

Abstract
Human replication protein A (RPA), composed of RPA70, RPA32, and RPA14 subunits, undergoes hyperphosphorylation in cells in response to DNA damage. Hyperphosphorylation that occurs predominately in the N-terminal region of RPA32 is believed to play a role in modulating the cellular activities of RPA essential for almost all DNA metabolic pathways. To understand how the hyperphosphorylation modulates the functions of RPA, we compared the structural characteristics of full-length native and hyperphosphorylated RPAs using mass spectrometric protein footprinting, fluorescence spectroscopy, and limited proteolysis. Our mass spectrometric data showed that of 24 lysines and 18 arginines readily susceptible to small chemical reagent modification in native RPA, the three residues Lys-343, Arg-335, and Arg-382, located in DNA binding domain B (DBD-B) of RPA70, were significantly shielded in the hyperphosphorylated protein. Tryptophan fluorescence studies indicated significant quenching of Trp-361, located in the DBD-B domain, induced by hyperphosphorylation of RPA. Consistently, DBD-B became more resistant to the limited proteolysis by chymotrypsin after RPA hyperphosphorylation. Taken together, our results indicate that upon hyperphosphorylation of RPA32 N terminus (RPA32N), RPA undergoes a conformational change involving the single-stranded DNA binding cleft of DBD-B. Comparison of the interactions of native and hyperphosphorylated RPAs with short single-stranded oligonucleotides or partial DNA duplexes with a short 5' or 3' single-stranded DNA tails showed reduced affinity for the latter protein. We propose that the hyperphosphorylation may play a role in modulating the cellular path-ways by altering the DBD-B-mediated RPA-DNA and RPA-protein interactions, hypothetically via the interaction of hyperphosphorylated RPA32N with DBD-B.
Replication protein A (RPA)2 is a eukaryotic single-stranded DNA (ssDNA)-binding protein essential for DNA replication, repair, recombination (1-3), and cellular DNA-damage checkpoints (4, 5). RPA is composed of three subunits, RPA70, RPA32, and RPA14, named after their molecular masses of 70, 32, and 14 kDa, respectively. Although the structure of full-length RPA trimer remains unsolved, x-ray crystallography, NMR, and biochemical studies reveal that RPA70 contains four domains, the N-terminal domain of RPA70 and DNA binding domains (DBDs) A, B, and C (6-8). The tandem DBD-A and DBD-B harbor the major ssDNA binding activity of RPA heterotrimer (9). DBD-C has a conserved zinc finger (10) and exhibits low affinity for ssDNA but interacts with the other two subunits to form a RPA trimerization core (11). RPA32 consists of three domains, including an unstructured N-terminal phosphorylation domain (RPA32N), a central DNA binding domain (DBD-D), and a C-terminal domain (RPA32C) largely involved in protein-protein interactions (12-14). RPA14 is referred to as DBD-E. All the DBDs in RPA have similar structures built around a central oligosaccharide/oligonucleotide binding fold (6, 7, 11, 12, 15). However, the N-terminal domain of RPA70 (also referred to as DBD-F or RPA70N) has only been implicated in protein-protein interactions in DNA metabolism (1-3, 6). In contrast, DBD-E (RPA14) shows no binding affinity for ssDNA but is structurally important for the formation of RPA trimerization core (11, 12). Slight differences in structure and residue compositions of the binding clefts of the DBDs may explain their differences in ssDNA binding (16, 17).
It has been reported that the N-terminal domain of RPA32 (RPA32N) becomes phosphorylated in a cell cycle-dependent manner (18-20) and hyperphosphorylated in response to DNA damage (21-24). The cell cycle-dependent phosphorylation is carried out by cyclin-dependent kinases and occurs at two consensus sites, Ser-23 and Ser-29, of RPA32 (19, 22, 25). More sites in RPA32N are phosphorylated in cells in response to DNA damage (22, 23, 26) or apoptosis (27). Phosphopeptide mapping has shown that at least five sites of RPA32N, including Thr-21, Ser-23, Ser-29, Ser-33, and either Ser-11, -12, or -13 and probably two additional sites, Ser-4 and Ser-8, can be phosphorylated in UV-irradiated HeLa cells (22, 25, 28). The DNA damage-induced hyperphosphorylation of RPA32 is believed to be carried out by the members of phosphatidylinositol 3-kinase-like serine/threonine protein kinase family, which includes DNA-dependent protein kinase (DNA-PK), ataxia-telangiectasia mutated kinase (ATM), and ATM- and Rad3-related kinase (ATR) (2, 4, 23, 24, 29-31). DNA-PK phosphorylates RPA32 in vitro at many of the same sites that are phosphorylated in vivo in HeLa cells after UV irradiation (22, 24, 25).
One of the most striking aspects of RPA is that the protein is involved in almost all DNA metabolic pathways in cells. It is believed that such broad cellular activities of RPA are mediated by its interactions with ssDNA and numerous proteins engaged in cellular processes (9, 14). RPA phosphorylation may play a role in regulation of these interactions and, thus, the cellular functions of RPA (24). It has been shown that RPA hyperphosphorylation down-regulates DNA replication (24, 32). In particular, hyperphosphorylation of RPA32 may modulate RPA interactions with DNA and proteins involved in the DNA repair and signaling pathways in response to DNA damage (2, 24). For instance, hyperphosphorylated RPA (hyp-RPA) has shown decreased interactions with simian virus 40 (SV40) T antigen, DNA polymerase α, DNA-PK, ATM, and p53, whereas hyperphosphorylation has no effects on RPA interactions with XPA, Rad51, and Rad52 (20, 24, 33-37). In addition, hyp-RPA binds double-stranded DNA with a reduced affinity (20, 38), whereas the effects of hyperphosphorylation on RPA interaction with ssDNA remain controversial (20, 38, 39). Despite the significant role of hyperphosphorylation of RPA in modulating its cellular functions, the bio-chemical basis of the effects is still poorly understood. A possible scenario is that RPA may change its structure or conformation upon hyperphosphorylation and, thus, alter the protein activities and functions. A synthetic peptide fragment with eight Ser/Thr residues substituted to Asp to mimic the hyperphosphorylated RPA32N was shown to interact with DBD-F (RPA70N) fragment (RPA701-168) (38). However, the structural characteristics of the hyperphosphorylated RPA trimer of full-length remain to be elucidated.
In the present study we determined the hyperphosphorylation-dependent structural alterations of human RPA protein using mass spectrometric protein footprinting, limited proteolysis, and tryptophan fluorescence spectroscopy. Of 42 basic residues readily susceptible to modification with small chemical reagents in the native protein, the three amino acids Lys-343, Arg-335, and Arg-382 in DBD-B of RPA70 were shielded in the context of the hyperphosphorylated RPA. Our results indicate that these residues were exclusively involved in a limited structural alteration of the protein upon hyperphosphorylation of the RPA32 N terminus. In particular, this local structural change showed effects on the RPA interactions with 8-mer ssDNA or partial DNA duplexes containing ssDNA tails. Considering the essential role of RPA in most of cellular DNA metabolic processes including those mediated with short ssDNA-associated partial DNA duplexes, our results provide the biochemical basis for the roles of hyperphosphorylation in regulation of RPA functions.
EXPERIMENTAL PROCEDURES
Protein Production and Purification—Recombinant human RPA was expressed in Escherichia coli BL21(DE3)-RP cells harboring the plasmid pTYB-RPA and purified as previously described (40). Two RPA mutants, Trp-107/528 and Trp-212/361, which had all the tryptophans of RPA replaced by phenylalanine except for residues 107 and 528 or residues 212 and 361, were expressed from BL21(DE3)-RP cells containing the plasmids constructed by site-directed mutagenesis. The mutant proteins were purified by the same procedures as for native RPA.
In Vitro Phosphorylation by DNA-PK—For hyperphosphorylation, the purified RPA or mutants were incubated with DNA-PK isolated from HeLa nuclear extracts as a complex consisting of a 400-kDa catalytic subunit and the DNA binding component of 85- and 70-kDa Ku subunits (Promega) at 30 °C for 30 min in the phosphorylation buffer (40 mM HEPES, pH 7.5, 10 mM MgCl2,1mM DTT, 200 μM ATP, and 10 μg/ml calf thymus DNA). A mock treatment of proteins was carried out in parallel under the exact same conditions except that no ATP was added in the phosphorylation buffer. The reaction mixtures were then loaded onto an HR10/30 Superdex 200 column equipped with an AKTA purifier system (Amersham Biosciences). To purify the hyperphosphorylated RPA, the column was pre-equilibrated with the fast protein liquid chromatography running buffer containing high concentrations of salt (40 mM HEPES, pH 7.5, 2 M NaCl, 10 m>M MgCl2,10 μM ZnCl2, and 1mM DTT) and run with the same buffer at 4 °C. The fractions containing hyperphosphorylated proteins or mock-treated proteins were pooled and dialyzed against RPA storage buffer (40 mM HEPES, pH 7.5, 50 mM NaCl, 10 mML> MgCl2,10 μM ZnCl2,1mM DTT, and 50% glycerol). Protein concentration was determined using Bio-Rad protein assay kit.
Chemical Modification and In-gel Proteolysis—Biotinylation of lysine residues of RPA was carried out as described previously (41). Briefly, 2 μM hyp-RPA or RPA was incubated with N-hydroxysuccinimidobiotin (NHS-biotin, Pierce) in 50 mM HEPES, pH 7.5, 50 mM NaCl at 25 °C for 30 min. The reactions were then quenched by the addition of 10 mM lysine. The three subunits of RPA were separated by SDS-PAGE and visualized by Coomassie Blue stain. The corresponding bands were excised, and the gel slices were destained, dehydrated, and digested with 1 μg of trypsin (Roche Applied Science) in 50 mM NH4HCO3 at 25 °C overnight. Proteolytic peptides were recovered and subjected to MS and MS/MS analysis.
Modification of arginine residues was performed by incubating 2 μM hyp-RPA or RPA with p-hydroxyphenylglyoxal (HPG, Pierce) in 50 mM HEPES/50 mM boric acid, pH 8.0, at 37 °C in the dark for 60 min. The modification was quenched by the addition of 100 mM arginine. Then samples were subjected to SDS-PAGE, and protein bands were excised and processed as described above.
MS and MS/MS Analysis—MS spectra were obtained using matrix-assisted laser desorption time of flight (MALDI-TOF) and quadrupole-time of flight (Q-TOF) techniques. MALDI-TOF experiments were performed using a Kratos Axima-CRF instrument (Kratos Analytical Instruments) with α-cyano-4-hydroxycinnamic acid matrix. MS and MS/MS analyses were performed on a Micromass (Manchester, UK) Q-TOF-II instrument equipped with an electrospray source and Micro-mass cap-LC. Peptides were separated with a Waters Symmetry300 5-μm precolumn (Waters, Milford, MA) and a Micro-Tech Scientific (Vista, CA) ZC-10-C18SBWX-150 column using two sequential gradients of 5-40% acetonitrile for 35 min and 40-90% acetonitrile for 20 min. MS/MS sequence data and the MASCOT automated peptide search engine (http://www.matrixscience.com) were used to identify RPA peptide peaks from the NCBInr primary sequence data base, and matched peaks were then located in the primary MS spectra. Protection events were qualitatively assigned as the appearance of a peak corresponding to a modified peptide in the modified protein spectrum and the absence of the modification peak in the modified hyperphosphorylated protein spectrum. A protection was considered to be significant only when the intensity of a modifiable peak was reduced by at least 85% in the hyp-RPA spectrum. To accurately identify protection events, at least two unmodified proteolytic peptide peaks present in all four spectra (unmodified protein, modified protein, unmodified hyp-protein, and modified hyp-protein) were used as controls. These control peaks served to standardize the peak intensities in each spectrum for accurate qualitative assignment of protection. Data were reproducibly compiled and analyzed from six independent experimental groups.
Fluorescence Spectroscopy Determination—The tryptophan fluorescence spectra of RPA, its mutants, and their hyperphosphorylated forms were recorded on a SPEX Fluorolog-3 fluorometer (Jobin Yvon, Inc.) with the excitation wavelength at 295 nm and the slit widths set at 3 nm for excitation and 5 nm for emission beams. After equilibration of 100 nM protein in RPA buffer (40 mM HEPES-KOH, pH 7.9, 75 mM KCl, 8 mM MgCl2,1mM DTT, and 5% glycerol) at 25 °C for 10 min, the fluorescence spectra were obtained by recording emission from 310 to 500 nm. For the constant wavelength analysis, the emission at 355 nm was measured with excitation at 295 nm.
Fluorescence anisotropy measurements were performed to compare the binding of RPA and hyp-RPA to an oligo(dT)8. The (dT)8 with a fluorescein labeled at 5' end was purchased from Qiagen in high performance liquid chromatography-purified quality. The anisotropy titrations were performed as described previously (42), and the data were processed using a one-site binding model and the non-linear least squares method for the determination of the binding constants (43).
Partial Proteolysis and Identification of Proteolytic Fragments—4 μg of recombinant human RPA or hyp-RPA was incubated with chymotrypsin (1:80) at room temperature for the indicated periods in a reaction buffer (10 μl) containing 40 mM HEPES, pH 7.8, 1 mM EDTA, 70 mM MgCl2, and 10 mM DTT. At each time point (5, 20, or 60 min), 2.5 μl of the reaction mixture was removed and terminated by the addition of Laemmli sample buffer and boiling for 10 min. The proteolytic products were resolved by 14% SDS-PAGE and stained with SYPRO Ruby protein gel stain (Bio-Rad). After destained with a solution containing 10% methanol and 7% acetic acid, the gel was imaged by phosphorimaging (FLA-5000, FUJIFILM) using a 473-nm laser line. For automated N-terminal sequencing, the proteolytic fragments separated on the 14% SDS-PAGE were transferred to a polyvinylidene difluoride membrane followed by staining with Coomassie Brilliant Blue G-250. The protein bands of interest were excised and sent to the protein chemistry laboratory at the University of Texas Medical Branch for sequencing.
Gel Mobility Shift Assays—ssDNA was radiolabeled with [γ-32P]ATP and T4 polynucleotide kinase. The labeled substrate (1 nM) was then incubated with the indicated amounts of hyp-RPA or RPA at room temperature for 15 min in 20 μl of the binding buffer (40 mM HEPES-KOH, pH 7.9, 75 mM KCl, 8 mM MgCl2,1mM DTT, 5% glycerol, and 100 μg/ml bovine serum albumin). After incubation, 2 μl of 80% (v/v) glycerol was added, and the mixture was immediately loaded onto a 4% native polyacrylamide gel in 1× TBE running buffer (89 mM Tris borate and 2 mM EDTA, pH 8.3) and electrophoresed at room temperature. The gel was then dried. The free and bound DNA was visualized using a PhosphorImager (FLA-5000, FUJIFILM).
Pull-down Assays—The binding of RPA to partial DNA duplexes containing 5'-protruding 11 nucleotides (DNA-11) or 3'-protruding 10 nucleotides (DNA-10) were investigated by a streptavidin-agarose pull-down assay. The sequences of the oligonucleotides used to construct the partial DNA duplexes are as follows: 55-mer, Biotin-GGACCTGAACACGTACGGAATTCGATATCCTCGAGCCAGATCTGCGCC AGCTGGC; 44-mer, GCCAGCTGGCGCAGATCTGGCTCGAGGATATCGAATTCCG TACG; 45-mer, GCAGATCTGGCTCGAGGATATCGAATTCCGTACGTGTTCAGGT CC. The 44- and 45-mer were 5'-32P-labeled and annealed to their complementary biotinylated 55-mer at a molar ratio of 1:1. The annealing mixture was then loaded onto an 8% native polyacrylamide gel in 1× TBE running buffer and electrophoresed at 80 V for 3 h at room temperature. Using the 5'-32P-labeled 44-, 45-, and 55-mer as controls, the bands identified as the partial DNA duplexes, which migrated slower than the single-stranded oligonucleotides in the gel, were excised, eluted, and precipitated with ethanol. After the purification 5 pmol of the partial DNA duplex were incubated with the indicated amounts of mixed hyp-RPA and RPA in 500 μl of the binding buffer at room temperature for 15 min. Then 20 μl of streptavidin-conjugated agarose beads (Invitrogen) pre-washed with the binding buffer was added. The mixture was placed on a rotating shaker with gentle mixing at room temperature for 30 min. The beads were collected by centrifugation and washed 3 times with the binding buffer. The proteins in the complex were assessed by Western blotting using an antibody specific for RPA32 as described below.
Western Blotting—Protein samples were separated by 10% SDS-PAGE and transferred to a polyvinylidene difluoride membrane in Tris-glycine buffer (0.375 M Tris, 0.192 M glycine, 20% methanol). The membrane was blocked with 5% nonfat milk at room temperature for 1 h and then treated with a monoclonal anti-RPA32 antibody (Kamiya Biomedical) at 0.5 μg/ml in phosphate-buffered saline supplemented with 5% nonfat milk at 4 °C overnight with shaking. The RPA32 protein bands were visualized using anti-mouse IgG conjugated with horseradish peroxidase as the secondary antibody (Santa Cruz) by following the protocol of the ECL Western blotting System (Amersham Biosciences).
RESULTS
In Vitro Hyperphosphorylation of RPA by DNA-PK—It has been well established that DNA-PK is involved in the DNA damage-induced RPA hyperphosphorylation in cells (23, 29-31, 39, 44-46). DNA-PK is composed of the Ku70/80 heterodimer and the catalytic subunit DNA-PKcs (47). The sites of RPA phosphorylated by DNA-PK in vitro are similar to those phosphorylated in vivo after UV irradiation of human cells (22, 25). Although RPA may also be hyperphosphorylated by other phosphatidylinositol 3-kinase-like serine/threonine protein kinase family members such as ATR and ATM in response to DNA damage in vivo, hyperphosphorylation of RPA by DNA-PK represents a typical one for study of the DNA damage-induced RPA hyperphosphorylation. To investigate the changes of RPA structure and activity upon hyperphosphorylation, we prepared the hyperphosphorylated RPA by incubating purified recombinant RPA with DNA-PK in the presence of calf thymus DNA (Promega). After a hyperphosphorylation reaction, the RPA (116 kDa) was separated from DNA-PK (three subunits: 400, 80, and 85 kDa) and DNA by gel filtration with the running buffer containing 2 M NaCl followed by dialysis against RPA storage buffer. A high salt concentration was used to dissociate RPA from the kinase and DNA, whereas the RPA remained in the form of trimer (Fig. 1A). The SDS-PAGE and Western blotting analyses of the RPA hyperphosphorylation showed that more than 85% of RPA32 was hyperphosphorylated by DNA-PK, as indicated by the band shifted from the native RPA32 (Fig. 1, A, lane 3,and B, lane 1). And this phosphorylation was fully reversed after the treatment of the hyp-RPA with calf intestinal phosphatase as the retardation was diminished (Fig. 1A, lane 4). No phosphorylation by DNA-PK was observed in RPA70 and RPA14 subunits, as confirmed by the ents with [γ-32P]ATP (data not shown), which is also in agreement with that of a previous report (48). It has been generally known that in vivo phosphorylation results in five isoforms of RPA, which represent different levels of phosphorylation as indicated by the mobility shifts of RPA32 on SDS-PAGE (20-22, 49) (Fig. 1B, lane 3). Of them, forms 2-3 occur in a cell cycle-dependent manner, whereas forms 4-5 are the hyperphosphorylated RPA32 and appear upon cellular DNA damage. Comparison of RPAs phosphorylated in vitro and in vivo by Western blotting revealed that most of the DNA-PK-hyperphosphorylated RPA had the same mobility shifts as did forms 4 and 5 of RPA from UV-irradiated HeLa cells.
FIGURE 1.
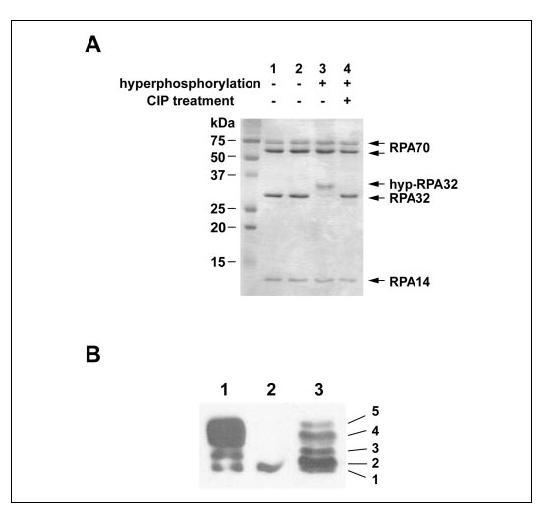
In vitro phosphorylation of RPA by DNA-PK. A, the phosphorylation reaction was conducted as described under “Experimental Procedures.” Proteins including untreated RPA (lane 1), mock-treated RPA (lane 2), hyp-RPA (lane 3), and calf intestinal phosphatase (CIP)-treated hyp-RPA (lane 4) were visualized by 14% SDS-PAGE and Coomassie Blue staining. B, immunoblot of RPA hyperphosphorylated by DNA-PK (lane 1), RPA (lane 2), and RPA in UV-irradiated HeLa cells (lane 3). To prepare the RPA in UV-irradiated HeLa cells, whole cell lysates were prepared from HeLa cells at 8 h after irradiation with 20 J/m2 UV. Proteins were separated on a 10% SDS-PAGE followed by Western blotting using an antibody specific for RPA32.
Surface Topology Analysis of Native and Hyperphosphorylated RPAs with Group-specific Reagents and Mass Spectrometry—Chemical modifications coupled with mass spectrometry have been used to probe the surface topology of proteins (50-52). Recently, a strategy of protein footprinting has been developed by Kvaratskhelia et al. (53) for accurate mapping of protein-nucleic acid interactions using mass spectrometry. Here we extend this method to probe conformational changes of RPA after hyperphosphorylation by monitoring the changes of surface accessibility of lysine and arginine residues susceptible to NHS-biotin and hydroxyphenylglyoxal (HPG) modifications, respectively. Comparison of surface topologies of native and hyperphosphorylated RPAs would enable us to identify conformational changes in the full-length protein induced by hyperphosphorylation.
Of note, maintenance of the structural integrity of proteins under the modification conditions is crucial for the success of these experiments. Therefore, we optimized concentrations of NHS-biotin and HPG. Our recent work indicated that upon treatment of the RPA-ssDNA complex with 400 μM NHS-biotin, the integrity of nucleoprotein complex was fully preserved (41). Therefore, in the present study we employed the same reaction conditions. A similar approach was used to optimize the HPG modification. We found that integrity of RPA-ssDNA complex was fully preserved with treatment of 1.25 mM HPG and employed this concentration in the present study.
NHS-biotin reacts specifically with primary amines on the surface of proteins, resulting in covalent addition of a biotin molecule (226 daltons) to lysine and release of an N-hydroxysuccinimide (53). The modified residues could be readily identified with subsequent MS and MS/MS analysis of the proteolytic fragments. We have recently reported an NHS-biotin modification pattern of the full-length RPA and RPA-ssDNA complex (41). In the present study we compared the biotinylation sites of the native and hyp-RPA proteins by MALDI-TOF. A total of 24 biotinylated tryptic peptides were identified for native RPA including 18 fragments from RPA70, 4 from RPA32, and 2 from RPA14 (TABLE ONE). The biotinylation patterns and intensities of the biotinylated peptides were almost identical between the native and hyp-RPA proteins, except that of the biotinylated peptide of amino acids 340-344 of DBD-B (Fig. 2). This peptide peak was readily detectable in the MS spectra of native RPA but was significantly diminished in hyp-RPA (Fig. 2A). Fig. 2B is a representative segment of MALDI-TOF spectra showing that Lys-88 and Lys-379, two others of the 24 modified lysines, were biotinylated equally well in both native and hyp-RPA proteins. MS/MS analysis of the amino acids 340-344 peptide peak, which exhibited protection in the context of hyp-RPA, indicated the biotinylation of Lys-343 (Fig. 2C). Given that 23 modified lysines distributed on the surfaces of almost all the domains of RPA trimer, the exhibited similar modification patterns in native and hyp-RPA suggest that no significant global conformational changes occurred in RPA after hyperphosphorylation. However, the inaccessibility of residue Lys-343 of RPA70 to NHS-biotin modification in hyp-RPA suggested occurrence of a limited structural re-arrangement involving the DBD-B domain containing Lys-343 and the hyperphosphorylated N terminus of RPA32. Because Lys-343 is located in the DNA binding cleft of DBD-B, adjacent to the L12 loop (amino acids 330-342), it is possible that the hyperphosphorylation resulted in at least partial shielding of the binding cleft of DBD-B from biotinylation via intramolecular interactions.
TABLE ONE.
Identification of modified lysines or arginines in RPA and hyp-RPAThe residues that were protected from modification in hyper-RPA are indicated in bold and underlined.
| RPA subunits | Biotin-modified lysines | HPG-modified arginines |
|---|---|---|
| RPA70 | 88, 163, 167, 183, 206, 220, 244, 263, 259, 324, 331, 343, 379, 489, 502, 577, 588, 595 | 91, 92, 202, 210, 216, 335, 382, 472, 573, 575, 586, 600, 604, 605, 611 |
| RPA32 | 38, 93, 138, 139 | 40, 133 |
| RPA14 | 33, 49 | 30 |
FIGURE 2.
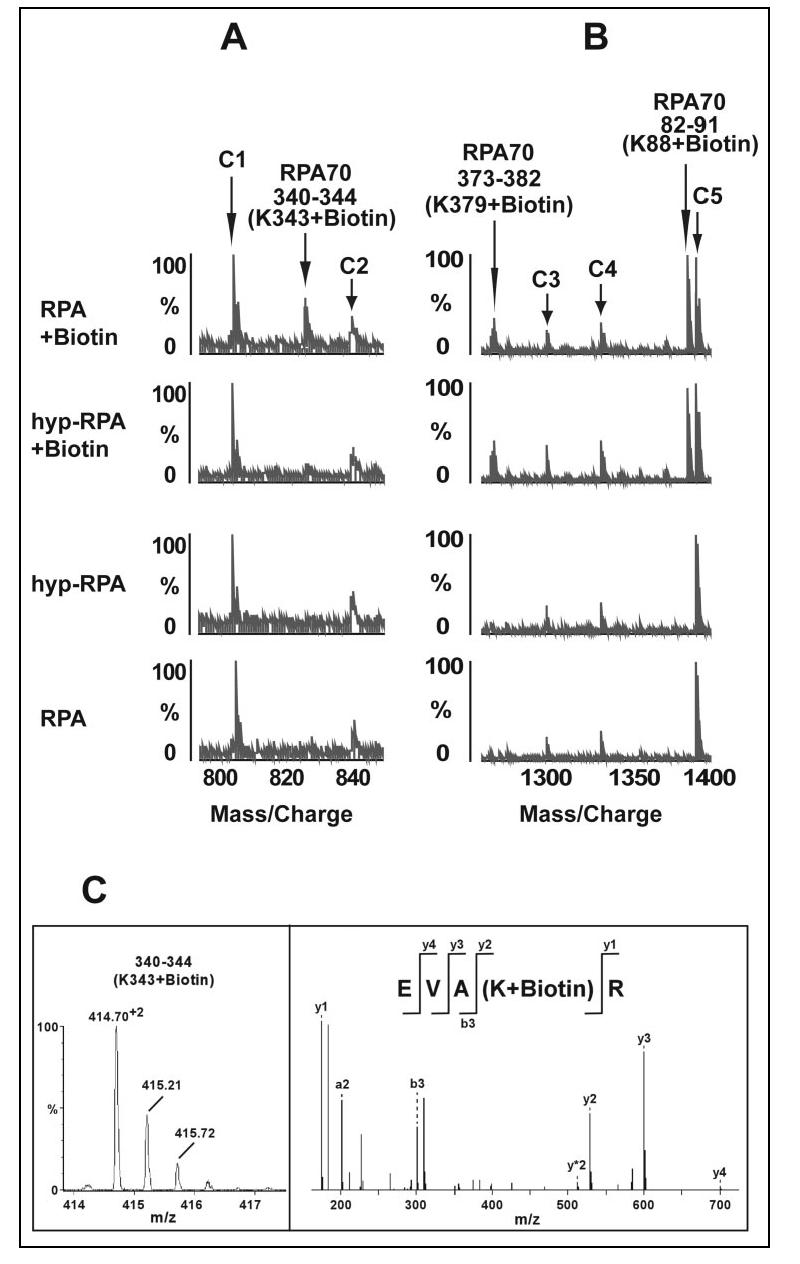
Mass spectrometric analysis of relative reactivity of lysines in RPA and hyp-RPA with NHS-biotin. A, representative segment of MALDI-TOF data showing that Lys-343 was readily biotinylated in native RPA and not in hyp-RPA. B, MALDI-TOF data illustrate the biotinylated peptide peaks (amino acids 373-382 and 82-91) were unchanged in RPA and hyp-RPA. C1-C5 were control peaks that serve as references for peak intensity. C, MS/MS analysis of the doubly charged ion 414.70 representing the modified peptide amino acids 340-344 with a Q-TOF instrument reveals biotinylated Lys-344. The left panel shows doubly charged parent ions; the right panel shows fragmentation results.
We next examined the arginine modifications of RPA and hyp-RPA with HPG. HPG reacts specifically with the guanidyl group of arginine under mild conditions (pH 7-9, 25 °C), resulting in the addition of a mass of 131 daltons to the modified molecule (54). We observed modification of 18 arginines in the context of native RPA, of which 16 residues were identically susceptible to modification in hyper-RPA (TABLE ONE). However, the following two peptide peaks, amino acids 332-339 (Arg-335 + HPG) and amino acids 380-389 (Arg-382 + HPG) of RPA70, were significantly diminished in hyper-RPA (Fig. 3A). Fig. 3B depicts representative segments of MALDI-TOF spectra to show equal intensities of mass signal for the modifications at Arg-133 of RPA32 in both RPA and hyp-RPA. Residue Arg-335 is located in the L12 loop of DBD-B, whereas Arg-382 is at the edge of the DNA binding cleft of DBD-B, adjacent to L45 (7, 15). These results are fully consistent with those of biotinylation studies above.
FIGURE 3.
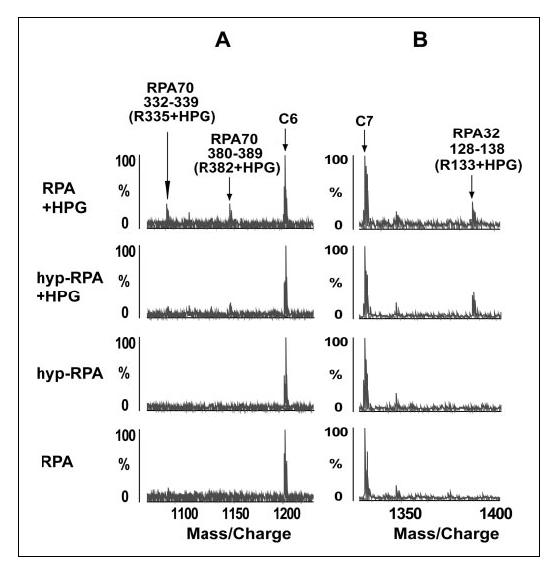
MALDI-TOF analysis of relative reactivity of arginines in RPA and hyp-RPA with HPG. A, representative segment of mass spectra showing that Arg-335 and Arg-382 in RPA70 were readily modified with HPG in native protein but were significantly protected from the modification in hyper-RPA. B, control data demonstrating that the peak corresponding to the peptide (128-138) with Arg-133 modified by HPG in RPA32 of RPA and hyp-RPA remained unchanged. Unmodified peptide peaks C6 and C7 serve as references for peak intensity.
Fluorescence Measurements—RPA has eight tryptophan residues. The locations of the tryptophans are shown in Fig. 4A: Trp-197 and Trp-212 in DBD-A, Trp-361 and Trp-414 in DBD-B, Trp-442 and Trp-528 in DBD-C, Trp-107 in DBD-D, and Trp-2 in the unstructured RPA32N. Trp-212 and Trp-361 have been shown to be located in the binding clefts of DBD-A and DBD-B, respectively, and to be involved in the interactions with ssDNA (7). Trp-528 and Trp-107 are located in the putative binding clefts of DBD-C and DBD-D, respectively, and were predicted to interact with ssDNA (11, 12). Indeed, we previously found that the intrinsic fluorescence of all these in-cleft tryptophans were quenchable upon ssDNA binding to RPA.3 If an intramolecular interaction involving any of the binding clefts occurs in hyp-RPA, the hyperphosphorylation could result in quenching of the tryptophan fluorescence in the clefts.
FIGURE 4.
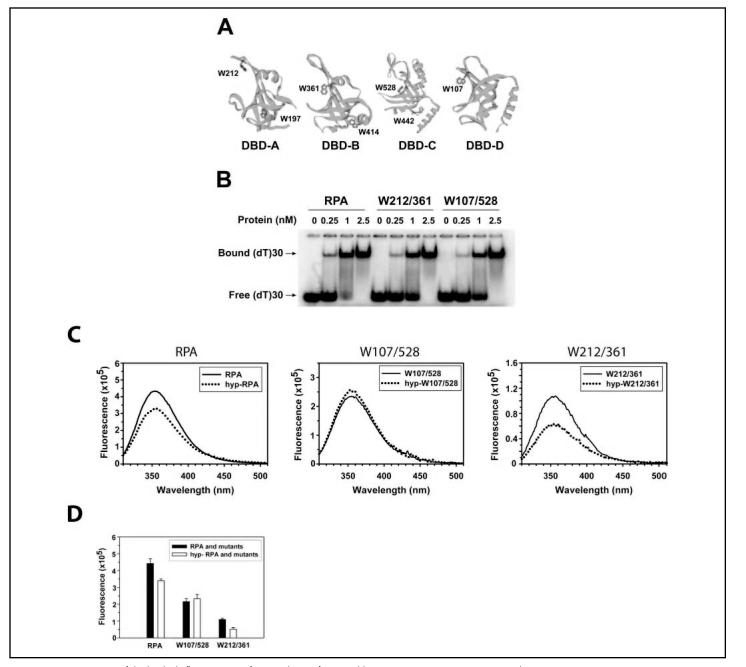
Measurements of the intrinsic fluorescence of tryptophans of RPA and its mutants. A, structural exhibition of the tryptophans in RPA. The DBDs (16, 18, 21) are positioned by projecting the ssDNA binding clefts or the putative clefts in the same direction. The tryptophans of each DBD are marked. B, binding of RPA and its mutants to oligo(dT)30. RPA or the mutants were incubated with 1 nM dT30 at increasing concentrations at 25 °C for 15 min in 20 μl of the binding buffer. The binding products were analyzed on a 4% native polyacrylamide gel. The positions of the bound and free oligonucleotides are indicated. C, emission spectra of RPA, its mutants, and their hyperphosphorylated forms. The tryptophan fluorescence of the proteins (100 nM) was recorded from 310 to 500 nm at 25 °C with excitation at 295 nm in 200 μl of RPA buffer. The slit widths were set at 3 and 5 nm for excitation and emission beams, respectively. D, constant wavelength analyses of RPA, its mutants, and their hyperphosphorylated forms. The intensity of tryptophan fluorescence of the proteins (100 nM) was recorded at 25 °C with excitation at 295 nm and emission at 355 nm in 200 μl of RPA buffer. Three data points with an integration time of 5 s and S.E. of 0.5% were collected for each measurement. Three measurements for each sample were performed independently.
To dissect the roles of individual tryptophans in these interactions we prepared the following two mutant proteins. The Trp-212/361 protein was obtained by preserving these two tryptophans and replacing the other 6 tryptophans with phenylalanines. Similarly, in the Trp-107/528 mutant the residues Trp-107 and Trp-528 were preserved, and the other tryptophans were substituted with phenylalanines. The binding of RPA mutants to ssDNA was tested to ensure that the mutants were active. Fig. 4B shows that both mutants bound a dT30 with an affinity comparable with that of native RPA, indicating that the substitutions of tryptophans with phenylalanines had minimum effects on the binding activity of RPA. Then these mutants together with wild type RPA were subjected to a series of fluorescence measurements. As shown in Figs. 4, C and D, the tryptophan fluorescence of the wild type RPA decreased upon hyperphosphorylation, suggesting that a structural re-arrangement in hyp-RPA occurred. However, no fluorescence changes were observed for the Trp-107/528 mutant before and after hyperphosphorylation. By contrast, about one-half of the fluorescence was quenched for hyp-Trp-212/361 (Figs. 4, C and D), suggesting that one of the two tryptophans was involved in the structural re-arrangement induced by hyperphosphorylation. Given that Trp-361 is located in the proximity of Lys-343 and Arg-335 residues that were inaccessible to small chemical modifications in hyp-RPA, we propose that the fluorescence of Trp-361 was quenched due to the intramolecular interaction involving hyperphosphorylated RPA32N and binding cleft of DBD-B. The fluorescence quenching for hyp-Trp-212/361 mutant was much more pronounced than that for wild type RPA. This suggests that more tryptophans in wild type RPA did not exhibit quenching upon hyperphosphorylation, resulting in a higher fluorescence background for the wild type protein.
Limited Proteolysis of RPA and hyp-RPA—Partial proteolysis could be employed for probing conformational changes of a protein upon ligand binding or macromolecular interactions via domain mapping (55-57). Because proteolytic cleavage sites are usually located in the linking regions or loops between domains on the surface of a protein (58), structural variation of the protein with domain rearrangement may change the accessibility of some of the cleavage sites, resulting in altered proteolytic profiles. Previously, proteolytic experiments were carried out to show that RPA changed its conformation upon binding to ssDNA (55, 56) or partial duplex DNA containing a 5'-protruding tail (56). Here we employed this method to compare the domain structures of native and hyperphosphorylated RPAs. The two proteins were partially digested with the fixed amount of chymotrypsin for the indicated time periods (Fig. 5). As shown in Fig. 5A, chymotrypsin digestion revealed several peptide fragments that were more resistant in hyp-RPA compared with native RPA. Amino acid sequencing enabled us to determine that all these fragments resulted from cleavages at the sites located in/near DBD-A and DBD-C of RPA70 (Fig. 5B> and TABLE TWO). In other words, these fragments contain no cleavages in DBD-B. Taken together, these partial hydrolysis data indicated that hyperphosphorylation of RPA32N led to conformational changes in RPA70 and relative resistance of DBD-B to partial proteolysis, which is consistent with the higher resolution results obtained from mass spectrometric footprinting and tryptophan fluorescence studies.
FIGURE 5.
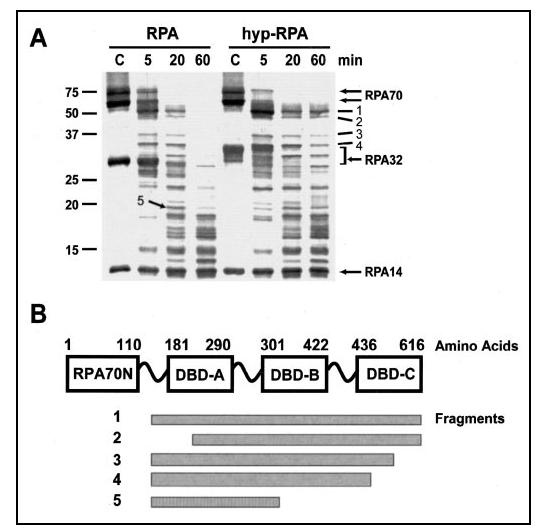
Partial proteolysis of RPA and hyp-RPA with chymotrypsin. A,4 μg of RPA or hyp-RPA was digested with chymotrypsin (1:80) at room temperature for the times indicated. The reactions were then terminated, resolved by 14% SDS-PAGE, and visualized using SYPRO Ruby stain. Untreated RPA and hyp-RPA were loaded as controls (the lanes marked as C). The molecular mass markers are shown on the left. Designations of individual fragments (dashes) and RPA subunits (arrows) are marked on the right. B, schematic map of proteolytic peptides generated upon RPA or hyp-RPA treatment with chymotrypsin. Domains are presented as boxes. The cleavage sites of chymotryptic peptide fragments are roughly shown.
TABLE TWO.
Identification of peptides of RPA or hyp-RPA digested by chymotrypsin
| Peptide fragment | Size | Origin | N-terminal sequence |
|---|---|---|---|
| kDa | |||
| 1 | 51 | RPA70 | 166GKAAG (major)a |
| 160GASKT (minor)a | |||
| 2 | 46 | RPA70 | 213SNSRG |
| 3 | 36 | RPA70 | 166GKAAG |
| 4 | 34 | RPA70 | 166GKAAG |
| 5 | 20 | RPA70 | 166GKAAG (major)a |
| 160GASKT (minor)a |
N-terminal sequencing gave two different sequences corresponding to two peptide fragments that migrated at the same position on SDS-PAGE. The amount of one peptide (major) was larger than the other (minor).
Binding of RPA and hyp-RPA to ssDNA and Partial DNA Duplex— How the structural changes of RPA affect its activities may define the role of RPA hyperphosphorylation in DNA metabolism. Our previous study showed that the Lys-343 residue in DBD-B was directly involved in the RPA interaction with ssDNA (41). Because the 8-10-nt binding mode of RPA-ssDNA interaction has been suggested to be important for the initiation of replication, replication lagging strand synthesis, and nucleotide excision repair (2, 59), here we examined the binding of hyp-RPA and RPA to an oligo(dT)8 by gel mobility shift assays (Fig. 6A) and fluorescence anisotropy measurements (Fig. 6B). Both assays revealed that hyp-RPA had a lower affinity for the (dT)8 compared with the native RPA. The anisotropy data were best fitted using a one-site binding model as described previously (42), and the resulting dissociation constants were 28.9 ± 3.3 and 60.8 ± 3.1 nM for native and hyp-RPAs, respectively. To test whether this affinity change was specific to DNA sequence, an 8-mer ssDNA with random sequences (CATCCTAC) also was used in our binding assays. Our results indicated similar reduction of the biding affinity for this DNA species (data not shown).
FIGURE 6.
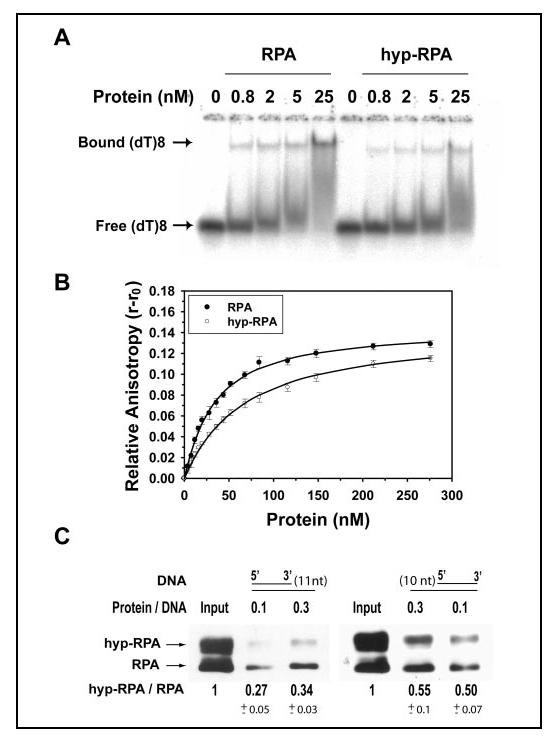
Binding of RPA and hyp-RPA to oligo(dT)8 and partial DNA duplexes. A, RPA was incubated with 1 nM (dT)8 at different molar ratios at 25 °C for 15 min in 20 μl of the binding buffer. The binding products were analyzed on a 4% native polyacrylamide gel. The positions of bound (dT)8 and free (dT)8 are indicated. B, 10 nM (dT)8 with a fluorescein labeled at 5' end was titrated with RPA or hyp-RPA. The anisotropy was measured at 520 nm with excitation at 492 nm. The binding isotherms were best fitted to obtain the equilibrium dissociation constants (Kd,obs). C, binding of RPA and hyp-RPA to partial DNA duplexes containing 5'-protruding 11 nucleotides (DNA-11, left) or 3'-protruding 10 nucleotides (DNA-10, right). DNA-11 and DNA-10 were constructed by annealing a biotinylated 55-mer with its complementary 44- and 45-mer, respectively. The biotinylated partial DNA duplex was incubated with a mixture of RPA and hyp-RPA. The protein bound to DNA was pulled down with streptavidin beads and detected by immunoblotting using an antibody against RPA32. The ratio of hyp-RPA to RPA in each binding was quantified.
To examine how hyp-RPA changes its interaction with physiologically relevant DNA substrates, a set of partial DNA duplexes that mimic in vivo intermediates which occur during DNA replication and DNA repair was subject to the binding assays. For this purpose, partial DNA duplexes containing 5'-protruding 11 nucleotides (DNA-11) or 3'-protruding 10 nucleotides (DNA-10) were constructed. Fig. 6C shows the binding of a mixture of hyp-RPA and RPA to biotin-labeled DNA-11 and DNA-10, as analyzed by the pull-down assay with streptavidin-agarose and by Western blotting with an antibody against RPA32. The pull-down assay allowed the binding of RPA and hyp-RPA to ssDNA to be monitored simultaneously and individually as the two forms of RPA protein were well separated on the SDS-PAGE. Thus, the binding of native RPA to ssDNA could serve as an internal control for determination of relative ssDNA binding efficiency of hyp-RPA. As shown in Fig. 6C, the hyperphosphorylation led to a significant decrease in binding of RPA to the DNA-11 and DNA-10 substrates by about 70 and 50%, respectively, demonstrating that hyp-RPA had significantly lower affinity for these partial DNA duplexes than native RPA.
DISCUSSION
As a single-stranded DNA-binding protein in eukaryotic cells, RPA interacts with a large variety of proteins required for DNA replication, repair, recombination, and DNA damage checkpoints, suggesting that RPA may play a regulatory role in the cellular DNA metabolic processes (2, 24). In response to DNA damage, RPA undergoes hyperphosphorylation in cells carried out by the protein kinases of phosphatidylinositol 3-kinase-like serine/threonine protein kinase family (24). The hyperphosphorylation may play a role in modulating the cellular activities of RPA via altering RPA ability to interact with DNA and proteins and is critical to the success of cellular responses to genotoxic stresses. In the present study we characterized the structural alterations of RPA upon hyperphosphorylation by DNA-PK. Our findings provide new and important structural information regarding the structural changes introduced in the full-length PRA trimer upon hyperphosphorylation of the protein.
Our mass spectrometric footprinting indicated that several basic residues were surface-inaccessible in DBD-B of RPA70 upon hyperphosphorylation of RPA32N. Tryptophan fluorescence studies suggested that Trp-361 located in DBD-B was significantly quenched in response to hyperphosphorylation. Limited proteolysis revealed structural changes in RPA70 and the relative resistance of DBD-B to the proteolytic digestion upon phosphorylation of RPA32N. Taken together, our findings suggest that a structural re-arrangement involving the RPA32N and a number of residues in the ssDNA binding cleft of DBD-B occurs after hyperphosphorylation of RPA (Fig. 7). Given that the N terminus of RPA32 becomes highly acidic (pI was estimated to be around 2) upon hyperphosphorylation, we propose that this structurally flexible protein segment of 40 residues could then be electrostatically attracted to the more positively charged basic region in the DNA binding cleft of DBD-B. This is supported by an estimation that the fragment amino acids 330-345 in the DNA binding cleft of DBD-B, where Lys-343 and Arg-335 were located, was more basic (pI ∼ 12) than the equivalent regions of other DBDs. It is obvious, however, that direct evidence is needed to confirm the interdomain interaction of hyperphosphorylated RPA32N with DBD-B in future studies.
FIGURE 7.
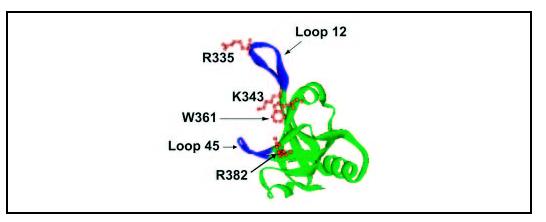
DBD-B structure (15) with highlighted residues. Arg-335, Lys-343, Trp-361, and Arg-382 are in brown that could directly interact with hyperphosphorylated RPA32N. Two loops, Leu-12 and Leu-45, are indicated in blue.
A previous NMR study showed that a synthesized acidic peptide mimicking hyperphosphorylated RPA32N was able to establish electrostatic interactions with the basic cleft of the purified DBD-F (RPA70N) domain fragment (RPA70 1-168). However, in the study protein fragments rather than the full-length RPA trimer were used (38). In the context of the full-length protein it is feasible that DBD-B is in the better spatial setting with RPA32N than DBD-F. Indeed, based on the direction of the backbone at amino acid 45 of RPA32 in a crystal structure of RPA trimerization core (DBD-C/DBD-D/RPA14), unphosphorylated RPA32N was predicted to be located in the vicinity of the putative ssDNA binding cleft of DBD-C (11). RPA32N in this position is closer to DBD-B than to DBD-F, and thus, hyperphosphorylation of RPA32N is more likely to have effects on the closely positioned DBD-B rather than on the more distant DBD-F.
A possible immediate effect of the hyperphosphorylation-induced protection of the ssDNA binding cleft of DBD-B from molecular access on RPA function was the inhibition of RPA binding to ssDNA, particularly short ssDNA. Indeed, we observed that hyperphosphorylation of RPA could affect the ability of the protein to bind short oligonucleotides. It has been generally accepted that RPA binds to ssDNA in three modes in terms of the ssDNA lengths, 8-10, 13-22, and 30 nt (2, 11). Of these modes, the binding to 8-10-nt (2, 7, 11, 60) or short ssDNA tails of partial DNA duplexes (56, 61, 62) were exclusively carried out by DBD-A and DBD-B, whereas the other two modes required the involvement of additional domains, DBD-C and DBD-C/DBD-D. Our results suggest that the potential intramolecular interaction induced by RPA32N hyperphosphorylation may compete with ssDNA binding to DBD-B. Such competition may provide a regulatory mechanism for modulating the RPA activities. However, the effect of the hyperphosphorylation of RPA appeared to be limited to the short ssDNA binding. It has been reported that the association constant for the interaction of RPA with 10-nt or shorter oligonucleotides was on the order of 107 M-1, whereas the association constant for the RPA interaction with ssDNAs longer than 15 nt was on the order of 109-10 M-1 (63). Because DBD-B is believed to participate in all modes of ssDNA binding, it is likely that the 2-3-order higher affinity of RPA to longer ssDNA may allow DBD-B to predominately bind ssDNA so that the effect of the hyperphosphorylation becomes minimal. Indeed, our results (data not shown) and also those from others showed that the hyperphosphorylation had a negligible effect on the RPA binding to the ssDNA of 30 nt or longer lengths (20, 38).
In contrast, our results indicate significant competition between hyperphosphorylation-elicited DBD-B shielding and DBD-B binding to (dT)8 or partial DNA duplexes with a short ssDNA tail. Such data suggest that the association constant for the possible intramolecular interaction causing DBD-B shielding could be on the order of 107 M-1. Interestingly, interactions of RPA with these short ssDNA or partial DNA duplexes with short ssDNA tails were believed to play a role in the initiation of DNA replication and replicative lagging strand synthesis (2, 38, 59, 64). Therefore, it is likely that the hyperphosphorylation of RPA could disrupt this process. Consistently, Vassin et al. (32) recently reported that a hyperphosphorylation-mimic RPA failed to associate with replication centers in vivo. Similarly, the DBD-B-engaged intramolecular interactions of RPA due to the hyperphosphorylation in the N terminus of RPA32 could also affect the RPA-protein interactions (with association constants on the order of 107 M-1) involved in various DNA metabolic pathways.
Acknowledgment—
We thank Dr. Lifeng Cai for help in producing the mutant RPA proteins used in this study.
Footnotes
This study was supported by NCI, National Institutes of Health Grant CA86927 (to Y. Z.).The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
- RPA
- replication protein A
- hyp-RPA
- hyperphosphorylated RPA
- HPG
- p-hydroxyphenylglyoxal
- NHS-biotin
- N-hydroxysuccinimidobiotin
- ssDNA
- single-stranded DNA
- DBD
- DNA binding domain
- DNA-PK
- DNA-dependent protein kinase
- ATM
- ataxia-telangiectasia mutated kinase
- ATR
- ATM- and Rad3-related kinase
- DTT
- dithiothreitol
- MS
- mass spectroscopy
- MALDI-TOF
- matrix-assisted laser desorption ionization time-of-flight
- Q-TOF
- quadrupole-TOF
- nt
- nucleotide(s
REFERENCES
- 1.Wold MS. Annu. Rev. Biochem. 1997;66:61–92. doi: 10.1146/annurev.biochem.66.1.61. [DOI] [PubMed] [Google Scholar]
- 2.Iftode C, Daniely Y, Borowiec JA. Crit. Rev. Biochem. Mol. Biol. 1999;34:141–180. doi: 10.1080/10409239991209255. [DOI] [PubMed] [Google Scholar]
- 3.Shivji MK, Podust VN, Hubscher U, Wood RD. Biochemistry. 1995;34:5011–5017. doi: 10.1021/bi00015a012. [DOI] [PubMed] [Google Scholar]
- 4.Barr SM, Leung CG, Chang EE, Cimprich KA. Curr. Biol. 2003;13:1047–1051. doi: 10.1016/s0960-9822(03)00376-2. [DOI] [PubMed] [Google Scholar]
- 5.Dart DA, Adams KE, Akerman I, Lakin ND. J. Biol. Chem. 2004;279:16433–16440. doi: 10.1074/jbc.M314212200. [DOI] [PubMed] [Google Scholar]
- 6.Jacobs DM, Lipton AS, Isern NG, Daughdrill GW, Lowry DF, Gomes X, Wold MS. J. Biomol. NMR. 1999;14:321–331. doi: 10.1023/a:1008373009786. [DOI] [PubMed] [Google Scholar]
- 7.Bochkarev A, Pfuetzner RA, Edwards AM, Frappier L. Nature. 1997;385:176–181. doi: 10.1038/385176a0. [DOI] [PubMed] [Google Scholar]
- 8.Brill SJ, Bastin-Shanower S. Mol. Cell. Biol. 1998;18:7225–7234. doi: 10.1128/mcb.18.12.7225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arunkumar AI, Stauffer ME, Bochkareva E, Bochkarev A, Chazin WJ. J. Biol. Chem. 2003;278:41077–41082. doi: 10.1074/jbc.M305871200. [DOI] [PubMed] [Google Scholar]
- 10.Bochkareva E, Korolev S, Bochkarev A. J. Biol. Chem. 2000;275:27332–27338. doi: 10.1074/jbc.M000620200. [DOI] [PubMed] [Google Scholar]
- 11.Bochkareva E, Korolev S, Lees-Miller SP, Bochkarev A. EMBO J. 2002;21:1855–1863. doi: 10.1093/emboj/21.7.1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bochkarev A, Bochkareva E, Frappier L, Edwards AM. EMBO J. 1999;18:4498–4504. doi: 10.1093/emboj/18.16.4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bochkareva E, Frappier L, Edwards AM, Bochkarev A. J. Biol. Chem. 1998;273:3932–3936. doi: 10.1074/jbc.273.7.3932. [DOI] [PubMed] [Google Scholar]
- 14.Mer G, Bochkarev A, Gupta R, Bochkareva E, Frappier L, Ingles CJ, Edwards AM, Chazin WJ. Cell. 2000;103:449–456. doi: 10.1016/s0092-8674(00)00136-7. [DOI] [PubMed] [Google Scholar]
- 15.Bochkareva E, Belegu V, Korolev S, Bochkarev A. EMBO J. 2001;20:612–618. doi: 10.1093/emboj/20.3.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bochkarev A, Bochkareva E. Curr. Opin. Struct. Biol. 2004;14:36–42. doi: 10.1016/j.sbi.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 17.Kerr ID, Wadsworth RI, Cubeddu L, Blankenfeldt W, Naismith JH, White MF. EMBO J. 2003;22:2561–2570. doi: 10.1093/emboj/cdg272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Din S, Brill SJ, Fairman MP, Stillman B. Genes Dev. 1990;4:968–977. doi: 10.1101/gad.4.6.968. [DOI] [PubMed] [Google Scholar]
- 19.Dutta A, Stillman B. EMBO J. 1992;11:2189–2199. doi: 10.1002/j.1460-2075.1992.tb05278.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oakley GG, Patrick SM, Yao J, Carty MP, Turchi JJ, Dixon K. Biochemistry. 2003;42:3255–3264. doi: 10.1021/bi026377u. [DOI] [PubMed] [Google Scholar]
- 21.Liu VF, Weaver DT. Mol. Cell. Biol. 1993;13:7222–7231. doi: 10.1128/mcb.13.12.7222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zernik-Kobak M, Vasunia K, Connelly M, Anderson CW, Dixon K. J. Biol. Chem. 1997;272:23896–23904. doi: 10.1074/jbc.272.38.23896. [DOI] [PubMed] [Google Scholar]
- 23.Block WD, Yu Y, Lees-Miller SP. Nucleic Acids Res. 2004;32:997–1005. doi: 10.1093/nar/gkh265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Binz SK, Sheehan AM, Wold MS. DNA Repair (Amst) 2004;3:1015–1024. doi: 10.1016/j.dnarep.2004.03.028. [DOI] [PubMed] [Google Scholar]
- 25.Niu H, Erdjument-Bromage H, Pan ZQ, Lee SH, Tempst P, Hurwitz J. J. Biol. Chem. 1997;272:12634–12641. doi: 10.1074/jbc.272.19.12634. [DOI] [PubMed] [Google Scholar]
- 26.Lee SH, Kim DK. J. Biol. Chem. 1995;270:12801–12807. doi: 10.1074/jbc.270.21.12801. [DOI] [PubMed] [Google Scholar]
- 27.Treuner K, Okuyama A, Knippers R, Fackelmayer FO. Nucleic Acids Res. 1999;27:1499–1504. doi: 10.1093/nar/27.6.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nuss JE, Patrick SM, Oakley GG, Alter GM, Robison JG, Dixon K, Turchi JJ. Biochemistry. 2005;44:8428–8437. doi: 10.1021/bi0480584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pan ZQ, Amin AA, Gibbs E, Niu H, Hurwitz J. Proc. Natl. Acad. Sci. U. S. A. 1994;91:8343–8347. doi: 10.1073/pnas.91.18.8343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shao RG, Cao CX, Zhang H, Kohn KW, Wold MS, Pommier Y. EMBO J. 1999;18:1397–1406. doi: 10.1093/emboj/18.5.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brush GS, Anderson CW, Kelly TJ. Proc. Natl. Acad. Sci. U. S. A. 1994;91:12520–12524. doi: 10.1073/pnas.91.26.12520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vassin VM, Wold MS, Borowiec JA. Mol. Cell. Biol. 2004;24:1930–1943. doi: 10.1128/MCB.24.5.1930-1943.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park JS, Park SJ, Peng X, Wang M, Yu MA, Lee SH. J. Biol. Chem. 1999;274:32520–32527. doi: 10.1074/jbc.274.45.32520. [DOI] [PubMed] [Google Scholar]
- 34.Jackson D, Dhar K, Wahl JK, Wold MS, Borgstahl GE. J. Mol. Biol. 2002;321:133–148. doi: 10.1016/s0022-2836(02)00541-7. [DOI] [PubMed] [Google Scholar]
- 35.Abramova NA, Russell J, Botchan M, Li R. Proc. Natl. Acad. Sci. U. S. A. 1997;94:7186–7191. doi: 10.1073/pnas.94.14.7186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu X, Shell SM, Zou Y. Oncogene. 2005;24:4728–4735. doi: 10.1038/sj.onc.1208674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patrick SM, Oakley GG, Dixon K, Turchi JJ. Biochemistry. 2005;44:8438–8448. doi: 10.1021/bi048057b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Binz SK, Lao Y, Lowry DF, Wold MS. J. Biol. Chem. 2003;278:35584–35591. doi: 10.1074/jbc.M305388200. [DOI] [PubMed] [Google Scholar]
- 39.Fried LM, Koumenis C, Peterson SR, Green SL, van Zijl P, Allalunis-Turner J, Chen DJ, Fishel R, Giaccia AJ, Brown JM, Kirchgessner CU. Proc. Natl. Acad. Sci. U. S. A. 1996;93:13825–13830. doi: 10.1073/pnas.93.24.13825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang ZG, Liu Y, Mao LY, Zhang JT, Zou Y. Biochemistry. 2002;41:13012–13020. doi: 10.1021/bi026064z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shell SM, Hess S, Kvaratskhelia M, Zou Y. Biochemistry. 2005;44:971–978. doi: 10.1021/bi048208a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Y, Yang Z, Utzat CD, Liu Y, Geacintov NE, Basu AK, Zou Y. Biochem. J. 2005;385:519–526. doi: 10.1042/BJ20041151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zou Y, Bassett H, Walker R, Bishop A, Amin S, Geacintov NE, Van Houten B. J. Mol. Biol. 1998;281:107–119. doi: 10.1006/jmbi.1998.1903. [DOI] [PubMed] [Google Scholar]
- 44.Boubnov NV, Weaver DT. Mol. Cell. Biol. 1995;15:5700–5706. doi: 10.1128/mcb.15.10.5700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gately DP, Hittle JC, Chan GK, Yen TJ. Mol. Biol. Cell. 1998;9:2361–2374. doi: 10.1091/mbc.9.9.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oakley GG, Loberg LI, Yao J, Risinger MA, Yunker RL, Zernik-Kobak M, Khanna KK, Lavin MF, Carty MP, Dixon K. Mol. Biol. Cell. 2001;12:1199–1213. doi: 10.1091/mbc.12.5.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith GC, Jackson SP. Genes Dev. 1999;13:916–934. doi: 10.1101/gad.13.8.916. [DOI] [PubMed] [Google Scholar]
- 48.Henricksen LA, Carter T, Dutta A, Wold MS. Nucleic Acids Res. 1996;24:3107–3112. doi: 10.1093/nar/24.15.3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carty MP, Zernik-Kobak M, McGrath S, Dixon K. EMBO J. 1994;13:2114–2123. doi: 10.1002/j.1460-2075.1994.tb06487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bennett KL, Matthiesen T, Roepstorff P. Methods Mol. Biol. 2000;146:113–131. doi: 10.1385/1-59259-045-4:113. [DOI] [PubMed] [Google Scholar]
- 51.Suckau D, Mak M, Przybylski M. Proc. Natl. Acad. Sci. U. S. A. 1992;89:5630–5634. doi: 10.1073/pnas.89.12.5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Leite JF, Cascio M. Biochemistry. 2002;41:6140–6148. doi: 10.1021/bi015895m. [DOI] [PubMed] [Google Scholar]
- 53.Kvaratskhelia M, Miller JT, Budihas SR, Pannell LK, Le Grice SF. Proc. Natl. Acad. Sci. U. S. A. 2002;99:15988–15993. doi: 10.1073/pnas.252550199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yamasaki RB, Vega A, Feeney RE. Anal. Biochem. 1980;109:32–40. doi: 10.1016/0003-2697(80)90006-8. [DOI] [PubMed] [Google Scholar]
- 55.Gomes XV, Henricksen LA, Wold MS. Biochemistry. 1996;35:5586–5595. doi: 10.1021/bi9526995. [DOI] [PubMed] [Google Scholar]
- 56.Pestryakov PE, Weisshart K, Schlott B, Khodyreva SN, Kremmer E, Grosse F, Lavrik OI, Nasheuer HP. J. Biol. Chem. 2003;278:17515–17524. doi: 10.1074/jbc.M301265200. [DOI] [PubMed] [Google Scholar]
- 57.Heyduk E, Heyduk T. Biochemistry. 1994;33:9643–9650. doi: 10.1021/bi00198a033. [DOI] [PubMed] [Google Scholar]
- 58.Zappacosta F, Pessi A, Bianchi E, Venturini S, Sollazzo M, Tramontano A, Marino G, Pucci P. Protein Sci. 1996;5:802–813. doi: 10.1002/pro.5560050502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mass G, Nethanel T, Lavrik OI, Wold MS, Kaufmann G. Nucleic Acids Res. 2001;29:3892–3899. doi: 10.1093/nar/29.18.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Blackwell LJ, Borowiec JA. Mol. Cell. Biol. 1994;14:3993–4001. doi: 10.1128/mcb.14.6.3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lavrik OI, Kolpashchikov DM, Weisshart K, Nasheuer HP, Khodyreva SN, Favre A. Nucleic. Acids Res. 1999;27:4235–4240. doi: 10.1093/nar/27.21.4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pestryakov PE, Khlimankov DY, Bochkareva E, Bochkarev A, Lavrik OI. Nucleic Acids Res. 2004;32:1894–1903. doi: 10.1093/nar/gkh346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim C, Paulus BF, Wold MS. Biochemistry. 1994;33:14197–14206. doi: 10.1021/bi00251a031. [DOI] [PubMed] [Google Scholar]
- 64.Lao Y, Gomes XV, Ren Y, Taylor JS, Wold MS. Biochemistry. 2000;39:850–859. doi: 10.1021/bi991704s. [DOI] [PubMed] [Google Scholar]