessions, and Rick Miller provided genomic DNA for four Ipomoea species. Alan Lloyd pointed out the importance of negatively charged, acidic amino acids in eukaryotic transcription-activation domains. This work was supported by National Science Foundation grants MCB 0110596 and DEB-0448889 to M.D.R.
APPENDIX
Data on accessions ofIpomoea trifida used in this study
| Accession no. | Collection area | Specific location |
|---|---|---|
| PI 540715 | Bolivar, Columbia | 10°, 25 min N; 75°, 30 min W |
| PI 540718 | Atlantico, Columbia | 10°, 40 min N; 74°, 58 min W |
| PI 540719 | Atlantico, Columbia | 10°, 58 min N; 74°, 50 min W |
| PI 540730 | Magdalena, CO | 9°, 50 min N; 74°, 50 min W |
| PI 561543 | Venezuela | Carabobo, Valencia, 10 km SE Valencia-Tocuyito |
| PI 561544 | Venezuela | Aragua, Maracay, La Morita-Maracay |
| PI 543815 | Costa Rica | 3 km E of entrance to Playa Ocotal |
| PI 543818 | Costa Rica | 8.7 km from Playa Naranjo near Santa Rosa National Park |
| PI 543819 | Costa Rica | 0.5 km from Laguna Escondida, near Santa Rosa National Park |
| PI 543828 | Canas, Costa Rica | |
| PI 561547 | Escuintla, Guatemala | 5 km SW Escuintla-Siquinala |
| PI 561548 | Escuintla, Guatemala | 15 km NE Escuintla-Siquinala |
| PI 618966 | Michoacan, Mexico | 19°, 21 min N; 102°, 15 min W |
| GRIF 6198 | Chiapas, Mexico | Villaflorea; 16°, 30 min N; 93°, 10 min W |
| GRIF 6199 | Oaxaca, Mexico | 3 km NW Pinotepa Nac-Acapulco; 16°, 21 min N; 98°, 16 min W |
| GRIF 6200 | Guerrero, Mexico | 10 km NW Poluta-Zorcua; 17°, 56 min N; 101°, 38 min W |
Accession numbers are as listed in the GRIN database of the National Plant Germplasm System maintained by the U.S. Department of Agriculture. Two plants were sampled from each accession except PI543819, from which only one plant was sampled.
Sequence data from this article have been deposited with the EMBL/GenBank Data Libraries under accession nos. AY986823, AY986824, AY986825, AY986826, AY986827, AY986828, AY986829, AY986830, AY986831, AY986832, AY986833, AY986834, AY986835, AY986836, AY986837, AY986838, AY986839, AY986840, AY986841, AY986842, AY986843, AY986844, AY986845, AY986846, AY986847, AY986848, AY986849, AY986850, AY986851, AY986852, AY986853, AY986854, AY986855, AY986856, AY986857, AY986858, AY986859, AY986860, AY986861, AY986862, AY986863, AY986864, AY986865, AY986866, AY986867, AY986868, AY986869, AY986870, AY986871, AY986872, AY986873, AY986874, AY986875, AY986876, AY986877, AY986878, AY986879, AY986880, AY986881, AY986882, AY986883, AY986884, AY986885, AY986886, AY986887, AY986888, AY986889, AY986890, AY986891, AY986892, AY986893, AY986894, AY986895, AY986896, AY986897, AY986898.
References
- Arthur, W., 2002. The emerging conceptual framework of evolutionary developmental biology. Nature 415: 757–764. [DOI] [PubMed] [Google Scholar]
- Barrier, M., R. H. Robichaux and M. D. Purugganan, 2001. Accelerated regulatory gene evolution in an adaptive radiation. Proc. Natl. Acad. Sci. USA 98: 10208–10213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bono, M., and J. Hodgkin, 1996. Evolution of sex determination in Caenorhabditis: unusually high divergence of tra-1 and its functional consequences. Genetics 144: 587–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry, L. M., S. M. Case and A. C. Wilson, 1978. Frog perspective on the morphological difference between humans and chimpanzees. Science 200: 209–211. [DOI] [PubMed] [Google Scholar]
- Cubas, P., C. Vincent and E. Coen, 1999. An epigenetic mutation responsible for natural variation in floral symmetry. Nature 401: 157–161. [DOI] [PubMed] [Google Scholar]
- Doebley, J., 1993. Genetics, development and plant evolution. Curr. Opin. Genet. Dev. 3: 865–872. [DOI] [PubMed] [Google Scholar]
- Doebley, J., and L. Lukens, 1998. Transcriptional regulators and the evolution of plant form. Plant Cell 10: 1075–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin, M. L., K. E. Lundy, P. L. Morrell, C. L. Torres-Martinez and M. T. Clegg, 2003. Genes that determine flower color: the role of regulatory changes in the evolution of phenotypic adaptations. Mol. Phylogenet. Evol. 29: 507–518. [DOI] [PubMed] [Google Scholar]
- Epperson, B. K., and M. T. Clegg, 1988. Genetics of flower color polymorphism in the common morning glory (Ipomoea purpurea). J. Hered. 79: 64–68. [DOI] [PubMed] [Google Scholar]
- Fares, M. A., D. Bezemer, A. Moya and I. Marin, 2003. Selection on coding regions determined Hox7 genes evolution. Mol. Biol. Evol. 20: 2104–2112. [DOI] [PubMed] [Google Scholar]
- Fu, Y.-X., and W.-H. Li, 1993. Statistical tests of neutrality of mutations. Genetics 133: 693–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukada-Tanaka, S., A. Hoshino, Y. Hisatomi, Y. Habu, M. Hasebe et al., 1997. Identification of new chalcone synthase genes for flower pigmentation in the Japanese and common morning glories. Plant Cell Physiol. 38: 88–96. [DOI] [PubMed] [Google Scholar]
- Goff, S. A., K. C. Cone and M. E. Fromm, 1991. Identification of functional domains in the maize transcriptional activator C1: comparison of wild-type and dominant inhibitor proteins. Genes Dev. 5: 298–309. [DOI] [PubMed] [Google Scholar]
- Goff, S. A., K. C. Cone and V. L. Chandler, 1992. Functional analysis of the transcription activator encoded by the maize B-gene: evidence for a direct functional interaction between two classes of regulatory proteins. Genes Dev. 6: 864–875. [DOI] [PubMed] [Google Scholar]
- Grantham, R., 1974. Amino acid difference formula to help explain protein evolution. Science 185: 862–864. [DOI] [PubMed] [Google Scholar]
- Grotewold, E., M. B. Sainz, L. Tagliani, J. M. Hernandez, B. Bowen et al., 2000. Identification of the residues in the myb domain of maize C1 that specify the interaction with the bHLH cofactor R. Proc. Natl. Acad. Sci. USA 97: 13579–13584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocking, D. R., 1985 The Analysis of Linear Models. Brooks/Cole, Monterey, CA.
- Hudson, R. R., M. Kreitman and M. Aguade, 1987. A test of neutral molecular evolution based on nucleotide data. Genetics 116: 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irani, N. G., J. M. Hernandez and E. Grotewold, 2003. Regulation of anthocyanin pigmentation. Rec. Adv. Phytochem. 37: 59–78. [Google Scholar]
- Jia, L., M. T. Clegg and T. Jiang, 2003. Excess nonsynonymous substitutions suggest that positive selection episodes operated in the DNA-binding domain evolution of Arabidopsis R2R3-MYB genes. Plant Mol. Biol. 52: 627–642. [DOI] [PubMed] [Google Scholar]
- Jia, L., M. T. Clegg and T. Jiang, 2004. Evolutionary dynamics of the DNA-binding domains in putative R2R3-myb genes identified from rice subspecies indica and japonica genomes. Plant Physiol. 134: 575–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King, M. C., and A. C. Wilson, 1975. Evolution at two levels in humans and chimpanzees. Science 188: 107–116. [DOI] [PubMed] [Google Scholar]
- Koes, R. E., F. Quattrocchio and J. N. M. Mol, 1994. The flavonoid biosynthetic pathway in plants: function and evolution. BioEssays 16: 123–132. [Google Scholar]
- Kroon, A. R., 2004 Transcription regulation of the anthocyanin pathway in Petunia hybrida. Ph.D. Thesis, Vrije Universiteit, Amsterdam.
- Manos, P. S., R. E. Miller and P. Wilkin, 2001. Phylogenetic analysis of Ipomoea, Argyreia, Stictocardia, and Turbina suggests a generalized model of morphological evolution in morning glories. Syst. Bot. 26: 585–602. [Google Scholar]
- Miller, R. E., M. D. Rausher and P. S. Manos, 1999. Phylogenetic systematics of Ipomoea (Convolvulaceae) based on ITS and waxy sequences. Syst. Bot. 24: 209–227. [Google Scholar]
- Mol, J., E. Grotewold and R. Koes, 1998. How genes paint flowers and seeds. Trends Plant Sci. 3: 212–217. [Google Scholar]
- Nielsen, R., and Z. Yang, 1998. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148: 929–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptashne, M., 1988. How eukaryotic transcriptional activators work. Nature 335: 683–689. [DOI] [PubMed] [Google Scholar]
- Purugganan, M. D., 1998. The molecular evolution of development. BioEssays 20: 700–711. [DOI] [PubMed] [Google Scholar]
- Purugganan, M. D., 2000. The molecular population genetics of regulatory genes. Mol. Ecol. 9: 1451–1461. [DOI] [PubMed] [Google Scholar]
- Purugganan, M. D., and S. R. Wessler, 1994. Molecular evolution of the plant R regulatory gene family. Genetics 138: 849–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purugganan, M. D., S. D. Rounsley, R. J. Schmidt and M. F. Yanofsky, 1995. Molecular evolution of flower development: diversification of the plant MADS-box regulatory gene family. Genetics 140: 345–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quattrocchio, F., J. Wing, K. van der Woude, E. Souer, N. de Vetten et al., 1999. Molecular analysis of the anthocyanin2 gene of Petunia and its role in the evolution of flower color. Plant Cell 11: 1433–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausher, M. D., R. E. Miller and P. Tiffin, 1999. Patterns of evolutionary rate variation among genes of the anthocyanin biosynthetic pathway. Mol. Biol. Evol. 16: 266–274. [DOI] [PubMed] [Google Scholar]
- Remington, D. L., and M. D. Purugganan, 2002. GAI homologues in the Hawaiian Silversword alliance (Asteraceae-Madiinae): molecular evolution of growth regulators in a rapidly diversifying plant lineage. Mol. Biol. Evol. 19: 1563–1574. [DOI] [PubMed] [Google Scholar]
- Romero, I., A. Fuertes, M. J. Benito, J. M. Malpica, A. Leyva et al., 1998. More than 80 R2R3-MYB regulatory genes in the genome of Arabidopsis thaliana. Plant J. 14: 273–284. [DOI] [PubMed] [Google Scholar]
- Rozas, J., and R. Rozas, 1999. DnaSP version 3: an integrated program for molecular population genetics and molecular evolution analysis. Bioinformatics 15: 174–175. [DOI] [PubMed] [Google Scholar]
- Sainz, M. B., S. A. Goff and V. L. Chandler, 1997. Extensive mutagenesis of a transcriptional activation domain identifies single hydrophobic and acidic amino acids important for activation in vivo. Mol. Cell. Biol. 17: 115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel, S., 1956 Nonparametric Statistics for the Behavioral Sciences. McGraw-Hill, New York.
- Sokal, R. R., and F. J. Rohlf, 1969 Biometry. W. H. Freeman, San Francisco.
- Stern, D., 1998. A role of ultrabithorax in morphological differences between Drosophila species. Nature 396: 463–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiffin, P., R. E. Miller and M. D. Rausher, 1998. Control of expression patterns of anthocyanin structural genes by two loci in the common morning glory. Genes Genet. Syst. 73: 105–110. [Google Scholar]
- Tucker, P. K., and B. Lundrigan, 1993. Rapid evolution of the sex-determinig loci in Old World mice and rats. Nature 364: 715–717. [DOI] [PubMed] [Google Scholar]
- Van De Peer, Y., J. S. Taylor, I. Braasch and A. Meyer, 2001. The ghost of selection past: rates of evolution and functional divergence of anciently duplicated genes. J. Mol. Evol. 53: 436–446. [DOI] [PubMed] [Google Scholar]
- Whitfield, L., R. Loveilbadge and P. Goodfellow, 1993. Rapid sequence evolution of the sex-determining gene SRY. Nature 364: 713–715. [DOI] [PubMed] [Google Scholar]
- Williams, C. E., and E. Grotewold, 1997. Differences between plant and animal Myb domains are fundamental for DNA binding activity and chimeric Myb domains have novel DNA-binding specifities. J. Biol. Chem. 272: 563–571. [DOI] [PubMed] [Google Scholar]
- Wray, G. A., M. W. Hahn, E. Abouheif, J. P. Balhoff, M. Pizer et al., 2003. The evolution of transcriptional regulation in eukaryotes. Mol. Biol. Evol. 20: 1377–1419. [DOI] [PubMed] [Google Scholar]
- Yang, Z., 1997. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13: 555–556. [DOI] [PubMed] [Google Scholar]
- Yang, Z., and R. Nielsen, 2000. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 17: 32–43. [DOI] [PubMed] [Google Scholar]
- Yang, Z., R. Nielsen, N. Goldman and A. M. Pedersen, 2000. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155: 431–449. [DOI] [PMC free article] [PubMed] [Google Scholar]