

Abstract
Maf family transcription factors are atypical basic region–leucine zipper (bZIP) proteins that contain a variant basic region and an ancillary DNA-binding region. These proteins recognize extended DNA sequence elements flanking the core recognition element bound by canonical bZIP proteins. We have investigated the causes for the differences in DNA recognition between Maf and other bZIP family proteins through studies of Maf secondary structure, trypsin sensitivity, binding affinity, dissociation rate and DNA contacts. Our results show that specific DNA binding by Maf is coupled to a conformational change involving both the basic and ancillary DNA-binding regions that depends on the extended DNA sequence elements. Two basic region amino acid residues that differ between Maf and canonical bZIP proteins facilitate the conformational change required for Maf recognition of the extended elements. Nucleotide base contacts made by Maf differ from those made by canonical bZIP proteins. Taken together, our results suggest that the unusual DNA binding specificity of Maf family proteins is mediated by concerted folding of structurally unrelated DNA recognition motifs.
Keywords: ancillary DNA-binding region/basic region/coil-to-helix transition/DNA contact mapping/protease cleavage
Introduction
Transcription regulatory proteins are classified based on the presence of conserved sequence motifs corresponding to structural domains that mediate specific DNA binding (Harrison, 1991). Members of a particular family of DNA-binding proteins generally contact DNA in a similar manner and often bind to related DNA sequences. Differences in DNA recognition specificities are typically mediated by amino acid substitutions within the interaction interface that result in altered base contacts but cause little change in the protein fold or positioning on DNA. In oligomeric DNA-binding proteins, changes in monomer arrangement can alter the spacing and orientations of individual recognition elements, but the fold of individual monomers is usually conserved among different family members.
The DNA-binding domain of basic region–leucine zipper (bZIP) family proteins represents perhaps the simplest structural motif for specific DNA recognition. Members of the bZIP family bind to DNA sequence elements that are typically 7–8 bp in length as homo- or heterodimeric complexes. The minimal dimerization and DNA-binding regions in most bZIP proteins consist of a leucine zipper coiled-coil interface and a basic DNA contact region. The basic regions contain many highly conserved amino acid residues that make direct contacts to nucleotide base pairs at AP-1 and core recognition element (CRE) sites (Ellenberger et al., 1992; Konig and Richmond, 1993; Glover and Harrison, 1995).
The basic DNA contact regions of bZIP family proteins undergo a conformational change to α-helical structure upon DNA binding (Patel et al., 1990; Weiss et al., 1990). Coupling of protein folding to DNA binding may contribute to the recognition specificity by linking interactions between different parts of the contact interface. Many bZIP family proteins can both activate and repress transcription at different promoters (Diamond et al., 1990; Li et al., 1999). Differences in protein conformation may contribute to these differences in transcriptional activity (Lefstin and Yamamoto, 1998).
The Maf family of transcription factors is classified as a subgroup of bZIP family proteins since they contain a leucine zipper dimerization interface adjacent to a region rich in basic amino acid residues (Nishizawa et al., 1989; Swaroop et al., 1992; Blank and Andrews, 1997). The prototype of this family is the product of the c-maf proto-oncogene (Nishizawa et al., 1989). Members of the Maf family differ in several respects from other bZIP proteins. The basic regions of all Maf family proteins contain non-conservative amino acid substitutions at positions that are highly conserved in other bZIP proteins (Figure 1A). In particular, an alanine that makes direct base contacts deep within the major groove in complexes formed by other bZIP proteins (Ellenberger et al., 1992; Konig and Richmond, 1993; Glover and Harrison, 1995) is substituted by a tyrosine in all Maf family proteins. The adjacent position in Maf family proteins is occupied by a glycine that is never found in the basic regions of other bZIP proteins, presumably because it disfavors the α-helical conformation required for DNA recognition by the basic region (Patel et al., 1990; Weiss et al., 1990). Maf family proteins also contain a highly conserved ancillary DNA-binding region on the N-terminal side of the basic region that is required for specific DNA recognition (Kerppola and Curran, 1994a).

Fig. 1. Differences between Maf family transcription factors and other bZIP proteins. (A) The conservation of amino acid residues among 27 bZIP proteins that bind to AP-1/CRE recognition sites was compared with that of 19 Maf family members using logo representations (Schneider, 1996) of each group of sequences constructed using the Blocks server (Henikoff et al., 1999). The height of each letter reflects the degree of conservation of the amino acid residue. Representative members of each group are aligned above and below the logo representations. The basic regions found in both groups and the ancillary DNA-binding region specific to Maf family proteins are indicated. The conserved G286 and Y287 residues in Maf, and the corresponding positions in the basic regions of other bZIP proteins are indicated by arrows. (B) Alignment of logo representations of DNA sequences bound by Fos and Jun (upper panel) versus Maf and Nrl (lower panel) (Kerppola and Curran, 1994a). The core recognition element bound by many bZIP family proteins and the extended recognition elements specific to binding sites for Maf family proteins are indicated. The numbering of the base pairs is indicated below the sequences. The [–1+1] base pairs are enclosed in brackets to indicate that different binding sites may contain either one or both of these base pairs.
Members of the Maf family recognize a longer DNA sequence element (13–14 bp) than other bZIP proteins (Figure 1B) (Kataoka et al., 1994; Kerppola and Curran, 1994a). This sequence contains extended recognition elements on both sides of the core recognition element. Whereas other bZIP proteins recognize primarily the core element, Maf family proteins recognize mainly the extended sequence elements (Kerppola and Curran, 1994a). The X-ray crystal structures of other bZIP proteins show that a straight α-helix in the major groove can directly contact a maximum of five adjacent base pairs (Ellenberger et al., 1992; Konig and Richmond, 1993; Glover and Harrison, 1995). Thus, recognition of the entire Maf-binding site requires a different mode of DNA binding.
The Maf family includes many proteins that control the differentiation of diverse cell types and regulate the expression of many gene products in terminally differentiated cells (Andrews et al., 1993; Ogino and Yusuda, 1998; Kelly et al., 2000). Several Maf family members are expressed in many different cell types and control the expression of different target genes in different cells (Ho et al., 1998; Kim et al., 1999; Li et al., 1999). The cell type-specific functions of Maf family members are determined by interactions with other transcription regulatory proteins, including heterodimerization with other bZIP proteins. DNA sequence elements that mediate transcription activation by Maf family proteins frequently differ from the consensus recognition sequence. This variation in the sequences of Maf regulatory elements may contribute to the cell type-specific activation of Maf target genes.
One group of bZIP proteins that can heterodimerize with Maf family members is the CNC family (Andrews et al., 1993; Igarashi et al., 1994; Itoh et al., 1995). CNC proteins also contain a conserved region on the N-terminal side of the basic region (Rupert et al., 1998), but recognize a DNA sequence element similar to that recognized by canonical bZIP proteins (Oyake et al., 1996; Johnsen et al., 1998). In Skn-1, which is a monomeric protein related to the CNC family, the N-terminal region contributes to the recognition of base pairs on the proximal side of the core recognition element (the side opposite to the extended recognition element) (Carroll et al., 1997; Kophengnavong et al., 1999). The X-ray crystal structure of the DNA-binding domain of Skn-1 revealed that the N-terminal region forms a four-helix bundle and a flexible arm that contacts the phosphodiester backbone on the proximal side of the core recognition element (Rupert et al., 1998).
To investigate the structural basis for the unusual DNA recognition specificity of Maf family proteins, we have examined complexes formed by Maf at different DNA sequence elements. The effects of single amino acid substitutions were analyzed to identify residues that were critical for Maf folding and for the extended DNA recognition specificity. DNA contacts made by Maf were probed by DNA footprinting and interference analysis. The results are interpreted in the context of a model for DNA recognition by Maf family proteins.
Results
The conformation of Maf varies at different DNA-binding sites
Many bZIP family members undergo a conformational change upon DNA binding that increases the α-helix content of their bZIP domains (Patel et al., 1990; Weiss et al., 1990). We examined the effect of DNA binding on the conformation of the Maf bZIP domain and ancillary DNA-binding region. Comparison of the circular dichroism (CD) spectra of free Maf and Maf in the presence of a consensus binding site oligonucleotide revealed a large change in ellipticity at 208 and 222 nm (Figure 2A). This change is consistent with an increase in the α-helicity of Maf upon binding to the optimal recognition sequence. The change in mean residue ellipticity was similar for Maf and Fos–Jun heterodimers (Figure 2E). Since the Maf protein used was larger than Fos or Jun, the number of amino acid residues that adopted an α-helical conformation was 40% larger in Maf than in Fos–Jun. In the absence of DNA, Fos–Jun heterodimers and Maf homodimers had a similar α-helix content that was consistent with folding of the leucine zipper prior to DNA binding. It is therefore likely that the ancillary DNA-binding region adopts a conformation of high α-helix content upon Maf binding to the consensus recognition sequence.

Fig. 2. Secondary structure of Maf at different DNA-binding sites and roles of atypical basic region amino acid residues. (A) Comparison of the CD spectra of free Maf (green), the oligonucleotide containing a consensus Maf-binding site M (blue) and the Maf–M complex (purple). The ellipticities of 10 µM Maf241-369, 10 µM M site oligonucleotide and 10 µM Maf241-369–M site complex were measured and plotted as molar ellipticity versus wavelength. Minima at 208 and 222 nm are characteristic of an α-helical conformation. The data represent averages of three independent experiments, and the standard deviations are indicated by vertical bars. (B) Comparison of Maf conformations at different recognition sites. The mean residue ellipticity of Maf241-369 bound to different recognition sites (solid symbols) was calculated by subtraction of the ellipticity of each oligonucleotide from the ellipticity of the complex formed by Maf at each site, and was compared with that of free Maf241-369 (open symbols). There were no significant differences between the spectra of the oligonucleotides alone. (C) Effects of substitution of the tyrosine residue in the basic region on Maf conformations at different recognition sites. The mean residue ellipticity of Maf241-369 Y287A free and bound to different recognition sites was plotted as in (B). (D) Effects of substitution of the glycine and tyrosine residues in the basic region on Maf conformation at different recognition sites. The mean residue ellipticity of Maf241-369 G286I Y287A free and bound to different recognition sites was plotted as in (B). (E) Comparison of Fos–Jun conformations at different recognition sites. The mean residue ellipticity of Fos139-200–Jun257-318 free and bound to different recognition sites was plotted as in (B). Each curve represents an average of five independent experiments, and the relative standard deviations of the data shown in (B), (C), (D) and (E) were comparable to those shown in (A).
Maf family members recognize a DNA sequence element that is almost twice as long as elements recognized by other bZIP proteins (Kataoka et al., 1994; Kerppola and Curran, 1994a). We examined the effects of base substitutions in both the core and extended recognition elements on the conformation of Maf bound to DNA (Figure 2B). Substitution of a single base pair in one of the extended recognition elements (position –6) reduced the change in Maf α-helicity induced by DNA binding by approximately half (Figure 2B, green line). This result is consistent with no induced folding of one of the subunits of the Maf homodimer or with partial folding of both subunits. At the high concentrations (10 µM) of both protein and DNA used in these experiments, >98% of Maf bound to the oligonucleotide based on both a shift in the mobility of the protein during non-denaturing gel electrophoresis (data not shown) as well as the measured binding affinity (see below). To ensure that all Maf molecules were bound to DNA, higher concentrations of oligonucleotide (up to 4-fold molar excess) were added to a constant amount of protein. There was no change in the CD spectrum of the protein at oligonucleotide concentrations above an equimolar ratio. Thus, the difference in the CD spectrum of Maf at the mutated binding site reflects a change in Maf conformation rather than reduced binding to the mutated oligonucleotide.
Substitution of the same base pair in both half-sites (positions ±6) abolished the induced α-helical conformation of Maf (Figure 2B, blue line). This base pair is therefore essential for Maf to undergo a conformational change upon binding to DNA. Quantitative binding of Maf to the oligonucleotide was observed under the experimental conditions (data not shown). There was only a small effect of these base substitutions on the α-helix content of Fos–Jun heterodimers (Figure 2E). Other base substitutions within the extended recognition elements (positions ±5 and positions ±7) had smaller effects on Maf conformation (data not shown). Base substitutions within the CRE (positions ±4) did not influence the α-helix content of Maf (Figure 2B, red line). In contrast, these base substitutions eliminated the coil-to-helix transition of Fos–Jun heterodimers (Figure 2E). No increase in the α-helix content of either Maf or Fos–Jun was observed in the presence of a CG co-polymer oligonucleotide (data not shown). Consequently, base substitutions in the core and extended recognition elements had reciprocal effects on the conformational changes of Maf and Fos–Jun (Figure 2B and E, compare the red and blue lines).
Two amino acid residues in the basic region influence the sequence dependence of Maf conformation
The basic regions of all Maf family members differ from those of all other bZIP proteins (Figure 1A). Initial studies with chimeric proteins demonstrated that the unique DNA recognition properties of Maf required the atypical basic region (data not shown). To identify amino acid residues that determine the DNA sequence dependence of Maf conformation, we replaced amino acid residues in the basic region of Maf by the corresponding amino acid residues from the canonical bZIP protein Jun. The effects of replacement of two amino acid residues (Y287A and G286I) were examined in detail (Figure 2C and D). Replacement of these residues had little effect on the CD spectra of the proteins in the absence of DNA. However, when these variants were bound to the consensus Maf recognition sequence, the proteins in which the tyrosine was replaced by an alanine had a higher α-helicity compared with wild-type Maf (compare black lines in Figure 2B, C and D). Replacement of the glycine by an isoleucine had little effect alone, but increased the effect of the tyrosine substitution. Thus, the tyrosine and glycine residues in wild-type Maf reduced its α-helix content at the consensus recognition element, presumably by preventing the basic region from adopting the fully α-helical conformation observed in other bZIP proteins.
The atypical structure of the basic region of Maf family proteins may contribute to their unusual DNA recognition properties. To test whether the amino acid residues in the basic region affected the sequence dependence of the change in Maf conformation, we compared the CD spectra of the mutant proteins at different DNA-binding sites (Figure 2C and D). Replacement of the tyrosine in the basic region of Maf by an alanine altered the influence of base substitutions in the extended recognition element on Maf conformation. The single base pair substitution (position –6) that had a large effect on the secondary structure of wild-type Maf caused only a small change in the secondary structures of proteins in which the tyrosine was replaced by alanine (compare green lines in Figure 2B, C and D). Substitution of this base pair in both half-sites (positions ±6) abolished the induced α-helical conformation of wild-type Maf, but had only a partial effect on proteins in which the tyrosine was replaced (compare blue lines in Figure 2B, C and D). Proteins in which both the tyrosine and glycine were replaced exhibited similar effects of base substitutions in the extended recognition elements on secondary structure. These results indicate that the tyrosine residue affected the conformation of Maf through interactions involving the extended recognition element.
Replacement of the atypical amino acid residues in the basic region of Maf also altered the effects of base substitutions in the CRE on Maf conformation (Figure 2C and D). Whereas base substitutions in the core recognition element (positions ±4) had no effect on the secondary structure of wild-type Maf, they reduced the α-helix content of proteins in which the glycine and tyrosine were replaced (compare red lines in Figure 2B and D). These substitutions had a small effect on proteins in which only the tyrosine was replaced. Base substitutions in the extended (positions ±6) and core (positions ±4) recognition elements had opposite effects on the secondary structures of Maf and Fos–Jun, but they had similar effects on proteins in which both the glycine and tyrosine were replaced (compare red and blue lines in Figure 2B, D and E). Hence, the tyrosine and glycine residues in the basic region are important, but not sole determinants of the differences in secondary structure between Maf and Fos–Jun at different recognition sites.
Effects of the sequence of the binding site and amino acid residues in the basic region on Maf cleavage by trypsin
To analyze changes in Maf conformation using an independent approach, we compared trypsin cleavage of Maf in the presence and absence of the consensus recognition sequence (Figure 3A). Maf was digested rapidly in the absence of DNA, producing a series of intermediates cleaved within the ancillary and bZIP domains. Binding of Maf to the optimal recognition sequence protected the bZIP domain and the ancillary DNA-binding region from trypsin digestion. The initial product (fragment I) was cleaved on the N-terminal side of the ancillary DNA-binding region (N-terminal sequence LHFDD). The second cleavage product (fragment II) had the same N-terminal sequence as fragment I, and the size of the product indicated that it was cleaved on the C-terminal side of the leucine zipper. This product was stable in the presence of an equimolar concentration of trypsin for up to 10 min. Thus, Maf binding to the consensus recognition sequence induced a conformational change involving the basic and ancillary DNA-binding regions and protected them from trypsin digestion.

Fig. 3. Trypsin cleavage of Maf bound to different oligonucleotides and effects of basic region substitutions. (A) Time course of trypsin cleavage of free Maf241–369 and Maf241–369 bound to the consensus recognition sequence. A 10 µM concentration of Maf241–369 with or without 20 µM M site oligonucleotide was incubated with trypsin at a 1:100 ratio (w:w) at 23°C. Aliquots (2.5 µg) were withdrawn at the times indicated above the lanes and the products were separated by 16% SDS–PAGE and visualized by Coomassie Blue staining. (B) Comparison of trypsin digestion of Maf bound to M, M±4, M±6 or M±7 oligonucleotides. A 10 µM concentration of Maf241–369 was incubated without trypsin (lanes 1, 4, 7 and 10) or with trypsin at 1:50 (lanes 2, 5, 8 and 11) or 1:100 (lanes 3, 6, 9 and 12) ratios (w:w) in the presence of 20 µM of the oligonucleotides indicated above the lanes for 10 min at 23°C. Lanes indicated by S contained molecular weight standards. (C) Comparison of the trypsin sensitivity of Maf241–369 G286I Y287A in the presence of oligonucleotides containing base substitutions in the core or extended recognition elements. A 10 µM concentration of Maf241–369 G286I Y287A was incubated with a 1:100 (w:w) ratio of trypsin in the presence of 20 µM of the oligonucleotides indicated above lanes 3–10 at 23°C for 10 min. Lanes 1 and 2 were controls with no trypsin and no oligonucleotide, respectively. (D) Comparison of intermediates during trypsin digestion of free Maf and Maf bound to the M±6 oligonucleotide. Trypsin digestion (1:200 ratio w:w) was stopped at the times indicated above the lanes, and the peptides were separated by 16% Tris-Tricine–PAGE. The reaction products from a gel run in parallel were transferred onto a PVDF membrane and subjected to five cycles of Edman degradation. The N-terminal amino acid sequences of cleavage products obtained in the absence (I*–VI*) and the presence (I–VII) of oligonucleotide-binding sites are shown on the left and the right sides, respectively. (E) Trypsin cleavage map of Maf in the presence (lower bar and arrows) and absence (upper bar and arrows) of the M±6 oligonucleotide. The peptides were sequenced from gels run under two different electrophoresis conditions to confirm the differences between the cleavage sites in the presence and absence of oligonucleotide and to resolve co-migrating peptides. Asterisks and bent arrows pointing to the right indicate the N-terminal ends of the peptides shown in (D) obtained by sequencing of the isolated peptides. Circles and bent arrows pointing to the left indicate C-terminal ends of the peptides inferred from mass spectrometric analysis. The maximum difference between the measured and predicted masses for the peptides was 0.7%.
The sequence of the binding site affected the overall secondary structure of Maf based on CD measurements. To determine the influence of the recognition sequence on the folding of individual regions of Maf, we examined trypsin cleavage of Maf at different binding sites (Figure 3B). Substitution of the base pairs at the ±6 positions dramatically reduced Maf protection from trypsin digestion (Figure 3B, lanes 8 and 9). In addition to fragments I and II, several peptides cleaved within the ancillary DNA-binding region and the bZIP domain were produced by trypsin digestion. These peptides were not observed following trypsin digestion in the absence of DNA or in the presence of a non-cognate oligonucleotide (see below). They suggest that Maf bound to this site in an altered conformation. Substitution of the base pairs at the ±4 positions did not affect the protection of wild-type Maf (Figure 3B, lanes 5 and 6). Base substitutions in the core and extended recognition elements therefore had parallel effects on the trypsin sensitivities and secondary structures of Maf–DNA complexes. Thus, both the α-helical conformation of Maf and the protection from trypsin digestion required specific interactions with the extended DNA recognition elements, but were unaffected by core substitutions.
The effects of the atypical amino acid residues within the basic region of Maf on the conformational change were examined by trypsin digestion in the presence of different oligonucleotide-binding sites (Figure 3C). Replacement of the glycine and tyrosine residues had no effect on trypsin cleavage of Maf bound to the consensus recognition site (Figure 3C, lane 3). Significantly, the base substitutions at the ±6 positions that eliminated protection of wild-type Maf did not reduce protection of the mutant protein (Figure 3C, lane 9). In contrast, substitution of the base pairs at the ±4 positions severely reduced the protection of the protein in which the glycine and tyrosine residues were replaced (Figure 3C, lane 7). The α-helicity (Figure 2B) as well as the binding affinity (see below) of the Maf G286I Y287A protein was similar at the M±4 and M±6 sites. Thus, the change in trypsin cleavage of this protein reflected a change in local structure or dynamics caused by the base substitutions at the ±4 positions. Replacement of the atypical amino acid residues in the basic region altered the DNA sequence dependence of both the α-helix content and the trypsin sensitivity of Maf.
Trypsin cleavage sites in free Maf and in Maf bound to a non-consensus recognition site
The trypsin digestion experiments were performed under conditions where >98% of Maf was bound to the oligonucleotides (data not shown). However, since the rate of dissociation of some of the complexes was faster than the rate of protease digestion, it was necessary to examine the possibility that the altered protease sensitivity reflected cleavage of proteins subsequent to dissociation from DNA. The protease cleavage pattern can also provide information about differences in local protein conformation. We therefore compared the trypsin cleavage sites of free Maf and Maf bound to the M±6 oligonucleotide (Figure 3D and E). High-resolution SDS–PAGE demonstrated that most of the peptide intermediates produced by trypsin digestion of Maf alone and Maf bound to the M±6 site migrated with different mobilities (Figure 3D). The peptides produced in the presence and absence of the oligonucleotide were analyzed by N-terminal sequencing and MALDI-TOF mass spectrometry (Figure 3E).
In the presence of oligonucleotide, Maf cleavage initiated on the N-terminal side of the ancillary DNA-binding region since no intermediates with an intact N-terminus were recovered. In the absence of oligonucleotide, Maf cleavage initiated from the C-terminal side of the leucine zipper. Two transient intermediates (II* and III*) were observed consistently in the absence, but not in the presence of the oligonucleotide. One of these (II*) is the product of two cleavage sites unique to the free Maf protein. There was insufficient material for identification of the other product (III*). A prominent cleavage product (IV*) also resulted from cleavage at a site unique to free Maf within the leucine zipper. In the presence of M±6 oligonucleotide, Maf exhibited a unique cleavage site within the basic region that corresponded to the N-terminal end of one peptide (III) and the C-terminal end of another (VII). This cleavage site supports the hypothesis that the basic region does not form a contiguous α-helix, and therefore does not penetrate deep within the major groove. In the presence of M±6 oligonucleotide, Maf also exhibited cleavage sites within the ancillary region and the leucine zipper that produced a peptide unique to the complex (V). Both free Maf and Maf in the presence of M±6 oligonucleotide shared a cleavage site at the junction between the basic region and the leucine zipper. Although we cannot distinguish between primary and secondary trypsin cleavage events in these experiments, the results clearly exclude the possibility that the altered trypsin sensitivity of Maf at the M±6 site is caused by dissociation of Maf from the oligonucleotide.
Replacement of amino acid residues in the basic region has distinct effects on Maf binding affinity at different recognition sites
To investigate the influence of the base substitutions that affected the conformation of Maf on the avidity of Maf binding to DNA, we compared the apparent binding affinities of complexes formed by Maf at sites containing base substitutions in the core and extended DNA recognition elements (Figure 4A, upper graph). Substitution of a single base pair (position –6) in the extended recognition element reduced the apparent binding affinity of Maf by one order of magnitude (Figure 4A, green line). Substitution of the same base pair in both half-sites (positions ±6) reduced the binding affinity by an additional 5-fold (Figure 4A, blue line). The effects of the base substitutions in the two half-sites were therefore relatively independent of each other. Substitution of other base pairs within the extended DNA recognition element (positions ±5 and ±7) had smaller effects on binding affinity (data not shown). Substitution of base pairs within the CRE (positions ±4) had little effect on Maf binding affinity (Figure 4A, red line). Thus, the same base pairs that influence the conformation of Maf upon binding to DNA also affect the apparent binding affinity.
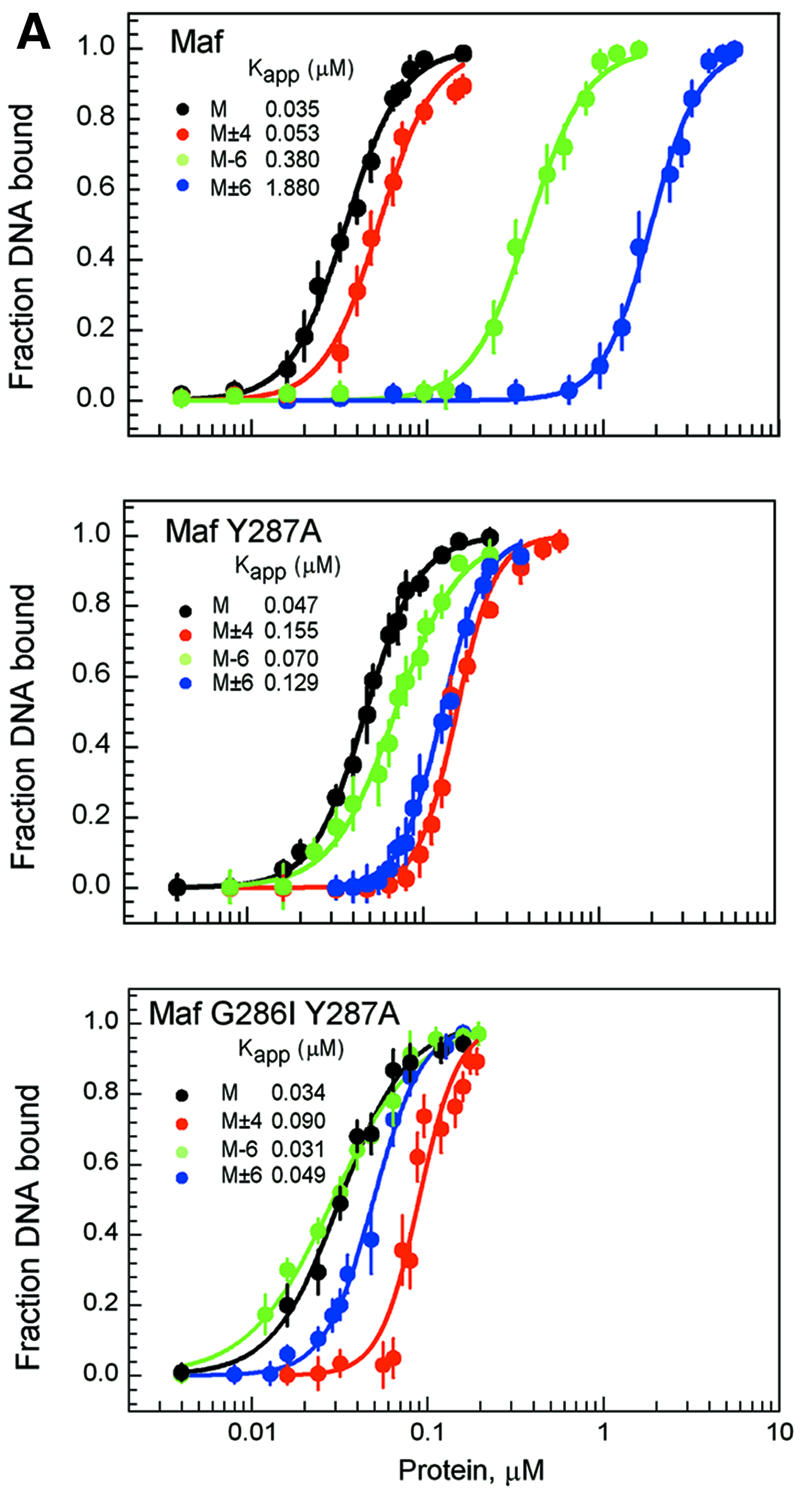
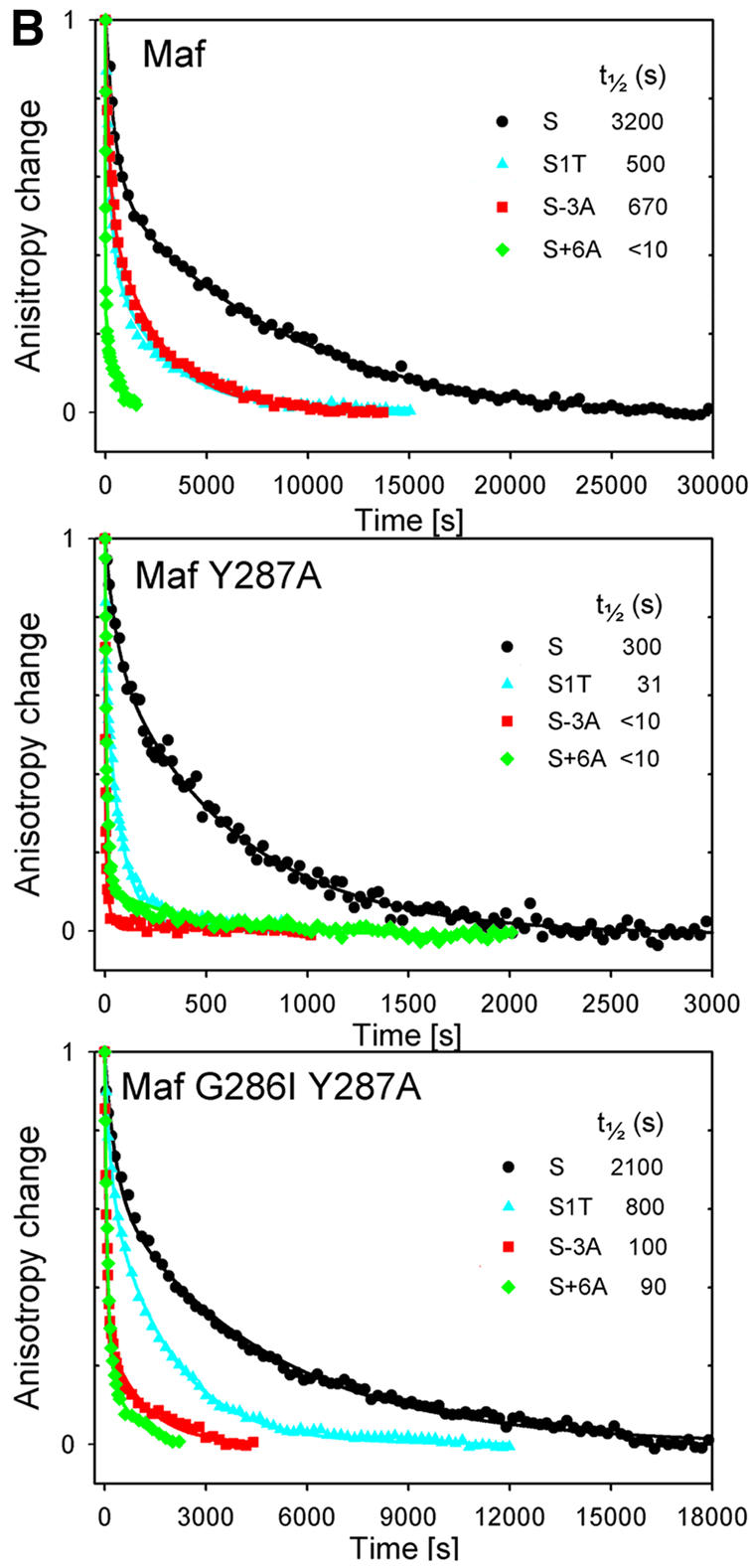
Fig. 4. Apparent binding affinities and dissociation rates of Maf complexes at different recognition sites and effects of atypical basic region amino acid residues. (A) Comparison of apparent binding affinities of Maf241-369 (upper graph), Maf241-369 Y287A (middle graph) and Maf241-369 G286I Y287A (lower graph) at the M, M–6, M±6 and M±4 sites. Different concentrations of the wild-type and mutant proteins were incubated for 30 min with 10 pM of each of the oligonucleotides indicated in the figure, and the fraction of the oligonucleotide bound in EMSAs was plotted versus protein concentration. The data represent the average of three independent experiments. Similar results were obtained when the complexes were incubated for 24 h prior to gel electrophoresis. The sigmoidal curves represent the best fit of the Hill equation to the data. The apparent binding affinity of each complex is shown. (B) Comparison of the dissociation rates of Maf241-369 (upper graph), Maf241-369 Y287A (middle graph) and Maf241-369 G286I (lower graph) at the S, S–1T, S–3T and S+6A sites. The dissociation rates of complexes formed by the proteins indicated in each panel at the binding sites indicated by the different symbols were measured by using fluorescence anisotropy to monitor complex dissociation in the presence of competitor DNA. The S site corresponds to a consensus Maf recognition sequence with symmetrical CTCTAGATTC flanking sequences on each end. The positions of base pair substitutions are indicated. The changes in anisotropy were normalized to facilitate comparison of the dissociation rates. The anisotropy changes for different proteins were plotted using different time scales to facilitate comparison of the effects of base substitutions on the stabilities of complexes formed by different homodimers.
The effects of the atypical amino acid residues in the basic region of Maf on the apparent binding affinities were examined. Replacement of the tyrosine residue within the basic region of Maf had converse effects on binding affinity at sites containing base substitutions in the core and extended recognition elements (Figure 4A, middle graph). Base substitutions in the extended recognition elements (positions ±6) had smaller effects on binding of MafY287A and Maf G286I Y287A. Moreover, substitution of base pairs in the CRE (positions ±4) reduced the apparent binding affinities of the mutant proteins. Hence, the changes in Maf structure caused by replacement of the atypical amino acid residues in the basic region altered the recognition of both the core and extended recognition elements.
The apparent binding affinities of Maf variants at the different binding sites do not necessarily reflect the equilibrium dissociation coefficients of the complexes. However, they provide a useful basis for comparison of the relative avidities of the various complexes. The large effects of base substitutions at the –6 and ±6 positions on the apparent binding affinities of Maf were corroborated by oligonucleotide competition analysis (data not shown). Furthermore, thermal denaturation studies of complexes formed by Maf and Maf Y287A at the consensus recognition site and sites containing base substitutions at the –6 and ±6 positions demonstrated that these base substitutions also had a larger effect on the thermal stabilities of complexes formed by wild-type Maf (data not shown). Substitution of two base pairs within the 13 bp recognition element therefore prevented the coil-to-helix transition of Maf and virtually eliminated specific recognition of the binding site.
The extended DNA contact interface of Maf mediates formation of exceptionally stable complexes
To investigate the influence of the unique DNA contact interface of Maf on the stabilities of Maf–DNA complexes, we determined the dissociation rates of complexes formed by Maf variants at different recognition sites (Figure 4B). The dissociation rates were measured in solution by monitoring the fluorescence anisotropy of fluorescein linked to one end of the oligonucleotide in Maf–DNA complexes following the addition of unlabeled competitor DNA. Maf formed an extraordinarily stable complex at the consensus recognition sequence with a half-life of almost 1 h (Figure 4B, upper panel). This exceptionally slow dissociation rate was also observed by monitoring complex dissociation by electrophoretic mobility shift analysis. Under identical conditions, Fos–Jun heterodimers dissociated from the same binding site with a half-life of <20 s. Substitution of either the central base pair or the base pair at the –3 position within the CRE caused a small (5- to 6-fold) decrease in the stability of the complex. In contrast, substitution of the base pair at the +6 position within the extended recognition element cause a large (>300-fold) decrease in complex stability. Thus, base pairs within the extended recognition elements were more important for the high stability of Maf–DNA complexes than base pairs within the CRE.
Replacement of the atypical tyrosine residue in the basic region of Maf reduced the stability of the complex at the consensus recognition sequence by >10-fold (Figure 4B, middle panel). Substitution of the central base pair or the base pair at the –3 position within the CRE had a larger effect on the stabilities of these complexes than on complexes formed by wild-type Maf. Replacement of both the tyrosine and glycine residues in the basic region of Maf had a small effect on complex stability at the consensus recognition sequence (Figure 4B, lower panel). Substitution of the base pair at position –3 in the CRE had a larger effect, whereas substitution of the base pair at position +6 in the extended recognition element had a smaller effect on the stabilities of complexes formed by Maf G286I Y287A than on the stabilities of complexes formed by wild-type Maf. Substitution of the central base pair had similar effects on the stabilities of both complexes. Consequently, the atypical amino acid residues in the basic region of Maf influence recognition of both the core and extended sequence elements, but are not essential for formation of highly stable complexes at the consensus recognition site.
Amino acid residues in the basic region of Maf influence recognition of several base pairs in both the core and extended elements
To investigate the effects of amino acid substitutions in the basic region of Maf on recognition of other base pairs in the core and extended sequence elements, we compared binding of wild-type and mutant Maf proteins at sites containing all base pair combinations at the 5 and 6 positions, and at sites with base substitutions in the CRE (a subset of the data is shown in Figure 5). There were large differences in Maf binding to sites that contained different base pairs at the ±6 positions, and smaller differences at binding sites containing different base pairs at the ±5 positions. Maf Y287A binding to oligonucleotides containing T/A base substitutions at the ±5 positions was reproducibly lower than that observed for wild-type Maf. Base substitutions in the CRE also had a larger effect on binding by Maf Y287A than on binding by wild-type Maf. Maf G286I binding to all sites containing base substitutions was reduced compared with wild-type Maf. Remarkably, replacement of the tyrosine by a phenylalanine (Y287F) markedly reduced binding to all sites containing base substitutions. The dissociation rate of complexes formed by this protein was more than an order of magnitude faster than that of wild-type Maf at the consensus recognition site (data not shown). The conserved glycine and tyrosine residues, and in particular the hydroxyl group on the tyrosine residue, are important for stabilization of Maf binding at both consensus and non-consensus recognition sites.

Fig. 5. Effects of amino acid substitutions in the basic region on the recognition of base pairs in the extended and core elements. The proteins indicated between the panels were incubated for 20 min with oligonucleotides containing the M site (*) with the base pair substitutions indicated below the lanes. Each set of three lanes contained different concentrations of the same protein (Maf, 14, 28 and 42 nM; Maf Y287A, 15, 30 and 45 nM; Maf G286I and Maf Y287F, 18, 36 and 54 nM). All lanes contained the same oligonucleotide concentration (11 nM). Unlabeled M site oligonucleotide was added to a 2.8 µM final concentration for 30 min, and the complexes were analyzed by 8% PAGE. Only the bands corresponding to protein–DNA complexes are shown.
The DNA contacts of Maf differ from those of other bZIP proteins
To identify DNA contacts that Maf makes at the consensus recognition site, dimethylsulfoxide (DMS) and hydroxyl radical footprinting as well as methylation interference and missing nucleoside analysis were performed (Figure 6). Both DMS footprinting and methylation interference demonstrated contacts to the N7 positions of the guanines at the –6 and +6 positions. Thus, Maf binds to the extended recognition elements within the major groove. In contrast, there was little protection from or interference by methylation of the guanines at the –3 or +3 positions. The guanines at these positions are protected by, and methylation of these guanines interferes with, binding by canonical bZIP proteins (Nakabeppu and Nathans, 1989). Thus, Maf contacts DNA in a manner distinct from that of other bZIP proteins.

Fig. 6. Footprinting and interference analysis of base contacts made by Maf. (A) Autoradiogram of cleavage products from DMS and ⋅OH probing of Maf complexes. The first five lanes show DMS reactions; the remaining lanes show ⋅OH probing. Lanes 1 and 6, cleavage of DNA in the absence of Maf; lanes 2 and 7, cleavage of free DNA fractions from binding reactions; lanes 3 and 8, footprinting of Maf–DNA complexes; lanes 4 and 9, cleavage products of free DNA fractions from methylation interference/missing nucleoside reactions; lanes 5 and 10, cleavage products of bound DNA fractions from methylation interference/missing nucleoside reactions. The Maf-binding site is indicated by a vertical line between lanes 5 and 6. The positions of the guanines in the site are marked on the left side of the gel according to the nomenclature in Figure 1B. Protected guanines are indicated by black dots. (B) Densitometric scan of the products of DMS cleavage of free DNA and Maf–DNA complexes (top two panels), and of ⋅OH cleavage of free and bound DNA (bottom two panels). The binding site is bordered by vertical lines. Guanines protected from DMS cleavage are indicated by black dots, which correspond to the positions marked in (A). (C) Densitometric scans of DMS interference data for Maf, Maf Y287A and MJM. The DMS-modified free DNA is shown above. Guanine positions are indicated above the free DNA trace. The differences in interference by guanine methylation within the core and extended recognition elements are shown by black dots above the traces.
The guanine at the center of the CRE (position 1) was protected from methylation, and methylation of this guanine interfered with binding by Maf, consistent with conservation of the arginine that contacts this base pair in Maf. The guanines at the –5 and +5 positions were not protected from methylation, and their methylation did not alter Maf binding detectably. The differences in methylation interference and DMS protection of the base pairs at the ±5 and ±6 positions emphasize the unique role of Maf contacts to the ±6 positions. Hydroxyl radical footprinting of Maf at the consensus binding site showed weak protection primarily within the 3′ half-site of each strand. This differs from the hydroxyl radical protection pattern for other bZIP proteins, which is centered on the CRE (Vinson et al., 1989). Consequently, the unique role of the ±6 positions in DNA recognition by Maf is at least partly due to direct contacts to the guanines at these positions, whereas the lesser importance of base pairs within the CRE is due to loss of contacts to some of these bases.
To investigate more directly the role of the basic region in the altered DNA contact interface of Maf, we examined the effect of replacing residues in the basic region of Maf with the corresponding residues from Jun (Figure 6C). Replacement of the entire basic region of Maf with the basic region of Jun (MJM) eliminated interference by methylation of the guanine at position –6. Moreover, methylation of the guanines at the –3 and +1 positions had a larger effect on binding by Maf containing the Jun basic region compared with wild-type Maf. Similar results were obtained from DMS footprinting analysis (data not shown), indicating that the basic region of Maf influences contacts with both the core and extended recognition elements. The Y287A substitution also increased the effect of methylation of the guanines at the –3 and +1 positions on complex formation. However, this substitution did not eliminate the influence of methylation of the guanine at position –6. This is consistent with the similar selectivity of Maf Y287A and wild-type Maf for base pairs at the ±6 positions (Figure 5), and indicates that this protein directly contacts the base pairs at the ±6 positions. Similar results were obtained from DMS footprinting analysis as well as from experiments using the Maf G286I Y287A protein. Consequently, the basic region of Maf influences contacts to both the core and extended recognition elements, and substitution of the tyrosine and glycine residues alters the relative importance of these DNA contacts.
Discussion
Maf family proteins differ from canonical bZIP proteins both in the amino acid residues required for DNA binding and in the lengths of their DNA recognition sites. Departing from the paradigm that different members of the same family of DNA-binding proteins have the same structural fold, Maf does not have the same secondary structure or DNA contact interface as canonical bZIP proteins. The atypical amino acid residues in the basic region of Maf alter interactions with both the core and extended recognition elements. Thus, Maf represents an unusual DNA-binding protein in which much of the DNA contact interface has been reorganized compared with other members of the bZIP family.
Maf undergoes a conformational change upon DNA binding as has been observed previously for other bZIP family proteins (Patel et al., 1990; Weiss et al., 1990). Both the basic region and the ancillary DNA-binding region participate in the conformational change. Our studies were performed with truncated proteins encompassing the regions conserved among members of the Maf family. This region corresponds to essentially the entire coding region of small Maf proteins (MafK, MafF and MafG). It is possible that large Maf family proteins (Maf, MafB, Nrl) contain additional regions that influence the conformation of the conserved domain, but they share the same DNA binding specificity and probably recognize DNA using a similar contact interface.
The sequence of the binding site has a dramatic effect on the conformation of Maf bound to DNA. In particular, the G:C base pairs at the ±6 positions are essential for stabilization of the α-helical conformation of Maf. Fos and Jun can also recognize base pairs flanking the AP-1 site through indirect recognition of differences in DNA structure (Leonard et al., 1997; Ramirez-Carrozzi and Kerppola, 2001). The extended recognition elements in the two half-sites have independent effects on both the α-helix content and the binding affinity of Maf. The complex in which only one half-site is mutated must therefore contain one subunit in an α-helical conformation that makes normal DNA contacts, and the other subunit in an unfolded conformation that does not contribute to recognition of base pairs outside the AP-1 core. Regulatory elements in which one half-site of the consensus recognition sequence is mutated are common in genes that are regulated by Maf (Ho et al., 1998; Ogino and Yusuda, 1998; Kim et al., 1999; Li et al., 1999). The difference in folding between the two subunits at these sites may influence the transcriptional activity of the Maf homodimer.
There was a close correspondence between the effects of base substitutions on Maf conformation and DNA binding affinity. The effects of base substitutions on Maf conformation were observed by both CD spectroscopy and proteolytic digestion, and did not reflect a reduced fraction of Maf bound to DNA. Substitution of base pairs in the extended recognition elements of both half-sites abolished both specific DNA binding and Maf folding. This bound, but not folded conformation of Maf may be important for efficient localization of regulatory elements in genomic DNA. Specific binding sites may be recognized initially through their steric compatibility with the folded conformation of Maf. The relationship between Maf conformation and binding affinity indicates that the conformational change is essential for specific high affinity DNA binding.
The atypical amino acid residues in the basic region of Maf affected recognition of both the core and extended sequence elements. Replacement of these residues with the corresponding residues from the canonical bZIP protein Jun altered the sequence dependence of Maf secondary structure, trypsin sensitivity and binding affinity. Molecular models of the basic region of Maf based on the structures of other bZIP family proteins exhibited unfavorable free energies, whereas models of the mutated proteins were satisfactory, consistent with the higher α-helicity of the mutated proteins at the consensus recognition site. The altered recognition of the core-binding site by Maf in comparison with other bZIP proteins is consistent with replacement of the alanine that makes direct base contacts in other bZIP protein complexes by tyrosine in Maf. Tyrosine residues rarely make direct base contacts in protein–DNA complexes. Direct contacts to the extended recognition elements would require a large change in protein or DNA structure since the corresponding amino acid residues in other bZIP proteins are >10 Å away from the ±6 base pairs. Alternatively, the atypical amino acid residues in the basic region may facilitate a conformational change required for recognition of the extended sequence elements. This is consistent with the change in Maf secondary structure caused by replacement of these amino acid residues. We favor the interpretation that the tyrosine and glycine residues influence DNA sequence recognition by breaking the basic region α-helix and thereby altering the conformation or position of the ancillary DNA-binding region.
The tyrosine and glycine residues serve an important function by preventing Maf binding to the many sites in the genome that contain AP-1 and CRE sites. Maf binds to an unusually long DNA sequence element compared with other bZIP family proteins. If each base pair in this sequence were recognized independently, single base pair substitutions would eliminate only a fraction of the binding energy. Coupling folding to DNA recognition provides a mechanism for excluding sites that do not stabilize the appropriate conformation of Maf.
To determine the possible structural basis for DNA recognition by the ancillary DNA-binding region, we performed threading analysis (Jones et al., 1992) and searched for structural motifs (Henikoff et al., 1999) compatible with the conserved amino acid residues in the ancillary DNA-binding regions of Maf family proteins. These methods identified similarities to the four-helix bundle of Skn-1 (Rupert et al., 1998) as well as the recognition helix of helix–turn–helix family proteins (Pabo et al., 1990) (Figure 7A). The similarity to Skn-1 is consistent with the high α-helix content of Maf bound to the consensus recognition sequence. However, the corresponding region in Skn-1 makes a single contact to the DNA backbone (Rupert et al., 1998), and the residue that makes this contact is not conserved in Maf. We therefore hypothesize that this region of Maf is repositioned by an altered secondary structure of the basic region to contact the extended recognition element (Figure 7B). Elimination of the break in the α-helix impeded recognition of the extended sequence element, consistent with this model. Additionally, we speculate that the helix preceding the basic region in Maf makes direct DNA contacts through a contact interface similar to the recognition helix of helix–turn–helix family proteins (Figure 7B). Whereas these results from database searches and threading analysis represent theoretical predictions that remain to be tested experimentally, it is clear that folding of the ancillary DNA-binding region into an α-helical conformation depends on contacts with the extended recognition element.

Fig. 7. Model for DNA recognition by Maf family proteins. (A) Sequence similarities between the ancillary DNA-binding region of Maf and structurally characterized DNA-binding proteins. The sequence of the ancillary DNA-binding region of Maf is aligned with the region on the N-terminal side of the basic region of Skn-1 (Rupert et al., 1998) and with a logo representation (Schneider, 1996) of the helix–turn–helix (HTH) motif (Pabo et al., 1990). Identical residues are connected by lines and conserved residues are connected by dashed lines (in the logo representation, the two most frequent residues were considered). The conservation of these residues in different Maf family members is shown in Figure 1. The structural significance of these sequence similarities was corroborated by threading analysis (Jones et al., 1992) (Z scores of 3.1 for P22 c2 repressor [1ADR], 2.6 for 434 Cro [2CRO], 2.4 for 434 repressor [1R69] and 9.1 for Skn-1 [1SKN]). (B) X-ray crystal structures of GCN4–AP-1 (Ellenberger et al., 1992), Skn-1–DNA (Rupert et al., 1998) and HTH–DNA (Fraenkel et al., 1998) complexes compared with a conceptual model of the Maf–DNA complex. Regions of predicted structural similarity are shown in the same color. The non-α-helical structure of the basic region is shown as a dashed line.
Our model for DNA recognition by Maf suggests that the protein follows the contour of the major groove over more than one full helical turn (Figure 7B). This is reminiscent of the original scissors grip model (Vinson et al., 1989), and we therefore refer to it as the pincer grip model for DNA binding by Maf family proteins. As a consequence of the pincer-like DNA contact interface, dissociation of Maf from DNA is predicted to require either a conformational change or disruption of the dimer interface. This is consistent with the unusually long half-life of Maf complexes at the consensus recognition site.
Both the Maf and CNC families of bZIP proteins contain basic and ancillary DNA-binding regions, but they recognize different DNA sequences (Kataoka et al., 1994; Kerppola and Curran, 1994a; Oyake et al., 1996; Johnsen et al., 1998). This difference in binding specificities is consistent with the proposed role of basic region conformation in control of DNA recognition by the ancillary region. Thus, the difference in DNA recognition specificities between these families of bZIP proteins is determined by differences in conformation that specify the protein domains that contact DNA.
Maf represents a new paradigm for elaboration of DNA binding specificity through combination of different classes of DNA recognition motif. The bZIP and helix– turn–helix motifs are found in many transcription regulatory proteins independently of each other. In Maf, neither of the regions corresponding to these motifs is functional on their own. Instead, folding of the two regions is coupled so that they recognize DNA as a cooperative unit. This provides a larger DNA contact interface that can recognize a longer DNA sequence. This mechanism is reminiscent of the cooperative DNA binding by different eukaryotic transcription regulatory proteins at composite regulatory elements. The diversification of DNA-binding proteins into distinct binding modules may represent an evolutionary adaptation to the regulation of genes in terminally differentiated cells. These cells express a more limited set of gene products and may therefore have less need for combinatorial regulation of gene expression by multiple independent transcription regulatory proteins.
Materials and methods
Protein expression and purification
The construction of expression vectors and purification of Maf241-369 have been described previously (Kerppola and Curran, 1994b). Plasmids encoding proteins containing the Y287A, G286I, Y287A G286I, Y287F and MJM (replacement of Maf residues 276-301 by residues from Jun) mutations were constructed by oligonucleotide replacement and verified by DNA sequencing. The wild-type protein is designated Maf, and amino acid substitutions are indicated by suffixes in the text. The construction and purification of Fos139-200 and Jun257-318 have been described (Leonard et al., 1997) and are designated Fos and Jun. The proteins were expressed in Escherichia coli and purified by nickel chelate affinity chromatography to >98% homogeneity. Protein concentrations were determined by the Bradford assay.
Oligonucleotide synthesis and purification
Oligonucleotides containing the consensus Maf recognition sequence (M) and variants thereof (Table I) were synthesized using standard phosphoramidite chemistry. For CD and proteolysis experiments, equimolar amounts of complementary strands were annealed in 10 mM Tris, 1 mM EDTA pH 8.0. The duplexes were purified by PAGE and dissolved in 25 mM sodium phosphate, 1 mM dithiothreitol (DTT) pH 7.5 buffer. For electrophoretic mobility shift and footprinting assays, the oligonucleotides were purified by denaturing PAGE, desalted and radiolabeled using T4 polynucleotide kinase and [γ-32P]ATP prior to annealing of complementary oligonucleotides.
Table I. Oligonucleotides used in studies of Maf conformation and binding affinity.
| Site | Sequence |
|---|---|
| 7654321234567 | |
| M | CCTCTAGATGCTGACTCAGCAGTCGACGC |
| M±4 | CCTCTAGATGCgGACTCcGCAGTCGACGC |
| M±5 | CCTCTAGATGaTGACTCAtCAGTCGACGC |
| M–6 | CCTCTAGATtCTGACTCAGCAGTCGACGC |
| M±6 | CCTCTAGATtCTGACTCAGaAGTCGACGC |
| M±7 | CCTCTAGAgGCTGACTCAGCcGTCGACGC |
| CG | CGCGCGCGCGCGCGCCGCGCGCGCGCGCG |
The consensus Maf-binding site is indicated by underlined letters and the extended recognition element is shown in bold. Mutations in the consensus binding site are indicated by lower case letters.
DNA binding assays
EMSAs were performed as described previously (Kerppola and Curran, 1994b). Binding affinity measurements were performed in the absence of dIdC competitor. The fraction of bound DNA was quantitated using a Molecular Dynamics PhosphorImager.
Circular dichroism studies
The proteins for CD experiments were dialyzed against 25 mM sodium phosphate, 1 mM DTT buffer pH 7.5. The CD spectra were recorded using a Jasco J-710 spectropolarimeter at 23°C. Protein samples at a final concentration of 5–20 µM were incubated with 5–40 µM oligonucleotide for 15 min prior to recording the spectra. The α-helix content was estimated based on the mean residue ellipticity [Θ]222 by assuming that a value of –30 723 deg cm2/dmol corresponds to an α-helix content of 100% at 23°C (Chen et al., 1974). Thermal denaturation was monitored by the change in ellipticity at 222 nm as a function of temperature.
Trypsin digestion
Maf (10 µM) or the indicated Maf derivative (10 µM) was pre-incubated with or without a 2-fold molar excess of oligonucleotide in 25 mM sodium phosphate, 1 mM DTT pH 7.5 for 20 min. Trypsin was added to the reaction mixture at the protease:substrate ratios indicated. The samples were incubated at 23°C and the digestion was terminated by the addition of SDS–PAGE loading buffer and heating at 95°C for 5 min. The proteolytic fragments were analyzed by SDS–PAGE using 16% Tris-glycine or 16% Tris-tricine gels. The peptides were transferred onto a PVDF membrane and analyzed by N-terminal amino acid sequencing and MALDI-TOF mass spectrometry.
Fluorescence anisotropy analysis
Oligonucleotides with fluorescein linked to their 5′ ends were synthesized by using the phosphoramidite derivative of fluorescein (Glen Research). The anisotropy of fluorescein emission was determined by measuring the vertical and horizontal components of fluorescence, correcting for the grating (g) factor. A 200 nM concentration of Maf (500 nM at the S+6 site) was added to 20 nM oligonucleotide in 20 mM HEPES pH 7.6, 100 mM KCl, 5% glycerol, 1 mM EDTA, 5 mM DTT and 0.5 mg/ml bovine serum albumin (BSA) at 25°C, and the complexes were allowed to equilibrate for 30 min. Sonicated herring DNA (1 mg/ml final concentration) was injected manually into a stirred cuvette with a mixing time of ∼2 s and the anisotropy change was monitored over time. The dissociation was considered complete when the anisotropy value decreased to within 10% of the value observed prior to Maf addition.
DNA footprinting and interference analysis
Protein and DNA concentrations for footprinting and interference assays were adjusted so that approximately half of the DNA was bound. A 2 µl aliquot of 10% DMS was added to Maf–DNA complexes in 50 µl of DNA binding buffer (Kerppola and Curran, 1994b), and the reactions were incubated for 4 min. The reactions were stopped by adding β-mercaptoethanol to 5 mM. Maf–DNA complexes were separated from free DNA on a 5% polyacrylamide gel. The free and bound DNA fragments were extracted from the gel and modified DNA was treated with hot piperidine for 20 min. Cleaved DNA was lyophilized and loaded on a denaturing 10% polyacrylamide gel. DMS interference was performed using the same procedure except that the DNA was modified prior to protein binding. Hydroxyl radical footprinting and missing nucleoside experiments were performed as described (Tullius and Dombroski, 1986).
Protein profile searches and fold recognition (threading)
The sequence of the conserved region in Maf family proteins was used as a query to search the Blocks database of protein structural motifs (Henikoff et al., 1999). Threading calculations were performed with THREADER (Jones et al., 1992) using default program parameters. Models of Maf were built using MODELLER (Sali et al., 1995), and the quality of each model was assessed based on its stereochemical parameters (Laskowski et al., 1993) and energy profile (Sippl, 1993).
Acknowledgments
Acknowledgements
We thank Kelli Biederman for help with mass spectrometry, and members of the Kerppola laboratory for helpful discussions and constructive criticism of the manuscript. M.D. is a Special Fellow of the Leukemia and Lymphoma Society, and was funded in part by the AACR-Sidney Kimmel Foundation for Cancer Research.
REFERENCES
- Andrews N.C., Erdjument,B.H., Davidson,M.B., Tempst,P. and Orkin,S.H. (1993) Erythroid transcription factor NF-E2 is a haematopoietic-specific basic–leucine zipper protein. Nature, 362, 722–728. [DOI] [PubMed] [Google Scholar]
- Blank V. and Andrews,N.C. (1997) The Maf transcription factors: regulators of differentiation. Trends Biochem. Sci., 22, 437–441. [DOI] [PubMed] [Google Scholar]
- Carroll A.S., Gilbert,D.E., Liu,X., Cheung,J.W., Michnowicz,J.E., Wagner,G., Ellenberer,T.E. and Blackwell,T.K. (1997) SKN-1 domain folding and basic region monomer stabilization upon DNA binding. Genes Dev., 11, 2227–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.H., Yang,J.T. and Chau,K.H. (1974) Determination of the helix and β form of proteins in aqueous solution by circular dichroism. Biochemistry, 13, 3350–3359. [DOI] [PubMed] [Google Scholar]
- Diamond M.I., Miner,J.N., Yoshinaga,S.K. and Yamamoto,K.R. (1990) Transcription factor interactions: selectors of positive or negative regulation from a single DNA element. Science, 249, 1266–1272. [DOI] [PubMed] [Google Scholar]
- Ellenberger T.E., Brandl,C.J., Struhl,K. and Harrison,S.C. (1992) The GCN4 basic region leucine zipper binds DNA as a dimer of uninterrupted α helices: crystal structure of the protein–DNA complex. Cell, 71, 1223–1237. [DOI] [PubMed] [Google Scholar]
- Fraenkel E., Rould,M.A., Chambers,K.A. and Pabo,C.O. (1998) Engrailed homeodomain–DNA complex at 2.2 Å resolution: a detailed view of the interface and comparison with other engrailed structures. J. Mol. Biol., 284, 351–361. [DOI] [PubMed] [Google Scholar]
- Glover J.N. and Harrison,S.C. (1995) Crystal structure of the heterodimeric bZIP transcription factor c-Fos–c-Jun bound to DNA. Nature, 373, 257–261. [DOI] [PubMed] [Google Scholar]
- Harrison S.C. (1991) A structural taxonomy of DNA-binding domains. Nature, 353, 715–719. [DOI] [PubMed] [Google Scholar]
- Henikoff J.G., Henikoff,S. and Pietrokovski,S. (1999) New features of the Blocks Database servers. Nucleic Acids Res., 27, 226–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho I.C., Lo,D. and Glimcher,L.H. (1998) c-Maf promotes T helper cell type 2 (Th2) and attenuates Th1 differentiation by both interleukin 4-dependent and -independent mechanisms. J. Exp. Med., 188, 1859–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi K., Kataoka,K., Itoh,K., Hayashi,N., Nishizawa,M. and Yamamoto,M. (1994) Regulation of transcription by dimerization of erythroid factor NF-E2 p45 with small Maf proteins. Nature, 367, 568–572. [DOI] [PubMed] [Google Scholar]
- Itoh K., Igarashi,K., Hayashi,N., Nishizawa,M. and Yamamoto,M. (1995) Cloning and characterization of a novel erythroid cell-derived CNC family transcription factor heterodimerizing with the small Maf family proteins. Mol. Cell. Biol., 15, 4184–4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnsen O., Murphy,P., Prydz,H. and Kolsto,A.B. (1998) Interaction of the CNC-bZIP factor TCF11/LCR-F1/Nrf1 with MafG: binding-site selection and regulation of transcription. Nucleic Acids Res., 26, 512–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D.T., Taylor,W.R. and Thornton,J.M. (1992) A new approach to protein fold recognition. Nature, 358, 86–89. [DOI] [PubMed] [Google Scholar]
- Kataoka K., Noda,M. and Nishizawa,M. (1994) Maf nuclear oncoprotein recognizes sequences related to an AP-1 site and forms heterodimers with both Fos and Jun. Mol. Cell. Biol., 14, 700–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly L.M., Englmeier,U., Lafon,I., Sieweke,M.H. and Graf,T. (2000) MafB is an inducer of monocytic differentiation. EMBO J., 19, 1987–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerppola T.K. and Curran,T. (1994a) A conserved region adjacent to the basic domain is required for recognition of an extended DNA binding site by Maf/Nrl family proteins. Oncogene, 9, 3149–3158. [PubMed] [Google Scholar]
- Kerppola T.K. and Curran,T. (1994b) Maf and Nrl can bind to AP-1 sites and form heterodimers with Fos and Jun. Oncogene, 9, 675–684. [PubMed] [Google Scholar]
- Kim J.I., Li,T., Ho,I., Grusby,M.J. and Glimcher,L.H. (1999) Requirement for the c-Maf transcription factor in crystallin gene regulation and lens development. Proc. Natl Acad. Sci. USA, 96, 3781–3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig P. and Richmond,T.J. (1993) The X-ray structure of the GCN4-bZIP bound to ATF/CREB site DNA shows the complex depends on DNA flexibility. J. Mol. Biol., 233, 139–154. [DOI] [PubMed] [Google Scholar]
- Kophengnavong T., Carroll,A.S. and Blackwell,T.K. (1999) The SKN-1 amino-terminal arm is a DNA specificity segment. Mol. Cell. Biol., 19, 3039–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R.A., MacArthur,M.W., Moss,D.S. and Thornton,J.M. (1993) PROCHECK: a program to check stereochemical quality of protein structures. J. Appl. Crystallogr., 26, 283–290. [Google Scholar]
- Lefstin J.A. and Yamamoto,K.R. (1998) Allosteric effects of DNA on transcriptional regulators. Nature, 392, 885–888. [DOI] [PubMed] [Google Scholar]
- Leonard D.A., Rajaram,N. and Kerppola,T.K. (1997) Structural basis of DNA bending and oriented heterodimer binding by the basic leucine zipper domains of Fos and Jun. Proc. Natl Acad. Sci. USA, 94, 4913–4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Tournier,C., Davis,R.J. and Flavell,R.A. (1999) Regulation of IL-4 expression by the transcription factor JunB during T helper cell differentiation. EMBO J., 18, 420–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakabeppu Y. and Nathans,D. (1989) The basic region of Fos mediates specific DNA binding. EMBO J., 8, 3833–3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizawa M., Kataoka,K., Goto,N., Fujiwara,K.T. and Kawai,S. (1989) v-maf, a viral oncogene that encodes a ‘leucine zipper’ motif. Proc. Natl Acad. Sci. USA, 86, 7711–7715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogino H. and Yusuda,K. (1998) Induction of lens differentiation by activation of a bZIP transcription factor, L-Maf. Science, 280, 115–118. [DOI] [PubMed] [Google Scholar]
- Oyake T., Itoh,K., Motohashi,H., Hayashi,N., Hoshino,H., Nishizawa,M., Yamamoto,M. and Igarashi,K. (1996) Bach proteins belong to a novel family of BTB-basic leucine zipper transcription factors that interact with MafK and regulate transcription through the NF-E2 site. Mol. Cell. Biol., 16, 6083–6095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabo C.O., Aggarwal,A.K., Jordan,S.R., Beamer,L.J., Obeysekare,U.R. and Harrison,S.C. (1990) Conserved residues make similar contacts in two repressor–operator complexes. Science, 247, 1210–1213. [DOI] [PubMed] [Google Scholar]
- Patel L., Abate,C. and Curran,T. (1990) Altered protein conformation on DNA binding by Fos and Jun. Nature, 347, 572–575. [DOI] [PubMed] [Google Scholar]
- Ramirez-Carrozzi V.R. and Kerppola,T.K. (2001) Long-range electrostatic interactions influence the orientation of Fos–Jun binding at AP-1 sites. J. Mol. Biol., 305, 411–427. [DOI] [PubMed] [Google Scholar]
- Rupert P.B., Daughdrill,G.W., Bowerman,B. and Matthews,B.W. (1998) A new DNA-binding motif in the Skn-1 binding domain–DNA complex. Nature Struct. Biol., 5, 484–491. [DOI] [PubMed] [Google Scholar]
- Sali A., Potterton,L., Yuan,F., van Vlijmen,H. and Karplus,M. (1995) Evaluation of comparative protein modeling by MODELLER. Proteins, 23, 318–326. [DOI] [PubMed] [Google Scholar]
- Schneider T.D. (1996) Reading of DNA sequence logos: prediction of major groove binding by information theory. Methods Enzymol., 274, 445–455. [DOI] [PubMed] [Google Scholar]
- Sippl M.J. (1993) Recognition of errors in three-dimensional structures of proteins. Proteins, 17, 355–362. [DOI] [PubMed] [Google Scholar]
- Swaroop A., Xu,J.Z., Pawar,H., Jackson,A., Skolnick,C. and Agarwal,N. (1992) A conserved retina-specific gene encodes a basic motif/leucine zipper domain. Proc. Natl Acad. Sci. USA, 89, 266–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tullius T.D. and Dombroski,B.A. (1986) Hydroxyl radical ‘footprinting’: high-resolution information about DNA–protein contacts and application to λ repressor and Cro protein. Proc. Natl Acad. Sci. USA, 83, 5469–5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinson C.R., Sigler,P.B. and McKnight,S.L. (1989) Scissors-grip model for DNA recognition by a family of leucine zipper proteins. Science, 246, 911–916. [DOI] [PubMed] [Google Scholar]
- Weiss M.A., Ellenberger,T., Wobbe,C.R., Lee,J.P., Harrison,S.C. and Struhl,K. (1990) Folding transition in the DNA-binding domain of GCN4 on specific binding to DNA. Nature, 347, 575–578. [DOI] [PubMed] [Google Scholar]