

Abstract
In 2002 Kim and Stephan proposed a promising composite-likelihood method for localizing and estimating the fitness advantage of a recently fixed beneficial mutation. Here, we demonstrate that their composite-likelihood-ratio (CLR) test comparing selective and neutral hypotheses is not robust to undetected population structure or a recent bottleneck, with some parameter combinations resulting in a false positive rate of nearly 90%. We also propose a goodness-of-fit test for discriminating rejections due to directional selection (true positive) from those due to population and demographic forces (false positives) and demonstrate that the new method has high sensitivity to differentiate the two classes of rejections.
THE substitution of a strongly selected advantageous mutation is expected to alter the frequencies of linked neutral variation (Maynard-Smith and Haigh 1974; Kaplan et al. 1989; Stephan et al. 1992). Several statistical tests have been proposed for inferring such a “selective sweep” event based on predicted effects relative to the standard neutral model. These include (1) a depression of expected heterozygosity relative to divergence at the target of selection (Hudson et al. 1987), (2) an excess of rare alleles compared to the standard neutral model (Tajima 1989; Braverman et al. 1995; Fu 1997), (3) an excess of high-frequency-derived alleles (Fay and Wu 2000), and (4) increased linkage disequilibrium (Przeworski 2002; Kim and Nielsen 2004). Since these signatures are localized to regions adjacent to the targets of selection, it seems reasonable to attempt to identify loci subject to recent directional selection by analyzing genomic patterns of presumably neutral polymorphism (e.g., Harr et al. 2002; Kim and Stephan 2002; Vigouroux et al. 2002).
A potential problem in this endeavor, however, is the low power to discriminate patterns expected under hitchhiking from similar patterns produced by chance under nonequilibrium conditions in the absence of selection. For example, recovery from a recent population bottleneck may result in an excess of rare alleles (Tajima 1989a,b) as can population expansion (Fu and Li 1993). More troubling is the fact that selection against linked deleterious mutation can also lead to an excess of rare alleles when effective population sizes are small (e.g., Charlesworth et al. 1993). More recently, Fay and Wu (2000) suggested that an excess of high-frequency-derived alleles in a sample is more likely due to hitchhiking than to other scenarios. However, they also pointed out that if there are many fixed differences between populations that exchange rare migrants, polymorphisms in the population would tend to be at very low or high frequencies. Furthermore, Przeworski (2002) demonstrated that a variety of demographic models have the same effect on Fay and Wu's H-statistic as a selective sweep. Recent bottlenecks and metapopulation structures (Wakeley and Alicar 2001) were also shown to result in high-frequency-derived alleles more often than would be expected under the standard neutral model. Despite these clear effects of nonselective forces, many have argued that one may still distinguish selective sweeps from demography, since the former generates a localized signature around the target of selection while the latter affects the entire genome equally. However, in the absence of selective sweeps, we may still observe local fluctuations of variation along a sequence, which are likely to be amplified by demographic forces and recombination that resemble the expected pattern of a selective sweep. Thus, while the pattern of variation along a chromosome produced by hitchhiking is quite predictable, it is often difficult to be certain that a given departure from neutrality is due to hitchhiking and not some stochastic effects manifested in the single realization of the evolutionary process.
Kim and Stephan (2002) present a composite-likelihood method for distinguishing selective sweeps from stochastic, neutral variation, assuming the sample of DNA sequences is drawn from a randomly mating population of constant size. They demonstrate that their method has considerable power to detect a recent selective sweep and yields unbiased estimates of the location and strength of the beneficial mutation. Here, we examine the extent to which bottlenecks and undetected population structure affect the type I error of their composite-likelihood-ratio (CLR) test. The CLR test was studied for two main reasons. First, it has been shown to have high power, indicating that it may be useful for whole-genome scans for adaptively evolving genes. Second, the test statistic (as is discussed below) is the ratio of the likelihood of the data given a recently completed selective sweep vs. an equilibrium neutral model. Therefore, one might predict that population processes that create large deviations from the latter model may lead to the spurious rejection of the null hypothesis of neutrality and thus to the erroneous inference of a recent selective sweep. Using coalescent simulations, we demonstrate that the CLR test as proposed by Kim and Stephan (2002) is not robust to the assumption of constant population size and random mating. However, through the use of the proposed goodness-of-fit test, it may be possible to distinguish data sets rejecting neutrality due to directional selection from those due to nonselective effects.
METHODS
Composite-likelihood analysis:
Kim and Stephan's (2002) CLR test uses the spatial distribution of mutation frequencies among a population sample of n DNA sequences to test for evidence of a selective sweep. Briefly, the method compares the ratio of the composite likelihood of the data under a null hypothesis (HN) of constant population size, neutral evolution, and random mating against an alternative hypothesis (HS) of a complete selective sweep. It is assumed that the beneficial mutation arose on a single chromosome in a population of constant size, drifted to frequency ε, changed deterministically to frequency 1 − ε, and then drifted to fixation. Formally, consider a stretch of DNA of length L in which S nucleotides are observed to be variable among a random sample of n sequences. Let yi for i = 1, … , L denote the observed count of the derived nucleotide at the ith site with corresponding random variable, Yi ∈ {0, 1, … , n − 1} (note that sites fixed for derived alleles are folded into the invariant class). Let α̂ and X̂ be the maximum-composite-likelihood estimates (MCLEs) of the strength of selection parameter (2Ns) and target of selection, respectively. These parameter estimates are found via maximization of the composite-likelihood function of Kim and Stephan (2002), so that
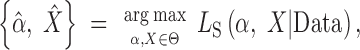 |
where
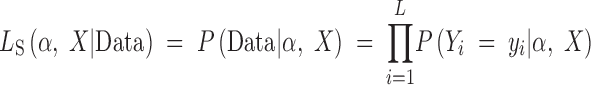 |
and P(Yi|α, X) is given by Equation 5 of Kim and Stephan (2002), using ε = (2α)−1. Throughout it is assumed that the neutral mutation rate for the region θ = 4Nμ (where N is the effective population size and μ is the mutation rate per locus per generation) and recombination rate between sites i and X are known. In practice, Watterson's (1975) estimate of θ is substituted in for the population mutation rate (i.e., corresponding to “test B” of Kim and Stephan 2002).
To discriminate between hypotheses HS and HN, the maximum composite likelihood of data under the model of a selective sweep, LS(α̂, X̂|Data), is compared to the composite likelihood of the data under a neutral equilibrium model, LN(Data). The latter quantity depends only on the mutation rate, which again is assumed known. The composite-likelihood-ratio test statistic employed is ΛKS = log LS(α̂, X̂|Data)/LN(Data). The null distribution of ΛKS is obtained by applying the CLR test to data sets obtained from simulations under the standard neutral model (Hudson 2002) with fixed θ. The neutral model is rejected at level γ when the observed ΛKS is greater than the 100(1 − γ) percentile of the null distribution (unless otherwise noted, we use a level of 5% for all tests in this study).
Neutral simulations and test of robustness:
A potential problem of the method outlined above is that the selective sweep hypothesis is compared to a null hypothesis in which the population is randomly mating and of constant size. Since the assumptions of this null hypothesis are frequently violated in natural populations, it is imperative to understand the robustness of the test to these assumptions. To quantify robustness, we simulated data under various neutral demographic scenarios that violate the panmixia and/or constant-size assumptions of equilibrium models and applied the CLR test. The proportion of neutral data sets that reject neutrality for each parameter combination is the realized type I error of the test.
Specifically, we simulated neutral data under bottleneck scenarios of varying intensity as well as under an island model of population subdivision using Hudson's (2002) ms program. We simulated a sample of 10-kb-long sequences with a scaled mutation rate of θ = 75 and 4Nr = 1000, where r is the probability per generation of crossover for the entire simulated region, values roughly corresponding to a typical Drosophila melanogaster data set. Bottlenecks are modeled in the following way: a population of constant size N is reduced to size βN at time tb (in units of 4N generations) in the past and then exponentially increases back to the same size. The rate of exponential growth is given by log β/tb. Population bottlenecks are simulated for various times since the reduction in units of 4N generations (tb = 0.0025, 0.0125, 0.025, 0.05, 0.125, 0.200, and 0.250) and severity (β = 0.01, 0.1, 0.2, and 0.5).
Simulations of population subdivision under an island model are performed with two subpopulations and scaled migration rate, M = 4Nm, where m is the fraction of migrants in each subpopulation in each generation. The sampling scheme is denoted by n = {n1, n2}, where n1 and n2 refer to the numbers of chromosomes sampled from the first and second subpopulations, respectively. To distinguish from bottlenecks and subdivisions, we refer to the model of neutral evolution under random mating and constant size as the “standard” neutral model.
Next, we conduct the CLR test using the simulated data and evaluate the type I error. Simulated data sets contain variable numbers of segregating sites (S), with Watterson's estimates of θ ranging from 2.8 to 100.9 per 10-kb region. For computational tractability, we use an approximate method to determine the cutoff values for rejecting the null hypothesis under the CLR test. We simulated 1000 replicate data sets under the standard neutral model for 20 values of θ ranging from 10 to 200 per region, denoted by θ1–θ20. For each θi, we obtained the corresponding cutoff value, ci, for ΛKS (95th percentile of the distribution). We use Watterson's estimate of θ, θ̂W, for each simulated data set to find the corresponding critical value that is interpolated by ci.
Composite-likelihood goodness-of-fit test:
In this section we derive a composite-likelihood goodness-of-fit (GOF) test for the Kim and Stephan (2002) inference scheme. A GOF test is employed to test if a random sample of data is drawn from a specific distribution of interest. In our case, the null hypothesis H0 is that the data are drawn from the Kim and Stephan (2002) model and the alternative hypothesis HA is that the data are not drawn from the Kim and Stephan model. To decide between H0 and HA, we compare the ratio of the probability of the data given the null, P(Data|H0), to the probability of the data given the alternative, P(Data|HA). Following Kim and Stephan (2002), we employ a composite-likelihood scheme to approximate these probabilities on the basis of the site-frequency spectrum and then simulate under the null hypothesis to find the critical value of our composite-likelihood-ratio goodness-of-fit statistic.
We calculate P(Data|H0) using the composite-likelihood function of Kim and Stephan (2002). For the alternative hypothesis, we model the number of sequences at each DNA site that carry the derived nucleotide as a binomially distributed random variable with unique unknown probability of success. Thus, as opposed to testing a specific demographic model, this approach is more general in that it posits that the data have been shaped by unidentified evolutionary and population processes that have affected the entire region under investigation. In this way, the issue of how well the data truly fit a selection model may be more directly addressed without having great concern regarding the appropriateness of the null. The likelihood function for the alternative model is
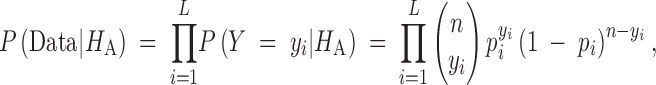 |
where yi is the number of sequences that carry the derived allele and pi is the unknown population frequency of the mutation at site i. The composite-maximum-likelihood estimates of pi can easily be shown to be the empirical frequency p̂i = yi/n.
The goodness-of-fit test statistic, ΛGOF, is defined as the ratio of the maximum probability of the data under the two hypotheses:
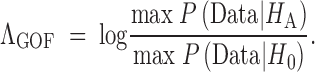 |
Calculating max log P(Data|HA) is straightforward, as p̂i is the same for all sites that have the same frequency. Therefore,
 |
where xj is the number of sites that have sample frequency j out of n. Calculating max log P(Data|H0) amounts to substituting in the maximum-composite-likelihood estimates of the location of the sweep and strength of selection in the Kim and Stephan (2002) composite-likelihood function: max P(Data|H0) = LS(α̂, X̂|Data).
Let Λ0GOF be the test statistic calculated from the observed data set. A large value of Λ0GOF will lead to the rejection of H0. To evaluate the significance of Λ0GOF, we need the distribution of this test statistic under the null model. An empirical distribution of Λ0GOF can be obtained from M replicate data sets that are generated by selective sweep simulations under the Kim and Stephan (2002) model (see below) with parameters α = α̂ and X = X̂. Let ΛiGOF be the test statistic calculated for the ith replicate data set. Then, we obtain the Monte Carlo estimate of the P-value:
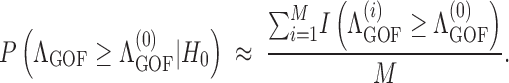 |
(Note that since the mutation rate is a nuisance parameter that must be estimated from the data, but is not part of the testing procedure, we simulate all data conditional on S, the total number of segregating sites in the observed data.) The C program used to calculate ΛGOF is available at http://www.mbg.cornell.edu/Aquadro_Lab.cfm.
Simulations with selection:
We simulated selective sweeps using a modification of the coalescent-with-recombination algorithm of Kim and Stephan (2002). The ancestral history of n chromosomes of L nucleotides is constructed into an ancestral recombination graph (Griffiths and Marjoram 1996a,b), from which marginal trees (coalescent trees corresponding to individual nucleotide sites) are extracted. Selective sweeps occur in a panmictic, constant-sized population. The fixation of the beneficial mutation occurs at the time of sampling (present). The construction of the graph depends on the following parameters: the intensity of selection (α = 2Ns), the scaled recombination rate (4Nr), and the location of the beneficial mutation (X). The mutations on the genealogy can be mapped, controlling either θ (proportional to branch lengths) or S, the number of segregating sites in the sample. Simulation with fixed S proceeds as follows: assume that the total branch length of the marginal tree obtained for site i is bi (i = 1, 2, 3, … , l). The cumulative total branch length up to site i is defined as  . We choose the smallest integer j that satisfies cj/cL > U, where U is a uniform random variable between 0 and 1. Then, a mutation is mapped on the tree corresponding to site j. The branch of the tree on which the mutation occurs is similarly chosen proportional to its branch length. Next, another mutation is placed at a new site using the same procedure (a new draw of U) except that the previously chosen site(s) is avoided. This is repeated until S mutations are mapped on the genealogy.
. We choose the smallest integer j that satisfies cj/cL > U, where U is a uniform random variable between 0 and 1. Then, a mutation is mapped on the tree corresponding to site j. The branch of the tree on which the mutation occurs is similarly chosen proportional to its branch length. Next, another mutation is placed at a new site using the same procedure (a new draw of U) except that the previously chosen site(s) is avoided. This is repeated until S mutations are mapped on the genealogy.
RESULTS
Robustness analysis:
Figure 1 summarizes the proportion of bottleneck data sets that reject neutrality (i.e., type I error of the CLR test) for various parameter combinations. We note that the pattern is complex and depends nonlinearly on both the severity (β) of the bottleneck and the time since the start of the bottleneck (tb). Even a modest bottleneck (e.g., β = 0.5) increases the false positive rate. If the bottleneck is very recent (tb = 0.0025), it has little effect on the type I error of the CLR test unless the bottleneck is extremely severe (e.g., 99% reduction). Weaker bottlenecks (e.g., β = 0.1) have a relatively greater effect if they occur deeper in the past while stronger (e.g., β = 0.01) bottlenecks have a greater effect when they occur more recently. For very recent bottlenecks (tb = 0.01) of strong effect (99%), close to 90% of the data sets reject the neutral model in favor of a model with a selective sweep. These results demonstrate that bottlenecks can frequently lead to spurious inference of a recent selective sweep, in the absence of further verification such as a goodness-of-fit test (discussed below). Our results are in general agreement with other studies that have demonstrated that many polymorphism-based tests of the equilibrium, neutral model have power to detect bottleneck events (Tajima 1989; Fu and Li 1993; Fay and Wu 2000; Wakeley and Alicar 2001; Przeworski 2002; Wakeley 2003).
Figure 1.—

Proportion of bottleneck simulations that rejected neutrality in favor of selection using the CLR test. The various lines (solid, dashed, etc.) denote the reduction in population size at the time of the bottleneck for the different scenarios considered. All simulations, unless otherwise specified, have fixed parameters n = 15, θ = 75, and 4Nr = 1000, as specified in methods.
In Figure 2, we plot measures of variation and summary statistics of the frequency spectrum across four simulated 10-kb regions that reject the CLR test. One can see from the sliding-window plots that all three estimators of θ [π (nucleotide diversity), θ̂W (Watterson 1975), and θ̂H (Fay and Wu 2000)] show large fluctuations along the sequence. Figure 2 also demonstrates that bottlenecks may produce data sets that reject neutrality via the CLR test and contain spatial patterns of nucleotide variation that are similar to those expected under a selective sweep. Shortly after a selective sweep, Tajima's D and Fu and Li's D-test statistics are expected to be negative for a region immediately adjacent to the target of selection as new mutations begin to accumulate. Fay and Wu's H-statistic (π − θ̂H) is also expected to be negative but the deepest “valleys” of this statistic are expected to flank the target of selection (Fay and Wu 2000; Kim and Stephan 2002). In these data sets, the predicted location of the sweep is typically within the deepest valley of Tajima's D-statistic. In all cases shown in Figure 2, this region also corresponds to the deepest valley in the sliding window of Fay and Wu's H-statistic. Relative to the other statistics, we observe a much greater tendency of Fay and Wu's H to be negative, indicating that high-frequency-derived alleles greatly influence the likelihood of the selective sweep model (LS).
Figure 2.—

Estimates of θ and neutrality test statistics across the 10-kb simulated sequence, depicting the frequency spectrum of variation for bottleneck simulations that rejected neutrality with the CLR test. Windows were 1000 bp long and shifted every 250 bp. Data were simulated under the following conditions and randomly selected: (a) β = 0.01, tb = 0.025; (b) β = 0.01, tb = 0.0125; (c) β = 0.1, tb = 0.125; (d) β = 0.1, tb = 0.05. Each arrow denotes the location of the predicted target of the putative selective sweep.
Interestingly, the average values of Tajima's D and Fu and Li's D for bottleneck data sets that generate sweep-like patterns are positive across the whole of the 10-kb sequence, indicating an excess of intermediate-frequency variants even under the most severe bottleneck scenarios. While this pattern differs from the prediction of an excess of rare alleles after a simple selective sweep, it is consistent with previous studies of population bottlenecks (e.g., Tajima 1989b), which showed that if a few divergent lineages survive the bottleneck, remaining segregating sites will tend to be in intermediate frequency immediately after the reduction in population size. In such a case, the CLR test may falsely reject neutrality due to an excess of derived alleles relative to the neutral expectation. Therefore, bottleneck simulations that generate false positive signals of a selective sweep may produce positive Tajima's D and negative Fay and Wu's H.
Next, we simulated two subpopulations with varying rates of symmetric migration between them (M = 0.1, 1, 4, and 10) and various sampling schemes [n = (15, 0), (10, 5) and (5, 0) and (50, 0)]. Figure 3 summarizes the type I error of the CLR test for data simulated under population substructure with two subpopulations. We note that for all sampling schemes considered, the highest false positive rate always occurs at the lowest level of migration. Likewise, the type I error decreases monotonically with increasing migration rate. By comparing n = (15, 0) and (10, 5), we infer that the sampling of all chromosomes from only one subpopulation results in a higher incidence of false positive signals of selective sweeps, as compared to sampling chromosomes from both subpopulations. Additionally, we observe that the variation in sample size we considered has little effect on the type I error.
Figure 3.—

The proportion of data sets that rejected neutrality in favor of selection using the CLR test on data simulated with subdivision and different levels of migration. The inset refers to how many alleles were sampled from each of the two subpopulations.
Figure 4 shows four randomly chosen data sets that reject an equilibrium neutral model in favor of a selective sweep model. Plotting the same three estimators of θ used above across the simulated region, we see that not only does the level of variation fluctuate across the region, but also, as with bottlenecks, the patterns expected to be produced by a selective sweep are replicated in a subdivided population with migration. Furthermore, while Tajima's D similarly fluctuates between positive and negative values across the region as observed under bottleneck scenarios, we continue to observe the most negative value near the putative target of selection. This region also corresponds with the most negative value of Fay and Wu's H, which tends to be strongly negative across the entire region (data not shown), indicating once again the heavy influence of high-frequency-derived alleles on the likelihood of the selective sweep model.
Figure 4.—

Estimates of θ and neutrality test statistics across the 10-kb simulated sequence, depicting the frequency spectrum of variation for population structure simulations that rejected neutrality with the CLR test. Data for each panel were simulated with n = (15, 0), M = 0.1. The particular simulation results shown were chosen at random. Note that Fay and Wu's H-statistic is strongly negative across these regions, ranging from −25 to −98, and is thus not plotted as it would be off scale. Each asterisk denotes the position of the most negative window for Fay and Wu's H-statistic and each arrow shows the predicted location of the putative sweep.
Goodness-of-fit test for simulated data:
As shown in Figures 2 and 4, the CLR test proposed by Kim and Stephan (2002) to detect selective sweeps is not robust to the effect of a strong, recent bottleneck or population structure with low rates of migration. We therefore sought to develop a method that might discriminate the sweep-like pattern caused by a demographic effect from the pattern caused by a “true” selective sweep. The goodness-of-fit test proposed is a logical approach to the problem, since it compares the relative likelihood of the data under a selective sweep hypothesis to a more general model with one parameter per nucleotide site.
Informally, one can reason as follows. A large value of ΛGOF indicates that the alternative model fits the data better than the sweep model, while a value of ΛGOF close to zero indicates a close fit between the selective sweep hypothesis and the observed data. If a recent selective sweep is the “real” reason a given data set rejects neutrality, the pattern of variation found in replicate data sets generated under the same selective scenario should be similar to the observed patterns in the original data. In fact, for data generated under this model, the distribution of P-values for the GOF test will be uniform by definition since a P-value is the probability of the data given the model. If, on the other hand, demography or other nonselective processes result in the rejection of the CLR test by generating a sweep-like pattern of variation, which is not completely compatible with the selective sweep model, the replicate GOF test statistics will be, on average, much smaller than the GOF test statistics for data generated under a sweep model (and consequently will have a low P-value; Figure 5).
Figure 5.—

Plots of the distribution of ΛGOF for two examples: (a) the “empirical” data set rejected the CLR test because of selection and (b) the empirical data set rejected the CLR test because of population structure with migration. The dotted line denotes the value of the test statistic for the observed data set. Thus, when the rejection is due to selection, the observed value falls within the distribution of the test statistic calculated for the replicate data sets; whereas, when the rejection is not due to selection, the observed value falls outside of the distribution. The corresponding P-values are given.
As a positive control of the GOF method, we performed selective sweep simulations with X = 5000, α = 1000. Using τ = 0.001, 0.01, and 0.1, the CLR test rejected the null hypothesis for 100, 88, and 62% of the data sets, respectively. We used increasing values of τ given that the calculation of LS assumes τ = 0 (Kim and Stephan 2002) and a failure of this assumption may lead to the failure of the GOF test. When the GOF test was applied to these data sets, the P-values were nearly uniform as expected (Figure 6).
Figure 6.—

The results of the goodness-of-fit approach for data sets that rejected the CLR test. For the “empirical” data set taken from selection simulations (a), equal numbers of points are taken from data sets simulated with τ = 0.001, 0.01, and 0.1. For the empirical data sets taken from migration simulations (b), equal numbers of points are taken from data sets simulated with M = 0.1, 1, 4, and 10. The reduction in population size at the time of the bottleneck is indicated as β = 0.1 (c) or β = 0.01 (d).
To evaluate the sensitivity of our tests, data sets were simulated under the models of population bottleneck, population subdivision, and a recent selective sweep. Data sets that rejected the null hypothesis in the CLR test were then analyzed using the proposed goodness-of-fit approach described above. The GOF test performed very well under both demographic models considered in detecting false positives of the CLR test (Figure 6). In cases of population subdivision, nearly all of the P-values were close to 0. When applied to the bottleneck simulations, the GOF yielded P-values near 0 in all but a small percentage of parameter combinations examined (Figure 6). Specifically, for β = 0.1, 18 and 28% of data sets had 0.1 < P < 0.5 for tb = 0.025 and tb = 0.05, respectively. For β = 0.01, 14, 17, and 19% of data sets had 0.1 < P < 0.5 for tb = 0.0125, tb = 0.025 and tb = 0.05, respectively, while 4 and 12% had P > 0.5 for tb = 0.025 and tb = 0.05, respectively. Taking these results together we note that the proposed GOF, when applied to data sets that rejected neutrality in favor of selection using the composite-likelihood analysis, may distinguish a selective sweep from other processes generating “sweep-like” patterns, with the exception of specific bottleneck scenarios. Namely, very severe bottlenecks appear to generate an effect very similar to a selective sweep at a single locus (99% reduction, tb = 0.025–0.05). This result is consistent with other work showing that a population bottleneck may indeed have an effect on the genealogy of a population that is indistinguishable from a selective sweep (Barton 1998; Depaulis et al. 2003).
Application to data:
We applied the proposed GOF test to six published polymorphism data sets that were argued to contain signatures of recent selective sweeps. The data sets and test results are listed in Table 1. We used recombination rates that were either suggested by the authors or known to be average for the species. The uncertainty in recombination rates appears to affect the CLR and GOF little, as different values of 4Nr gave similar results (see janus/ocnus and sweep regions 1 and 2 in Table 1). Two data sets (janus/ocnus region in D. simulans and jingwei gene in D. teissieri) show evidence of partial selective sweeps. In these cases, we took only subsets of sampled chromosomes that exhibit strong evidence of linkage to the putative beneficial mutation (haplotype group I of janus/ocnus and intron-absent sequences of jingwei). The resulting pattern of polymorphism due to hitchhiking in these subsets should be identical to that of a complete selective sweep (Meiklejohn et al. 2004).
TABLE 1 .
Analysis of published data
| Data set | 4Nr (per base) | α̂ | CLR test: ΛKS (P-value) | GOF test: ΛGOF (P-value) |
|---|---|---|---|---|
| Acp26Aa | 0.04 | 29.4 | 7.76 (0.033) | 294.5 (0.159) |
| Duffy locusb | 0.0015 | 90.9 | 8.84 (0.024) | 260.7 (0.602) |
| janus/ocnus regionc | 0.02 | 109.6 | 16.60 (0.001) | 1107 (0.017) |
| 0.065 | 446.6 | 16.61 (<0.001) | 1107 (0.022) | |
| 0.13 | 1009.4 | 16.61 (0.002) | 1107 (0.029) | |
| jingwei gened | 0.034 | 25.3e | 3.84 (0.120)e | NA |
| Sweep region 1f | 0.005 | 129.4 | 14.00 (0.005) | 675.7 (0.081) |
| 0.015 | 444.4 | 14.01 (0.003) | 675.7 (0.110) | |
| Sweep region 2f | 0.005 | 22.3 | 4.24 (0.145) | NA |
| 0.015 | 81.1 | 4.24 (0.123) | NA |
P-values are based on 1000 replicates of simulations under null models.
North Carolina population of Drosophila melanogaster (Aguadé et al. 1992; Kim and Nielsen 2004).
Human Duffy blood group locus from Hausa population (Hamblin et al. 2002).
Haplotype group I sequences of janus/ocnus region sequences of D. simulans (Meiklejohn et al. 2004), analyzed for three different rates of recombination (4Nr).
Intron-absent sequences of jingwei gene in D. teissieri (Llopart et al. 2002).
Likelihood is calculated without ancestral/derived allele information (option 2 of Kim and Stephan 2002).
Sweep regions 1 and 2 of Harr et al. (2002) (D. melanogaster), each analyzed for two different rates of recombination (4Nr).
We first conducted the CLR test. Of the six data sets, two failed to reject neutrality (“sweep region 2” of Harr et al. 2002 and the jingwei gene from Llopart et al. 2002). The four remaining data sets that showed significantly large ΛKS were subsequently analyzed using the proposed GOF test. Only the janus/ocnus data yielded a significantly large ΛGOF, with P-values between 0.017 and 0.029, thus indicating a poor fit to the selective sweep model of Kim and Stephan (2002). However, these data are not likely to represent a “false positive” sweep pattern caused by demography or population structure. Quesada et al. (2003) independently found the same pattern for a partial selective sweep spanning a much wider region surrounding the janus/ocnus region. Thus, we suggest that the large ΛGOF for these data is more likely caused by a deviation from the simple model of directional selection in a random-mating population as assumed by Kim and Stephan (2002): the haplotype group I sequences were sampled from many geographic regions, thus reflecting the complex spread of the beneficial allele across the worldwide population structure of D. simulans.
The remaining three data sets did not reject the selective sweep model, although the corresponding P-value for sweep region 1 falls in a range in which selection appears to be indistinguishable from certain bottleneck scenarios (P = 0.081–0.110; Table 1 and Figure 6). The failure to reject the sweep model for the Duffy locus and Acp26A may be surprising given that these data sets are likely to similarly violate the assumptions of the Kim and Stephan model (population in equilibrium). This result suggests, however, that the original rejection of neutrality by the CLR test is more likely to be due to a selective sweep than to demography alone.
DISCUSSION
Simulations were used to investigate the effects of population history and structure on the composite-likelihood-ratio test proposed by Kim and Stephan (2002) to detect signatures of hitchhiking along a recombining chromosome. As with standard tests of neutrality based on the site-frequency spectrum (e.g., Tajima's D, Fu and Li's D, Fay and Wu's H), the CLR test was found to be sensitive to past and present nonequilibrium demographies. For example, when sampling is done across an unknown population structure where rare migrants are symmetrically exchanged between subpopulations, we found that the CLR test rejects neutrality in favor of the selection alternative nearly 90% of the time. The test has a similarly high false positive rate for severe bottlenecks.
An ideal approach to this problem would be to directly compare the likelihood of the data given selection to the likelihoods of the data under various demographic scenarios, in the manner in which selection is compared with neutrality under the existing method of Kim and Stephan. However, given the enormous parameter space that would need to be explored to calculate these likelihoods, the number of models becomes intractable. As an alternative and computationally feasible approach to this problem, we have proposed a goodness-of-fit test. If a given data set rejects the standard CLR test, the maximum-likelihood parameter estimates derived from that analysis, as well as the number of segregating sites in the empirical data set, are then used to simulate replicates under a selective sweep model. Each of these replicates is subsequently analyzed via a modification of the standard GOF statistic, and the P-value of the observed data is estimated via Monte Carlo simulations.
The utility of methods such as this becomes evident when considering species such as humans and fruit flies for which there has been interest in detecting positive selection, yet for which demographic histories are known to include both population bottlenecks and population structure. For example, D. melanogaster is believed to have had an ancestral range in sub-Saharan Africa and to have recently dispersed worldwide. This range expansion appears to have involved at least some contraction in population size associated with the founding of new continents. A major bottleneck is estimated to have occurred ∼6000 years ago (e.g., Baudry et al. 2004), which, given 10 generations per year and an effective population size of 106, would correspond to tb = 0.015. Sequences simulated with similar parameters rejected neutrality in favor of selection with the CLR test in the great majority of cases for the strongest bottleneck scenario. However, Figure 6 suggests that the GOF test would successfully distinguish this particular demographic event from a selective sweep.
Thus, while the proposed GOF test offers some encouragement that positive selection may in fact be teased apart from the nonequilibrium effects investigated, further questions have been raised that will be the subject of future investigation. Perhaps foremost among these issues is the desire to consider a wider breadth of relevant demographic scenarios. Additionally, the performance of both the proposed and existing methods of detecting selection when a sweep has occurred in a nonequilibrium population presents a much more realistic scenario that is yet to be investigated. More importantly, however, the relevant ranges of demographic parameters for species of interest need to be considered, as this quantification may potentially allow for the rejection of parameter combinations that have been shown to be difficult to distinguish from selection.
Acknowledgments
We appreciate fruitful discussions with Rasmus Nielsen, Floyd Reed, and the Aquadro and Bustamante labs and comments on the article from Andy Clark and the reviewers. This research was supported by National Institutes of Health (NIH) grant GM36431 to C.F.A.; National Science Foundation (NSF) grant DMS-0201037 to R. Durrett, C. F. Aquadro, and R. Nielsen; U.S. Department of Agriculture-Hatch grant NY 151411 to C.D.B.; and NSF grant DBI-0319553 to M. Purugganan, C.D.B., S. McCouch, and R. Nielsen. Y.K. was supported by NSF grant DEB-0449851. J.D.J. was also partially supported by a NIH training grant in genetics and development awarded to Cornell University.
References
- Aguadé, M., N. Miyashita and C. H. Langley, 1992. Polymorphism and divergence in the Mst26a male accessory gland gene region. Genetics 132 755–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton, N. H., 1998. The effect of hitch-hiking on neutral genealogies. Genet. Res. 72 123–133. [Google Scholar]
- Baudry, E., B. Viginier and M. Veuille, 2004. Non-African populations of Drosophila melanogaster have a unique origin. Mol. Biol. Evol. 21 1482–1491. [DOI] [PubMed] [Google Scholar]
- Braverman, J. M., R. R. Hudson, N. L. Kaplan, C. H. Langley and W. Stephan, 1995. The hitchhiking effect on the site frequency spectrum of DNA polymorphisms. Genetics 140 783–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth, B., M. T. Morgan and D. Charlesworth, 1993. The effect of deleterious mutations on neutral molecular variation. Genetics 134 1289–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depaulis, F., S. Mousset and M. Veuille, 2003. Power of neutrality tests to detect bottlenecks and hitchhiking. J. Mol. Evol. 57(Suppl. 1): S190–S200. [DOI] [PubMed] [Google Scholar]
- Fay, J., and C.-I Wu, 2000. Hitchhiking under positive Darwinian selection. Genetics 155 1405–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y.-X., 1997. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147 915–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y.-X., and W.-H. Li, 1993. Statistical tests of neutral mutations. Genetics 133 693–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths, R. C., and P. Marjoram, 1996. a Ancestral inference from samples of DNA sequences with recombination. J. Comput. Biol. 3 479–502. [DOI] [PubMed] [Google Scholar]
- Griffiths, R. C., and P. Marjoram, 1996b An ancestral recombination graph, pp. 257–270 in IMA Volume on Mathematical Population Genetics, edited by P. Donnelly and S. Tavaré. Springer-Verlag, Berlin/Heidelberg, Germany/New York.
- Hamblin, M. T., E. E. Thompson and A. Di Rienzo, 2002. Complex signatures of natural selection at the Duffy blood group locus. Am. J. Hum. Genet. 70 369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harr, B., M. Kauer and C. Schlötterer, 2002. Hitchhiking mapping: a population-based fine-mapping strategy for adaptive mutations in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 99 12949–12954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., 2002. Generating samples under a Wright-Fisher neutral model. Bioinformatics 18 337–338. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., M. Kreitman and M. Aguadé, 1987. A test of neutral molecular evolution based on nucleotide data. Genetics 116 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan, N. L., R. R. Hudson and C. H. Langley, 1989. “The hitchhiking effect” revisited. Genetics 123 887–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y., and R. Nielsen, 2004. Linkage disequilibrium as a signature of selective sweeps. Genetics 167 1513–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y., and W. Stephan, 2002. Detecting a local signature of genetic hitchhiking along a recombining chromosome. Genetics 160 765–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llopart, A., J. M. Comeron, F. G. Brunet, D. Lachaise and M. Long, 2002. Intron presence-absence polymorphism in Drosophila driven by positive Darwinian selection. Proc. Natl. Acad. Sci. USA 99 8121–8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard Smith, J., and J. Haigh, 1974. The hitch-hiking effect of a favorable gene. Genet. Res. 23 23–35. [PubMed] [Google Scholar]
- Meiklejohn, C. D., Y. Kim, D. L. Hartl and J. Parsch, 2004. Identification of a locus under complex positive selection in Drosophila simulans by haplotype mapping and composite-likelihood estimation. Genetics 168 265–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przeworski, M., 2002. The signature of positive selection at randomly chosen loci. Genetics 160 1179–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesada, H., U. E. Ramirez, J. Rozas and M. Aguadé, 2003. Large-scale adaptive hitchhiking upon high recombination in Drosophila simulans. Genetics 165 895–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan, W., T. H. E. Wiehe and M. W. Lenz, 1992. The effect of strongly selected substitutions on neutral polymorphism: analytical results based on diffusion theory. Theor. Popul. Biol. 41 237–254. [Google Scholar]
- Tajima, F., 1989. a Statistical method for testing the neutral mutation hypothesis. Genetics 123 437–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, F., 1989. b The effect of change in population size on DNA polymorphism. Genetics 123 597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigouroux, Y., M. McMullen, C. T. Hittinger, K. Houchins, L. Schulz et al., 2002. Identifying genes of agronomic importance in maize by screening microsatellites for evidence of selection during domestication. Proc. Natl. Acad. Sci. USA 99 9650–9655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeley, J., 2003. Polymorphism and divergence for island-model species. Genetics 163 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeley, J., and N. Alicar, 2001. Gene genealogies in a metapopulation. Genetics 159 893–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson, G. A., 1975. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7 256–276. [DOI] [PubMed] [Google Scholar]