

Abstract
In multivalent polyploids, simultaneous pairings among homologous chromosomes at meiosis result in a unique cytological phenomenon—double reduction. Double reduction casts an impact on chromosome evolution in higher plants, but because of its confounded effect on the pattern of gene cosegregation, it complicates linkage analysis and map construction with polymorphic molecular markers. In this article, we have proposed a general statistical model for simultaneously estimating the frequencies of double reduction, the recombination fraction, and optimal parental linkage phases between any types of markers, both fully and partially informative, or dominant and codominant, for a tetraploid species that undergoes only multivalent pairing. This model provides an in-depth extension of our earlier linkage model that was built upon Fisher's classifications for different gamete formation modes during the polysomic inheritance of a multivalent polyploid. By implementing a two-stage hierarchical EM algorithm, we derived a closed-form solution for estimating the frequencies of double reduction through the estimation of gamete mode frequencies and the recombination fraction. We performed different settings of simulation studies to demonstrate the statistical properties of our model for estimating and testing double reduction and the linkage in multivalent tetraploids. As shown by a comparative analysis, our model provides a general framework that covers existing statistical approaches for linkage mapping in polyploids that are predominantly multivalent. The model will have great implications for understanding the genome structure and organization of polyploid species.
BECAUSE of their biological and economic importance, polyploids have long been a focus of genetic and evolutionary analyses (Bever and Felber 1992; Soltis and Soltis 2000). One of the most useful tools for these analyses is provided by genetic linkage maps constructed from molecular markers, which allow not only for comparative studies of genome structure and organization across different polyploids (da Silva et al. 1995; Ming et al. 1998), but also for the characterization of specific loci affecting quantitatively inherited traits (Meyer et al. 1998; Ming et al. 2001). However, compared to diploid species, linkage analysis in polyploids is complicated by their underlying meiotic processes. For bivalent polyploids in which only two chromosomes pair during meiosis, there are higher pairing probabilities between more similar chromosomes than between less similar chromosomes. Whereas many models assume random pairings (Hackett et al. 1998; Ripol et al. 1999; Luo et al. 2001), we have derived a host of statistical models that integrate the so-called chromosomal pairing preference (Sybenga 1994) within a linkage analysis framework (Ma et al. 2002; Wu et al. 2002a).
Different from bivalent polyploids, multivalent polyploids pair their chromosomes among more than two homologous copies at meiosis. The consequence of this multivalent pairing is the formation of double reduction; i.e., two sister chromatids of a chromosome sort into the same gamete (Darlington 1929). Fisher (1947) proposed a conceptual model for characterizing the probabilities of 11 different modes of gamete formation for a quadrivalent polyploid in terms of the recombination fraction between two different loci and their double reductions. Although Fisher's model did not have the capacity to estimate the linkage and double reduction, it provides a theoretical foundation for S. S. Wu et al. (2001) to successfully derive a closed-form solution for estimating these parameters within the maximum-likelihood context. To clearly describe the idea, Wu et al. derived their EM-implemented algorithm on the basis of fully informative markers that display completely different alleles between two parents. Because each multilocus genotype observed in a full-sib family is formed with a predictable mechanism (see S. S. Wu et al. 2001 for a detailed description of this), it can be made possible to derive the closed-form solution for estimating the recombination fraction and double reduction.
While fully informative markers represent only a subset of polymorphic markers in polyploids, it is essential to develop a more general model that has power to analyze those partially informative markers, such as dominant markers or markers with multiple dosages. Luo et al. (2004) proposed a statistical model that attempts to consider different marker types. A key step of Luo et al.'s model is the derivation of the frequency of each gamete formation mode as a function of the recombination fraction and double reduction at one marker (see their Table 1). To show Luo et al.'s deriving process, we suppose there are two fully informative markers, ℳ and 𝒩, that are linked with recombination fraction r. Assume that marker ℳ is closer to the centromere than marker 𝒩 so that the coefficient of double reduction at marker 𝒩 is larger than the coefficient of double reduction (α) at marker ℳ (Darlington 1929; Fisher 1947). Denote four different alleles by M1, · · · , M4 for marker ℳ and by N1, · · · , N4 for marker 𝒩. Thus, we have a total of 24 allelic configurations or linkage phase assignments between the two markers, one of which is schematically expressed as 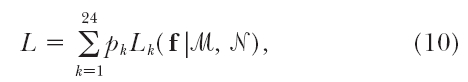 where lines indicate the individual homologous chromosomes on which the two markers are located. This configuration generates 16 chromatids during meiosis, involving four nonrecombinants, M1N1, M2N2, M3N3, and M4N4, each with frequency (1 − r), and 12 recombinants, M1N2, · · · , M4N3, each with frequency r. All these chromatids are randomly combined to form diploid gametes. Consider the first four modes of gamete formation in Luo et al. (2004)(Table 1), for which only marker ℳ undergoes the double reduction. The first mode includes four gametes, M1M1/N1N1, M2M2/N2N2, M3M3/N3N3, and M4M4/N4N4, for which there is no recombinant for each chromatid. If the occurrence of double reduction at marker 𝒩 is due completely to the double reduction at marker ℳ, the frequency of this gamete mode is simply expressed as α(1 − r)2. The third mode is composed of 12 gametes each with only one recombinant, i.e., M1M1/N1N2, M1M1/N1N3, · · · , M4M4/N4N3, and the frequency of this mode is expressed as 2αr(1 − r), where the coefficient 2 accounts for the combination between different chromatids. The second and fourth modes each include the gametes that contain two recombinants, with a total mode frequency of αr2. The second mode contains 12 gametes M1M1/N2N2, M1M1/N3N3, · · · , M4M4/N3N3, whereas the fourth mode contains 12 gametes M1M1/N2N3, M1M1/N2N4, · · · , M4M4/N3N2. It can be seen that the ratio of the second and fourth mode frequencies is 1:2 because the former contains the same chromatid and the latter contains two different chromatids. This provided a clue for Luo et al. to derive their probability distributions of the first four gamete formation modes. A similar idea can be used to derive the frequencies of the other seven gamete modes in which marker ℳ has no double reduction. Ultimately, Luo et al. was able to derive the formula for calculating the coefficient of double reduction at marker 𝒩.
where lines indicate the individual homologous chromosomes on which the two markers are located. This configuration generates 16 chromatids during meiosis, involving four nonrecombinants, M1N1, M2N2, M3N3, and M4N4, each with frequency (1 − r), and 12 recombinants, M1N2, · · · , M4N3, each with frequency r. All these chromatids are randomly combined to form diploid gametes. Consider the first four modes of gamete formation in Luo et al. (2004)(Table 1), for which only marker ℳ undergoes the double reduction. The first mode includes four gametes, M1M1/N1N1, M2M2/N2N2, M3M3/N3N3, and M4M4/N4N4, for which there is no recombinant for each chromatid. If the occurrence of double reduction at marker 𝒩 is due completely to the double reduction at marker ℳ, the frequency of this gamete mode is simply expressed as α(1 − r)2. The third mode is composed of 12 gametes each with only one recombinant, i.e., M1M1/N1N2, M1M1/N1N3, · · · , M4M4/N4N3, and the frequency of this mode is expressed as 2αr(1 − r), where the coefficient 2 accounts for the combination between different chromatids. The second and fourth modes each include the gametes that contain two recombinants, with a total mode frequency of αr2. The second mode contains 12 gametes M1M1/N2N2, M1M1/N3N3, · · · , M4M4/N3N3, whereas the fourth mode contains 12 gametes M1M1/N2N3, M1M1/N2N4, · · · , M4M4/N3N2. It can be seen that the ratio of the second and fourth mode frequencies is 1:2 because the former contains the same chromatid and the latter contains two different chromatids. This provided a clue for Luo et al. to derive their probability distributions of the first four gamete formation modes. A similar idea can be used to derive the frequencies of the other seven gamete modes in which marker ℳ has no double reduction. Ultimately, Luo et al. was able to derive the formula for calculating the coefficient of double reduction at marker 𝒩.
The above derivation has been strictly based on the assumption that the frequency of double reduction at a marker is determined by the frequency of double reduction at its linked marker and the recombination fraction between these two markers. However, as revealed by cytological and molecular experiments, this assumption that has facilitated Luo et al.'s linkage analysis is hardly met in practice. The values of double reduction are observed to range from 0 to almost 30% (Haynes and Douches 1993) and are likely to be species, chromosome, and position dependent (Butruille and Boiteux 2000). In a genetic mapping study of cultivated tetraploid alfalfa with microsatellite and AFLP markers, 20 loci that display significant double reduction are sporadically distributed throughout the genome (Julier et al. 2003). For example, on linkage group 6 composed of nine loci, only markers MTIC153 and MTIC14 have significant double reductions of similar values (0.15 and 0.16), while the two markers, separated by 39 cM, flank three intermediate markers. It thus can be seen that it is unreasonable to employ a fixed function of the recombination fraction to model the change of double reduction across different loci.
In this article, we generalize our multivalent pairing model for fully informative markers (S. S. Wu et al. 2001) to take into account complexities due to the segregation of less informative or dominant marker types. For partially informative markers, the same zygote genotype can be formed due to the combination between different gametes with double reduction or with no double reduction. A two-stage hierarchical model is derived to estimate the probabilities of gamete formation modes and therefore double reduction in the upper hierarchy and estimate the recombination fraction in the lower hierarchy within the maximum-likelihood context implemented with the EM algorithm. We undertake extensive simulation studies to investigate statistical properties of our model and demonstrate its analytical and biological advantages over existing models.
The derivation of our model centers on tetraploid species in which multivalent pairing is only one mechanism for chromosomal pairings during meioses. This consideration should be relevant for two reasons. First, many polyploids are multivalent in practice. Second, this allows us to investigate better the impacts of the assumption on the estimation of linkage about two-marker gamete formation modes expressed as a function of the recombination fraction and double reduction at one marker in multivalent tetraploids. Indeed, our model can be extended to consider any tetraploids in which both bivalent and multivalent pairings occur as pointed out by Luo et al. (2004). Different from Luo et al.'s treatment, however, we model these pairings as two cytogenetically related meiotic processes using Sybenga's (1994) theory (Wu et al. 2004).
THE GAMETE MODEL
Fully informative markers:
Our model focuses on a tetraploid that undergoes only multivalent pairings. Let a heterozygous multivalent tetraploid line be crossed with a homozygous line to generate a so-called pseudo-test backcross population. For such a population, the genotypes of progeny are consistent with the genotypes of gametes produced by the heterozygous parent and, therefore, the derivation of our linkage analysis can be based on the segregation of gametes. Not only is it useful for the pseudo-test backcross design, but also this gamete-based model provides an analytical clue for a more complicated linkage analysis based on progeny genotypes.
We use M1, · · ·, M4 and N1, · · · , N4 to denote different alleles for markers ℳ and 𝒩, respectively. There are 24 allelic configurations between the two markers, one of which is shown in display (1). For simplicity, we use 1, · · · , 4 to denote alleles, M1, · · · , M4 or N1, · · · , N4, at a marker. The recombination fraction (r) between these two markers is estimated on the basis of the segregation of recombinant and nonrecombinant gametes observed in the offspring of the family under a particular linkage phase. However, as seen below, some gametes can be generated from different unobservable mechanisms between which there are different numbers of recombinant events. S. S. Wu et al. (2001) implemented the EM algorithm to separate these different mechanisms, which makes it possible to estimate the recombination fraction.
For a phase-known multivalent tetraploid, as shown in display (1), that undergoes double reduction, a total of 10 gametes, arrayed by (11, 22, 33, 44, 12, 13, 14, 23, 24, 34), for each of the two markers will be produced. The first 4 gametes in the gamete array for each marker are those due to double reduction, whereas the remaining 6 gametes are derived from nondouble reduction. The proportion of the double-reduction-derived gametes to all the gametes is defined as the frequency of double reduction indexed by α for marker ℳ and by β for marker 𝒩. The frequency of double reduction is a constant for any given locus, with the value depending on its distance from the centromere.
When two linked markers are segregating in a multivalent tetraploid, a total of 136 diploid gamete formation mechanisms are generated although only 100 gamete genotypes are observable. On the basis of the presence/absence of double reduction and the number of recombinant events, Fisher (1947) classified these 136 formation mechanisms into 11 gamete modes. Of these 11 gamete modes, however, only 9 can be observed, each with a frequency denoted by fi (i = 1, · · · 9). These 9 observable gamete modes were rearranged by S. S. Wu et al. (2001) in matrix form expressed as  where f1 and f2 are associated with double reductions at both markers, f3 and f4 with double reductions only at marker M, f5 and f6 with double reductions only at marker N, and f7–f9 with nondouble reductions. From matrix (2), we see that there are no, one, and two recombinant events in the cells (f1), (f3, f5), and (f2, f4, f6, f9), respectively. The cells (f7) and (f8) are each a mixture of two different gamete formation mechanisms or configurations (A and B), i.e., f7 = f7A + f7B and f8 = f8A + f8B, with relative proportions determined by r. Because different configurations contain different numbers of recombination events, the expected number of recombination events in each cell, i.e., an observable gamete genotype, should be the weighted average of the number of recombination events for each configuration. S. S. Wu et al. (2001) used a matrix form (e) to count the expected number of recombination events for each observable gamete genotype expressed as
where f1 and f2 are associated with double reductions at both markers, f3 and f4 with double reductions only at marker M, f5 and f6 with double reductions only at marker N, and f7–f9 with nondouble reductions. From matrix (2), we see that there are no, one, and two recombinant events in the cells (f1), (f3, f5), and (f2, f4, f6, f9), respectively. The cells (f7) and (f8) are each a mixture of two different gamete formation mechanisms or configurations (A and B), i.e., f7 = f7A + f7B and f8 = f8A + f8B, with relative proportions determined by r. Because different configurations contain different numbers of recombination events, the expected number of recombination events in each cell, i.e., an observable gamete genotype, should be the weighted average of the number of recombination events for each configuration. S. S. Wu et al. (2001) used a matrix form (e) to count the expected number of recombination events for each observable gamete genotype expressed as 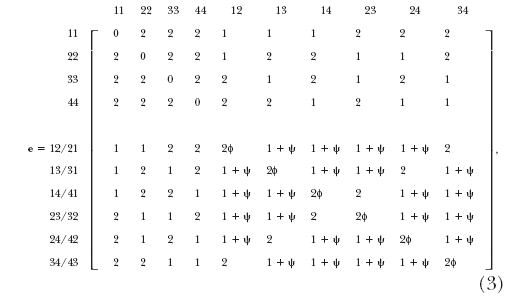 where
where
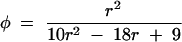 |
4 |
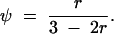 |
5 |
On the basis of matrices (2) and (3), we give the expressions for the frequencies of double reduction (α and β) and the recombination fraction r in terms of fi as follows:
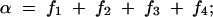 |
6 |
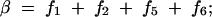 |
7 |
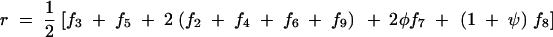 |
8 |
It can be seen that, to estimate the frequencies of double reduction and the recombination fraction, we need to first estimate the nine gamete mode frequencies. For fully informative markers, every cell in matrix (2) is distinguishable. Thus, corresponding to the nine observable gamete mode frequencies arrayed by f = (f1, · · · , f9) shown in matrix (2), there are nine offspring observations n1, · · · , n9 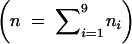 in a full-sib family. It is straightforward to derive the explicit expressions of the maximum-likelihood estimates (MLEs) for the frequencies of these nine formation modes on the basis of the following likelihood function given the observed marker data (ℳ and 𝒩),
in a full-sib family. It is straightforward to derive the explicit expressions of the maximum-likelihood estimates (MLEs) for the frequencies of these nine formation modes on the basis of the following likelihood function given the observed marker data (ℳ and 𝒩),
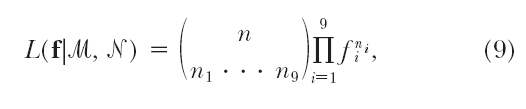 |
9 |
from which we have
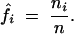 |
By substituting the MLEs of f into Equations 6–8, we derive a closed-form solution for the EM algorithm to estimate α, β, and r. In the E-step, calculate the expected number of recombination events for each cell in matrix 3 using Equations 4 and 5. In the M-step, use the updated values from the E-step to estimate the MLEs of the parameters based on Equations 6–8. The E- and M-steps are iterated until the estimates converge to stable values.
For a given data set, we need to estimate these parameters under all possible parental linkage phases and choose a most likely phase on the basis of the likelihood values calculated by Equation 9. However, because of the symmetrical effect, some different linkage phases may have the same likelihood value. In this case, the recombination fraction r should be used as a criterion; the linkage phase corresponding to a small estimate of r is a more correct one.
We take a further step to obtain the MLE of the probability with which a linkage phase occurs in the heterozygous tetraploid. Let 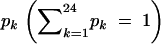 be the probability of the kth phase for the tetraploid parent. For observed marker data with unknown linkage phase, we formulate the likelihood function on the basis of a polynomial mixture expressed as
be the probability of the kth phase for the tetraploid parent. For observed marker data with unknown linkage phase, we formulate the likelihood function on the basis of a polynomial mixture expressed as
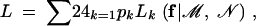 |
10 |
which leads to
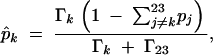 |
where
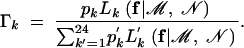 |
The MLE of pk can be used to determine an optimal linkage phase.
Partially informative markers:
Whereas a fully informative marker with four distinct alleles 1, 2, 3, 4 produces 10 observable gametes in a multivalent tetraploid, a partially informative markers produces <10 observable gamete genotypes because some of the four alleles are identical. For example, genotype 1122 produces an array of 10 gametes (11, 11, 22, 22, 11, 12, 12, 12, 12, and 22) that are collapsed into three observable genotypes 11, 12, and 22. The marker genotype that produces these three observable gamete genotypes can also be 1222 or 1112. However, because these three marker genotypes 1222, 1122, and 1112 are phenotypically identical although they produce different frequencies of gamete genotypes 11, 12, and 22, they will provide different pieces of information for linkage analysis. R. L. Wu et al. (2001) provided an algorithm to characterize marker genotypes on the basis of their segregation patterns in a progeny.
To clearly describe statistical algorithms for linkage analysis using partially informative markers, we start with a specific example, from which a more general algorithm can be derived. Suppose we have marker genotypes 1122 for marker ℳ and 1122 for marker 𝒩 for a heterozygous multivalent tetraploid. There are three possible linkage phases between these two markers expressed, in order, as
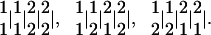 |
11 |
Each of these two markers has 3 gamete genotypes (11, 12, and 22), which form 9 joint gamete genotypes with observations denoted in matrix form as
 |
with 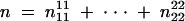 , where the superscripts and subscripts denote the gamete phenotypes at markers ℳ and 𝒩, respectively. Under the assumption of the first linkage phase, the 100 two-marker gamete genotypes shown in the h matrix (2) are collapsed to 9 observable genotypes with the gamete frequency matrix expressed as
, where the superscripts and subscripts denote the gamete phenotypes at markers ℳ and 𝒩, respectively. Under the assumption of the first linkage phase, the 100 two-marker gamete genotypes shown in the h matrix (2) are collapsed to 9 observable genotypes with the gamete frequency matrix expressed as 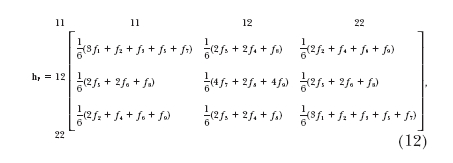 where each cell is indexed by Fi1…iL, that is, the sum of the corresponding gamete mode frequencies, fi1, · · · , fiL, multiplied by the coefficients specified in the matrix. For example, F12357 = 3f1 + f2 + f3 + f5 + f7; the same is held for the rest.
where each cell is indexed by Fi1…iL, that is, the sum of the corresponding gamete mode frequencies, fi1, · · · , fiL, multiplied by the coefficients specified in the matrix. For example, F12357 = 3f1 + f2 + f3 + f5 + f7; the same is held for the rest.
We derive a two-stage hierarchic model for the EM algorithm to estimate the MLEs of the gamete mode frequencies and the recombination fraction. In the E-step at the upper hierarchy, we calculate the expected proportion of one particular gamete mode il (shown by the superscript) to all the L possible gamete modes in each cell of matrix (12), i.e.,
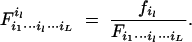 |
These proportions are used to provide the MLEs of the gamete mode frequency fi in the M-step on the basis of
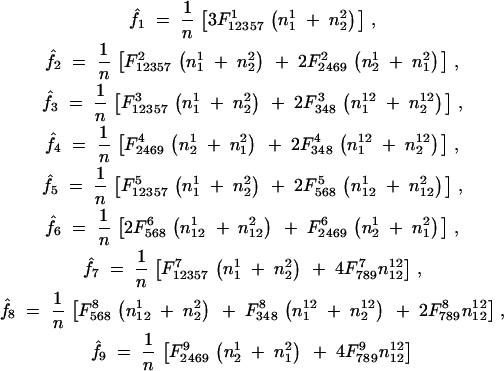 |
With the MLEs of these fi's, the frequencies of double reduction are then estimated using Equations 6 and 7. At the lower hierarchy, the EM algorithm is implemented to estimate the MLE of the recombination fraction on the basis of the updated fi values using Equations 4, 5, and 8. As in the case for fully informative markers, the most likely linkage phase for two partially informative markers should be determined on the basis of the likelihood values calculated under each phase. Meanwhile, the linkage probabilities can be calculated.
The algorithm described above for two particular markers can be generalized to any partially informative markers. To do so, we need an automatic procedure for deriving the collapsed gamete probability matrix (hP) as shown in matrix (12) as an example. This can be done by designing a left (dL) and right matrix (dR) that aims to reduce the gamete frequency matrix (2) for fully informative markers. Assume the second linkage phase in display (11), and then we have
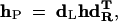 |
where
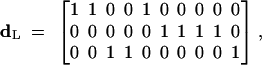 |
and
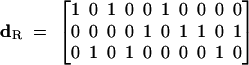 |
For any particular pair of markers, left and right design matrices can be determined on the basis of marker genotypes and linkage phases.
THE ZYGOTE MODEL
Consider two outbred multivalent tetraploids that are crossed to generate a full-sib family for the construction of a linkage map. For two fully informative markers at each of which there are eight different alleles between the two parents, the zygote genotype for an offspring can be uniquely determined by the genotypes of two gametes each derived from a different parent. Because each parent produces (10 × 10) gamete genotypes as shown in matrix (2), we have a total of 10,000 observable zygote genotypes that can be expressed in a (10 × 10) ⊗ (10 × 10) matrix, where ⊗ is the Kronecker product. All these observed zygote genotypes can be sorted into nine distinct gamete modes for each of the two parents, from which we can obtain the MLEs of various gamete mode frequencies for one parent (fi) and the second parent (gi). These estimated f̂i and ĝi values are then used to estimate the frequencies of double reductions and the recombination fraction between the two linked markers with the EM algorithm described in the gamete model.
The most challenging aspect for linkage analysis in a full-sib family is the development of statistical models with partially informative markers. This is due to the fact that the same genotypes will be collapsed at both the gamete and zygote levels. Consider two crossed parents with a combination of parental linkage phases between two linked markers expressed as
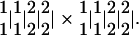 |
13 |
We have provided the collapsed matrix (12) for observed gamete mode frequencies for one parent under this linkage phase. Let  and
and 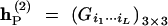 be such collapsed matrices for the two parents, respectively, with Fi1···iL and Gi1···iL defined in matrix (12). Thus, the frequencies of distinct zygote genotypes between the two parents are the Kronecker product of these two collapsed matrices, i.e.,
be such collapsed matrices for the two parents, respectively, with Fi1···iL and Gi1···iL defined in matrix (12). Thus, the frequencies of distinct zygote genotypes between the two parents are the Kronecker product of these two collapsed matrices, i.e.,
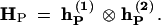 |
14 |
The above distinct zygote genotype frequencies will further be collapsed according to three marker types. For the first marker type, an exact correspondence between marker phenotypes and marker genotypes exists. As a result of this correspondence, marker genotypes with different allele dosages, e.g., 1112, 1122, and 1222, are phenotypically different. According to this genotype model, we will have five phenotypically observable zygote genotypes 1111, 1112, 1122, 1222, and 2222 for marker cross type 1122 × 1122. For a two-marker cross type shown in display (13), we will obtain a 5 × 5 matrix for observable zygote genotype frequencies collapsed from the 9 × 9 matrix of Equation 14. The second marker type is that in which marker genotype is not dependent on allele dosage. Such noncorrespondence makes marker genotypes 1112, 1122, and 1222 phenotypically identical. Using 1, 12, and 2 to denote three possible zygote phenotypes for a marker, this so-called phenotype model has a 3 × 3 collapsed zygote genotype frequency matrix. The last marker type is dominant markers for any of which there are only two phenotypes for a marker. Thus, the dominant model has a 2 × 2 zygote genotype frequency matrix collapsed from matrix (14). The detailed structures of zygote genotype frequency matrix under the genotype, phenotype, and dominant models will be provided upon request. With these known structures, it is not difficult to derive the EM algorithm to estimate observed gamete mode frequencies for two parents (fi and gi).
RESULTS
After the model parameters are estimated under a most likely linkage phase, we need to make statistical hypothesis tests for the frequencies of double reduction (α̂ and β̂) and the recombination fraction (r̂) between two linked markers. In each case, traditional log-likelihoodratio test statistics can be appropriately formulated and calculated under two alternative hypotheses. S. S. Wu et al. (2001) mentioned that the difference of double reduction between the two markers is bounded by two times the recombination fraction in tetraploid. We can also test whether position-dependent double reductions follow a particular pattern (Butruille and Boiteux 2000).
It is imperative that our linkage model is examined in terms of its estimation precision and power. The statistical behavior of a model can be investigated through two approaches, one based on the calculation of information amount and the other based on simulation studies. The information matrix of parameter estimates can be estimated by calculating the asymptotic (co)variance matrix of the MLEs of the model parameters that include the frequencies of double reduction and recombination fraction. The standard statistical methods based on the expectation of the second derivatives of the likelihood function can be used to estimate such an asymptotic (co)variance matrix. As the frequencies of double reduction are estimated from the gamete mode frequencies, the sampling variances of the MLEs of these parameters are also estimated through the gamete mode frequencies.
We performed extensive simulation studies to investigate the statistical behavior of our model for linkage analysis in multivalent tetraploids. We simulate a full-sib family of different sample sizes (n = 100, 200, and 400) by crossing two multivalent tetraploid parents, in a range of double reduction (0.05–0.30) and recombination fraction (0.05 and 0.25). The means of the MLEs of these parameters and their standard errors based on 1000 simulation replicates under the genotype model are illustrated in Table 1. Our model provides reasonable estimates of all the model parameters. The precision of the MLEs, as assessed by the standard errors, increases for all the parameters with increased sample sizes and with increased degrees of linkage. Different degrees of double reduction can be precisely estimated. The percentage of correctly determining a parental linkage phase is high, increasing with sample size and the degree of linkage.
TABLE 1.
The MLEs of the frequencies of double reduction (α, β) and the recombination fraction (r) and their standard errors (in parentheses) estimated from the genotype model for a simulated full-sib family with a sample size ofn = 100, 200, and 400
| n | α | β | r | α̂ | β̂ | r̂ | %1 | %2 | %3 |
|---|---|---|---|---|---|---|---|---|---|
| Parental cross type 1122/1122 × 1111/1111 | |||||||||
| 100 | 0.05 | 0.1 | 0.05 | 0.047 (0.0259) | 0.098 (0.0283) | 0.057 (0.0251) | 95.4 | 0 | 4.6 |
| 200 | 0.05 | 0.1 | 0.05 | 0.048 (0.0244) | 0.099 (0.0264) | 0.056 (0.0183) | 97.8 | 0 | 2.2 |
| 400 | 0.05 | 0.1 | 0.05 | 0.047 (0.0184) | 0.098 (0.0198) | 0.054 (0.0132) | 99.4 | 0 | 0.6 |
| 100 | 0.10 | 0.3 | 0.25 | 0.111 (0.0598) | 0.307 (0.0654) | 0.271 (0.0682) | 87.6 | 3.8 | 8.6 |
| 200 | 0.10 | 0.3 | 0.25 | 0.104 (0.0458) | 0.300 (0.0492) | 0.264 (0.0498) | 88.0 | 4.4 | 7.6 |
| 400 | 0.10 | 0.3 | 0.25 | 0.103 (0.0343) | 0.303 (0.0365) | 0.261 (0.0669) | 86.8 | 3.2 | 10.0 |
| Parental cross type 1122/1122 × 1122/1122 | |||||||||
| 100 | 0.05 | 0.1 | 0.05 | 0.049 (0.0276) | 0.102 (0.0405) | 0.068 (0.0397) | 81.0 | 0 | 19.0 |
| 200 | 0.05 | 0.1 | 0.05 | 0.048 (0.0217) | 0.096 (0.0312) | 0.064 (0.0308) | 88.6 | 0 | 11.0 |
| 400 | 0.05 | 0.1 | 0.05 | 0.049 (0.0155) | 0.095 (0.0240) | 0.060 (0.0219) | 94.7 | 0 | 5.3 |
| 100 | 0.10 | 0.3 | 0.25 | 0.115 (0.0598) | 0.305 (0.0721) | 0.273 (0.0620) | 88.0 | 2.6 | 9.4 |
| 200 | 0.10 | 0.3 | 0.25 | 0.102 (0.0472) | 0.298 (0.0552) | 0.268 (0.0729) | 87.2 | 3.4 | 9.4 |
| 400 | 0.10 | 0.3 | 0.25 | 0.105 (0.0365) | 0.302 (0.0422) | 0.260 (0.0311) | 85.2 | 2.5 | 12.3 |
| Parental cross type 1122/1122 × 1122/1122 | |||||||||
| 100 | 0.05 | 0.1 | 0.05 | 0.049 (0.0297) | 0.101 (0.0325) | 0.056 (0.0200) | 96.3 | 0 | 3.7 |
| 200 | 0.05 | 0.1 | 0.05 | 0.048 (0.0228) | 0.100 (0.0252) | 0.055 (0.0141) | 98.7 | 0 | 1.3 |
| 400 | 0.05 | 0.1 | 0.05 | 0.048 (0.0186) | 0.099 (0.0193) | 0.053 (0.0096) | 99.5 | 0 | 0.5 |
| 100 | 0.10 | 0.3 | 0.25 | 0.108 (0.0603) | 0.277 (0.0891) | 0.279 (0.1076) | 75.3 | 0 | 24.7 |
| 200 | 0.10 | 0.3 | 0.25 | 0.105 (0.0416) | 0.288 (0.0661) | 0.275 (0.0732) | 80.6 | 0 | 19.4 |
| 400 | 0.10 | 0.3 | 0.25 | 0.101 (0.0310) | 0.292 (0.0439) | 0.272 (0.0565) | 88.2 | 0.1 | 11.7 |
%1, %2, and %3 are the percentages of the simulation replicates that correctly determine, cannot determine, and incorrectly determine the linkage phase for a given parent cross type, respectively, among 1000 simulation replicates.
As expected, the estimation precision of the model parameters is a function of marker type. As compared to fully informative markers (S. S. Wu et al. 2001), the recombination fraction between partially informative markers is more difficult to estimate for the same sample size and linkage degree. Of the three parent cross types simulated in this study, 1122/1122 × 1111/1111 is the most informative, providing the best estimates of the recombination fraction (Table 1). Parent cross type 1122/1122 × 1122/1122 is better in estimating a stronger linkage (r = 0.05) than 1122/1112 × 1122/1112, but the inverse seems true in estimating a looser linkage (r = 0.25). For parent cross type 1122/1112 × 1122/1112, double reduction can be better estimated for marker 1122 than for 1112.
The same simulation scheme was also used to test our statistical methods under the phenotype and dominant models (results not shown). Compared to the genotype model, these two models have markers that are less informative. But our model is still able to provide reliable estimates of all the model parameters for these markers, although the estimation precision is reduced especially for loosely linked markers and smaller sample sizes.
Comparison with Luo et al.'s (2004) model:
Luo et al. proposed a simplified model for characterizing nine gamete mode frequencies (see their Table 1). To demonstrate the advantage of our model over Luo et al.'s model, we performed an additional simulation study based on the simulation scheme by these authors. They used a parameter, λ, to specify the relative proportion of bivalent and quadrivalent pairings. When λ is zero, all chromosomal pairings are quadrivalent. On the basis of λ = 0, α = 0.10, and r = 0.10, we used the Luo et al. model to simulate 200 full-sib progeny from two tetraploid parents, 1220/1222 and 2355/1130. Table 2 describes the means and standard errors of the MLEs from our model, compared with those from Luo et al.'s model. The consistency of the results between the two models suggests that our model can deal well with the case on which Luo et al. derived their model.
TABLE 2.
Results from a reciprocal analysis
| Analytical model | α | β | r | α̂ | β̂ | r̂ |
|
%1 | %2 | %3 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Data simulated by the Luo et al. model | |||||||||||
| Wu-Ma | 0.10 | 0.14 | 0.10 | 0.101 (0.028) | 0.139 (0.027) | 0.105 (0.028) | −687 | 100 | 0 | 0 | |
| Luo et al. | 0.10 | 0.14 | 0.10 | 0.100 (0.026) | 0.139 (0.020) | 0.103 (0.029) | −688 | 100 | 0 | 0 | |
| Data simulated by the Wu-Ma model | |||||||||||
| Wu-Ma | 0.10 | 0.14 | 0.10 | 0.097 (0.031) | 0.141 (0.028) | 0.100 (0.025) | −742 | 100 | 0 | 0 | |
| Luo et al. | 0.10 | 0.14 | 0.10 | 0.089 (0.026) | 0.134 (0.021) | 0.113 (0.034) | −765 | 100 | 0 | 0 | |
| Wu-Ma | 0.10 | 0.30 | 0.25 | 0.096 (0.023) | 0.295 (0.034) | 0.256 (0.036) | −683 | 100 | 0 | 0 | |
| Luo et al. | 0.10 | 0.30 | 0.25 | 0.109 (0.035) | 0.209 (0.092) | 0.347 (0.112) | −691 | 97 | 0 | 3 | |
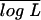
The data simulated by our (Wu-Ma) and Luo et al.'s models are analyzed by the two models, respectively. The marker cross type for a full-sib family of size n = 200 is the one used by Luo et al., expressed as 1220/1222 × 3455/1130.  is the mean of the log-likelihood values obtained from 200 simulations. See Table 1 for explanations of the other parameters.
is the mean of the log-likelihood values obtained from 200 simulations. See Table 1 for explanations of the other parameters.
We used the same values of parameters (λ, α, and r) to simulate a second data set based on our model. This data set was analyzed simultaneously by our and Luo et al.'s models, with the results shown in Table 2. Our model provides precise estimates of all the parameters for the case on which our model was derived. But the estimates from Luo et al.'s model were biased for the three parameters. The estimation of r from their model has a larger sampling error than our estimation. For the two markers with a looser linkage (r = 0.25) and larger double reduction, Luo et al.'s model provided highly biased and imprecise estimates for the recombination fraction and double reduction (Table 2). For example, Luo et al.'s model estimated r = 0.25 as r̂ = 0.347 with a sampling error of 0.112, whereas these two estimated values from our model were 0.256 and 0.036. Our reciprocal-simulation studies suggest that our model has broader adaptation to different situations than Luo et al.'s model. In practice, Luo et al.'s model can be viewed as a special case of our model, although a biological justification of their model is unclear.
DISCUSSION
Double reduction is an important cytological characteristic of polyploids that undergo multivalent chromosomal pairings during meiosis. Double reduction may play a significant role in plant evolution and the maintenance of genetic polymorphism in natural populations. Moreover, coupled with the crossing-over events between different chromosomes, double reduction affects the patterns of gene cosegregation and therefore linkage analysis. S. S. Wu et al. (2001) have, for the first time, devised a maximum-likelihood approach, implemented with the EM algorithm, to integrate double reduction into linkage analysis in tetraploids. Wu et al.'s model made use of 11 different classifications of two-locus gamete formations, derived by Fisher (1947), during tetraploid meiosis and has proven to be powerful for simultaneous estimation of the frequencies of double reduction and the recombination fraction between different loci.
To clearly present their ideas, S. S. Wu et al.'s (2001) model was derived on the basis of fully informative markers in which alleles are all different among homologous chromosomes between two parents used for a cross. For a polyploid genome project, although fully informative markers are the important target for extracting the information for genetic polymorphisms, some other less informative markers are also widely used in part because their inclusion can reflect a more comprehensive picture of the structure and organization of the polyploid genome and they are economically cheaper to obtain. In this article, a more general model has been developed for linkage analysis between any type of partially informative markers in a multivalent tetraploid. This new model preserves the advantages of the earlier model by S. S. Wu et al. (2001), but will be more broadly useful in practice.
To better describe our model, we have taken two subsequent steps toward a complete understanding of linkage analysis in multivalent tetraploids. Our work in the first step is based on a simpler pseudo-test backcross design in which one parent is heterozygous whereas the other is null. Thus, our modeling process of gene cosegregation in the progeny can take advantage of simpler gamete segregation patterns. With clear analytical lines, our model is readily expanded to a full-sib family derived from two heterozygous parents in the second step. Extensive simulation studies were carried out to investigate the statistical behaviors of our model. It is robust in that the frequencies of double reduction and the recombination fraction can be reasonably estimated in a range of sample sizes and parameter values. The model, incorporated within a mixture-likelihood function based on a multinomial distribution, has power to estimate the probabilities of any possible parental linkage phases between two given markers and further determine the most likely one for the estimation of their linkage.
Although our presentation was based on a particular parent cross type, the model has been extended to several practically important situations. These, implemented in a more complete computer package, include an unknown dosage of alleles, the existence of null alleles, ambiguous parent cross type, and the mixture of bivalent and multivalent pairings. This package allows for the test of various hypotheses regarding the degree of double reduction and the recombination fraction and has the capacity to calculate the sampling errors of the MLEs of the model parameters through the asymptotic (co)variance of their MLEs.
We have focused the model derivation on a tetraploid that undergoes only multivalent pairing. Because many species belong to such multivalent tetraploids, this model will find its immediate application in practice. However, there are also some species that display both bivalent and multivalent pairings. Luo et al. (2004) attempted to model the mixed bivalent and multivalent pairings by defining an additional proportion parameter λ. While statistically reasonable, their approach seems not to be founded on solid cytological mechanisms for meiosis in a polyploid. They treated bivalent and multivalent pairings as two totally different meiotic processes. This may not always be correct if the degree of multivalent pairings depends on the relative relatedness among different chromosomes in a set, which also determines the pattern of bivalent pairings. Sybenga (1994) proposed a series of cytological models to understand the chromosome relatedness by estimating the so-called preferential pairing factor defined as the probability with which more identical chromosomes pair more frequently than less identical chromosomes. To better perform linkage analysis in polyploids that undergo the mixture of bivalent and multivalent pairings, Sybenga's preferential pairing factor that specifies meiotic mechanisms for such polyploids should be incorporated into our mapping model (see Wu et al. 2004).
Our model based on pairwise analysis has provided a first step toward multipoint analysis and map construction in multivalent tetraploids. Subsequent work is needed to develop an efficient algorithm for grouping different markers and ordering those that are assigned within the same group. Some useful algorithms include the hidden Markov chain model advocated by Lander and Green (1987) for diploid populations and the evolutionary strategy algorithm proposed by Mester et al. (2003). A better approach for ordering markers may be based on three-point analysis. In diploids, three-point analysis has been detected to be more powerful and precise than two-point analysis (Wu et al. 2002b; Lu et al. 2004). It is thus worthwhile to implement three-point analysis into an algorithm for map construction in polyploids. With a powerful marker-ordering algorithm available for polyploids at different levels of ploidy, we will be in a better position to understand the genetic differentiation among polyploid genomes and characterize the genetic architecture of quantitatively inherited traits for this unique group of species.
Acknowledgments
We thank two anonymous referees for their constructive comments on this article. This work is partially supported by an Outstanding Young Investigators Award (no. 30128017) of the National Natural Science Foundation of China and the University of Florida Research Opportunity Fund (no. 02050259) to R.W. The publication of this article is approved as journal series no. R-10578 by the Florida Agricultural Experimental Station.
References
- Bever, J. D., and F. Felber, 1992. The theoretical population genetics of autopolyploidy. Oxf. Surv. Evol. Biol. 8: 185–217. [Google Scholar]
- Butruille, D. V., and L. S. Boiteux, 2000. Selection-mutation balance in polysomic tetraploids: impact of double reduction and gametophytic selection on the frequency and subchromosomal localization of deleterious mutations. Proc. Natl. Acad. Sci. USA 97: 6608–6613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darlington, C. D., 1929. Chromosome behaviour and structural hybridity in the Tradescantiae. J. Genet. 21: 207–286. [Google Scholar]
- da Silva, J., M. E. Sorrells, W. L. Burnquist and S. D. Tanksley, 1995. RFLP linkage map and genome analysis of Saccharum spontaneum. Genome 36: 782–791. [DOI] [PubMed] [Google Scholar]
- Fisher, R. A., 1947. The theory of linkage in polysomic inheritance. Philos. Trans. R. Soc. Ser. B 233: 55–87. [Google Scholar]
- Hackett, C. A., J. E. Bradshaw, R. C. Meyer, J. W. McNicol, D. Milbourne et al., 1998. Linkage analysis in tetraploid species: a simulation study. Genet. Res. 71: 143–154. [Google Scholar]
- Haynes, K. G., and D. S. Douches, 1993. Estimation of the coefficient of double reduction in the cultivated tetraploid potato. Theor. Appl. Genet. 85: 857–862. [DOI] [PubMed] [Google Scholar]
- Julier, B., S. Flajoulot, P. Barre, G. Cardinet, S. Santoni et al., 2003. Construction of two genetic linkage maps in cultivated tetraploid alfalfa (Medicago sativa) using microsatellite and AFLP markers. BMC Plant Biol. 3 (1): 9.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander, E. S., and P. Green, 1987. Construction of multilocus genetic linkage maps in humans. Proc. Natl. Acad. Sci. USA 84: 2363–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, Q., Y. H. Cui and R. L. Wu, 2004. A multilocus likelihood approach to joint modeling of linkage, parental diplotype and gene order in a full-sib family. BMC Genet. 5: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, Z. W, C. A. Hackett, J. E. Bradshaw, J. W. McNicol and D. Milbourne, 2001. Construction of a genetic linkage map in tetraploid species using molecular markers. Genetics 157: 1369–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, Z. W., R. M. Zhang and M. J. Kearsey, 2004. Theoretical basis for genetic linkage analysis in autotetraploid species. Proc. Natl. Acad. Sci. USA 101: 7040–7045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, C.-X., G. Casella, T. C. Osborn and R. L. Wu, 2002. A unified framework for mapping quantitative trait loci in bivalent tetraploids using single-dose restriction fragments: a case study from alfalfa. Genome Res. 12: 1974–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mester, D., Y. Ronin, D. Minkov, E. Nevo and A. Korol, 2003. Constructing large-scale genetic maps using an evolutionary strategy algorithm. Genetics 165: 2269–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, R. C., D. Milbourne, C. A. Hackett, J. E. Bradshaw, J. W. McNichol et al., 1998. Linkage analysis in tetraploid potato and association of markers with quantitative resistance to late blight (Phytophthora infestans). Mol. Gen. Genet. 259: 150–160. [DOI] [PubMed] [Google Scholar]
- Ming, R., S. C. Liu, Y. R. Lin, J. da Silva, W. Wilson et al., 1998. Detailed alignment of Saccharum and Sorghum chromosomes: comparative organization of closely related diploid and polyploid genomes. Genetics 150: 1663–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming, R., S.-C. Liu, J. E. Irvine and A. H. Paterson, 2001. Comparative QTL analysis in a complex autopolyploid: candidate genes for determinants of sugar content in sugarcane. Genome Res. 11: 2075–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripol, M. I., G. A. Churchill, J. A. G. da Silva and M. Sorrells, 1999. Statistical aspects of genetic mapping in autopolyploids. Gene 235: 31–41. [DOI] [PubMed] [Google Scholar]
- Soltis, P. S., and D. E. Soltis, 2000. The role of genetic and genomic attributes in the success of polyploids. Proc. Natl. Acad. Sci. USA 97: 7051–7057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sybenga, J., 1994. Preferential pairing estimates from multivalent frequencies in tetraploids. Genome 37: 1045–1055. [DOI] [PubMed] [Google Scholar]
- Wu, R. L., M. Gallo-Meagher, R. C. Littell and Z-B. Zeng, 2001. A general polyploid model for analyzing gene segregation in outcrossing tetraploid species. Genetics 159: 869–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, R. L., C.-X. Ma and G. Casella, 2002. a A bivalent polyploid model for linkage analysis in outcrossing tetraploid species. Theor. Popul. Biol. 62: 129–151. [DOI] [PubMed] [Google Scholar]
- Wu, R. L., C.-X. Ma, I. Painter and Z-B. Zeng, 2002. b Simultaneous maximum likelihood estimation of linkage and linkage phases in outcrossing species. Theor. Popul. Biol. 61: 349–363. [DOI] [PubMed] [Google Scholar]
- Wu, R. L., C. X. Ma and G. Casella, 2004. A mixed polyploid model for linkage analysis in tetraploids. J. Comput. Biol. 11: 562–580. [DOI] [PubMed] [Google Scholar]
- Wu, S. S., R. L. Wu, C. X. Ma, Z-B. Zeng, M. Yang et al., 2001. A multivalent pairing model of linkage analysis in autotetraploids. Genetics 159: 1339–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]