

Abstract
We report here the breakpoint structure and sequences of the Drosophila melanogaster cosmopolitan chromosomal inversion In(3R)P. Combining in situ hybridization to polytene chromosomes and long-range PCR, we have identified and sequenced the distal and proximal breakpoints. The breakpoints are not simple cut-and-paste structures; gene fragments and small duplications of DNA are associated with both breaks. The distal breakpoint breaks the tolkin (tok) gene and the proximal breakpoint breaks CG31279 and the tolloid (tld) gene. Functional copies of all three genes are found at the opposite breakpoints. We sequenced a representative sample of standard (St) and In(3R)P karyotypes for a 2-kb portion of the tok gene, as well as the same 2 kb from the pseudogene tok fragment found at the distal breakpoint of In(3R)P chromosomes. The tok gene in St arrangements possesses levels of polymorphism typical of D. melanogaster genes. The functional tok gene associated with In(3R)P shows little polymorphism. Numerous single-base changes, as well as deletions and duplications, are associated with the truncated copy of tok. The overall pattern of polymorphism is consistent with a recent origin of In(3R)P, on the order of Ne generations. The identification of these breakpoint sequences permits a simple PCR-based screen for In(3R)P.
THROUGH the classic work of Dobzhansky on Drosophila pseudoobscura, chromosome inversions claim an important place in the early study of genetic variation in natural populations (Powell 1997). Because inversions reduce recombination in heterokaryotypes, they are a genomic feature with potential to come under natural selection and play a role in evolution. A common belief is that inversion polymorphisms are maintained by balancing selection (Dobzhansky 1970). If so, individual inversions might be ancient, that is, old relative to a hypothetical neutral arrangement (Andolfatto et al. 2001). This age hypothesis can be examined by comparing patterns and levels of sequence variation between and within inverted and standard (St) arrangements. Inverted regions are subject to reduced recombination. On a timescale relevant to the question of balancing selection, however, recombination is often not sufficiently suppressed by the inversions to prevent exchanges (see Ashburner 1989; Hasson and Eanes 1996). Any assessment of inversion age using a sequence-derived estimate of gene genealogy will, therefore, be confounded by crossing over and gene conversion operating within the inverted arrangement (Hasson and Eanes 1996). However, topological constraint on homolog pairing increases with increasing proximity to the inversion breakpoints (Novitski and Braver 1954). This increase in constraint results in decreasing crossing over with increasing proximity to the breakpoints. For this reason, recovering nucleotide sequences as close to the breakpoints as possible offers the most informative way to study the population genetics of inversion polymorphisms. Finally, the structural features at the breakpoints may also offer insight into the molecular nature and mutational origin of the inversion (e.g., transposable elements), the potential for genetic damage by disrupting gene function, and the genealogical uniqueness of the arrangement.
Drosophila melanogaster possesses well-studied cosmopolitan inversion polymorphisms on all four autosomal arms, as well as a common X-linked inversion in east African populations (Krimbas and Powell 1992). In 1994, using a mixed strategy of chromosome microdissection, nonspecific PCR, and reciprocal hybridization to a St arrangement genomic library, Wesley and Eanes (1994) reported the molecular characterization of the breakpoints of the In(3L)Payne inversion in D. melanogaster. The In(3L)P breakpoints are basic cut-and-paste structures. No novel features, such as transposable elements or other arrangements, were associated with the breakpoints. A population study of sequence variation indicated that the arrangement, while not ancient, also appeared to be not recent; it emerged from the base of the inferred genealogy of St arrangements and possessed several fixed differences from the St arrangement. In 1999, the In(2L)t inversion was extensively studied by Andolfatto and colleagues (Andolfatto et al. 1999; Andolfatto and Kreitman 2000). They recovered a sequence spanning the proximal breakpoint and reported on a large population-based sample of sequences >5 kb in length. They predicted that the inversion was not old and estimated the minimum age to be 0.3N generations or ∼100,000 years. There was an unusual haplotype structure associated with the St chromosomes in this region, as well as an apparent excess of molecular polymorphism in the breakpoint region, all suggesting balancing selection.
In this report, we describe the breakpoints of a third D. melanogaster cosmopolitan inversion In(3R)Payne, using the known cytological position and D. melanogaster genome sequence as a starting reference point for a long-range PCR walk. We find breakpoints with complex arrangements, including duplication and fragmentation of the tolkin (tok) gene. Sequence data for the St arrangement copies of tok, and the inverted copy of tok and its partial pseudogene, predict a relatively recent origin of this cosmopolitan inversion.
MATERIALS AND METHODS
Long-range PCR and in situ hybridization:
Earlier cytological studies predicted that the breakpoints of In(3R)P are at 89C2–3 (proximal breakpoint) and 96A18–19 (distal breakpoint, Krimbas and Powell 1992). To identify the precise molecular breakpoints we used long-range PCR and in situ hybridization of amplified fragments to In(3R)P-bearing chromosomes and focused on the 89C2–3 breakpoint. Primers were synthesized using the sequence of the D. melanogaster genome, release 3.2.2 (Adams et al. 2000; http://www.fruitfly.org), and starting from a site in the region of the predicted (cytological) breakpoint. The strategy was to move successively closer to the breakpoint until the long-range, and eventually short-range PCRs failed. We utilized two lab-created lines homozygous for In(3R)P and a single-standard line (Bloomington Drosophila Stock Center line 2057). Under this strategy the breakpoint sequence would eventually be recovered via inverse PCR using standard methods (Ochman et al. 1988). Briefly, a region of known sequence was used to design reverse orientation PCR primers and to identify suitable restriction endonuclease sites. Genomic DNA from a In(3R)P line was digested with various endonucleases, self-ligated to form loops, and used as template for PCR reactions. This method is efficient at recovering regions of unknown sequence that are contiguous with regions of known sequence. Long-range PCR was carried out using the Expand Long Template PCR system (Roche Applied Science, Mannheim, Germany). Primers were designed to the specifications of the polymerase enzyme system. In situ hybridization of polytene chromosomes was performed following a modified version of the Berkley Drosophila Genome Project protocol (http://www.fruitfly.org/aout/methods/cytogenetics.html). PCR fragments (∼10 kb) were labeled with digoxigenin (Roche Applied Science) and in situ hybridization carried out on both the standard line and a line homozygous for In(3R)Payne.
Populations:
Iso-third chromosome lines were collected in 1997 and have been characterized in other studies (Duvernell and Eanes 2000; Verrelli and Eanes 2001; Sezgin et al. 2004). In the population study reported here, lines with In(3R)P were identified by a PCR-based screen using nested primers. A set of three PCR primers was designed from the above work to screen for the inversion: 12253917+ (ACT AGC GTT GAG AAT GCA AAG TCC AAC), 12254223− (AAA TGC TGC ACG TAA TTG TAA GTT ATG AGC), and 20560888− (TTT GTT TGT GTC TGT GTG AGC TGC). The numbering again refers to the annotated D. melanogaster genome sequence, release 3.2.2 (Adams et al. 2000). Given the duplication associated with the inversion (see results), primer pair 12253917+/12254223− will amplify a 306-bp fragment in both St and inverted chromosomes, while primer pair 12254223−/20560888− will amplify a 663-bp fragment only from inverted chromosomes. The accuracy of this method was tested by PCR screening 10 St and 10 inverted isochromosomal lines previously karyotyped using the salivary gland preparations. All chromosome karyotypes were correctly scored using the PCR screen.
We amplified and sequenced an ∼2-kb fragment, starting at the distal breakpoint, from both St and In(3R)P chromosomes. All In(3R)P lines available from the earlier studies were sequenced (Sezgin et al. 2004). Sequenced St lines were selected to match the number and location of the In(3R)P lines used. Samples spanned collection sites from southern Florida to Maryland. The amplified ∼2-kb fragment covers a segment of the tolkin gene (tok) in the standard chromosomes and the beginning of the tok pseudogene in In(3R)P chromosomes. We also sequenced the same ∼2-kb fragment from the functional copy of the tok gene in In(3R)P chromosomes. PCR primers designated 20560313+ (TTG GCC TAA TCG AAT TGC TAT G) and 20562431− (CGC CAC CGC AAA CAA CTA TGG) were used to amplify a 2.1-kb fragment from both the St orientation chromosomes and the functional copy on the In(3R)P chromosomes. PCR primers 12254223− and 20562431− amplified a similarly sized fragment of the tok gene broken by the In(3R)P inversion. Six lines of Drosophila simulans, initially collected on Long Island, New York, full-sibling inbred for 20 generations, and the not-yet-annotated D. simulans genome sequence (http://www.genome.wustl.edu/projects/simulans/index.php) were used for sequence comparison to root the phylogenetic analysis and to determine the likely ancestral state of polymorphic sites.
Sequencing and data analysis:
PCR amplification products were cleaned using a QIAGEN PCR purification kit and sequenced directly using both the initial PCR primers and the internal primers. All sequencing was done by the DBS sequencing facility, University of California at Davis, using Big Dye Terminator version 3.1 (ABI) and sequenced on a 3730 DNA analyzer (ABI). Base calls from the chromatograms were checked, and the 2-kb fragments were assembled, using Sequencher (Gene Codes, Ann Arbor, MI). The St tok and In(3R)P inverted tok and pseudogene tok fragment were aligned using Clustal X (Thompson et al. 1997), with minor adjustments done by eye, resulting in a final aligned sequence of 2053 bases. All sequences have been deposited in GenBank (accession nos. AY881252, AY881253, AY881254, AY881255, AY881256, AY881257, AY881258, AY881259, AY881260, AY881261, AY881262, AY881263, AY881264, AY881265, AY881266, AY881267, AY881268, AY881269, AY881270, AY881271, AY881272, AY881273, AY881274, AY881275, AY881276, AY881277, AY881278, AY881279, AY881280, AY881281, AY881282, AY881283, AY881284, AY881285, AY881286, AY881287, AY881288, AY881289, AY881290, AY881291, AY881292).
The effective number of synonymous and nonsynonymous sites was estimated using DnaSP 4.0 (Rozas and Rozas 1997). This program was also used to calculate θ and π and carry out the Tajima (1989) and Fu and Li (1993) tests. A phylogeny was constructed using the neighbor-joining method (Saitou and Nei 1987) as implemented in MEGA version 2.1 (Kumar et al. 2001) and the Kimura two-parameter model (Kimura 1980) for distance estimation. Gaps in the aligned sequence were deleted in a pairwise manor and 1000 bootstrap replicates (Felsenstein 1985) performed to evaluate support for each node in the tree.
RESULTS
Defining the breakpoints:
Localization of breakpoints:
Figure 1 shows the region of the proximal breakpoint, its predicted genes, and the genomic fragments (between 3 and 14 kb) amplified by long-range PCR in the initial search for the In(3R)P inversion breakpoints. The numbering refers to the annotated D. melanogaster genome sequence, release 3.2.2 (Adams et al. 2000; http://www.fruitfly.org). In situ hybridization of the 90 and 120 fragments (Figure 1) to polytene chromosomes of In(3R)P confirmed that the breakpoint fell between 12,243 and 12,264 kb in the D. melanogaster genome sequence, but all long-range PCR amplifications across this 20-kb region worked on both St and In(3R)P samples. One explanation for this paradox was a possible duplication in the In(3R)P chromosome of the region containing the positive primers for fragments G and F and/or the negative primers for E and C. An ∼14-kb fragment (marked D) failed to amplify in inverted arrangements, but amplified in St chromosomes. Our approach was to carry out subsequent smaller PCR reactions, while maintaining one of the primers of D constant. Using the same negative primer as in D (12261570−, CAT TAT ACC AGC CAC CAC CGC GTT GAG CAG G), we designed positive primers that would amplify smaller pieces. None of these smaller PCR reactions should work in an inverted sample unless the positive primer was within the duplicated region (all PCR reaction will amplify from a standard line). We observed that PCR amplification failed when using primer 12253902+ (ACT AGC GTT GAG AAT GCA AAG TCC AAC), but was successful when utilizing 12254192+ (GTT GCT CAT AAC TTA CAA TTA CGT GCA GC). This suggested that the end of the duplicated region (the breakpoint) was between these two positive primers (Figure 1; black bar and arrow).
Figure 1.—

Proximal region of chromosomal inversion In(3R)P (89C2–7). The ruler indicates the position in arm 3R according to the D. melanogaster genome. The numbers in parentheses indicate the numbering scheme used for the primers. Coding genes immediately flanking the inversion breakpoints (see Figure 2) are illustrated in green; other coding genes covered by the in situ and long-range PCR analysis are shown in yellow. Blue bars indicate the locations of PCR amplifications that worked in both inverted and standard lines. The red bar indicates the location of the PCR amplification that worked only in standard lines. Orange bars indicate the locations of the in situ probes. The black arrow indicates the 289-bp region implicated in our long-range PCR screen to contain the proximal breakpoint. Primers used for A were (12233497+, 1240696−), B (12240514+, 12247904−), C (12240514+, 12254800−), D (12247448+, 12261570−), E (12247448+, 12254800−), F (12254500+, 12261570−), G (12254500+, 12268649−), and H (12258824+, 12261719−). The 90 and 120 in situ probes were created using primers 12233497+/12243306 and 12264288+/12273660, respectively.
To localize the distal breakpoint, reverse orientation primers, 12254224− (AAA TGC TGC ACG TAA TTG TAA GTT ATG AGC) and 12254500+ (TGA TGC AGT CCG ACG ACA ACA ACA CAA GCA GC) were designed next to this 289-bp region to allow inverse PCR amplification of the flanking DNA. These primers amplified an ∼500-bp fragment in TaqI-digested and self-ligated DNA from a In(3R)P line. Sequencing this fragment we found sequence from coordinate 12,254 kb contiguous with exon 4 of the tolkin gene (tok; Nguyen et al. 1994; Finelli et al. 1995). The tok gene lies near the predicted (cytological) distal breakpoint, while 12,254 kb is near the predicted proximal breakpoint. A second inverse PCR, using MspI digested In(3R)P DNA, yielded an ∼400-bp fragment. Sequencing this fragment we again found bases from 12,254 kb contiguous with the tok fourth exon. The junction of sequences from the region of the cytological predicted proximal and distal breakpoints appears to have identified the distal breakpoint of In(3R)P. This breakpoint appears to break the tok gene. To further clarify and confirm the distal breakpoint, we attempted to PCR amplify sequence between a primer in the CG31279 gene, the nearest coding gene to the genomic sequence recovered in the inverse PCR (i.e., inside the inversion), 12255485− (CAG CAG CAG CTA CTT GGC TTT TAT TTA TT), and a primer located distal to the predicted distal breakpoint, 500-bp past the 3′-end of the tok gene 20565305− (CCT TAG GCA TCT TAG CAT AAG TCA ATG GGT GGC). As predicted, this amplification failed in St lines, but produced an ∼6-kb fragment in In(3R)P lines. This fragment was completely sequenced from three lines. The fragment contained bases 12,255,485–12,254,041 of the D. melanogaster genome sequence contiguous with 4471 bp of the tok gene and 434 bases past the 3′-end of the tok gene. The tok sequence included 269 bases of exon 4, and all of the downstream region (3′-end) of the tok gene; exons 1–3 and the first 18 bases of exon 4 are not present. This tok fragment is most likely a pseudogene (ψtok, block 7 in Figures 2 and 3). The tok end of this fragment matches the St chromosome sequence and it appears that this 7-kb amplification crosses the distal breakpoint.
Figure 2.—

Structure of breakpoints for Standard and In(3R)P chromosomes. Both the distal and proximal In(3R)P breakpoints split, and presumably inactivate, known coding genes. Each “split” gene is asymmetrically duplicated at the opposite breakpoint. The boxed text indicates the numbering and color-coding scheme. (A and B) Standard orientation chromosomes showing the inversion breakpoints and putative pattern of the inversion event. (C and D) Inverted chromosomes. The chromosome in D contains a small deletion at the proximal breakpoint and is presumably derived from C. See results and Figure 3 for a detailed description of the breakpoints. This figure is not to scale.
Figure 3.—
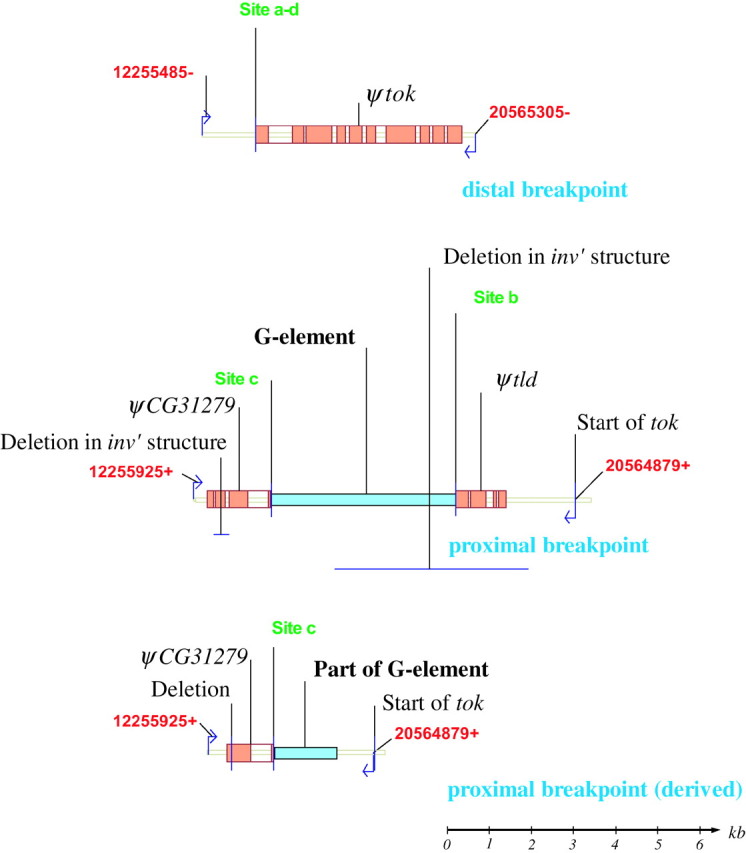
Detail of the distal, proximal, and derived proximal breakpoints from the In(3R)P chromosomes. Numbering and color coding are as in Figure 2. Primers and primer sites from the long-range PCR used to characterize each breakpoint are indicated. See results for details. A scale bar is shown in the bottom right-hand corner.
Characterization and sequencing of the breakpoints:
We next attempted to amplify and sequence the proximal breakpoint. To begin, we conducted inverse PCR on In(3R)P and St samples using primers designed to match the portion of the tok gene missing at the distal breakpoint of In(3R)P chromosomes. If the proximal breakpoint was simply a cut-and-paste match to the distal breakpoint, these primers would amplify a fragment of unknown size across the proximal breakpoint of the In(3R)P samples. The primers were expected to also amplify a 750-bp fragment from the functional copy of tok in St chromosome. This amplification, however, produced a 750-bp fragment from TaqI-digested DNA from both In(3R)P and St samples, suggesting that a second copy of the tok gene existed in the In(3R)P chromosome.
Since our initial long-range PCR results suggested the possibility of a duplication in the region of the inversion breakpoints, a second copy of the tok gene was not unexpected. Furthermore, loss-of-function mutations of the tok gene are homozygous lethal (Finelli et al. 1995), yet In(3R)P homozygotes are viable. Our finding that the distal breakpoint of In(3R)P fragmented the tok gene suggested that either the tok gene was duplicated elsewhere or some compensatory change in another functionally connected gene was involved. If, however, a duplicate copy of tok did exist it would have to be closely linked to either breakpoint to keep the inversion from being homozygous lethal. To test whether a second copy of the tok gene was associated with the proximal breakpoint, we attempted to amplify from a primer at the 3′-end of the tok sequence, 20564879+ (TCA GTG GTT CCA CCA TAG CC), to a primer in the CG31279 gene, 12255925+ (TGA GTT TCG GCA TAA ATT ACG A). The primers were oriented such that amplification would be possible only if a duplicated copy of tok was in an inverted orientation (our expectation if it were a product of the inversion that led to In(3R)P) and located near a copy of CG31279 (in the St orientation). The primers were designed such that amplification was not possible from the copy of CG31279 and ψtok fragment found at the distal breakpoint.
This PCR amplification yielded an ∼7-kb fragment, both ends of which were sequenced. The fragment crossed from the 3′-end of the tok gene into the 5′-end of CG31279, presumably crossing the proximal breakpoint (Figure 2, blocks 6 and 8; Figure 3). Sequencing 2.5 kb of the tok end of the fragment revealed 1339 bp of the intergenic region between tok and tld and exons 1–4 of the tld gene, suggesting that a functional copy of the tok gene is associated with the proximal breakpoint. Continuing along the fragment, the tld sequence is contiguous with sequence that appears to be from a G-element-like transposable element (Di Nocera et al. 1986; Kaminker et al. 2002). Sequencing 2 kb of the “CG31279 end” of this 7-kb fragment, we found all of the exons 1–3, and 18 bp of exon 4, of CG31279. The CG31279 sequence is then contiguous with sequence from a G-element-like transposable element. The truncated tld and CG31279 are most likely pseudogenes (designated ψtld and ψCG31279). The CG31279 end of the fragment matches the St chromosome sequence and we are confident that this 7-kb amplification crosses the proximal breakpoint. The presence of G-element-like sequence at the ends of both ψtld and ψCG31279 suggests that such an element separates these two fragments in the proximal breakpoint of In(3R)P. The size and restriction digest of a fragment amplified from this putative element also match those expected of a G element (see below). The duplicated primer sites that confounded the initial long-range PCR walk are found in the regions of the ψCG31279, ψtld, and tok sequences at the proximal breakpoint. Further characterization of both breakpoints is diagramed in Figure 3.
The mutational event that resulted in the In(3R)P arrangement left a complicated genomic structure consisting of duplicated coding sequences and pseudogene fragments at each breakpoint. We hypothesize that In(3R)P might have arisen by inversion of a region of a St chromosome and replacement of a smaller chromosomal region of its sister chromosome. This would have resulted in duplication of the regions flanking both breakpoints and prevented the fragmentation of genes at the breakpoints from being lethal mutations. This model is diagramed in Figure 2; coding genes are shown in green, and pseudogenes are shown in orange. The red broken-line box outlines the region of the St chromosome that was inverted, the proximal and distal ends of the inverted region are denoted a and b, respectively (Figure 2A). The insertion sites are indicated in Figure 2B and denoted c and d, and the arrows between Figures 2A and 2B indicate the inversion. The In(3R)P structure is shown in Figure 2C, and the likely origin of the pseudogene fragments created by inversion and replacement are indicated by the arrows between Figures 2B and 2C. In the In(3R)P chromosome, one copy of CG31279 is broken at site c leaving a 1391-bp fragment of exons 1–4 (ψCG31279, block 6 in Figure 2; Figure 3). The inversion also breaks the tld gene at site b, leaving 1114 bp of exons 1–4 (ψtld, block 8 in Figures 2 and 3).
When we sequenced the proximal breakpoint in different In(3R)P lines we observed two slightly different breakpoint structures. In one structure (Figure 2C; Figure 3), a G-element-like sequence is found between ψCG31279 and ψtld (between blocks 6 and 8). In the other structure (Figure 2D; Figure 3, inv′), which is likely a derivate of the first structure, the ψtld gene was not present and only 1352 bp of the 5′-end of the G element remained. It appears that this second structure resulted from an imprecise excision of the G element from the breakpoint resulting in the concurrent deletion of the ψtld fragment. The inverted (functional) copy of the tok gene is still intact in this derived structure. Eight of the 13 In(3R)P proximal breakpoints sequenced had the longer sequence structure (Figure 3, inv′), the remaining five had the shorter, apparently derived, structure (Figure 3, inv′). All five lines with the inv′ proximal breakpoint structure also had a 354-bp deletion that covers all of the second exon of their ψCG31279 sequence (Figure 3). The sequences of the distal, proximal, and derived-proximal breakpoints have been submitted to GenBank (accession nos. AY886890, AY886891, AY886892).
We did not sequence the entire ∼3.5 kb of the G-element region of the In(3R)P structure, just ∼300 bp of the ends. These ends exactly match both ends of a transposable element sequence on the X chromosome (bases 44576–47033 of GenBank submission AC011704) and are 73% identical to the canonical G-element sequence (Di Nocera et al. 1986; Kaminker et al. 2002). Additionally, the length of the long-range PCR amplification fragment, from ψCG31279 to ψtld (between blocks 6 and 8, Figure 2), implied that the element size of the insertion is approximately the same as the X chromosome G-element-like sequence. Restriction endonuclease digestions with EcoRI and BspHI of a 4-kb fragment amplified from this putative element matches the expected digestion pattern of this X chromosome G-element-like sequence. There is no transposable element insertion at the distal breakpoint of In(3R)P in any of the lines in our study.
Sequence variation:
We amplified and sequenced 2050 bases of the single tok in St lines and both the functional tok and ψtok from In(3R)P lines. Two earlier studies characterized the genomic structure of the tolkin gene (Nguyen et al. 1994; Finelli et al. 1995); our fragment begins at the nineteenth base of exon 4 (this is the first base after the distal breakpoint and defines the 5′-end of ψtok). The first 519 amino acids of the TOK protein form an N-terminal proregion that is apparently proteolytically cleaved to produce a functional TOK protein (Nguyen et al. 1994; Finelli et al. 1995). In our fragment, bases 1–270 and bases 792–834 code for the last 104 amino acids of this proregion. We have separated these bases from the rest of the coding regions in our analysis of sequence polymorphism because such regions are known to evolve under very different constraints than “typical” coding regions (Garcia-Maroto et al. 1991). Our analysis also covers all of exons 5–7, introns 4–6, and 84 bases of intron 7. We chose to study population level variation in the tok gene because it is better annotated than CG31279 and the pseudogene fragment was present in a greater number of In(3R)P lines than the tld fragment.
We sequenced representative regions of the St tok gene and the functional tok and ψtok copies found in In(3R)P using lines from our eastern seaboard populations (Duvernell and Eanes 2000; Verrelli and Eanes 2001; Sezgin et al. 2004). The region included 1790 bp common to 13 copies of the St arrangement tok gene, 13 copies of the functional In(3R)P tok gene, and 9 ψtok copies from In(3R)P. Four copies of In(3R)P would not amplify for the ψtok region and are presumed to bear large insertions. The variable sites are listed in Figure 4, and Figure 5 depicts a neighbor-joining tree (Saitou and Nei 1987) to show general relationships among the sequences. Table 1 summarizes the statistics associated with these regions. The tok gene from St arrangements shows normal levels of synonymous polymorphism (S = 15; θ = 0.022) for D. melanogaster genes (Moriyama and Powell 1996). The inverted (functional) tok gene in the In(3R)P arrangement shows very little synonymous polymorphism (S = 1; θ = 0.001). Between the St and In(3R)P there are five fixations in the functional tok gene, but these are not the result of acquired mutations since origination. All five changes are present in both functional tok and ψtok sequences in In(3R)P and therefore reflect differences in the founding tok allele at the time of duplication and origin of the inversion. Furthermore, the functional tok gene copy from line MFL61 is exceptional. It possesses several polymorphisms that are common to the St arrangement and appears to be a recombination (gene conversion) product. This apparent gene conversion event further inflates the estimate of S in the In(3R)P copies of tok. The truncated ψtok copy associated with the inversion breakpoint shows many features expected of a nonfunctional copy (Figure 4). Of the 10 copies, 5 possess insertions and deletions that would disrupt function. All of the Tajima (1989) and Fu and Li (1993) tests conducted on the tok sequences from constructed sets of St and In(3R)P chromosomes were statistically nonsignificant.
Figure 4.—
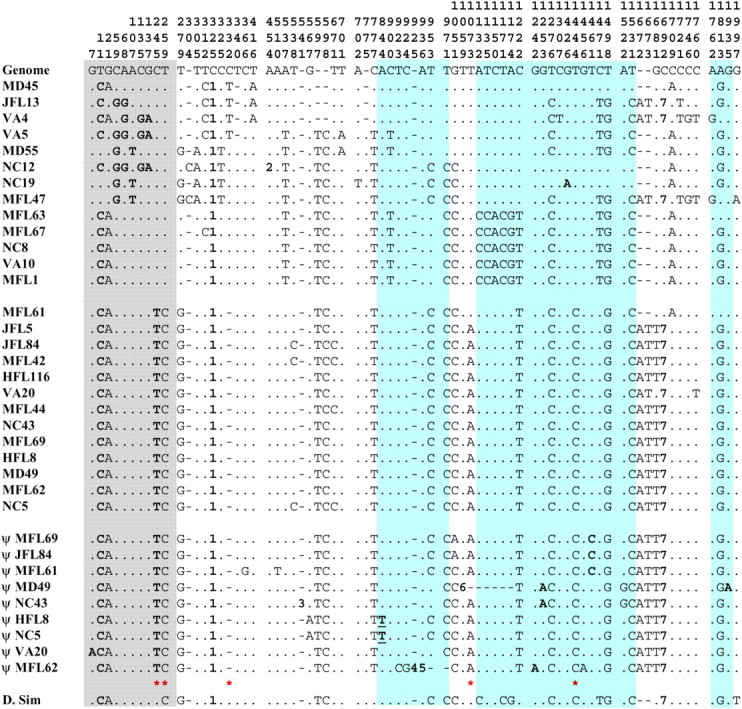
Variable sites for a 2053-bp fragment of the tolkin gene from the St chromosomes (first 13 sequences) and the corresponding regions from both the functional (second 13 sequences) and inactivated (ψ) tok genes in In(3R)P chromosomes (see Figures 2 and 3). Base 1 of this fragment is the first base distal to the distal breakpoint. Coding sites are highlighted: polymorphic sites in the proregion of the protein are highlighted gray; those in the rest of the coding sequence are highlighted blue. Nonsynonymous changes are shown in boldface type. The underlined change is to a stop codon. The boldface numbers refer to insertion/deletion events: 1, a variable length poly(C) string; 2, a 12-bp deletion; 3, a 17-bp insertion; 4, an 11-bp insertion; 5, a 46-bp deletion; 6, a 142-bp deletion; 7, an 8-bp deletion. Red stars indicate the sites unique to the In(3R)P sequences. The reference sequence (“Genome”) is from the annotated D. melanogaster genome, release 3.2.2 (Adams et al. 2000; http://www.fruitfly.org).
Figure 5.—

Neighbor-joining tree of D. melanogaster tolkin sequences rooted with the sequences from D. simulans. Numbers at key nodes represent the proportion of 1000 bootstrap replicates supporting that node.
TABLE 1 .
Comparison of levels of polymorphism (π and θ) within and betweenSt andIn(3R)P chromosomes for the three regions of thetolkin locus studied
| Basesa: | |||
|---|---|---|---|
| 312 Proregionb |
725 Introns |
955 Exons |
|
| Standard | |||
| S | 7 | 17 | 16 |
| Non | 8 | NA | 1 |
| Syn | 2 | NA | 15 |
| No. of sites | 66.15 | 725 | 215.74 |
| π | 0.012 | 0.008 | 0.031 |
| θ | 0.007 | 0.007 | 0.022 |
| Indels | 0 | 7 | 0 |
| Inverted | |||
| S | 0 | 6 | 1 |
| Non | 0 | NA | 0 |
| Syn | 0 | NA | 1 |
| No. of sites | 66.33 | 725 | 215.83 |
| π | 0 | 0.002 | 0.001 |
| θ | 0 | 0.003 | 0.001 |
| Indels | 0 | 3 | 0 |
| Pseudogene | |||
| S | 1 | 4 | 7 |
| Non | 1 | NA | 5 |
| Syn | 0 | NA | 2 |
| No. of sites | 66.33 | 725 | 181.04 |
| π | 0 | 0.002 | 0.004 |
| θ | 0 | 0.002 | 0.004 |
| Indels | 0 | 3 | 2 |
Non, nonsynonymous changes; syn, synonymous changes.
The number of sites used in each calculation, excluding positions with gaps. For exons, the number of sites, π, and θ were all calculated for synonymous sites only.
The first 519 amino acids of the TOK protein, 104 of which are included in our analysis, are proteolytically cleaved from the functional protein.
DISCUSSION
The breakpoints of In(3R)P are not the simple two cut-and-paste structures such as observed in the In(3L)P inversion (Wesley and Eanes 1994). The duplication of several kilobases adjacent to the breakpoints was unexpected and confounded our initial attempt to PCR walk across the distal breakpoint. We would not have recognized that we had passed over the breakpoint had it not been for the parallel confirmation of the breakpoint region using in situ chromosome hybridization with flanking probes. The placement of our long-range primers coincidently landed in the duplicated regions. The precise molecular mechanism for the inversion event is not apparent, although unequal exchange between two sister chromosomes seems to be a reasonable explanation. It is noteworthy that a G element is found at the proximal breakpoint in all the inverted copies. Whether the G element existed before the inversion event, or has inserted after the fact, cannot be determined from our data. The associated duplications add another twist to the story of inversion breakpoints and the origin of inversions in Drosophila and Anopheles, where previously both simple cut-and-paste and transposable elements have been implicated in breakpoints (Cirera et al. 1995; Mathiopoulos et al. 1998; Caceres et al. 2001; Casals et al. 2003).
One inverted tok sequence MFL61 contributes entirely to the sharing of any polymorphisms between St and In(3R)P arrangements. This shared tract probably represents a gene conversion event. While reciprocal exchange recombination will be severely reduced in such close proximity to the breakpoint, gene conversion is not (Rozas and Aguadé 1994). We also observed short-gene conversion tracts in our study of the Pgm gene, which is located just inside the proximal breakpoint of In(3L)P (Verrelli and Eanes 2000).
The relative low level of polymorphism of functional tok in In(3R)P could also be the signature of a young age for the inversion, although the possibility that In(3R)P has historically been low in frequency would also explain this observation. The observation of extensive polymorphism in the ψtok copies, which are linked to the functional tok sequences, argues against recent strong bottlenecks or adaptive sweeps. The frequency of In(3R)P varies widely; it is absent from most northern populations, yet can be the majority arrangement in some tropical and subtropical populations (Krimbas and Powell 1992; Sezgin et al. 2004). We can estimate the historical population frequency and subsequent minimum age of the inversion (in units of Ne generations) using the ratio of polymorphisms as in Andolfatto et al. (1999). This estimation assumes that upon origination the In(3R)P inversion rapidly moved to its current frequency and has subsequently reached mutation-drift neutral equilibrium. Ignoring the clear case of MFL61 as a gene conversion event, we observe only four polymorphic mutations in the functional tok sequences from our sample of 12 In(3R)P chromosomes and 40 in our sample of 13 St chromosomes. From these numbers and associated sample sizes we estimate the historical frequency of In(3R)P to be 0.091, and the minimum age to be 0.33Ne generations. This is the time back to the most recent common ancestor (MRCA) of the In(3R)P sequences. Failure to reach mutation-drift equilibrium will cause us to underestimate the historical frequency; however, this estimate of historical frequency is consistent with the global frequency of the inversion, so the low polymorphism per se is not necessarily an indication of recent origin.
The failure to find fixed de novo differences in the tok sequence variation among the St and inverted sequences strongly suggests that In(3R)P is not an ancient inversion polymorphism (predating the MRCA ancestor of St chromosomes). The number of fixed differences between arrangements can also be used to provide an independent estimate of age. There are five fixed differences, but the sharing of all these differences between tok and ψtok shows that none have arisen since the inversion event. From a coalescence perspective, fixations within the inversion lineage must have occurred prior to the MRCA of In(3R)P and the MRCA with the St arrangement. There are no true fixed differences, so this defines the lower time of In(3R)P origination to In(3R)P MRCA as simply “recent.” The upper time interval depends on the maximum time interval over which there is a probability of no fixations having occurred by chance under Poisson sampling. We can put some confidence on this upper interval of time.
The average level of divergence in the D. melanogaster lineage (since the MRCA with D. simulans) is 0.0852 changes per silent site (Dunn et al. 2001), and Tamura et al. (2004) estimate the time of divergence between D. melanogaster and D. simulans as 5.4 million years. For a region with 231 silent sites the expected divergence would be ∼(0.085 × 231) = 19.68 changes or 19.68/2.7 = 7.288 changes/region/million years. Assuming a Poisson distribution of changes per unit time, a time interval with expected mean of 3.0 mutations has a 5% chance of realizing a sample of no fixed mutations under Poisson sampling. Therefore, an upper 95% age confidence interval for the age of the inversion would be 3.00/7.288 = 411,000 years. This translates into 4.11 × 106 generations, using a reasonable estimate for D. melanogaster of ∼10 generations/year. With the widely used estimate of Ne ≈ 106 for D. melanogaster (Kreitman 1983) this suggests a total upper age limit of 4.11 Ne and a lower age limit of 0.33Ne generations (see above). Therefore, it is possible that the In(3R)P inversion dates to the MRCA of the St arrangements. It is also worth noting that of the five “fixed” mutations captured by the inversion, two are the ancestral state (shared with D. simulans), suggesting that the inversion is old enough for those states to now not be represented in the contemporary St arrangements. Nevertheless, our observations for In(3R)P are consistent with the assertion by Andolfatto et al. (2001) that most inversion polymorphisms have ages on the order of Ne generations and are not ancient.
Acknowledgments
We thank Mike Ippolito for his help in the early attempts to clone the breakpoint positions. This study was supported by U.S. Public Health Service Grant GM-45247 to W.F.E. This is contribution no. 1126 from the Graduate Program in Ecology and Evolution, State University of New York, Stony Brook, New York.
Sequence data from this article have been deposited with the EMBL/GenBank Data Libraries under accession nos. AY881252, AY881253, AY881254, AY881255, AY881256, AY881257, AY881258, AY881259, AY881260, AY881261, AY881262, AY881263, AY881264, AY881265, AY881266, AY881267, AY881268, AY881269, AY881270, AY881271, AY881272, AY881273, AY881274, AY881275, AY881276, AY881277, AY881278, AY881279, AY881280, AY881281, AY881282, AY881283, AY881284, AY881285, AY881286, AY881287, AY881288, AY881289, AY881290, AY881291, AY881292 and AY886890, AY886891, AY886892.
References
- Adams, M. D., S. E. Celniker, R. A. Holt, C. A. Evans, J. D. Gocayne et al., 2000. The genome sequence of Drosophila melanogaster. Science 287 2185–2195. [DOI] [PubMed] [Google Scholar]
- Andolfatto, P., and M. Kreitman, 2000. Molecular variation at the In(2L)t proximal breakpoint site in natural populations of Drosophila melanogaster and D. simulans. Genetics 154 1681–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andolfatto, P., J. D. Wall and M. Kreitman, 1999. Unusual haplotype structure at the proximal breakpoint of In(2L)t in a natural population of Drosophila melanogaster. Genetics 153 1297–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andolfatto, P., F. Depaulis and A. Navarro, 2001. Inversion polymorphisms and nucleotide variability in Drosophila. Genet. Res. 77 1–8. [DOI] [PubMed] [Google Scholar]
- Ashburner, M., 1989 Drosophila: A Laboratory Handbook. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Caceres, M., M. Puig and A. Ruiz, 2001. Molecular characterization of two natural hotspots in the Drosophila buzzatii genome induced by transposon insertions. Genome Res. 11 1353–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casals, F., M. Caceres and A. Ruiz, 2003. The foldback-like transposon Galileo is involved in the generation of two different natural chromosomal inversions of Drosophila buzzatii. Mol. Biol. Evol. 20 674–685. [DOI] [PubMed] [Google Scholar]
- Cirera, S., J. M. Martincampos, C. Segarra and M. Aguadé, 1995. Molecular characterization of the breakpoints of an inversion fixed between Drosophila melanogaster and D. subobscura. Genetics 139 321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Nocera, P. P., F. Graziani and G. Lavorgna, 1986. Genomic and structural organization of the Drosophila melanogaster G elements. Nucleic Acids Res. 14 675–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobzhansky, Th., 1970 Genetics of the Evolutionary Process. Columbia University Press, New York.
- Dunn, K. A., J. P. Bielawski and Z. Yang, 2001. Substitution rates in Drosophila nuclear genes: implications for translational selection. Genetics 157 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvernell, D. D., and W. F. Eanes, 2000. Contrasting molecular population genetics of four hexokinases in Drosophila melanogaster, D. simulans and D. yakuba. Genetics 156 1191–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein, J., 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39 783–791. [DOI] [PubMed] [Google Scholar]
- Finelli, A. L., T. Xie, C. A. Bossie, R. K. Blackman and R. W. Padgett, 1995. The tolkin gene is a tolloid/BMP-1 homologue that is essential for Drosophila development. Genetics 141 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y. X., and W. H. Li, 1993. Statistical tests of neutrality of mutations. Genetics 133 693–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Maroto, F., A. Castangnaro, P. Sanchez de la Hoz, C. Marana, P. Carbonera et al., 1991. Extreme variations in the ratios of non-synonymous to synonymous nucleotide substitution rates in signal peptide evolution. FEBS Lett. 287 67–70. [DOI] [PubMed] [Google Scholar]
- Hasson, E., and W. F. Eanes, 1996. Contrasting histories of three gene regions associated with In(3L)Payne of Drosophila melanogaster. Genetics 144 1565–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminker, J. S., C. M. Bergman, B. Kronmiller, J. Carlson, R. Svirskas et al., 2002 The transposable elements of Drosophila melanogaster: a genomics perspective. Genome Biol. 3: 0084.1–0084.20. [DOI] [PMC free article] [PubMed]
- Kimura, M., 1980. A simple method for estimating the evolutionary rates of base substitution through comparative studies of nucleotide sequences. J. Mol. Evol. 16 111–120. [DOI] [PubMed] [Google Scholar]
- Kreitman, M., 1983. Nucleotide polymorphism at the alcohol dehydrogenase locus of Drosophila melanogaster. Nature 304 412–417. [DOI] [PubMed] [Google Scholar]
- Krimbas, C. B., and J. R. Powell, 1992 Drosophila Inversion Polymorphisms. CRC Press, Cleveland.
- Kumar, S., K. Tamura, I. B. Jakobsen and M. Nei, 2001 MEGA2: molecular evolutionary genetics analysis software. Arizona State University, Tempe, Arizona. [DOI] [PubMed]
- Mathiopoulos, K. D., A. Dellatorre, V. Predazzi, V. Petrarca and M. Coluzzi, 1998. Cloning of inversion breakpoints in the Anopheles gambiae complex traces a transposable element at the inversion junction. Proc. Natl. Acad. Sci. USA 95 12444–12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama, E. N., and J. R. Powell, 1996. Intraspecific nuclear DNA variation in Drosophila. Mol. Biol. Evol. 13 261–277. [DOI] [PubMed] [Google Scholar]
- Nguyen, T., J. Jamal, M. J. Shimell, K. Arora and M. B. O'Connor, 1994. Characterization of tolloid-related-1: a BMP-1-like product that is required during larval and pupal stages of Drosophila development. Dev. Biol. 166 569–586. [DOI] [PubMed] [Google Scholar]
- Novitski, E., and G. Braver, 1954. An analysis of crossing over within a heterozygous inversion in Drosophila melanogaster. Genetics 39 197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman, H., A. S. Gerber, and D. L. Hartl, 1988. Genetic applications of an inverse polymerase chain reaction. Genetics 120 621–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell, J. R., 1997 Progress and Prospects in Evolutionary Biology: The Drosophila Model. Oxford University Press, New York.
- Rozas, J., and M. Aguadé, 1994. Gene conversion is involved in the transfer of genetic information between naturally occurring inversions of Drosophila. Proc. Natl. Acad. Sci. USA 91 11517–11521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas, J., and R. Rozas, 1997. DnaSP version 2.0: a novel software package for extensive molecular population genetics analysis. Comput. Appl. Biosci. 13 307–311. [PubMed] [Google Scholar]
- Saitou, N., and M. Nei, 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4 406–425. [DOI] [PubMed] [Google Scholar]
- Sezgin, E., D. D. Duvernell, L. M. Matzkin, Y. Duan, C.-T. Zhu et al., 2004. Single-locus latitudinal clines and their relationship to temperate adaptation in metabolic genes and derived alleles in Drosophila melanogaster. Genetics 168 923–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura, K., S. Subramanian and S. Kumar, 2004. Temporal patterns of fruit fly (Drosophila) evolution revealed by mutation clocks. Mol. Biol. Evol. 21 36–44. [DOI] [PubMed] [Google Scholar]
- Thompson, J. D., T. J. Gibson, F. Plewniak, F. Jeanmougin and D. G. Higgins, 1997. The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verrelli, B. C., and W. F. Eanes, 2000. Extensive amino acid polymorphism at the Pgm locus is consistent with adaptive protein evolution in Drosophila melanogaster. Genetics 156 1737–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verrelli, B. C., and W. F. Eanes, 2001. Clinal variation for amino acid polymorphisms at the Pgm locus in Drosophila melanogaster. Genetics 157 1649–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesley, C. S., and W. F. Eanes, 1994. Isolation and analysis of the breakpoint sequences of chromosome inversion In(3L)Payne in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 91 3132–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]