

Abstract
SR proteins purified from uninfected HeLa cells inhibit adenovirus IIIa pre-mRNA splicing by binding to the intronic IIIa repressor element (3RE). In contrast, SR proteins purified from late adenovirus-infected cells are functionally inactivated as splicing repressor proteins by a virus-induced dephosphorylation. We have shown that the adenovirus E4-ORF4 protein, which binds the cellular protein phos phatase 2A (PP2A) and activates IIIa splicing in vitro and in vivo, induces SR protein dephosphorylation. Here we show that E4-ORF4 interacts with only a subset of SR proteins present in HeLa cells. Thus, E4-ORF4 interacts efficiently with SF2/ASF and SRp30c, but not with other SR proteins. Interestingly, E4-ORF4 interacts with SF2/ASF through the latter’s RNA recognition motifs. Furthermore, E4-ORF4 interacts preferentially with the hyperphosphorylated form of SR proteins found in uninfected HeLa cells. E4-ORF4 mutant proteins that fail to bind strongly to PP2A or SF2/ASF do not relieve the repressive effect of HeLa SR proteins on IIIa pre-mRNA splicing in transient transfection experiments, suggesting that an interaction between all three proteins is required for E4-ORF4-induced SR protein dephosphorylation.
Keywords: adenovirus/E4-ORF protein/protein phosphatase 2A/splicing enhancer/SR proteins
Introduction
The adenovirus E4-ORF4 protein is a 14 kDa protein that binds to the B55 and B56 subunits of protein phosphatase 2A (PP2A) (Kleinberger and Shenk, 1993; Shtrichman et al., 2000). PP2A is a serine/threonine phosphatase consisting of a core dimer of the A subunit and the catalytic C subunit, which interacts with a family of regulatory B subunits (Kamibayashi et al., 1994; McCright and Virshup, 1995; Csortos et al., 1996). A wealth of data implicate PP2A in the regulation of a number of cellular functions, but the mechanisms by which B subunits target the core dimer to different substrates are not well understood (reviewed in Wera and Hemmings, 1995; Millward et al., 1999). The E4-ORF4–PP2A complex reduces E1A activation of AP-1 transcription factor activity (Muller et al., 1992). This reduction in transcription factor activity correlates with an increased accumulation of underphosphorylated forms of E1A and c-Fos. E4-ORF4 also inhibits E1A activation of junB (Kleinberger and Shenk, 1993), and adenovirus E2 (Mannervik et al., 1999) and E4 transcription (Bondesson et al., 1996). E4-ORF4 has also been shown to induce p53-independent apoptosis (Lavoie et al., 1998; Marcellus et al., 1998; Shtrichman and Kleinberger, 1998) by a mechanism requiring E4-ORF4 binding to PP2A (Marcellus et al., 2000; Shtrichman et al., 2000). We have shown further that E4-ORF4 regulates adenovirus L1 splicing, and induces SR protein dephosphorylation (Kanopka et al., 1998). Collectively, available evidence suggests that E4-ORF4 regulates transcription, splicing and apoptosis through its binding to PP2A. Potentially the effect of E4-ORF4 on apoptosis is the result of its regulatory role on transcription factor and/or splicing factor activity.
The adenovirus L1 unit is an example of a gene whose last intron is spliced using a common 5′ splice site and two competing 3′ splice sites, generating two predominant cytoplasmic L1 mRNAs: the 52,55K (proximal 3′ splice site) or the IIIa (distal 3′ splice site) mRNAs, respectively (reviewed in Imperiale et al., 1995). L1 splicing is regulated during the infectious cycle. Thus, the 52,55K mRNA is the only L1 mRNA produced early after infection, whereas the IIIa mRNA becomes the predominant L1 mRNA accumulating late after infection (Chow et al., 1979; Akusjärvi and Persson, 1981; Nevins and Wilson, 1981; Svensson and Akusjärvi, 1986). We have shown previously that SR proteins purified from HeLa cells (SR-HeLa) inhibit IIIa splicing by binding to a purine-rich intronic repressor element, the 3RE, located immediately upstream of the IIIa branch site (Kanopka et al., 1996). SR protein binding to the 3RE blocks splicing by inhibiting U2 snRNP recruitment to the IIIa branch site.
SR proteins are a family of essential splicing factors required for constitutive splicing and functioning as regulators of alternative splicing (reviewed in Fu, 1995; Manley and Tacke, 1996; Graveley, 2000). SR proteins have one or two N-terminal RNA recognition motifs (RRMs) and a C-terminus rich in arginine/serine dipeptide repeats (the RS domain), which mediates protein interactions with other components of the splicing machinery (Zahler et al., 1992; Caceres and Krainer, 1993; Wu and Maniatis, 1993; Zuo and Manley, 1993; Kohtz et al., 1994).
SR proteins are highly phosphorylated in vivo (Roth et al., 1990), mainly in the RS domain (Colwill et al., 1996), a modification that is required for their function in spliceosome assembly (Mermoud et al., 1994; Roscigno and Garcia-Blanco, 1995; Xiao and Manley, 1997). Thus, phosphorylated SR proteins are needed for initiation of spliceosome assembly, and SR protein dephosphorylation appears to be required for splicing catalysis (Cao et al., 1997). On the other hand, the level of SR protein phosphorylation appears to be critical, since both hyperphosphorylation and hypophosphorylation inhibit their activity as activators of splicing (Kanopka et al., 1998; Prasad et al., 1999), in both constitutive and activated alternative splicing. The phosphorylated status of SR proteins has also been shown to control their localization and transport in the nucleus between speckles and sites of active transcription and splicing. Thus, phosphorylation appears to release SR proteins from their storage sites and target them to sites of transcription and splicing in the nucleus (Colwill et al., 1996; Caceres et al., 1998; Misteli et al., 1998).
We have shown previously that, during an adenovirus infection, SR proteins become functionally inactivated as IIIa splicing repressor proteins by a virus-induced dephosphorylation (Kanopka et al., 1998). This post-translational modification reduces their capacity to bind to the 3RE and, as a consequence, alleviates their repressive effect on IIIa splicing. We further showed that the virus-encoded E4-ORF4 protein induces SR protein dephosphorylation, and has the capacity to activate IIIa pre-mRNA splicing, both in vitro and in transiently transfected cells (Kanopka et al., 1998).
Here we determine the binding specificity of E4-ORF4 interaction with SR proteins. Much to our surprise, we find that E4-ORF4 interacts specifically with only a subset of the SR proteins expressed in HeLa cells. Thus, E4-ORF4 interacts efficiently with SF2/ASF (also named ASF/SF2) and SRp30c, but not with SRp20, SC35, SRp40, SRp55 or SRp75. The specificity appears to result from the fact that E4-ORF4 associates with SF2/ASF through the latter’s RRMs. Furthermore, E4-ORF4 selectively interacts with the hyperphosphorylated form of SR proteins. Previously we showed that the 3RE is necessary for E4-ORF4 activation of IIIa splicing (Kanopka et al., 1998). Here we extend this finding by demonstrating that the 3RE is sufficient for E4-ORF4-mediated activation of splicing. Moreover, we show that E4-ORF4 mutant proteins that show reduced binding to SF2/ASF or inefficient recruitment of an active PP2A phosphatase complex do not activate IIIa splicing in transient transfection experiments.
Results
E4-ORF4 interacts selectively with SF2/ASF and SRp30c
To study the specificity of E4-ORF4 interaction with SR proteins, we incubated GST–E4-ORF4 with 35S-labeled SR proteins produced by coupled in vitro transcription– translation. As shown in Figure 1, glutathione S-transferase (GST)–E4-ORF4 interacts efficiently with SF2/ASF and SRp30c, but only weakly, if at all, with SRp20, SC35, SRp40, SRp55 or SRp75 in a GST pull-down assay. Interestingly, the RS domain of SF2/ASF is not important for this interaction, since SF2-ΔRS interacts at least as efficiently as wild-type SF2/ASF with E4-ORF4 (Figure 2B, lanes 1–6). In contrast, deletion of RRM1 of SF2/ASF abolished binding (Figure 2B, lanes 7–9).

Fig. 1. E4-ORF4 interacts selectively with SF2/ASF and SRp30c. GST or GST–E4-ORF4 was incubated with the indicated 35S-labeled SR proteins. Bound proteins were separated by gel electrophoresis and visualized by autoradiography.

Fig. 2. E4-ORF4 interacts with SF2/ASF through its RNA recognition motifs. (A) Schematic illustration showing the structure of SF2/ASF mutant proteins used in (B) and (C). (B) An in vitro binding assay using GST–E4-ORF4 and 35S-labeled SF2/ASF wild-type and mutant proteins. In lanes 13–15, the in vitro transcribed/translated 35S-labeled SF2/ASF extract was treated with 5 µg of RNase A for 15 min at 37°C before incubation with GST–E4-ORF4. (C) Extracts prepared from 293 cells co-transfected with pCMV-E4-ORF4 and plasmids encoding T7-tagged wild-type or mutant SF2/ASF proteins or SC35 were immunoprecipitated with an anti-E4-ORF4 antibody. Proteins were separated by gel electrophoresis and transferred to a membrane. SF2/ASF and SC35 proteins were detected using an anti-T7 antibody in a western blot assay. In lanes 11 and 12, the cell extract was treated with RNase A before immunoprecipitation with E4-ORF4.
The requirement for the RRMs of SF2/ASF for E4-ORF4 interaction was confirmed further in vivo. For this experiment, 293 cells were co-transfected with plasmids encoding wild-type E4-ORF4 and T7-tagged SF2/ASF mutant proteins. As shown in Figure 2C, immunoprecipitation with an anti-E4-ORF4 antibody co-immunoprecipitated wild-type SF2/ASF and SF2-ΔRS, but not SF2-RRM1/RS or SF2-RRM2/RS, confirming the in vitro binding data (Figure 2B). Furthermore, E4-ORF4 did not interact with SC35 in vivo (Figure 2C, lanes 13 and 14), reinforcing the conclusion from the in vitro binding studies (Figure 1) suggesting that E4-ORF4 interaction with SF2/ASF is specific. Probing the western blot with anti-E4-ORF4 demonstrated that E4-ORF4 was expressed similarly in all transfections (data not shown).
The observation that the RRMs of SF2/ASF are required for interaction with E4-ORF4 could indicate that E4-ORF4 becomes tethered indirectly to SF2/ASF through simultaneous, but independent, interaction with contaminating RNA in the extract. However, RNase treatment of the in vitro translated 35S-labeled SF2/ASF did not disrupt the interaction with E4-ORF4 in vitro (Figure 2B, lanes 13–15) or in vivo (Figure 2C, lanes 11–12), suggesting that the SF2/ASF interaction with E4-ORF4 is direct. Also, the observation that SRp20, SC35, SRp40, SRp55 and SRp75, which would also be expected to bind RNA in the TNT extract, did not interact with E4-ORF4, further supports our conclusion that RNA does not unspecifically mediate the E4-ORF4 interaction with SF2/ASF.
Collectively, our results show that E4-ORF4 interacts efficiently with a subset of HeLa SR proteins: SF2/ASF and SRp30c. Moreover, E4-ORF4 appears to associate specifically with SF2/ASF through its RRMs.
E4-ORF4 interacts with hyperphosphorylated SR proteins
SR proteins are highly phosphorylated in vivo, a modification that is important for their function in spliceosome assembly and splicing catalysis (reviewed in Misteli, 1999). Since E4-ORF4 induces SR protein dephosphorylation, E4-ORF4 might be expected to interact preferentially with hyperphosphorylated SR proteins. To test this hypothesis, we compared the capacity of E4-ORF4 to interact with SR-HeLa and SR proteins purified from late adenovirus-infected cells (SR-Ad). For this experiment, equal amounts of SR-HeLa and SR-Ad were separated by gel electrophoresis and transferred to a membrane. As shown in Figure 3A, probing the filter with the phospho-epitope-specific monoclonal antibody 104 (mAb104; Zahler et al., 1992) demonstrated that SR-HeLa proteins were recognized efficiently by mAb104, whereas SR-Ad were not (lanes 1 and 4). Interestingly, incubating SR-Ad with a small amount of HeLa cell nuclear extract (HeLa-NE) restored the phospho-epitope(s) recognized by mAb104 to SR-Ad (lane 5), whereas incubation in HeLa-NE pre-treated with hexokinase and glucose to deplete the small ATP pool present in a dialyzed extract did not restore the phospho-epitope(s) (lane 6). The small amount of HeLa-NE used in this phosphorylation assay did not contribute detectable amounts of SR proteins (data not shown). Note that the proteins recognized by mAb104 in SR-Ad migrate slightly faster than SR-HeLa (compare the SRp30 fraction in lanes 1 and 4), a result that is compatible with the conclusion that the SR proteins recognizable by mAb104 in SR-Ad are hypophosphorylated. Also, incubating SR-Ad in HeLa-NE resulted in a reduction in mobility such that SR-Ad migration was identical to that of SR-HeLa (lane 5). Taken together, these results support our previous conclusion that SR-Ad proteins are hypophosphorylated (Kanopka et al., 1998), and furthermore suggest that SR-Ad can be reconverted to SR proteins with the same electrophoretic mobility and mAb104-recognizable phospho-epitope(s) as SR-HeLa by in vitro phosphorylation.

Fig. 3. E4-ORF4 interacts preferentially with hyperphosphorylated SR proteins. (A) SR proteins prepared from HeLa cells (SR-HeLa, lane 1) and adenovirus-infected HeLa cells (SR-Ad, lane 4) were pre-incubated in nuclear extract (HeLa-NE) or nuclear extract depleted of ATP [HeLa-NE(–ATP)] (lanes 2, 3, 5 and 6). Samples were separated on a 10% SDS–polyacrylamide gel, transferred to a membrane and probed with mAb104 (an SR protein-specific antibody that detects phospho-epitopes in the RS domain) in a western blot assay. (B) The filter was probed with mAb96 (an SF2/ASF-specific antibody that detects the RRM1 of SF2/ASF) in a western blot assay. (C) The filter was probed with GST–E4-ORF4 in a far western blot assay. E4-ORF4-interacting proteins were visualized by chemiluminescence using an anti-E4-ORF4 antibody. Note that the autoradiograms shown are taken from different experiments.
To measure total SF2/ASF levels, we reprobed the filter with mAb96 (Hanamura et al., 1998), which recognizes RRM1 of SF2/ASF. As shown in Figure 3B, two distinct populations of SF2/ASF, with clear differences in migration, can be detected in SR-HeLa and SR-Ad, respectively (compare lanes 1 and 4). The slower migrating species is recognized by mAb104. Treatment of SR-HeLa with calf intestinal alkaline phosphatase resulted in an accumulation of the faster migrating species observed in SR-Ad (data not shown). We conclude that the two species represent hyperphosphorylated and unphosphorylated forms of SF2/ASF. Interestingly, the weak mAb104 signal seen in the SRp30 fraction in SR-Ad (Figure 3A, lane 4) migrates at a position between the highly and unphosphorylated SF2ASF, a result strengthening the conclusion that SR-Ad is hypophosphorylated (Kanopka et al., 1998). The results extend our previous results by suggesting that most of SF2/ASF is unphosphorylated in late adenovirus-infected cells. Furthermore, the results show that incubation of SR-Ad in HeLa-NE produces a quantitative shift in mobility of SF2/ASF to the slower migrating species seen in SR-HeLa (compare lanes 4 and 5).
Interestingly, reprobing the filter with GST–E4-ORF4 in a far western blot assay showed that E4-ORF4 interacts efficiently only with the SRp30 fraction in SR-HeLa but not in SR-Ad (Figure 3C, lanes 1 and 4). Importantly, in vitro phosphorylation of SR-Ad restored E4-ORF4 interaction to the slower, mAb104-recognizable species (Figure 3C, lane 5). Collectively, these results suggest that E4-ORF4 interacts preferentially with hyperphosphorylated SR proteins, and support our binding studies (Figure 1) by indicating that E4-ORF4 interacts selectively with the SRp30 fraction, which contains SF2/ASF and probably SRp30c. It is noteworthy that E4-ORF4 also interacts with a cluster of slower migrating proteins, in a phosphorylation-dependent manner (Figure 3C). However, these interactions are only seen in reactions containing HeLa-NE (Figure 3C, lanes 2, 3, 5 and 6), suggesting that they represent E4-ORF4 interactions with other host cell proteins. Coomassie Blue staining of identical gels showed that equal amounts of SR proteins were analyzed in each sample (data not shown).
The 3RE is necessary and sufficient for E4-ORF4 activation of splicing
We have shown previously that the 3RE is necessary for E4-ORF4 activation of IIIa pre-mRNA splicing in a transient transfection assay (Kanopka et al., 1998). Thus, replacing the 49 nucleotide 3RE with the corresponding sequence from the rabbit β-globin first intron abolished E4-ORF4 stimulation of IIIa splicing. More recently we have shown that IIIa splicing is also regulated by a second viral element, the IIIa virus infection-dependent splicing enhancer (the 3VDE; Mühlemann et al., 2000). This finding prompted us to re-examine whether the 3RE was the only element required for E4-ORF4 activation of splicing. For this experiment, we reconstructed eukaryotic expression vectors for some of the IIIa–β-globin hybrid constructs previously used in our in vitro studies (Kanopka et al., 1996). As shown in Figure 4, transfer of the 3RE to β-globin significantly reduced β-globin splicing [transcript glob (3RE), compare lanes 1 and 3]. This finding is in agreement with our in vitro experiments showing that the 3RE is the minimal sequence element required for SR protein inhibition of splicing (Kanopka et al., 1996). Importantly, co-transfection of a plasmid encoding E4-ORF4 stimulated β-globin splicing and annulled the repressive effect of the 3RE (compare lanes 1 and 4). In agreement with our previous demonstration that the 3VDE only functions efficiently in the context of virus-infected cells (Mühlemann et al., 2000), transfer of the 3VDE to β-globin resulted in a severe inhibition of β-globin splicing [transcript glob (3VDE), lanes 5]. However, compared with glob (3RE) splicing, glob (3VDE) splicing was only marginally activated by E4-ORF4 co-transfection (lane 6). Collectively, our data show that the 3RE is the primary element conferring an E4-ORF4-enhanced splicing phenotype, and show furthermore that the 3RE is both necessary (Kanopka et al., 1998) and sufficient for E4-ORF4 activation of splicing (Figure 4).
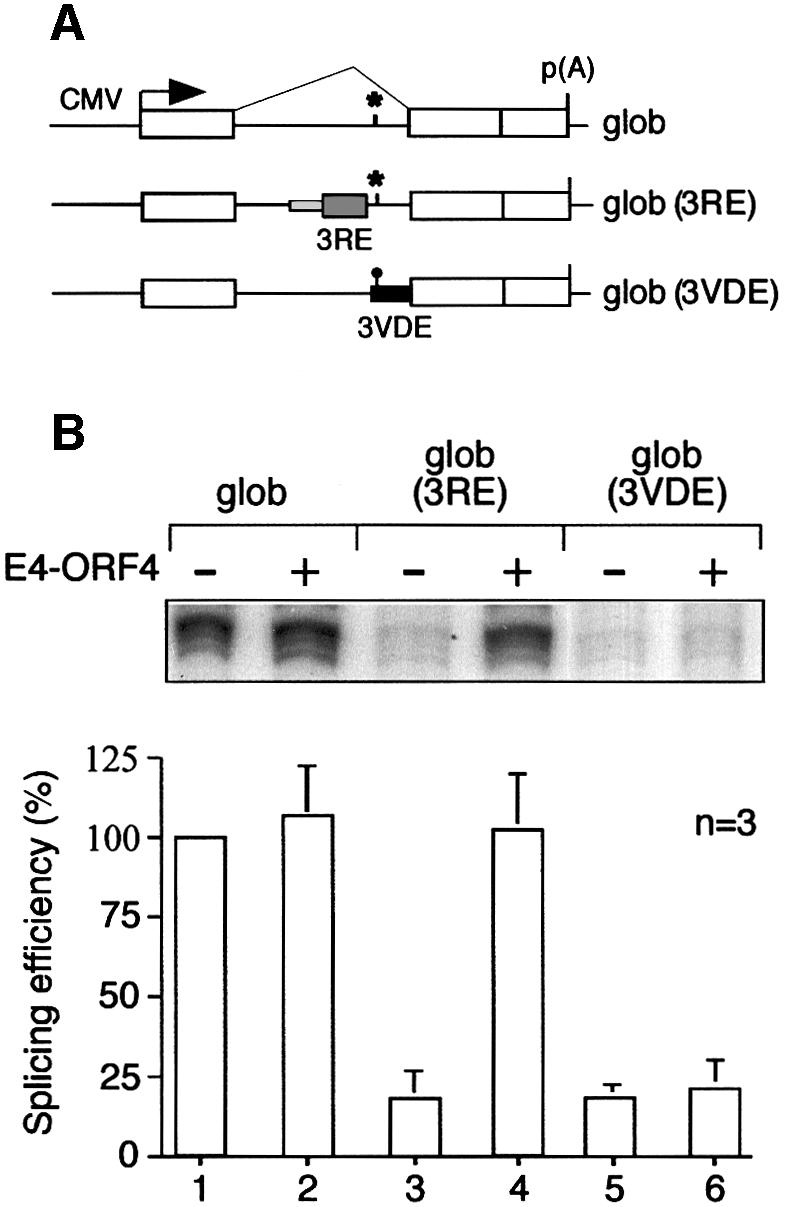
Fig. 4. The 3RE is sufficient to confer an E4-ORF4-enhanced splicing phenotype to β-globin. (A) Schematic representation of the IIIa–β-globin hybrid reporter transcripts used. (B) Quantitative S1 analysis of IIIa–β-globin hybrid mRNA levels in HeLa cells co-transfected with pCMV-E4-ORF4 and the reporter plasmids depicted in (A). Average splicing efficiencies and standard deviations are shown (three experiments).
Strong E4-ORF4 interaction with SF2/ASF and efficient E4-ORF4 recruitment of a functional phosphatase complex correlate with activation of IIIa pre-mRNA splicing
Our working hypothesis predicts that E4-ORF4 functions as a specificity factor that targets the SR protein SF2/ASF for PP2A dephosphorylation. To test this model, we analyzed the capacity of E4-ORF4 mutant proteins to interact with SF2/ASF in vitro and to activate IIIa pre-mRNA splicing in a transient transfection assay.
In a recent study, Shtrichman et al. (1999) established the capacity of E4-ORF4 mutant proteins to bind PP2A and co-immunoprecipitate an active phosphatase complex. Here we used this collection of E4-ORF4 mutant plasmids (Figure 5A) to study E4-ORF4 interaction with SF2/ASF in vitro (Figure 5B) and, furthermore, their capacity to activate IIIa pre-mRNA splicing in transiently transfected HeLa cells (Figure 5C). Care was taken to ensure that equal amounts of E4-ORF4 mutant proteins were used in the assays (data not shown).

Fig. 5. Capacity of E4-ORF4 mutant proteins to bind to SF2/ASF and activate IIIa pre-mRNA splicing. (A) Schematic representation of E4-ORF4 showing the position of mutated sequences in the E4-ORF4 mutant proteins. The interactions are summarized at the top. Gray boxes indicate regions essential for strong interaction of E4-ORF4 with SF2/ASF [see (B)], and efficient E4-ORF4 recruitment of a functional PP2A phosphatase complex (Shtrichman et al., 1999). Thin lines indicate regions significantly affecting SF2/ASF binding and phosphatase recruitment. (B) In vitro binding assay showing the interaction capacity of 35S-labeled SF2/ASF and GST–E4-ORF4 mutant proteins. (C) E4-ORF4 activation of IIIa pre-mRNA splicing. Quantitative S1 nuclease analysis of IIIa mRNA levels in HeLa cells co-transfected with pCMV plasmids encoding E4-ORF4 mutant proteins and the reporter pAdCMV-IIIa (schematically shown at the top). Average splicing efficiencies and standard deviations are shown (three experiments).
As shown in Figure 5B, wild-type E4-ORF4, and mutants R2, A11, R3-9 and A10 interact efficiently with 35S-labeled SF2/ASF in a GST pull-down assay. E4-ORF4 mutants p20, R4 and R3-2 show reduced binding affinity, whereas mutants R1, 12 and A3 lost almost all of the binding capacity. This analysis suggests that E4-ORF4 sequences critical for SF2/ASF binding map to three non-overlapping E4-ORF4 sequence elements (Figure 5A). Thus, the N-terminus and regions located around amino acids 61 and 95 are essential for E4-ORF4–SF2/ASF interaction. The lack of a simple primary amino acid sequence determinant of binding indicates that the E4-ORF4–SF2/ASF interaction may depend on E4-ORF4 protein conformation.
Strong E4-ORF4 interaction with SF2/ASF is not sufficient for activated IIIa splicing. Thus, mutants R2, A11, R3-9 and A10 interact efficiently with SF2/ASF (Figure 5B), whereas only mutant A11 enhances IIIa splicing (Figure 5C). However, comparing the binding and splicing activation results with the capacity of the E4-ORF4 mutant proteins to interact functionally with PP2A (Shtrichman et al., 1999) shows that strong interaction of E4-ORF4 with both SF2/ASF and PP2A correlates with IIIa splicing activation. Thus, mutants that show a strong interaction with one component were weak activators of IIIa splicing (Figure 5C; mutants R2, 12, R4 and A10), whereas mutants that did not either interact efficiently with SF2/ASF or recruit an active phosphatase (Figure 5C; mutants R1, p20, R3-2 and A3) slightly repressed IIIa splicing. In contrast, mutant A11, which shows strong interaction with SF2/ASF and is efficient in recruiting an active phosphatase, was almost as efficient as the wild-type E4-ORF4 protein in activating IIIa splicing. Mutant R3-9 is the only E4-ORF4 protein that does not obey this general rule (see Discussion).
Discussion
We have shown previously that E4-ORF4 activates IIIa pre-mRNA splicing both in vivo and in vitro and, furthermore, induces SR protein dephosphorylation (Kanopka et al., 1998). The results presented here reveal several novel aspects of the mechanisms by which E4-ORF4 regulates SR protein activity and RNA splicing. (i) The SR protein-responsive 3RE is sufficient to confer an E4-ORF4-activated splicing phenotype to a heterologous pre-mRNA (Figure 4). (ii) E4-ORF4 interacts selectively with a subset of HeLa SR proteins, namely SF2/ASF and SRp30c (Figure 1). (iii) E4-ORF4 interacts preferentially with the hyperphosphorylated form of SR proteins (Figure 3). (iv) E4-ORF4 activation of IIIa pre-mRNA splicing correlates with strong interaction of E4-ORF4 with an active PP2A and SF2/ASF (Figures 5 and 6).

Fig. 6. Strong E4-ORF4 interaction with SF2/ASF, and E4-ORF4 recruitment of a functional phosphatase complex correlate with E4-ORF4 activation of IIIa pre-mRNA splicing. The figure presents a summary of E4-ORF4 mutant protein interactions with SF2/ASF (Figure 5B), E4-ORF4 recruitment of a functional phosphatase complex (Shtrichman et al., 1999) and the capacity of E4-ORF4 mutant proteins to activate IIIa pre-mRNA splicing in a transient transfection assay (Figure 5C). *The percentage binding or recruitment are as follows:–, 0–30%; (+), 30–60%; and +, 60–100%.
The E4-ORF4 mutant analysis shows an almost perfect correlation between strong E4-ORF4 interaction with SF2/ASF, E4-ORF4 recruitment of an active phosphatase complex and IIIa splicing activation (Figure 6). Thus, strong interaction between E4-ORF4 and only one component is not enough to activate IIIa splicing in a transient transfection assay. For example, mutant 12 recruits an active phosphatase complex but does not interact efficiently with SF2/ASF, and is defective in IIIa pre-mRNA splicing activation. Mutant R2 shows the opposite binding specificity, i.e. it binds SF2/ASF but does not recruit an active phosphatase, and it is similarly defective in activation of splicing. Moreover, mutants with reduced binding affinity for SF2/ASF or reduced phosphatase recruitment do not stimulate IIIa pre-mRNA splicing, whereas mutant A11, which interacts efficiently with both SF2/ASF and PP2A, is a strong activator of IIIa pre-mRNA splicing. The only E4-ORF4 mutant protein that does not conform to this general rule is mutant R3-9, which does not activate IIIa splicing despite the fact that it interacts efficiently with SF2/ASF in vitro and recruits an active phosphatase complex (Figure 6). We do not know the significance of this observation. However, we note that E4-ORF4 binding to PP2A and E4-ORF4 recruitment of an active phosphatase complex similarly did not show a perfect match (Shtrichman et al., 1999). Thus, E4-ORF4 binding to PP2A is not synonymous with active dephosphorylation. The simplest explanation for this discrepancy between binding and dephosphorylation is that the E4-ORF4–PP2A interaction requires a higher order structure that correctly presents the target protein to the PP2A catalytic subunit. Thus, mutant R3-9 interaction with SF2/ASF and PP2A may be non-productive and fail to present the SR protein correctly for catalysis. However, we cannot exclude the possibility that E4-ORF4 targets another important splicing factor that is required for E4-ORF4 function in splicing.
The good correlation between E4-ORF4–PP2A–SF2/ASF interaction and IIIa pre-mRNA splicing activation (Figure 6) suggests that SF2/ASF is a major SR protein targeted by E4-ORF4. Although E4-ORF4 also interacts with SRp30c, this interaction does not show such a strict correlation with IIIa splicing activation. Thus, all tested E4-ORF4 mutant proteins (Figure 5A) bind with essentially the same efficiency to SRp30c (data not shown). Taken together, these results indicate that SRp30c, which is probably a minor species compared with the abundant SF2/ASF (Hanamura et al., 1998), does not make a significant contribution to E4-ORF4-induced activation of IIIa pre-mRNA splicing, at least not in HeLa cells.
The observation that E4-ORF4 interacts specifically with SF2/ASF through its RRMs (Figure 2) provides an explanation for the observed selective interaction of E4-ORF4 with only a subset of SR proteins (Figure 1). However, the observation that E4-ORF4 interacts preferentially with hyperphosphorylated SR proteins (Figure 3) may seem paradoxical. Why does E4-ORF4 interact efficiently with SF2-ΔRS, which lacks the RS domain containing the serines that are phosphorylated? The answer to this problem appears to lie in the fact that SF2/ASF is also phosphorylated at residues located in the RRMs in HeLa-NE (Petersen-Mahrt et al., 1999). Also, Clk/Sty phosphorylate SF2/ASF outside the RS domain (Colwill et al., 1996), further corroborating the idea that SF2/ASF is not only phosphorylated in the RS domain. Thus, E4-ORF4 may induce RS domain dephosphorylation, but binding to SF2/ASF may be mediated through phosphorylated residues in the SF2/ASF RRMs. Alternatively, RS domain dephosphorylation may perturb the structure of SF2/ASF such that E4-ORF4 cannot bind.
We have shown previously that the cellular protein p32, similarly to E4-ORF4, regulates IIIa pre-mRNA splicing, and inhibits SF2/ASF as a splicing factor (Petersen-Mahrt et al., 1999). Interestingly, both E4-ORF4 and p32 appear to interact with SF2/ASF through residues located in the RRMs and, ‘at a distance’, affect RS domain phosphorylation. Thus, E4-ORF4 and p32 both induce accumulation of hypophosphorylated SR proteins. However, they do so by opposite mechanisms. E4-ORF4 activates dephosphorylation while p32 accomplishes the same by inhibiting phosphorylation (reviewed in Akusjärvi, 1999).
Collectively, our results suggest that two proteins regulating SF2/ASF function interact with SF2/ASF through its RRMs and regulate RS domain function. Hypothetically, this observation may provide an explanation for the finding that SF2/ASF chimeric proteins, created by fusing RS domains from other SR proteins to the SF2/ASF RRMs, functionally substitute for SF2/ASF in vivo (Wang et al., 1998). Thus, if regulatory proteins controlling SF2/ASF phosphorylation specifically interact with SF2/ASF through its RRMs, the origin of the RS domain may be secondary for SF2/ASF regulation.
The observation that E4-ORF4 interacts selectively with only some HeLa SR proteins was surprising, and is potentially very significant. Thus, we showed previously that all SR proteins, except SRp20, are underphosphorylated in late adenovirus-infected cells (Kanopka et al., 1998). The finding that E4-ORF4 does not interact with all SR proteins, therefore, suggests that there must be other viral proteins that contribute to the accumulation of hypophosphorylated SR proteins in late virus-infected cells. In this respect, it is interesting to note that we have shown recently that the viral E4-ORF6 protein, which functions as an alternative splicing factor in vivo (Nordqvist et al., 1994), activates IIIa pre-mRNA splicing in vitro, in a reaction requiring the SR-responsive 3RE (S.Fan and G.Akusjärvi, in preparation). Potentially, the E4-ORF6 protein may be the missing viral factor controlling SR protein phosphorylation. However, the mechanistic details of how E4-ORF6 activates IIIa splicing remain to be established.
Materials and methods
Plasmids and proteins
Detailed plasmid maps and sequences are available upon request, and some are found at http://www.bmc.uu.se/IMIM/res/GA.html. Plasmids for transfection (pCMV variants) and in vitro binding studies (pGST variants) of E4-ORF4 and derivatives encoding mutant E4-ORF4 proteins have been described (Kleinberger and Shenk, 1993; Shtrichman et al., 1999). Reporter plasmids pAdCMV-glob, pAdCMV-glob (3RE) and pAdCMV-glob (3VDE) were generated by recloning the relevant fragments from previously described plasmids 1, 3 and 4 (Figure 1 in Kanopka et al., 1996) into the BamHI site in plasmid pAdCMV-BamHI vector (Molin et al., 1998). Reporter plasmid pAdCMV-IIIa has been described (Kanopka et al., 1998). Plasmids encoding SRp20, SRp30c, SRp40, SRp55 and SRp75 and those encoding T7-tagged SF2/ASF, SF2-ΔRS, SF2-RRM2/RS, SF2-RRM1/RS, SF2-RS and SC35 have been described (Caceres and Krainer, 1993; Screaton et al., 1995; Caceres et al., 1997). 35S-labeled SR proteins were made by in vitro transcription–translation using the TNT system (Promega), from PCR templates amplified using a 5′ primer, containing a T7 promoter and the first 15 bases of the respective SR protein gene, and a common 3′ primer, complementary to β-globin sequences in the plasmid sequence downstream of the protein insert. GST and GST fusion proteins were expressed and purified from Escherichia coli as described by the manufacturer (Pharmacia Biotech). SR proteins were purified from HeLa cells (SR-HeLa) and late adenovirus-infected HeLa cells (SR-Ad) as described (Zahler et al., 1992; Kanopka et al., 1998).
In vitro binding assay
In vitro translated 35S-labeled SR proteins were incubated with GST, GST–E4-ORF4 or GST–E4-ORF4 mutant proteins on glutathione–Sepharose beads for 2 h at 4°C in TBS buffer (20mM Tris–HCl pH 7.5, 139 mM NaCl) containing 0.15% Tween-20, washed twice in the binding buffer, and separated on a 12% SDS–polyacrylamide gel under reducing conditions. As a standard, half of the 35S-labeled SR protein extract used in the GST binding assay was run in parallel on the gel.
Cells and transient transfections
HeLa and 293 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies) supplemented with 10% newborn calf serum and 100 U/ml penicillin–streptomycin (Life Technologies). Subconfluent monolayers of cells grown on 60 mm dishes were transfected using the FuGene cocktail (Boehringer-Mannheim) or the calcium phosphate co-precipitation technique (Ausubel et al., 1987). Cells were harvested 46 h post-transfection and lysed in IsoB-NP40 (10 mM Tris–HCl pH 7.9, 0.15 M NaCl, 1.5 mM MgCl2, 0.5% NP-40) on ice. Cytoplasmic RNA was prepared by phenol/chloroform/isoamylalcohol (25:24:1) extractions. To prepare soluble extracts for immunoprecipitation, cells were incubated for 30 min on ice in lysis buffer [50 mM Tris–HCl pH 7.4, 250 mM NaCl, 5 mM EDTA, 0.1% Triton X-100, 0.5% NP-40, protease inhibitor mix (Boehringer Mannheim)]. Cell debris was removed by centrifugation at 13 500 g for 10 min.
Immunoprecipitations and western blotting
A 3 µg aliquot of pCMV-E4-ORF4 or empty vector (pCMV) and 1 µg of plasmids encoding T7-tagged SF2/ASF, SF2-ΔRS, SF2-RRM1/RS, SF2-RRM2/RS or SC35 were co-transfected into 293 cells. Soluble cell lysates were incubated with anti-E4-ORF4 antibody covalently coupled to protein A–Sepharose (Pharmacia Biotech) at 4°C overnight. Immuno precipitates were washed three times in lysis buffer and separated on a 12% SDS–polyacrylamide gel. Proteins were transferred to a nitrocellulose membrane by electroblotting and probed with anti-E4-ORF4 or anti-T7 (Novagen) antibodies in a standard western blot assay (Ausubel et al., 1987). Antibody interaction with proteins on the membrane was visualized by chemiluminescence (Amersham).
Western and far western analysis of SR protein
SR-HeLa or SR-Ad proteins (0.5 µg) were incubated for 10 min at room temperature in either the absence or presence of 5 µg of HeLa cell nuclear extract (HeLa-NE) or HeLa-NE depleted of ATP [HeLa-NE(–ATP)], separated on a 10% SDS–polyacrylamide gel and electroblotted to a nitrocellulose membrane. The filter was blocked in TBS buffer containing 0.05% Tween-20, 0.5 mM β-glycerophosphate and 5% milk for 30 min at room temperature. The filter was then incubated with 2 µg/ml GST–E4-ORF4 in TBS buffer containing 0.05% Tween-20 and 1% bovine serum albumin (BSA) for 30 min at room temperature. The filter was washed in TBS containing 0.05% Tween-20, and probed in the first round with an anti-E4-ORF4 antibody (Shtrichman et al., 1999), which was visualized by chemiluminescence as described by the manufacturer (Amersham). The filter was stripped by incubation in 0.78% mercaptoethanol, 62.5 mM Tris–HCl, 2% SDS for 20 min at 50°C and reprobed in the second round with mAb104 (Zahler et al., 1992), and in a third round with mAb96 (Hanamura et al., 1998).
ATP depletion
HeLa-NE (5 µg) was mixed with 2 mM glucose, 2.5 mM MgCl2 and 0.7 U of hexokinase (Boehringer Mannheim) in a total volume of 20 µl, and incubated for 10 min at 30°C. The amount of hexokinase and glucose required for complete depletion of the ATP pool in dialyzed nuclear extracts was determined in control experiments containing 1 µCi of [γ-32P]ATP.
S1 nuclease assays
A 3 µg aliquot of reporter plasmids pAdCMV-IIIa, pAdCMV-glob, pAdCMV-glob (3RE) or pAdCMV-glob (3VDE) and 0.2 µg of pCMV plasmids encoding wild-type or mutant E4-ORF4 proteins were co-transfected into HeLa cells. Total cytoplasmic RNA was isolated 46 h post-transfection and 10 µg subjected to S1 analysis (Ausubel et al., 1987) using the IIIa-specific 5′-end-labeled DNA probe previously described (Kanopka et al., 1998), or a β-globin-specific 5′-end-labeled DNA probe. After S1 nuclease digestion, the products were precipitated and separated on an 8% denaturing polyacrylamide gel and the protected fragments visualized and quantitated by PhosphorImager scanning.
Acknowledgments
Acknowledgements
This work was supported by the Swedish Cancer Society and the Göran Gustafsson foundation for Natural and Medical Research. A.R.K. was supported in part by grant CA13106 from the National Cancer Institute. T.K. was supported by the Israel Science Foundation.
REFERENCES
- Akusjärvi G. (1999) Remodelling of the host cell RNA splicing machinery during an adenovirus infection. Recent Res. Dev. Virol., 1, 621–630. [DOI] [PubMed] [Google Scholar]
- Akusjärvi G. and Persson,H. (1981) Controls of RNA splicing and termination in the major late adenovirus transcription unit. Nature, 292, 420–426. [DOI] [PubMed] [Google Scholar]
- Ausubel F.M., Brent,R., Kingston,R.E., Moore,D.D., Seidman,J.G., Smith,J.A. and Struhl,K. (1987) Current Protocols in Molecular Biology. John Wiley & Sons, Inc., Boston, MA.
- Bondesson M., Öhman,K., Mannervik,M., Fan,S. and Akusjärvi,G. (1996) Adenovirus E4 open reading frame 4 protein autoregulates E4 transcription by inhibiting E1A transactivation of the E4 promoter. J. Virol., 70, 3844–3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caceres J.F. and Krainer,A.R. (1993) Functional analysis of pre-mRNA splicing factor SF2/ASF structural domains. EMBO J., 12, 4715–4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caceres J.F., Misteli,T., Screaton,G.R., Spector,D.L. and Krainer,A.R. (1997) Role of the modular domains of SR proteins in subnuclear localization and alternative splicing specificity. J. Cell Biol., 138, 225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caceres J.F., Screaton,G.R. and Krainer,A.R. (1998) A specific subset of SR proteins shuttles continuously between the nucleus and the cytoplasm. Genes Dev., 12, 55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao W., Jamison,S.F. and Garcia-Blanco,M.A. (1997) Both phosphorylation and dephosphorylation of ASF/SF2 are required for pre-mRNA splicing in vitro. RNA, 3, 1456–1467. [PMC free article] [PubMed] [Google Scholar]
- Chow L.T., Broker,T.R. and Lewis,J.B. (1979) Complex splicing patterns of RNAs from the early regions of adenovirus-2. J. Mol. Biol., 134, 265–303. [DOI] [PubMed] [Google Scholar]
- Colwill K., Pawson,T., Andrews,B., Prasad,J., Manley,J.L., Bell,J.C. and Duncan,P.I. (1996) The Clk/Sty protein kinase phosphorylates SR splicing factors and regulates their intranuclear distribution. EMBO J., 15, 265–275. [PMC free article] [PubMed] [Google Scholar]
- Csortos C., Zolnierowicz,S., Bako,E., Durbin,S.D. and DePaoli-Roach,A.A. (1996) High complexity in the expression of the B′ subunit of protein phosphatase 2A0. Evidence for the existence of at least seven novel isoforms. J. Biol. Chem., 271, 2578–2588. [DOI] [PubMed] [Google Scholar]
- Fu X.D. (1995) The superfamily of arginine/serine-rich splicing factors. RNA, 1, 663–680. [PMC free article] [PubMed] [Google Scholar]
- Graveley B.B. (2000) Sorting out the complexity of SR protein functions. RNA, 6, 1197–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanamura A., Caceres,J.F., Mayeda,A., Franza,B.R.,Jr and Krainer,A.R. (1998) Regulated tissue-specific expression of antagonistic pre-mRNA splicing factors. RNA, 4, 430–444. [PMC free article] [PubMed] [Google Scholar]
- Imperiale M., Akusjärvi,G. and Leppard,K. (1995) Post-transcriptional control of adenovirus gene expression. Curr. Top. Microbiol., 199, 139–171. [DOI] [PubMed] [Google Scholar]
- Kamibayashi C., Estes,R., Lickteig,R.L., Yang,S.I., Craft,C. and Mumby,M.C. (1994) Comparison of heterotrimeric protein phosphatase 2A containing different B subunits. J. Biol. Chem., 269, 20139–20148. [PubMed] [Google Scholar]
- Kanopka A., Mühlemann,O. and Akusjärvi,G. (1996) Inhibition by SR proteins of splicing of a regulated adenovirus pre-mRNA. Nature, 381, 535–538. [DOI] [PubMed] [Google Scholar]
- Kanopka A., Mühlemann,O., Petersen-Mahrt,S., Estmer,C., Öhrmalm,C. and Akusjärvi,G. (1998) Regulation of adenovirus alternative RNA splicing by dephosphorylation of SR proteins. Nature, 393, 185–187. [DOI] [PubMed] [Google Scholar]
- Kleinberger T. and Shenk,T. (1993) Adenovirus E4orf4 protein binds to protein phosphatase 2A and the complex down regulates E1A-enhanced junB transcription. J. Virol., 67, 7556–7560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohtz J.D., Jamison,S.F., Will,C.L., Zuo,P., Luhrmann,R., Garcia-Blanco,M.A. and Manley,J.L. (1994) Protein–protein interactions and 5′-splice-site recognition in mammalian mRNA precursors. Nature, 368, 119–124. [DOI] [PubMed] [Google Scholar]
- Lavoie J.N., Nguyen,M., Marcellus,R.C., Branton,P.E. and Shore,G.C. (1998) E4orf4, a novel adenovirus death factor that induces p53-independent apoptosis by a pathway that is not inhibited by zVAD-fmk. J. Cell Biol., 140, 637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manley J.L. and Tacke,R. (1996) SR proteins and splicing control. Genes Dev., 10, 1569–1579. [DOI] [PubMed] [Google Scholar]
- Mannervik M., Fan,S., Ström,A.C., Helin,K. and Akusjärvi,G. (1999) Adenovirus E4 open reading frame 4-induced dephosphorylation inhibits E1A activation of the E2 promoter and E2F-1-mediated transactivation independently of the retinoblastoma tumor suppressor protein. Virology, 256, 313–321. [DOI] [PubMed] [Google Scholar]
- Marcellus R.C., Lavoie,J.N., Boivin,D., Shore,G.C., Ketner,G. and Branton,P.E. (1998) The early region 4 orf4 protein of human adenovirus type 5 induces p53-independent cell death by apoptosis. J. Virol., 72, 7144–7153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcellus R.C., Chan,H., Paquette,D., Thirlwell,S., Boivin,D. and Branton,P.E. (2000) Induction of p53-independent apoptosis by the adenovirus E4orf4 protein requires binding to the Bα subunit of protein phosphatase 2A. J. Virol., 74, 7869–7877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCright B. and Virshup,D.M. (1995) Identification of a new family of protein phosphatase 2A regulatory subunits. J. Biol. Chem., 270, 26123–26128. [DOI] [PubMed] [Google Scholar]
- Mermoud J.E., Cohen,P.T. and Lamond,A.I. (1994) Regulation of mammalian spliceosome assembly by a protein phosphorylation mechanism. EMBO J., 13, 5679–5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millward T.A., Zolnierowicz,S. and Hemmings,B.A. (1999) Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem. Sci., 24, 186–191. [DOI] [PubMed] [Google Scholar]
- Misteli T. (1999) RNA splicing: what has phosphorylation got to do with it? Curr. Biol., 9, R198–R200. [DOI] [PubMed] [Google Scholar]
- Misteli T., Caceres,J.F., Clement,J.Q., Krainer,A.R., Wilkinson,M.F. and Spector,D.L. (1998) Serine phosphorylation of SR proteins is required for their recruitment to sites of transcription in vivo.J. Cell Biol., 143, 297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molin M., Shoshan,M.C., Öhman-Forslund,K., Linder,S. and Akusjärvi,G. (1998) Two novel adenovirus vector systems permitting regulated protein expression in gene transfer experiments. J. Virol., 72, 8358–8361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mühlemann O., Yue,B.G., Petersen-Mahrt,S. and Akusjärvi,G. (2000) A novel type of splicing enhancer regulating adenovirus pre-mRNA splicing. Mol. Cell. Biol., 20, 2317–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller U., Kleinberger,T. and Shenk,T. (1992) Adenovirus E4orf4 protein reduces phosphorylation of c-Fos and E1A proteins while simultaneously reducing the level of AP-1. J. Virol., 66, 5867–5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevins J.R. and Wilson,M.C. (1981) Regulation of adenovirus-2 gene expression at the level of transcriptional termination and RNA processing. Nature, 290, 113–118. [DOI] [PubMed] [Google Scholar]
- Nordqvist K., Öhman,K. and Akusjärvi,G. (1994) Human adenovirus encodes two proteins which have opposite effects on accumulation of alternatively spliced mRNAs. Mol. Cell. Biol., 14, 437–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen-Mahrt S.K., Estmer,C., Öhrmalm,C., Matthews,D.A., Russell,W.C. and Akusjärvi,G. (1999) The splicing factor-associated protein, p32, regulates RNA splicing by inhibiting ASF/SF2 RNA binding and phosphorylation. EMBO J., 18, 1014–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad J., Colwill,K., Pawson,T. and Manley,J.L. (1999) The protein kinase Clk/Sty directly modulates SR protein activity: both hyper- and hypophosphorylation inhibit splicing. Mol. Cell. Biol., 19, 6991–7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roscigno R.F. and Garcia-Blanco,M.A. (1995) SR proteins escort the U4/U6⋅U5 tri-snRNP to the spliceosome. RNA, 1, 692–706. [PMC free article] [PubMed] [Google Scholar]
- Roth M.B., Murphy,C. and Gall,J.G. (1990) A monoclonal antibody that recognizes a phosphorylated epitope stains lampbrush chromosome loops and small granules in the amphibian germinal vesicle. J. Cell Biol., 111, 2217–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Screaton G.R., Caceres,J.F., Mayeda,A., Bell,M.V., Plebanski,M., Jackson,D.G., Bell,J.I. and Krainer,A.R. (1995) Identification and characterization of three members of the human SR family of pre-mRNA splicing factors. EMBO J., 14, 4336–4349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtrichman R. and Kleinberger,T. (1998) Adenovirus type 5 E4 open reading frame 4 protein induces apoptosis in transformed cells. J. Virol., 72, 2975–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtrichman R., Sharf,R., Barr,H., Dobner,T. and Kleinberger,T. (1999) Induction of apoptosis by adenovirus E4orf4 protein is specific to transformed cells and requires an interaction with protein phosphatase 2A. Proc. Natl Acad. Sci. USA, 96, 10080–10085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtrichman R., Sharf,R. and Kleinberger,T. (2000) Adenovirus E4orf4 protein interacts with both Ba and B′ subunits of protein phosphatase 2A, but E4orf4-induced apoptosis is mediated only by Ba. Oncogene, 19, 3757–3765. [DOI] [PubMed] [Google Scholar]
- Svensson C. and Akusjärvi,G. (1986) Defective RNA splicing in the absence of adenovirus-associated RNAI. Proc. Natl Acad. Sci. USA, 83, 4690–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Xiao,S.H. and Manley,J.L. (1998) Genetic analysis of the SR protein ASF/SF2: interchangeability of RS domains and negative control of splicing. Genes Dev., 12, 2222–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wera S. and Hemmings,B.A. (1995) Serine/threonine protein phosphatases. Biochem. J., 311, 17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J.Y. and Maniatis,T. (1993) Specific interactions between proteins implicated in splice site selection and regulated alternative splicing. Cell, 75, 1061–1070. [DOI] [PubMed] [Google Scholar]
- Xiao S.H. and Manley,J.L. (1997) Phosphorylation of the ASF/SF2 RS domain affects both protein–protein and protein–RNA interactions and is necessary for splicing. Genes Dev., 11, 334–344. [DOI] [PubMed] [Google Scholar]
- Zahler A.M., Lane,W.S., Stolk,J.A. and Roth,M.B. (1992) SR proteins: a conserved family of pre-mRNA splicing factors. Genes Dev., 6, 837–847. [DOI] [PubMed] [Google Scholar]
- Zuo P. and Manley,J.L. (1993) Functional domains of the human splicing factor ASF/SF2. EMBO J., 12, 4727–4737. [DOI] [PMC free article] [PubMed] [Google Scholar]