

Abstract
The protein phosphatase 2A (PP2A) regulatory subunit Tap42 is essential for target of rapamycin (TOR)-mediated signaling in yeast, but its role in higher eukaryotes has not been established. Here we show that Tap42 does not contribute significantly to TOR signaling in Drosophila, as disruption of the Tap42 gene does not cause defects in cell growth, metabolism, or S6-kinase activity characteristic of TOR inactivation. In addition, Tap42 is not required for increased cell growth in response to activation of TOR signaling. Instead, we find that Tap42 mutations cause disorganization of spindle microtubules in larval neuroblasts, leading to a preanaphase mitotic arrest in these cells. Loss of Tap42 ultimately results in increased JNK signaling, caspase activation, and cell death. These phenotypes are associated with increased accumulation and nuclear localization of PP2A in Tap42 mutant cells. Our results demonstrate that the role of Tap42 in TOR signaling has not been conserved in higher eukaryotes, indicating fundamental differences in the mechanisms of TOR signaling between yeast and higher eukaryotes.
THE target of rapamycin (TOR) protein kinase promotes cell growth in response to favorable nutrient and environmental conditions. By regulating the phosphorylation of its downstream effector proteins, TOR controls a number of metabolic processes important for cell growth, including ribosome biogenesis and translation, nutrient import, transcription, and autophagy. In response to nutrient withdrawal or treatment with rapamycin, a specific inhibitor of TOR, downstream targets of TOR are rapidly dephosphorylated, leading to a reversible growth arrest.
In addition to direct phosphorylation of its substrates, TOR may also control the phosphorylation status of these proteins by modifying the activity of protein phosphatases. In yeast, TOR promotes the association of the type 2A-related phosphatases Pph21/Pph22 and Sit4 with the 42-kDa regulatory subunit Tap42 (Di Como and Arndt 1996). Inactivation of TOR signaling results in dephosphorylation of Tap42 and dissociation of Tap42/PP2A complexes. Mutations in Tap42 block the TOR-dependent phosphorylation of a number of proteins and mimic many of the effects of rapamycin treatment, including growth arrest, reduced translation, nutrient transporter degradation, and induction of stress response genes (Di Como and Arndt 1996; Schmidt et al. 1998; Beck and Hall 1999; Duvel et al. 2003). Moreover, specific Tap42 point mutations confer dominant rapamycin resistance (Di Como and Arndt 1996; Duvel et al. 2003). Tap42 thus behaves overall as a positive effector of TOR signaling, although some TOR-regulated processes such as ribosomal protein gene transcription and autophagy are independent of or antagonized by Tap42 (Noda and Ohsumi 1998; Duvel et al. 2003). Current models posit that Tap42 may act as a modifier of PP2A substrate specificity or that phosphorylation of Tap42 may convert it from an activator to a repressor of PP2A activity (Duvel and Broach 2004).
PP2A-related phosphatases may also play an important role in TOR signaling in mammalian cells, although their role is less well defined than in yeast. Inhibitors of type I and type 2A phosphatases such as calyculin and okadaic acid antagonize rapamycin-induced dephosphorylation of the TOR substrates S6K and 4E-BP1 and promote S6K activation and cell growth (Leicht et al. 1996; Lin and Lawrence 1997; Hara et al. 1998; Peterson et al. 1999; Krause et al. 2002). Furthermore, rapamycin treatment increases the activity of PP2A toward 4E-BP1 in vitro (Peterson et al. 1999), suggesting that TOR may promote phosphorylation of its substrates in part by restraining PP2A activity. Consistent with this idea, PP2A can directly bind and inhibit wild-type S6K, whereas a truncated version of S6K has reduced affinity for PP2A and does not require TOR for its activity (Peterson et al. 1999; Westphal et al. 1999; Gonzalez-Garcia et al. 2002). The potential function of Tap42 homologs in TOR signaling in higher eukaryotes is also less clear than in yeast. The mammalian homolog of Tap42, α4, binds to type 2A-related phosphatases, and some studies have shown that rapamycin disrupts this association (Murata et al. 1997; Inui et al. 1998), although others found that it does not (Chen et al. 1998; Nanahoshi et al. 1998; Kloeker et al. 2003). As in yeast, α4 may act to modify PP2A specificity, as it has been shown to inhibit in vitro phosphatase activity toward 4E-BP1 and increase it toward other substrates (Murata et al. 1997; Inui et al. 1998; Nanahoshi et al. 1998). T or B lymphocytes carrying a conditional disruption of α4 show a severe defect in antigen-induced proliferation, yet remain sensitive to rapamycin, suggesting a partial overlap in function between TOR and α4 (Inui et al. 2002; Hua et al. 2003).
In addition to a potential role in TOR signaling, α4 has been identified in several independent studies as a binding partner for the tripartite motif (TRIM) family protein MID1 (Liu et al. 2001; Reymond et al. 2001; Trockenbacher et al. 2001; Short et al. 2002; Schweiger and Schneider 2003). Mutations in the MID1 gene are responsible for the X-linked form of Opitz G/BBB syndrome, characterized by developmental defects of the ventral midline (Schweiger and Schneider 2003). MID1 is a microtubule-associated protein (Schweiger et al. 1999) and has been shown to possess ubiquitin ligase activity toward PP2A (Trockenbacher et al. 2001). By acting as an adaptor to promote MID1/PP2A association, α4 may thus function to direct PP2A activity to microtubules and/or to regulate PP2A stability. Whether and how these functions of Tap42/α4 relate to TOR signaling is not clear.
In Drosophila, the functions of both TOR and PP2A have been well characterized genetically, but a role for Tap42 has not been described. Here we report the isolation of mutations in the Drosophila Tap42 gene. Although Tap42 is essential for cell viability, we demonstrate by a number of molecular and phenotypic criteria that it does not play an appreciable role in TOR signaling. Our results reveal that a major function of Tap42 is to regulate the abundance and intracellular localization of PP2A.
MATERIALS AND METHODS
Generation of a Tap42 deletion mutant:
Tap42 sequence alignments and E-values were determined from pairwise BLAST comparisons using the BLOSUM62 matrix (Tatusova and Madden 1999). P[lacW]l(2)k07826, a P-element insertion in the 5′-UTR of Nnp-1, was mobilized through standard crosses to strains expressing P transposase. Resulting w− flies that failed to complement the original l(2)k07826 line were tested for imprecise excision events in PCR reactions with primers flanking the P insertion site and the Tap42 transcription unit. From ∼400 lines generated, a single line Δ305b was identified with a unidirectional deletion that extends into the Tap42 gene. The extent of this deletion was determined by sequencing PCR products generated with oligos flanking the deletion.
Tap42 rescue and expression constructs:
A 12-kb ClaI/EcoRV fragment containing the Tap42 genomic region was excised from P1 phage DS05554 and ligated into ClaI/EcoRV-digested pBluescript KS II. Two DNA fragments from the resulting plasmid were used to generate rescue constructs: a 5.1-kb KpnI/BsaHI fragment, which encodes Nnp-1, CG6523 but not Tap42, and a 6.6-kb KpnI/AvrII fragment, which encodes Nnp-1, CG6523, and Tap42. These fragments were ligated into pP[CaSpeR-4] digested with KpnI/StuI and KpnI/XbaI, respectively. Rescue constructs were injected according to standard procedures into balanced Δ305b mutant embryos. For construction of UAS-Tap42, EST LP01001 (Research Genetics, Birmingham, AL) was digested partially with XhoI and to completion with EcoRI. The 1.4-kb full-length Tap42 cDNA was ligated into EcoRI/XhoI-cut pUAST and injected into yw embryos.
Histochemistry:
Seventy-two 96-hr control and homozygous Tap42 mutant larvae were dissected and fixed 30 min in 3.7% formaldehyde/PBS. Antibodies used were mouse anti-tubulin E7 (Developmental Studies Hybridoma Bank; 1:400 dilution), rabbit anti-phospho-H3 (Upstate Biotechnology; 1:400), mouse anti-MPM2 (Upstate Biotechnology; 1:200), anti-PP2A clone 1D6 (Upstate Biotechnology; 1:400), rabbit anti-active Caspase-3 CM1 (BD Biosciences, 1:10,000), and rabbit anti-β-galactosidase (Cappel; 1:10,000). Anti-Tap42 antibodies were raised in guinea pigs against a glutathione agarose-purified Drosophila Tap42/GST fusion protein, generated in Escherichia coli from a plasmid consisting of the PCR-amplified coding region of EST LP0100 (Research Genetics) ligated into BamHI/EcoRI-digested pGEX-2T (Amersham, Arlington Heights, IL). Pretreatment and lysotracker staining of fat body for autophagy was performed as described (Scott et al. 2004). For phalloidin staining, fixed imaginal discs were stained in 0.165 μm Texas Red-phalloidin (Molecular Probes, Eugene, OR) in PBS for 30 min.
Flow cytometry:
Fluorescence-activated cell sorting (FACS) analysis of wing imaginal discs containing Tap42 and PTEN Tap42 mutant clones was performed as described (Neufeld et al. 1998).
S2 cells:
Drosophila cell culture, RNAi, and Western blotting were performed as described (Gao et al. 2002). The phospho-S6K antibody against Thr 398 (corresponding to Thr 389 of human S6K) was from Cell Signaling Technology.
RESULTS AND DISCUSSION
Generation of Drosophila Tap42 mutants:
BLAST searches of the Drosophila genome revealed a single gene CG31852 with significant identity to Tap42/α4 (Figure 1, A and B). The protein encoded by CG31852 displays 29 and 24% overall identity to human α4 and yeast Tap42, respectively, similar to the 24% identity between the human and yeast proteins (Di Como and Arndt 1996). The central region of α4 shown to mediate interactions with PP2A (Liu et al. 2001; Trockenbacher et al. 2001) is particularly well conserved (35% identity). Consistent with this, antibodies raised against a CG31852-GST fusion recognized a protein of ∼42 kDa and were able to co-immunoprecipitate the PP2A catalytic subunit (PP2Ac) from larval extracts (Figure 1C). We therefore follow the yeast nomenclature and refer to CG31852 as Tap42, for two A-associated protein of 42 kDa.
Figure 1.—
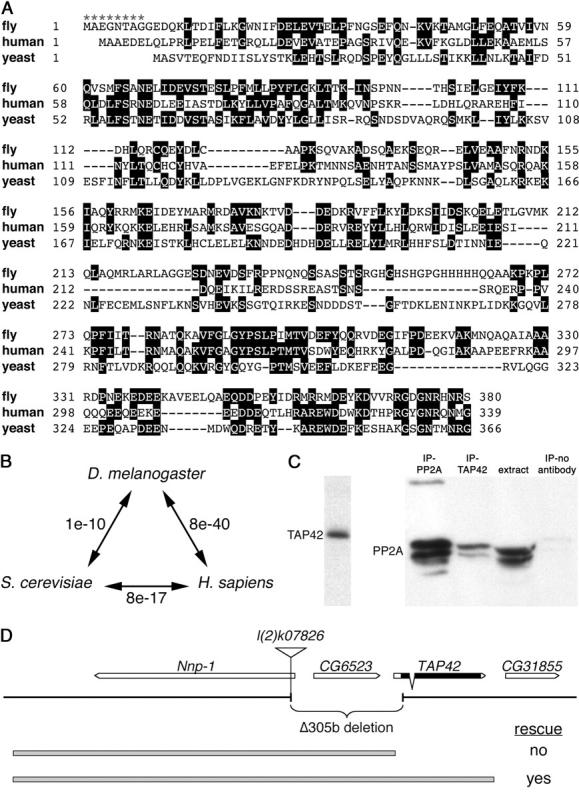
The Drosophila genome encodes a single protein with significant identity to yeast Tap42 and mammalian α4. (A) Alignment of Tap42/α4 protein sequences from D. melanogaster, Homo sapiens, and Saccharomyces cerevisiae. Identical residues are boxed in black. Asterisks indicate the extent of the Δ305b deletion into the Tap42 coding region. (B) E-values of pairwise BLAST comparisons between fly, human, and yeast Tap42/α4 protein sequences. (C) Antibodies raised against Drosophila Tap42 recognize a single band of ∼42 kDa (right) and co-immunoprecipitate PP2A from larval extracts (right, lane 2). (D) Tap42 genomic region, showing the orientation of transcription units, the extent of the Δ305b deletion generated from excision of the l(2)k07826 P element, and the 5.1- and 6.6-kb genomic fragments used for transgenic rescue (shaded bars). Arrows indicate the direction of transcription of each gene. Exon/intron structure and coding region (solid boxes) are shown for Tap42 only. Ability of the rescue constructs to restore viability to flies homozygous for the Δ305b deletion is indicated.
To initiate a genetic analysis of Drosophila Tap42, we made use of a strain harboring a lethal P-element insertion in the Nnp-1 locus ∼1.5 kb upstream of the start of the Tap42 transcription unit (Figure 1D). Through imprecise excision of this P element, a single deletion Δ305b that removed the P element and 1.6 kb of sequences downstream was recovered. The Δ305b deletion extends through the transcription and translation start sites of Tap42 and removes the first eight codons of the Tap42 coding region, thus resulting in a presumptive null allele. As the deletion disrupts both Nnp-1 and Tap42, as well as a small intervening gene CG6523, we generated two genomic rescue constructs: a 6.6-kb genomic fragment that spans all three genes and a 5.1-kb fragment encompassing the Nnp-1 and CG6523 genes but not Tap42 (Figure 1D). Whereas the Δ305b deletion resulted in recessive embryonic lethality, a single copy of the 6.6-kb genomic construct provided full rescue to adult viability and fertility. In contrast, the lethality of Δ305b homozygous animals was not rescued by either one or two copies of the 5.1-kb rescue construct (designated Tap42Δ305b+5.1).
To ask whether the lethality of Tap42Δ305b+5.1 animals was due solely to loss of Tap42, we tested whether ubiquitous expression of Tap42 could rescue these mutants, by introducing a tubulin-GAL4 driver and UAS-Tap42 insertion into the Tap42Δ305b+5.1 mutant background. This combination of rescue constructs allowed development to adulthood, confirming that Tap42Δ305b+5.1 mutants are specifically defective for Tap42. These animals are henceforth referred to as Tap42 mutants. Tap42 mutant animals progressed to the early third instar larval stage with a slight developmental delay and died shortly thereafter, between 96 and 120 hr after egg laying. Thus, Tap42 is an essential gene required for larval development.
Tap42 is not an essential component of the TOR pathway:
In Drosophila, mutations in TOR result in a severe growth arrest during larval development, and mutant animals display an extended larval period of up to 4 weeks (Oldham et al. 2000; Zhang et al. 2000). As this phenotype is rather distinct from that of Tap42- animals, which grow larger but die sooner than TOR mutants, we compared the cellular consequences of Tap42 and TOR mutations. Loss of TOR has been shown to result in reduced cell size, arrest in the G1 phase of the cell cycle, and induction of autophagy (Oldham et al. 2000; Zhang et al. 2000; Scott et al. 2004). In contrast, confocal and FACS analysis revealed no change in the size of Tap42 mutant cells, and the cell cycle phasing of these cells was indistinguishable from that of wild-type controls (Figure 2, A and A′). The size of Tap42 mutant cells in specific G1, S, or G2 cell cycle phases was also indistinguishable from that of wild type. Overexpression of Tap42 using the flip-out GAL4 method (Neufeld et al. 1998) with a UAS-Tap42 transgene also had no measurable effects by FACS analysis (data not shown). As shown previously, loss of TOR signaling results in a marked accumulation of large lysotracker-positive vesicles in the larval fat body, indicative of autophagy induction (Scott et al. 2004). In contrast, lysotracker staining was not observed in Tap42 mutants (Figure 2D). Thus, loss or overexpression of Tap42 does not result in cell growth or metabolism defects typical of bona fide components of the TOR pathway.
Figure 2.—
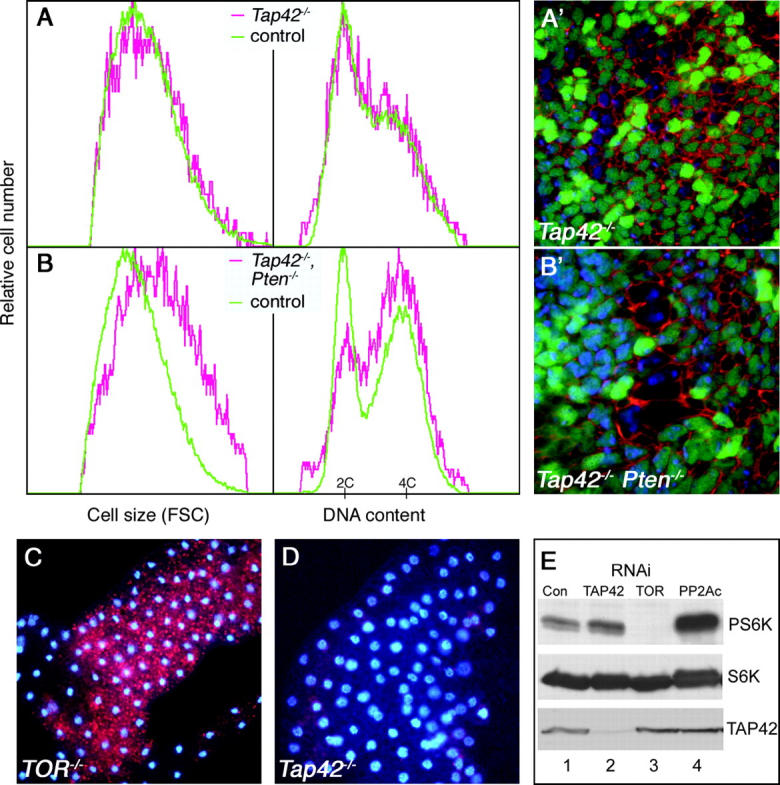
TOR signaling is not disrupted in Tap42 mutant cells. (A) FACS analysis of wing imaginal discs containing Tap42 loss-of-function clones. Tap42- homozygous cells (red traces) do not display the reduced cell size and S/G2 content characteristic of TOR inactivation. Cell size and cell cycle phasing is similar to that of control cells (green traces) from the same discs. (A′) A confocal image of a wing disc containing GFP-marked Tap42 mutant clones, stained with TR-phalloidin to outline cell boundaries. No effect on cell size is observed in the Tap42 mutant cells. (B) FACS and confocal (B′) analysis of cells doubly mutant for Tap42 and PTEN. The pronounced increase in cell size and S/G2 content of PTEN mutants is not blocked by deletion of Tap42. (C and D) LysoTracker Red staining of larval fat body. TOR mutant animals (C) accumulate significant numbers of lysotracker-positive autolysosomes due to induction of autophagy, whereas Tap42 mutants (D) do not stain with lysotracker under normal feeding conditions. (E) Lysates of S2 cells treated with the indicated dsRNAs were probed with anti-phospho-Thr 398-S6K to detect active S6K (top), anti-S6K (middle), and anti-Tap42 (bottom). Control cells were treated with dsRNA against the mammalian CYP7A1 gene as described (Gao et al. 2002). Genotypes: (A) hsflp; Tap42Δ305b FRT40A/UbiGFP FRT40A; P[5.1]-3/+; (B) hsflp; PTENDJ1 Tap42Δ305b FRT40A/UbiGFP FRT40A; P[5.1]-3/+; (C) TORΔP; (D) Tap42Δ305b P[5.1]-2.
TOR promotes cell growth in large part through activation of S6K, as S6K activity is greatly reduced in TOR- cells, and expression of constitutively active S6K can rescue the growth defects of TOR mutants (Oldham et al. 2000; Zhang et al. 2000; Gao et al. 2002; Scott et al. 2004). TOR is also required for stimulation of cell growth in response to PI3K overexpression or loss of the tumor suppressor PTEN (Zhang et al. 2000; Neufeld 2004). To further test the role of Tap42 in TOR signaling, we examined whether Tap42 influences the activity or effects of these molecules. Whereas RNAi-mediated depletion of TOR in S2 cells resulted in complete inactivation and dephosphorylation of S6K (Figure 2E, lane 3), depletion of Tap42 had no effect or in some cases slightly increased S6K phosphorylation (Figure 2E, lane 2), again indicating that TOR signaling is not disrupted in these cells. Depletion of PP2Ac resulted in a marked increase in S6K phosphorylation (Figure 2E, lane 4). Thus, TOR and PP2A have opposing effects on S6K activation, independent of Tap42.
Consistent with these observations, ubiquitous expression of activated S6K did not bypass the arrest of Tap42 mutants (not shown). Furthermore, while TOR mutants block the effects of PTEN inactivation (Zhang et al. 2000), Tap42- cells showed a normal response to disruption of PTEN, as Tap42- PTEN- double-mutant cells displayed the characteristic increase in cell size and S/G2 content of PTEN- single mutants (Figure 2, B and B′). Thus, Tap42 is not required for TOR-dependent growth responses to PI3K/PTEN. Taken together, these data indicate that Tap42 does not contribute significantly to TOR signaling in Drosophila.
Tap42 is required for progression through mitosis:
Given their lack of effect on TOR-mediated processes, we asked whether Tap42 mutants displayed other cellular phenotypes that could account for their lethality. In Drosophila, mutations in PP2Ac or the PP2A-related phosphatase PP4 have been shown to result in specific mitotic defects, including a preanaphase arrest with highly condensed DNA, missing or abnormal mitotic spindles, and multiple centromeres (Snaith et al. 1996; Helps et al. 1998). Mutations in twins, which encodes a B/PR55 regulatory subunit of PP2A, also result in delayed or aberrant progression through anaphase (Gomes et al. 1993). These phosphatases likely antagonize the activity of multiple kinases active during mitosis, including Cdc2, aurora, and polo-like kinases.
To examine the effect of Tap42 on mitotic progression, we stained whole-mount brain preparations from Tap42 mutant larvae with an antibody against phosphorylated histone H3 (PH3), which labels chromosomes of mitotic cells. In control animals, we observed PH3-marked chromosomes with varying configurations and degrees of condensation (Figure 3A), reflecting progression from prophase through late anaphase. In contrast, in Tap42 mutant brains nearly all PH3-positive cells displayed highly condensed chromosomes (Figure 3B). Whereas 14.8% of PH3-marked control cells were in anaphase (n = 682 cells in seven brains), we were unable to identify any PH3-marked cells in anaphase or telophase in Tap42- animals (0/338 cells in 10 brains; Figure 3C). In addition, mitotic spindles were less organized and more diffuse in appearance in Tap42- than in control brains (Figure 3B). We also examined the staining pattern of MPM2, a phospho-epitope found on a number of components of the mitotic apparatus, including regions of the centromere and centrosome. In Tap42- cells, centromeric labeling of MPM2 was absent or reduced, while staining surrounding centrosomes was increased and more diffuse than that in controls (Figure 3E), indicating that Tap42 may differentially affect MPM2 phosphorylation in different cellular compartments. In summary, mutations in Tap42 disrupt spindle morphology and arrest mitosis prior to anaphase entry, phenotypes similar to those caused by mutations in PP2A catalytic and regulatory subunits. While this phenotypic similarity is consistent with a positive role for Tap42 in regulating PP2A activity during mitosis, it may also reflect engagement of a common mitotic checkpoint despite distinct or even opposing primary defects.
Figure 3.—
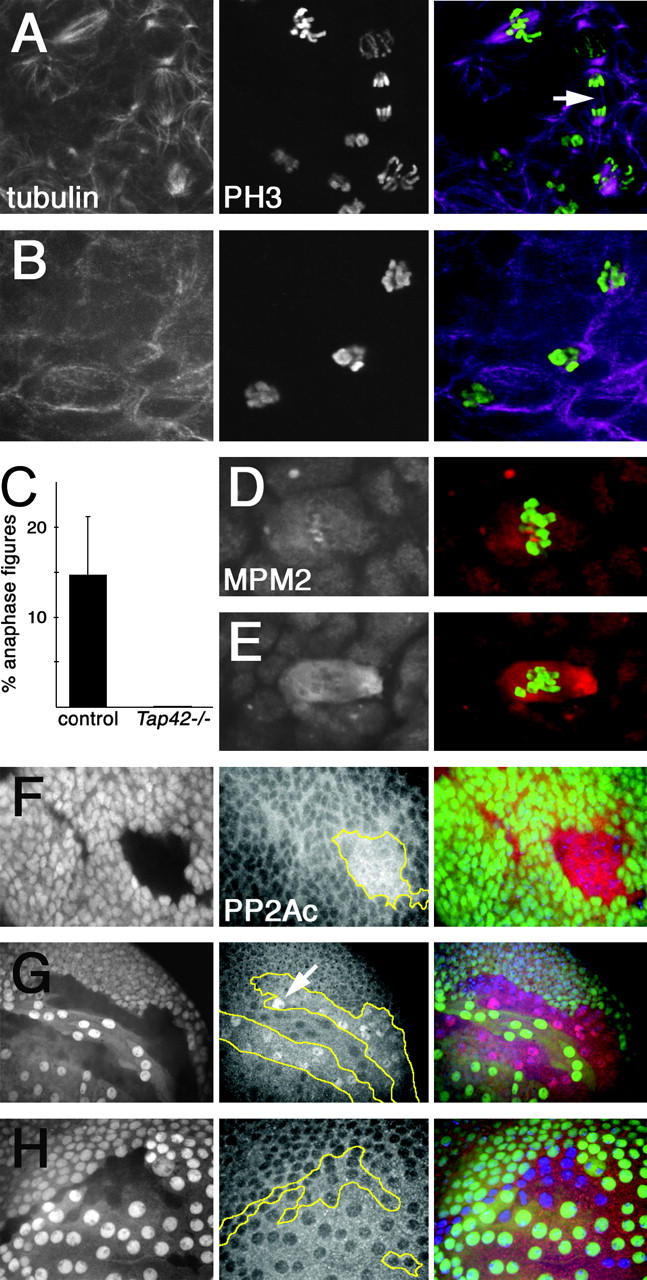
Loss of Tap42 causes metaphase arrest and PP2A accumulation. (A–C) Larval central nervous systems stained with anti-β-tubulin (left) and anti-phospho-H3 (center); right side shows pseudocolor merged images. Control animals (A) display well-defined mitotic spindles and contain PH3-positive chromosomes in all phases of mitosis. Arrow indicates a cell in anaphase. In Tap42 mutants (B), mitotic spindles are smaller and poorly organized, and most PH3-positive chromosomes are in a highly condensed metaphase configuration. Quantitation of the percentage of PH3-positive cells in anaphase in control and Tap42 mutant brains is shown. (D and E) Anti-MPM2 (left) and phospho-H3 (shown in green in merged images, right) staining of larval brains. MPM2 antigen localizes to centrosome and centromere regions in control cells (D). In Tap42 mutants (E), MPM2 staining is lost from centromeres and is stronger and more diffuse near the centrosomes. (F–H) Anti-PP2A staining in wing imaginal discs containing GFP-marked mitotic clones. Left, GFP (which labels wild-type cells); center, PP2A staining; right, the merge. Clonal boundaries are outlined in yellow. Loss of Tap42 results in increased expression of PP2A in both the disc proper (F) and peripodial membrane cells (G). Note the specific accumulation of PP2A in cell nuclei (arrow in G). PP2A levels are not altered in TOR loss-of-function clones (H). Genotypes: (A and D) Tap42Δ305b P[6.6]-2; (B and E) Tap42Δ305b P[5.1]-2; (F and G) hsflp; Tap42Δ305b FRT40A/UbiGFP FRT40A; P[5.1]-3/+; (H) hsflp; TORΔP FRT40A/UbiGFP FRT40A.
Tap42 mutations alter the levels and cellular localization of PP2A:
The defects in spindle microtubules in Tap42 mutants are consistent with prior observations of a specific microtubule-associated pool of PP2A (Sontag et al. 1995). In mammalian cells, Tap42/α4 has been suggested to play a role in targeting PP2Ac to microtubules through its interactions with the microtubule-associated protein MID1 (Cainarca et al. 1999; Schweiger et al. 1999; Liu et al. 2001; Short et al. 2002). Although the consequences of these interactions are unclear, MID1 has been shown to stabilize microtubules when overexpressed (Schweiger et al. 1999). In addition, MID1 acts as an E3 ubiquitin ligase to target microtubule-associated PP2Ac for degragradation, and fibroblasts with mutations in MID1 have elevated PP2A levels and reduced phosphorylation of microtubule-associated proteins (Trockenbacher et al. 2001). We therefore examined PP2Ac expression in cells lacking Tap42. Consistent with a role for Tap42 in contributing to PP2Ac degradation, PP2Ac levels were markedly increased in Tap42- clones in the wing imaginal disc (Figure 3F). Interestingly, this is not a feature shared with other PP2A regulatory subunits, as disruption of A or B subunits results in destabilization of PP2Ac (Liu et al. 2001; Silverstein et al. 2002). In addition, we found that loss of Tap42 altered the intracellular localization of PP2A. Whereas PP2Ac was predominantly cytoplasmic in wild-type cells, loss of Tap42 resulted in increased nuclear localization of PP2Ac; this effect was most noticeable in the large squamous cells of the wing disc peripodial membrane (Figure 3G). In contrast, loss of TOR had no effect on PP2Ac localization (Figure 3H). Thus, Tap42 acts independently of TOR to limit PP2Ac expression levels and to restrict its localization to the cytoplasm. Tap42 may effect the nuclear exclusion of PP2Ac through a direct anchoring mechanism, perhaps to the cytoplasmic microtubule network, or may regulate nuclear permeability or transport rates. Alternatively, loss of Tap42 may lead to nuclear membrane disruption, allowing diffusion and accumulation of PP2A in the nucleus.
Although several MID1-related genes can be identified in Drosophila, none is an obvious MID1 homolog, as each displays significantly higher identity with other TRIM family members. For example, the Drosophila gene most similar to human MID1, CG31721 (blast e-value 1e-41), is a significantly better match to TRIM9 (blast e-value e-177), which localizes not to microtubules but to discrete cytoplasmic bodies of variable size (Reymond et al. 2001). A P-element insertion in CG31721 did not display genetic interactions with Tap42 (data not shown).
Disruption of Tap42 leads to activation of Jun N-terminal kinase signaling and cell death:
In the course of analyzing the phenotypes of Tap42- wing imaginal disc cell clones, we noted that, whereas such clones could be readily identified 48 hr after induction, by 72 hr they were no longer present (Figure 4A). In the eye imaginal disc, Tap42 mutant clones were found to survive beyond 72 hr in the mitotically quiescent region posterior to the morphogenetic furrow but not in the mitotically active anterior region (not shown), suggesting that progression into mitosis may contribute to the death of Tap42 mutant cells.
Figure 4.—

Tap42 mutant cells activate JNK signaling and die. (A) Wing imaginal disc 72 hr after induction of GFP-marked Tap42 mutant clones. At this timepoint no cells lacking GFP are observed, indicating elimination of Tap42 mutant cells; wild-type twin spots with 2× levels of GFP (left) mark the sites of clone induction. Nuclei stained with Höechst are shown at center, and the merge is shown at right. See C and D for comparison to 48-hr clones. (B) Seventy-two hour Tap42- clones induced in trans to a Minute. Use of the Minute technique allows Tap42- clones to persist for 72 hr (Tap42- cells are marked by absence of GFP, left). However, the mutant cells contain highly condensed pyknotic nuclei (Höechst staining at center; Tap42- clones are outlined in yellow). (C) Anti-active caspase-3 staining of 48-hr Tap42 mutant clones (caspase staining shown at center, with clone outlined in yellow). (D) At 48 hr after induction, Tap42- cells express high levels of the puckered-lacZ marker, indicating activation of JNK signaling (lacZ staining shown at center, with mutant clone outlined in yellow). Genotypes: (A and C) hsflp; Tap42Δ305b FRT40A/UbiGFP FRT40A; P[5.1]-3/+; (B) hsflp; Tap42Δ305b FRT40A/M(2)32A UbiGFP FRT40A; P[5.1]-3/+; (D) hsflp; Tap42Δ305b FRT40A/UbiGFP FRT40A; P[5.1]-3/P[puc-lacZ]e69.
As cells with a growth disadvantage can be eliminated through a process of cell competition (Morata and Ripoll 1975), we used the Minute technique to impair the growth of wild-type cells surrounding Tap42 mutant clones. Although this enabled Tap42- cells to persist to 72 hr, these cells showed signs of massive cell death, with highly condensed, pyknotic nuclei (Figure 4B), and they were eliminated at later timepoints. Pyknotic cells were also present in third instar larval brains of Tap42 mutant animals (data not shown). Caspase activation was observed in Tap42 mutant clones at 48 hr and beyond (Figure 4C), as well as in the basal regions of discs in which Tap42 clones had been eliminated (not shown). That Tap42- cells were eliminated even when given a growth advantage and in a nonclonal background suggests that their death is not a result of cell competition and implies that Tap42 is required autonomously for cell viability. We note that clones of TOR- cells, which are severely compromised for growth, are able to survive and contribute to adult tissues (Oldham et al. 2000; Zhang et al. 2000).
Previous studies have shown that both the MAPK and Jun N-terminal kinase (JNK) pathways, which regulate cell survival in Drosophila (Dominguez et al. 1998; Adachi-Yamada et al. 1999), are targets of PP2A activity (Wassarman et al. 1996; Silverstein et al. 2002; Kins et al. 2003). Using an antibody that recognizes dual-phosphorylated ERK as a marker for MAPK activation, we observed no changes in MAPK activity in Tap42- clones, even in those containing dying cells (data not shown). In contrast, JNK signaling was markedly activated in Tap42 mutant clones, as evidenced by induction of a lacZ marker in the puckered locus (Figure 4D), a transcriptional target of JNK signaling (Martin-Blanco et al. 1998). Tap42 may thus promote cell survival in part by regulating the phosphorylation of one or more components of the JNK pathway. However, we note that activation of JNK signaling is required for a number of cell functions in addition to apoptosis, including cell migration and morphogenesis. Furthermore, mutations in hemipterous, which reduce JNK signaling, did not suppress the lethality of Tap42 mutants (not shown), indicating that other factors are likely to contribute to the death of Tap42 mutant cells.
In summary, our results demonstrate that Tap42 controls the levels and localization of the PP2A catalytic subunit, and in so doing it regulates essential functions including cell division and survival. Tap42 may directly control the association of PP2A with specific cellular compartments and substrates, or it may indirectly alter substrate specificity by preventing specific pools of PP2A from forming heterotrimeric complexes with A and B subunits. In contrast to yeast, where Tap42 plays an essential role downstream of TOR, our results indicate that this is not the case in Drosophila. Tap42 is not required for TOR-dependent growth, and two independent branches of TOR signaling, activation of S6K and suppression of autophagy, are not affected by loss of Tap42. While it remains possible that additional effects of TOR not examined here are mediated through Tap42, our results demonstrate clear differences in the mechanisms of TOR signaling in yeast and metazoans.
Acknowledgments
We thank Andrea McElhone for technical assistance and members of the Neufeld lab for fruitful discussions. This work was supported by National Institutes of Health grant RO1 GM62509 (T.P.N.). X.S.G. is a Special Fellow of Leukemia and Lymphoma Society (3643-04).
References
- Adachi-Yamada, T., K. Fujimura-Kamada, Y. Nishida and K. Matsumoto, 1999. Distortion of proximodistal information causes JNK-dependent apoptosis in Drosophila wing. Nature 400: 166–169. [DOI] [PubMed] [Google Scholar]
- Beck, T., and M. N. Hall, 1999. The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature 402: 689–692. [DOI] [PubMed] [Google Scholar]
- Cainarca, S., S. Messali, A. Ballabio and G. Meroni, 1999. Functional characterization of the Opitz syndrome gene product (midin): evidence for homodimerization and association with microtubules throughout the cell cycle. Hum. Mol. Genet. 8: 1387–1396. [DOI] [PubMed] [Google Scholar]
- Chen, J., R. T. Peterson and S. L. Schreiber, 1998. Alpha 4 associates with protein phosphatases 2A, 4, and 6. Biochem. Biophys. Res. Commun. 247: 827–832. [DOI] [PubMed] [Google Scholar]
- Di Como, C. J., and K. T. Arndt, 1996. Nutrients, via the Tor proteins, stimulate the association of Tap42 with type 2A phosphatases. Genes Dev. 10: 1904–1916. [DOI] [PubMed] [Google Scholar]
- Dominguez, M., J. D. Wasserman and M. Freeman, 1998. Multiple functions of the EGF receptor in Drosophila eye development. Curr. Biol. 8: 1039–1048. [DOI] [PubMed] [Google Scholar]
- Duvel, K., and J. R. Broach, 2004. The role of phosphatases in TOR signaling in yeast. Curr. Top. Microbiol. Immunol. 279: 19–38. [DOI] [PubMed] [Google Scholar]
- Duvel, K., A. Santhanam, S. Garrett, L. Schneper and J. R. Broach, 2003. Multiple roles of Tap42 in mediating rapamycin-induced transcriptional changes in yeast. Mol. Cell 11: 1467–1478. [DOI] [PubMed] [Google Scholar]
- Gao, X., Y. Zhang, P. Arrazola, O. Hino, T. Kobayashi et al., 2002. Tsc tumour suppressor proteins antagonize amino-acid-TOR signalling. Nat. Cell Biol. 4: 699–704. [DOI] [PubMed] [Google Scholar]
- Gomes, R., R. E. Karess, H. Ohkura, D. M. Glover and C. E. Sunkel, 1993. Abnormal anaphase resolution (aar): a locus required for progression through mitosis in Drosophila. J. Cell Sci. 104: 583–593. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Garcia, A., E. Garrido, C. Hernandez, B. Alvarez, C. Jimenez et al., 2002. A new role for the p85-phosphatidylinositol 3-kinase regulatory subunit linking FRAP to p70 S6 kinase activation. J. Biol. Chem. 277: 1500–1508. [DOI] [PubMed] [Google Scholar]
- Hara, K., K. Yonezawa, Q. P. Weng, M. T. Kozlowski, C. Belham et al., 1998. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J. Biol. Chem. 273: 14484–14494. [DOI] [PubMed] [Google Scholar]
- Helps, N. R., N. D. Brewis, K. Lineruth, T. Davis, K. Kaiser et al., 1998. Protein phosphatase 4 is an essential enzyme required for organisation of microtubules at centrosomes in Drosophila embryos. J. Cell Sci. 111: 1331–1340. [DOI] [PubMed] [Google Scholar]
- Hua, D. R., S. Inui, T. Yamashita, K. Maeda, K. Takagi et al., 2003. T cell-specific gene targeting reveals that alpha4 is required for early T cell development. Eur. J. Immunol. 33: 1899–1906. [DOI] [PubMed] [Google Scholar]
- Inui, S., H. Sanjo, K. Maeda, H. Yamamoto, E. Miyamoto et al., 1998. Ig receptor binding protein 1 (alpha4) is associated with a rapamycin-sensitive signal transduction in lymphocytes through direct binding to the catalytic subunit of protein phosphatase 2A. Blood 92: 539–546. [PubMed] [Google Scholar]
- Inui, S., K. Maeda, D. R. Hua, T. Yamashita, H. Yamamoto et al., 2002. BCR signal through alpha 4 is involved in S6 kinase activation and required for B cell maturation including isotype switching and V region somatic hypermutation. Int. Immunol. 14: 177–187. [DOI] [PubMed] [Google Scholar]
- Kins, S., P. Kurosinski, R. M. Nitsch and J. Gotz, 2003. Activation of the ERK and JNK signaling pathways caused by neuron-specific inhibition of PP2A in transgenic mice. Am. J. Pathol. 163: 833–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloeker, S., R. Reed, J. L. McConnell, D. Chang, K. Tran et al., 2003. Parallel purification of three catalytic subunits of the protein serine/threonine phosphatase 2A family (PP2A(C), PP4(C), and PP6(C)) and analysis of the interaction of PP2A(C) with alpha4 protein. Protein Expr. Purif. 31: 19–33. [DOI] [PubMed] [Google Scholar]
- Krause, U., L. Bertrand and L. Hue, 2002. Control of p70 ribosomal protein S6 kinase and acetyl-CoA carboxylase by AMP-activated protein kinase and protein phosphatases in isolated hepatocytes. Eur. J. Biochem. 269: 3751–3759. [DOI] [PubMed] [Google Scholar]
- Leicht, M., A. Simm, G. Bertsch and J. Hoppe, 1996. Okadaic acid induces cellular hypertrophy in AKR-2B fibroblasts: involvement of the p70S6 kinase in the onset of protein and rRNA synthesis. Cell Growth Differ. 7: 1199–1209. [PubMed] [Google Scholar]
- Lin, T. A., and J. C. Lawrence, Jr., 1997. Control of PHAS-I phosphorylation in 3T3–L1 adipocytes: effects of inhibiting protein phosphatases and the p70S6K signalling pathway. Diabetologia 40: S18–S24. [DOI] [PubMed] [Google Scholar]
- Liu, J., T. D. Prickett, E. Elliott, G. Meroni and D. L. Brautigan, 2001. Phosphorylation and microtubule association of the Opitz syndrome protein mid-1 is regulated by protein phosphatase 2A via binding to the regulatory subunit alpha 4. Proc. Natl. Acad. Sci. USA 98: 6650–6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Blanco, E., A. Gampel, J. Ring, K. Virdee, N. Kirov et al., 1998. puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes Dev. 12: 557–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morata, G., and P. Ripoll, 1975. Minutes: mutants of drosophila autonomously affecting cell division rate. Dev. Biol. 42: 211–221. [DOI] [PubMed] [Google Scholar]
- Murata, K., J. Wu and D. L. Brautigan, 1997. B cell receptor-associated protein alpha4 displays rapamycin-sensitive binding directly to the catalytic subunit of protein phosphatase 2A. Proc. Natl. Acad. Sci. USA 94: 10624–10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanahoshi, M., T. Nishiuma, Y. Tsujishita, K. Hara, S. Inui et al., 1998. Regulation of protein phosphatase 2A catalytic activity by alpha4 protein and its yeast homolog Tap42. Biochem. Biophys. Res. Commun. 251: 520–526. [DOI] [PubMed] [Google Scholar]
- Neufeld, T. P., 2004. Genetic analysis of TOR signaling in Drosophila. Curr. Top. Microbiol. Immunol. 279: 139–152. [DOI] [PubMed] [Google Scholar]
- Neufeld, T. P., A. F. de la Cruz, L. A. Johnston and B. A. Edgar, 1998. Coordination of growth and cell division in the Drosophila wing. Cell 93: 1183–1193. [DOI] [PubMed] [Google Scholar]
- Noda, T., and Y. Ohsumi, 1998. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem. 273: 3963–3966. [DOI] [PubMed] [Google Scholar]
- Oldham, S., J. Montagne, T. Radimerski, G. Thomas and E. Hafen, 2000. Genetic and biochemical characterization of dTOR, the Drosophila homolog of the target of rapamycin. Genes Dev. 14: 2689–2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson, R. T., B. N. Desai, J. S. Hardwick and S. L. Schreiber, 1999. Protein phosphatase 2A interacts with the 70-kDa S6 kinase and is activated by inhibition of FKBP12-rapamycin-associated protein. Proc. Natl. Acad. Sci. USA 96: 4438–4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reymond, A., G. Meroni, A. Fantozzi, G. Merla, S. Cairo et al., 2001. The tripartite motif family identifies cell compartments. EMBO J. 20: 2140–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt, A., T. Beck, A. Koller, J. Kunz and M. N. Hall, 1998. The TOR nutrient signalling pathway phosphorylates NPR1 and inhibits turnover of the tryptophan permease. EMBO J. 17: 6924–6931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweiger, S., and R. Schneider, 2003. The MID1/PP2A complex: a key to the pathogenesis of Opitz BBB/G syndrome. BioEssays 25: 356–366. [DOI] [PubMed] [Google Scholar]
- Schweiger, S., J. Foerster, T. Lehmann, V. Suckow, Y. A. Muller et al., 1999. The Opitz syndrome gene product, MID1, associates with microtubules. Proc. Natl. Acad. Sci. USA 96: 2794–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott, R. C., O. Schuldiner and T. P. Neufeld, 2004. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev. Cell 7: 167–178. [DOI] [PubMed] [Google Scholar]
- Short, K. M., B. Hopwood, Z. Yi and T. C. Cox, 2002. MID1 and MID2 homo- and heterodimerise to tether the rapamycin-sensitive PP2A regulatory subunit, alpha 4, to microtubules: implications for the clinical variability of X-linked Opitz GBBB syndrome and other developmental disorders. BMC Cell Biol. 3: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverstein, A. M., C. A. Barrow, A. J. Davis and M. C. Mumby, 2002. Actions of PP2A on the MAP kinase pathway and apoptosis are mediated by distinct regulatory subunits. Proc. Natl. Acad. Sci. USA 99: 4221–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snaith, H. A., C. G. Armstrong, Y. Guo, K. Kaiser and P. T. Cohen, 1996. Deficiency of protein phosphatase 2A uncouples the nuclear and centrosome cycles and prevents attachment of microtubules to the kinetochore in Drosophila microtubule star (mts) embryos. J. Cell Sci. 109: 3001–3012. [DOI] [PubMed] [Google Scholar]
- Sontag, E., V. Nunbhakdi-Craig, G. S. Bloom and M. C. Mumby, 1995. A novel pool of protein phosphatase 2A is associated with microtubules and is regulated during the cell cycle. J. Cell Biol. 128: 1131–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusova, T. A., and T. L. Madden, 1999. BLAST 2 Sequences, a new tool for comparing protein and nucleotide sequences. FEMS Microbiol. Lett. 174: 247–250. [DOI] [PubMed] [Google Scholar]
- Trockenbacher, A., V. Suckow, J. Foerster, J. Winter, S. Krauss et al., 2001. MID1, mutated in Opitz syndrome, encodes an ubiquitin ligase that targets phosphatase 2A for degradation. Nat. Genet. 29: 287–294. [DOI] [PubMed] [Google Scholar]
- Wassarman, D. A., N. M. Solomon, H. C. Chang, F. D. Karim, M. Therrien et al., 1996. Protein phosphatase 2A positively and negatively regulates Ras1-mediated photoreceptor development in Drosophila. Genes Dev. 10: 272–278. [DOI] [PubMed] [Google Scholar]
- Westphal, R. S., R. L. Coffee, Jr., A. Marotta, S. L. Pelech and B. E. Wadzinski, 1999. Identification of kinase-phosphatase signaling modules composed of p70 S6 kinase-protein phosphatase 2A (PP2A) and p21-activated kinase-PP2A. J. Biol. Chem. 274: 687–692. [DOI] [PubMed] [Google Scholar]
- Zhang, H., J. P. Stallock, J. C. Ng, C. Reinhard and T. P. Neufeld, 2000. Regulation of cellular growth by the Drosophila target of rapamycin dTOR. Genes Dev. 14: 2712–2724. [DOI] [PMC free article] [PubMed] [Google Scholar]