

Abstract
Stimulation of T cells via the CD3–T-cell receptor (TCR) complex results in rapid increases in β1 integrin-mediated adhesion via poorly defined intracellular signaling events. We demonstrate that TCR-mediated activation of β1 integrins requires activation of the Tec family tyrosine kinase Itk and phosphatidylinositol 3-kinase (PI 3-K)-dependent recruitment of Itk to detergent-insoluble glycosphingolipid-enriched microdomains (DIGs) via binding of the pleckstrin homology domain of Itk to the PI 3-K product PI(3,4,5)-P3. Activation of PI 3-K and the src family kinase Lck, via stimulation of the CD4 co-receptor, can initiate β1 integrin activation that is dependent on Itk function. Targeting of Itk specifically to DIGs, coupled with CD4 stimulation, can also activate β1 integrin function independently of TCR stimulation. Changes in β1 integrin function mediated by TCR activation of Itk are also accompanied by Itk-dependent modulation of the actin cytoskeleton. Thus, TCR-mediated activation of β1 integrins involves membrane relocalization and activation of Itk via coordinate action of PI 3-K and a src family tyrosine kinase.
Keywords: integrin/Itk/Lck/phosphatidylinositol 3-kinase/T lymphocyte
Introduction
Efficient recognition of foreign pathogens by the immune system requires the systemic trafficking of a pool of potentially antigen-reactive T lymphocytes through secondary lymphoid organs and peripheral tissue sites. Integrin adhesion receptors mediate critical interactions of T cells with other cells and extracellular matrix components during trafficking, as well as during antigen-specific recognition events in tissue (Shimizu et al., 1999). Consequently, the functional activity of integrin receptors on T cells is dynamically regulated by external cues provided by other cell surface receptors. Stimulation of the antigen-specific CD3–T-cell receptor (TCR) complex results in increased T-cell adhesion mediated by β1 or β2 integrins that does not require an increase in overall levels of integrins on the cell surface (Dustin and Springer, 1989; van Kooyk et al., 1989; Shimizu et al., 1990). This change in T-cell adhesion upon antigen receptor stimulation occurs within minutes of stimulation and represents one of the earliest functional responses of T lymphocytes to activation.
Stimulation of the CD3–TCR complex initiates a complex array of intracellular signaling events, beginning with src family tyrosine kinase-mediated phosphorylation of cytoplasmic immunoreceptor tyrosine-based activation motifs and recruitment of the Syk family tyrosine kinase ZAP-70 to the CD3–TCR complex (Kane et al., 2000). Activated ZAP-70 subsequently phosphorylates substrates such as the adapter proteins LAT and SLP-76, resulting in the formation of protein–protein signaling complexes that initiate downstream signaling events, such as phospholipase C-γ1 (PLC-γ1) activation and subsequent production of inositol 1,4,5-trisphosphate (IP3) and diacylglycerol. Recent studies have highlighted the importance of the nucleation of these signaling complexes in specialized microdomains at the T-cell plasma membrane, where critical signaling molecules, such as src family kinases and LAT, are preferentially localized due to acylation or palmitoylation (Xavier et al., 1998; Zhang et al., 1998; Janes et al., 1999; Lin et al., 1999; Langlet et al., 2000). Although the importance of these signaling cascades to CD3–TCR-mediated transcriptional activation of cytokine genes such as interleukin-2 is well established, the signaling pathways by which the CD3–TCR complex regulates integrin-mediated T-cell adhesion are less clear. Studies with ZAP-70-deficient T cells have demonstrated an essential role for ZAP-70 in CD3–TCR-mediated increases in β1 integrin function (Epler et al., 2000). Phorbol ester stimulation can also enhance integrin function (Dustin and Springer, 1989; van Kooyk et al., 1989; Shimizu et al., 1990), and protein kinase C inhibitors can partially block CD3–TCR-mediated activation of β1 and β2 integrins (Dustin and Springer, 1989; van Kooyk et al., 1989; Mobley et al., 1994). Integrin function can also be modulated by activation of various GTPases, including H-ras, R-ras and Rap1 (Zhang et al., 1996; Hughes et al., 1997; A.M.O’Rourke et al., 1998; Caron et al., 2000; Katagiri et al., 2000; Reedquist et al., 2000). However, while GTPase activation is often sufficient to activate integrins, the role that these GTPases play in the signaling pathways by which CD3–TCR and other receptors regulate integrin activity is less clearly established. Modulation of the actin cytoskeleton is likely to play a key role in activation-dependent integrin regulation, as integrin-dependent cell adhesion is sensitive to cytochalasin D, and CD3–TCR stimulation can induce β2 integrin clustering (Stewart et al., 1996). While activation-dependent changes in β1 integrin affinity, as assessed by soluble ligand binding and induction of integrin activation epitopes, have been observed following CD3–TCR stimulation, T-cell adhesion to fibronectin (FN) is not inhibited by excess soluble ligand (Woods et al., 2000).
Several recent studies have demonstrated a critical role for the lipid kinase phosphatidylinositol 3-kinase (PI 3-K) in the regulation of integrin activity by CD3–TCR as well as other PI 3-K-coupled cell surface receptors (Shimizu and Hunt, 1996; Zell et al., 1996; Chan et al., 1997; Kivens et al., 1998; Nagel et al., 1998; Kinashi et al., 1999; Woods et al., 2000). However, the identification of molecules downstream of PI 3-K that regulate integrin functional activity has remained elusive. Although the β2 integrin-binding protein cytohesin-1 has been proposed to regulate lymphocyte function-associated antigen-1 (LFA-1) function downstream of PI 3-K (Nagel et al., 1998), cytohesin-1 does not bind to the β1 integrin cytoplasmic domain or regulate its functional activity (Kolanus et al., 1996).
PI 3-K activation results in the generation of D-3-phosphorylated lipid products at the cell membrane, which results in membrane recruitment of proteins containing pleckstrin homology (PH) domains via PH domain binding to the PI 3-K-generated lipid products (Klarlund et al., 1997; Lemmon et al., 1997). We reasoned that CD3–TCR-mediated regulation of β1 integrins may involve PI 3-K-dependent recruitment of an effector with such a PH domain. Members of the Tec family of tyrosine kinases represent potential candidate effectors (Schaeffer and Schwartzberg, 2000). The Tec tyrosine kinase Itk (also Emt or Tsk) is regulated in a PI 3-K-dependent manner and has been implicated in phosphorylation of PLC-γ1, calcium flux and mitogen-activated protein (MAP) kinase activation (Liu et al., 1998; Perez-Villar and Kanner, 1999; Schaeffer et al., 1999). Itk plays a role in T-cell development (Liao and Littman, 1995) and mutations in the PH domain of the Tec family kinase Btk can result in B-cell immunodeficiency (Sideras and Smith, 1995). In this report, we identify a novel function for Itk in the regulation of β1 integrin function by CD3–TCR in a manner that is dependent on coordinate upstream activation of src family kinases, PI 3-K, and the specific recruitment of Itk to detergent-insoluble membrane microdomains.
Results
CD3–TCR stimulation results in PI 3-K-dependent changes in the intracellular localization of Itk in T cells
Recent studies have highlighted the critical role of recruitment to and assembly of protein–protein complexes in detergent-insoluble glycosphingolipid-enriched membrane microdomains (DIGs) for efficient T-cell activation (Moran and Miceli, 1998; Xavier et al., 1998; Xavier and Seed, 1999). We examined the potential role of PI 3-K in the CD3–TCR-dependent recruitment of Itk to DIGs (Xavier et al., 1998). Cytosolic and membrane fractions and DIGs were prepared from unstimulated and CD3-stimulated Jurkat T cells. Western blotting was utilized to examine the presence of Itk in these fractions (Figure 1A). While a basal level of Itk could be found in both cytosolic and membrane fractions, CD3 stimulation resulted in increased levels of Itk in T-cell membrane fractions. More significantly, CD3 stimulation of Jurkat T cells resulted in increased localization of Itk in DIGs (Figure 1A). The src family kinase p56lck (Lck), which can phosphorylate Itk and thereby regulate Itk kinase activity (August et al., 1997; Heyeck et al., 1997), was also enriched in DIGs, although CD3 stimulation did not appreciably increase Lck localization in DIGs when compared with unstimulated T cells. The cytoplasmic kinase extracellular regulated kinase (ERK1), the CD45 cell surface receptor and the GM1 glycosphingolipid were used as markers to verify the integrity of our cytosolic, membrane and DIG preparations, respectively (Figure 1A). In the presence of the PI 3-K inhibitors wortmannin or LY294,002, CD3-induced increases in the localization of Itk to both the membrane and DIGs were dramatically inhibited (Figure 1B and data not shown). Thus, CD3 stimulation results in PI 3-K-dependent recruitment of Itk to DIGs, where it co-localizes with Lck.
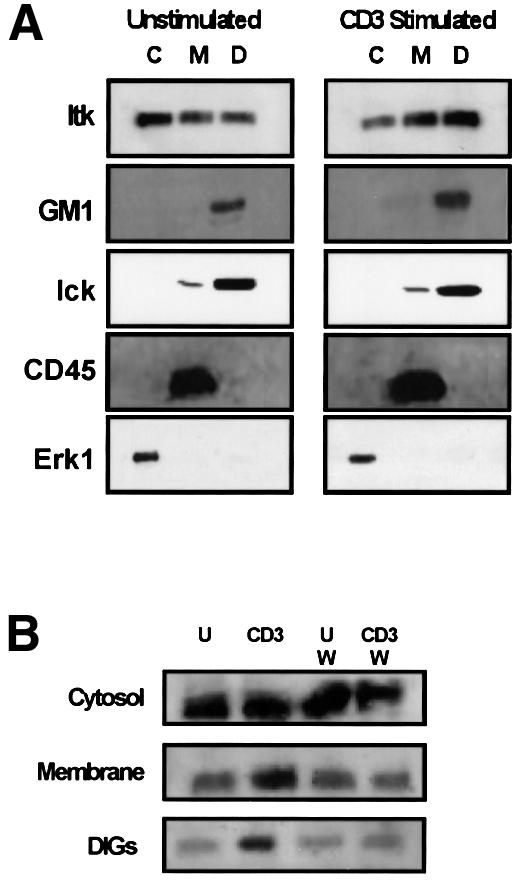
Fig. 1. CD3–TCR stimulation results in PI 3-K-dependent increases in Itk localization in DIGs. (A) Jurkat T cells were left unstimulated (left panel) or were CD3 stimulated (right panel) for 5 min at 37°C. Cytosolic (C), membrane (M) and DIG (D) preparations containing 2 × 106 cell equivalents from cytosolic fractions or 6 × 106 cell equivalents from the membrane or DIG fractions were separated by SDS–PAGE, transferred to PVDF membranes, and immunoblotted for Itk, Lck, CD45 and Erk1 using specific antibodies. The presence of the GM1 ganglioside was detected with cholera toxin B subunit as described in Materials and methods. (B) Jurkat T cells were either left unstimulated (U) or CD3 stimulated (CD3) as in (A) in the presence or absence of 100 nM wortmannin (W). Cytosolic, membrane and DIG preparations were prepared as in (A) and analyzed for the presence of Itk. Results are representative of a minimum of three different experiments performed with fractions prepared on separate days.
The PH domain of Itk binds to PI(3,4,5)-P3 and mediates CD3-dependent relocalization of Itk
We measured phospholipid production in unstimulated and CD3-stimulated T cells by activating permeabilized Jurkat T cells in the presence of [32P]ATP, followed by the analysis of labeled phospholipids by TLC. CD3 stimulation of T cells resulted in an increase in the relative amounts of both PI(3,4)-P2 and PI(3,4,5)-P3; this increase in D-3-phosphorylated lipids was inhibited by the PI 3-K inhibitors LY294,002 and wortmannin (Figure 2A and data not shown). Our results using this approach are consistent with previous studies of PI production in Jurkat T cells (Ward et al., 1992). Since both PI(3,4)-P2 and PI(3,4,5)-P3 can bind to specific PH domains, we analyzed the ability of a glutathione S-transferase (GST) fusion protein expressing Itk to bind to various amounts of purified phospholipids immobilized on a nitrocellulose membrane (Stevenson et al., 1998). Nitrocellulose membranes spotted with phospholipids were incubated with GST–Itk fusion proteins and binding of the fusion protein was detected by an anti-GST antibody and enhanced chemiluminescence (ECL). The GST–wild-type (wt) Itk fusion protein bound in a dose-dependent manner specifically to PI(3,4,5)-P3 (Figure 2B). No binding was detectable to PI(3)-P, PI(4)-P or PI(4,5)-P2, and only very low binding between GST–wt Itk and PI(3,4)-P2 was observed (Figure 2B). All lipids were bound on the membrane at comparable levels, as detected by iodine visualization of the lipids following blotting (data not shown). Binding of Itk to PI(3,4,5)-P3 was mediated by the PH domain of Itk, since: (i) binding to PI(3,4,5)-P3 was lost when the PH domain of Itk was deleted in the GST–Itk fusion protein (GST–ΔPH Itk); and (ii) a GST fusion protein expressing only the PH domain of Itk (GST–PH Itk) exhibited binding to PI(3,4,5)-P3 similar to GST–Itk (Figure 2B).
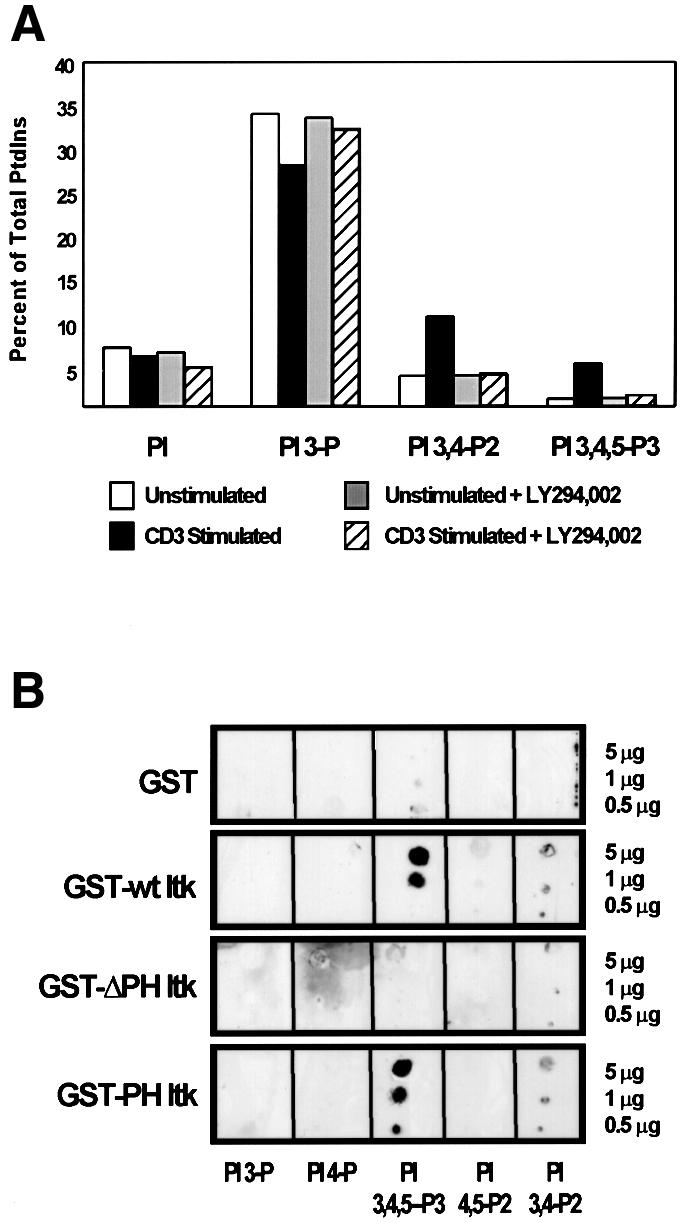
Fig. 2. The PH domain of Itk mediates binding of Itk to the PI 3-K product PI(3,4,5)-P3. (A) [32P]ATP-labeled Jurkat cells were left unstimulated (open and shaded bars) or were CD3 stimulated (solid and hatched bars) for 10 min at 37°C in the presence (shaded and hatched bars) or absence (open and solid bars) of 50 µM LY294,002. Phospholipids were extracted and analyzed as described in Materials and methods. Incorporation of 32P into specific phosphatidylinositols was quantitated on a phosphoimager. Relative levels of PI, PI(3)-P, PI(3,4)-P2 and PI(3,4,5)-P3 were determined by dividing the volume intensity (as determined by the phosphoimager) of each individual phospholipid spot by the total volume intensity of all phospholipid spots and multiplying by 100. Results shown are from one representative experiment out of a minimum of three separate experiments. (B) The binding of GST, GST–wild-type (wt) Itk, GST–PH Itk and GST–ΔPH Itk fusion proteins to different amounts of PI(3)-P, PI(4)-P, PI(4,5)-P2, PI(3,4)-P2 and PI(3,4,5)-P3 immobilized on nitrocellulose membranes was determined by FAT western blotting as described in Materials and methods. Following immunoblotting, membranes were exposed to iodine to verify that comparable amounts of each lipid were immobilized on the membranes (data not shown).
To determine the role of the Itk PH domain in CD3-mediated relocalization of Itk, unstimulated and CD3-stimulated Jurkat T cells that were transiently transfected with green fluorescent protein (GFP)-tagged Itk constructs (Figure 3) were examined by confocal microscopy. Representative images of cells expressing varying levels of GFP are shown in Figure 4A. In unstimulated Jurkat T cells, GFP–wt Itk was found in both the cytoplasm and at the membrane. CD3 stimulation led to increased punctated distribution of GFP–wt Itk specifically at the membrane. In contrast, Jurkat T cells expressing the GFP–ΔPH Itk fusion protein exhibited cytoplasmic GFP expression, with little membrane localization. Furthermore, CD3 stimulation did not lead to any changes in this pattern of expression. In cells expressing the GFP–PH Itk fusion protein, predominant membrane localization of this fusion protein was observed, even in unstimulated Jurkat T cells. CD3 stimulation of these cells resulted in accentuated punctate GFP expression at the membrane. Similar results were observed when assessing the intracellular localization of endogenous Itk in unstimulated Jurkat T cells (Figure 4B). CD3 stimulation of Jurkat T cells led to membrane localization of endogenous Itk and co-localization of endogenous Itk with DIGs, which were detected in these confocal microscopy experiments with fluorescein isothiocyanate (FITC)-conjugated cholera toxin B subunit (Figure 4B).

Fig. 3. Diagram of GFP–Itk fusion protein constructs utilized in this study.

Fig. 4. The PH domain of Itk is required for Itk membrane redistribution upon CD3 stimulation of human T cells. (A) Jurkat T cells transiently transfected with GFP–wt Itk, GFP–ΔPH Itk and GFP–PH Itk were left unstimulated or CD3 stimulated for 10 min at 37°C. The cells were then visualized by confocal microscopy. Arrows highlight areas of punctate membrane localization of Itk. A minimum of 100 cells for each transfectant/stimulation condition were examined and three representative cells for each construct and stimulation condition are shown. (B) Jurkat T cells were left unstimulated or CD3 stimulated for 10 min at 37°C. Cells were stained with anti-Itk, biotin-conjugated rabbit anti-goat IgG and streptavidin–APC, followed by staining with FITC-conjugated cholera toxin B subunit. Representative cell images are shown, and a merged image demonstrating co-localization of endogenous Itk in DIGs following CD3 stimulation (in yellow) is shown. (C) Jurkat cells transiently transfected with GFP–wt Itk, GFP–ΔPH Itk or GFP–ΔPH Itk with a farnesylation sequence tag (F-GFP–ΔPH Itk) were left unstimulated (U) or CD3 stimulated (CD3) for 5 min at 37°C. Cytosolic, membrane and DIG fractions were prepared as in Figure 1. Anti-HA immunoprecipitations were performed with lysates containing 5 × 106 GFP+ cell equivalents. Immunoprecipitates were separated on a 7.5% SDS–polyacrylamide gel, transferred to PVDF and immunoblotted with an anti-HA antibody. (D) Jurkat cells transiently transfected with GFP–control, GFP–wt Itk, GFP–KN Itk or GFP–PH Itk were sorted and left unstimulated (U) or CD3 stimulated (CD3) for 5 min at 37°C. DIG fractions were prepared as in Figure 1. Anti-Itk immunoprecipitations were performed with lysates containing 3 × 106 cells. Immunoprecipitates were separated on a 7.5% SDS–polyacrylamide gel, transferred to PVDF and immunoblotted with an anti-phosphotyrosine mAb, stripped and reprobed with an anti-Itk antibody.
Similar to endogenous Itk, CD3 stimulation resulted in increased localization of the GFP–Itk fusion protein in membrane fractions and DIGs of Jurkat T cells (Figure 4C). Low levels of GFP–wt Itk were observed in DIGs of unstimulated transfectants with overexposure of the blot shown in Figure 4C (data not shown). In contrast, the GFP–ΔPH Itk fusion protein was found exclusively in the cytosolic fraction in both unstimulated and CD3-stimulated T cells, providing further evidence that the PH domain of Itk is critical for membrane localization of Itk.
We also determined the tyrosine phosphorylation status of endogenous Itk and GFP–Itk constructs localized to DIGs in unstimulated and CD3-stimulated Jurkat T cells (Figure 4D). Sorted Jurkat T cells expressing GFP only, GFP–wt Itk, GFP–kinase-inactive Itk (GFP–KN Itk) or the GFP–PH Itk fusion protein were CD3 stimulated and Itk was immunoprecipitated from DIGs and analyzed by western blotting. While unstimulated Jurkat T cells exhibited minimal tyrosine phosphorylation of Itk found in DIGs, CD3 stimulation resulted in tyrosine phosphorylation of endogenous Itk, as well as increased localization of Itk in DIGs (Figure 4D). CD3 stimulation also resulted in tyrosine phosphorylation of the GFP–wt Itk construct. In contrast, expression of GFP–KN Itk or the GFP–PH Itk fusion protein inhibited CD3-induced tyrosine phosphorylation of endogenous Itk in DIGs and CD3-induced increases in localization of endogenous Itk to DIGs (Figure 4D).
CD3-induced activation of β1 integrin-mediated T-cell adhesion to FN requires the kinase activity of Itk
Since CD3 stimulation of T cells results in increased β1 integrin functional activity that is sensitive to PI 3-K inhibition (Woods et al., 2000), we explored a potential role for Itk in this functional response of T cells. Jurkat T cells were transiently transfected with constructs expressing either GFP or various GFP–Itk fusion proteins (Figure 3). GFP expression was obtained in ∼20–35% of the total number of cells recovered (data not shown). Following adhesion of this heterogeneous population of transfectants to FN under various stimulation conditions, the adherent cells were collected and quantitated by flow cytometric analysis (Kivens and Shimizu, 1998; Kivens et al., 1998). Post-acquisition gating was used to quantitate the percentage adhesion of the GFP-negative and -positive subpopulations in each sample. In all of the transfectants analyzed, CD3 stimulation or stimulation with the phorbol ester phorbol 12-myristate 13-acetate (PMA) for 10 min at 37°C resulted in enhanced adhesion to FN of the GFP-negative cells (Figure 5). Adhesion of unstimulated and stimulated transfectants expressing GFP or the GFP–Itk fusion protein was comparable to that of GFP-negative cells in the same sample. In contrast, transfectants expressing a GFP fusion expressing the kinase-inactive form of Itk (GFP–KN/Itk) exhibited impaired adhesion to FN following CD3 stimulation (Figure 5). This defect in CD3-induced activation of β1 integrins was not due to global defects in β1 integrin function, since transfectants expressing kinase-inactive Itk still exhibited increased adhesion to FN following PMA stimulation. Furthermore, CD3-induced increases in T-cell adhesion to FN were not inhibited by expression of a GFP fusion protein expressing a kinase-inactive form of the related Tec family tyrosine kinase Etk (GFP–KN/Etk) (Figure 5), even though Etk is expressed in Jurkat T cells (data not shown). These results demonstrate a specific role for Itk tyrosine kinase activity in CD3-mediated regulation of β1 integrin function.

Fig. 5. Expression of kinase-inactive Itk or the PH domain of Itk inhibits CD3-mediated increases in β1 integrin-mediated T-cell adhesion to FN. Adhesion of transiently transfected Jurkat cells expressing either GFP, GFP–Itk, GFP–KN/Itk, GFP–PH Itk, GFP–KN/Etk or GFP–PH Etk to FN following no stimulation (UNSTIM.) or after stimulation with PMA (PMA STIM.) or CD3 stimulation (CD3 STIM.) for 10 min at 37°C was assessed. Adhesion was quantitated by flow cytometry and the results indicate the percentage adhesion of GFP-negative cells (open bars) and GFP-positive cells (closed bars) under each stimulation condition with each transfected population. Results shown are from one of three independent replicate experiments.
CD3-induced regulation of β1 integrin function involves membrane localization of Itk
Similar to the result obtained with cells expressing the kinase-inactive GFP–Itk fusion protein, expression of the GFP fusion protein expressing just the PH domain of Itk also resulted in specific inhibition of CD3-induced adhesion to FN (Figure 5). This effect on adhesion was specific to the PH domain of Itk, as expression of a GFP fusion protein expressing the PH domain of Etk did not inhibit either PMA- or CD3-induced adhesion to FN (Figure 5). This suggests a critical role for PH domain-dependent membrane localization of Itk in CD3–TCR regulation of β1 integrin function. To determine whether membrane targeting of Itk was sufficient to induce increased β1 integrin function, we created a GFP fusion protein containing a membrane-targeting farnesylation sequence (F-GFP) and Itk lacking the PH domain (F-GFP–ΔPH Itk). Western blotting analysis of cell fractions demonstrated that the F-GFP–ΔPH Itk fusion protein was constitutively localized to the T-cell membrane, even in unstimulated T cells (Figure 4C). In addition, CD3 stimulation dramatically increased localization of F-GFP–ΔPH Itk to DIGs (Figure 4C). In adhesion experiments, expression of either F-GFP or F-GFP–ΔPH Itk did not enhance the adhesion of unstimulated, PMA- or CD3-stimulated Jurkat T cells (Figure 6 and data not shown), suggesting that membrane targeting of Itk in the absence of additional stimulation is not sufficient to induce increased β1 integrin function. However, stimulation of the CD4 co-receptor, which activates Lck (Turner et al., 1990; Baldari et al., 1995), resulted in enhanced adhesion of transfectants expressing F-GFP–ΔPH Itk but not F-GFP. Itk kinase activity in the membrane-targeted Itk construct was critical for this response, since CD4 stimulation of Jurkat T cells expressing F-GFP–ΔPH Itk containing the kinase-inactive mutation K391R did not result in enhanced adhesion to FN (Figure 6). Increased adhesion induced by CD4 stimulation and membrane-targeted Itk was inhibited by the anti-β1 integrin antibody AIIB2 (data not shown). These results suggest that Itk-dependent regulation of β1 integrin function requires Lck activation and membrane targeting of Itk.

Fig. 6. Membrane-targeted Itk requires an additional activation signal provided by CD4 co-receptor stimulation in order to activate β1 integrins. Jurkat cells were transiently transfected with the farnesylated GFP expression vectors encoding F-GFP, F-GFP–ΔPH Itk or F-GFP–ΔPH KN/Itk and analyzed for adhesion as in Figure 5 following no stimulation (UNSTIM.) or stimulation for 10 min at 37°C with PMA (PMA STIM.) or a CD4-specific antibody (CD4 STIM.). Adhesion was quantitated by flow cytometry and the results indicate the percentage adhesion of GFP-negative cells (open bars) and GFP-positive cells (closed bars) under each stimulation condition with each transfected population. Results shown are from one of three independent replicate experiments.
Increased β1 integrin function mediated by PI 3-K and Lck is dependent on Itk
Since our results suggest that PI 3-K-dependent membrane localization of Itk is critical to CD3-mediated activation of β1 integrins, we tested whether expression of a constitutively active catalytic subunit of PI 3-K (ACT.p110) would be sufficient to induce β1 integrin activation. Similar to the results obtained with F-GFP–ΔPH Itk, expression of active PI 3-K in the absence of additional signals was insufficient to enhance basal adhesion of Jurkat T cells to FN (Figure 7A). However, expression of active PI 3-K together with CD4 cross-linking led to increased adhesion similar to that observed with PMA stimulation (Figure 7A). Increased T-cell adhesion induced by active PI 3-K and CD4 stimulation was dependent on Itk kinase activity, since adhesion was inhibited by kinase-inactive GFP–Itk, but not by wild-type GFP–Itk (Figure 7A).


Fig. 7. Enhanced basal β1 integrin-mediated adhesion of human T cells induced by activation of PI 3-K and Lck can be inhibited by kinase-inactive Itk. (A) Jurkat T cells were transiently transfected with vectors encoding either GFP, GFP plus constitutively active PI 3-K (ACT.p110), GFP–Itk plus ACT.p110, or GFP–KN/Itk plus ACT.p110, and analyzed for adhesion as in Figure 5 following no stimulation (UNSTIM.) or stimulation for 10 min at 37°C with PMA (PMA STIM.) or the CD4-specific antibody OKT4 (CD4 STIM.). Adhesion was quantitated by flow cytometry and the results indicate the percentage adhesion of GFP-negative cells (open bars) and GFP-positive cells (closed bars) under each stimulation condition with each transfected population. Results shown are from one of three independent replicate experiments. (B) J.CaM1 cells and stable transfectants of J.CaM1 expressing Lck (J.CaM1/lck+) were transiently transfected with vectors encoding GFP plus constitutively active PI 3-K (ACT.p110), and analyzed for adhesion as in Figure 5 following no stimulation (UNSTIM.) or stimulation for 10 min at 37°C with PMA (PMA STIM.), the CD3-specific antibody OKT3 (CD3 STIM.) or the CD4-specific antibody OKT4 (CD4 STIM.). Adhesion was quantitated by flow cytometry and the results indicate the percentage adhesion of GFP-negative cells (open bars) and GFP-positive cells (closed bars) under each stimulation condition with each transfected population. Results shown are from one of three independent replicate experiments.
The effect of CD4 stimulation on T-cell adhesion in the presence of active PI 3-K requires Lck, as CD4 stimulation of Lck-deficient J.CaM1 cells expressing active PI 3-K did not result in enhanced adhesion to FN (Figure 7B). However, adhesion was enhanced following CD4 stimulation of stable Lck+ J.CaM1 transfectants (Denny et al., 2000) expressing active PI 3-K. In addition, CD3-induced adhesion was defective in J.CaM1 cells but not in Lck+ J.CaM1 transfectants (Figure 7B), illustrating a critical role for Lck in CD3-mediated increases in T-cell adhesion to FN. Flow cytometric analysis revealed comparable levels of expression of CD3, α4β1 integrin and α5β1 integrin on wild-type Jurkat T cells and J.CaM1 cells. However, CD4 expression was slightly lower on J.CaM1 cells (data not shown).
Kinase-inactive Itk inhibits CD3-induced β1 integrin activation in PTEN+ peripheral human T cells
Levels of PI(3,4,5)-P3 in the T-cell plasma membrane are regulated not only by PI 3-K, which produces PI(3,4,5)-P3 when active, but also by phosphatases that dephosphorylate PI(3,4,5)-P3. One such phosphatase is the dual specificity phosphatase PTEN (phosphatase and tensin homolog deleted on chromosome 10) (Di Cristofano and Pandolfi, 2000). Recent studies indicate that PTEN expression is impaired in Jurkat T cells (Shan et al., 2000; Wang et al., 2000), resulting in elevated basal levels of membrane-associated Itk in this cell line when compared with PTEN+ peripheral T cells (Shan et al., 2000). Thus, we also assessed the role of Itk in β1 integrin regulation in phytohemagglutinin (PHA)-stimulated human T-cell blasts, which we have previously shown exhibit CD3-inducible increases in adhesion to FN via β1 integrins (Epler et al., 2000). Western blotting analysis indicated that human T-cell blasts express PTEN (Figure 8A). Consistent with previous studies (Shan et al., 2000; Wang et al., 2000), minimal levels of PTEN were detected in Jurkat T-cell lysates (Figure 8A). Localization of Itk to DIGs upon CD3 stimulation of human T-cell blasts was also assessed (Figure 8B). In contrast to Jurkat T cells, there was less Itk found in DIGs isolated from unstimulated T-cell blasts (Figure 8B). However, similar to the results observed with Jurkat T cells, CD3 stimulation resulted in a wortmannin-sensitive increase in Itk localized to DIGs in human T-cell blasts (Figure 8B). In adhesion assays, basal adhesion of human T-cell blasts to FN was slightly lower than that observed with Jurkat T cells. However, expression of GFP–KN/Itk, but not GFP–Itk or GFP, also inhibited CD3-mediated enhancement of the adhesion of human T-cell blasts to FN (Figure 8C). Kinase-inactive Itk did not affect PMA-induced adhesion of human T-cell blasts to FN.
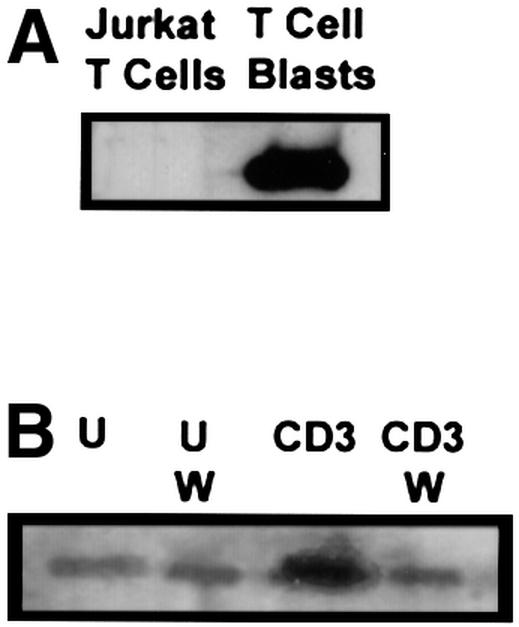

Fig. 8. Kinase-inactive Itk inhibits CD3-mediated activation of β1 integrins expressed on peripheral human T-cell blasts. PHA-stimulated human T-cell blasts were prepared as described in Materials and methods. (A) Whole-cell lysates from Jurkat T cells (3 × 106 cell equivalents) and human T-cell blasts (3 × 106 cell equivalents) were separated by SDS–PAGE, transferred to PVDF membranes and immunoblotted for PTEN with an anti-PTEN polyclonal antibody. (B) DIGs were prepared from unstimulated (U) and CD3-stimulated (CD3) human T-cell blasts (5 × 106 cell equivalents) in the presence or absence of 100 nM wortmannin (W) and immunoblotted for the presence of Itk as in Figure 1. (C) Adhesion of transiently transfected human T-cell blasts expressing either GFP, GFP–Itk or GFP–KN/Itk to FN following no stimulation (UNSTIM.) or after stimulation with PMA (PMA STIM.) or CD3 (CD3 STIM.) for 10 min at 37°C was assessed as in Figure 5. Adhesion was quantitated by flow cytometry and the results indicate the percentage adhesion of GFP-negative cells (open bars) and GFP-positive cells (closed bars) under each stimulation condition with each transfected population. Results shown are from one of three independent replicate experiments using human T-cell blasts isolated from different donors.
Itk regulates CD3-induced actin polymerization
Regulation of integrin function can be controlled by changes in the actin cytoskeleton. Since Tec family tyrosine kinases associate with proteins implicated in cytoskeletal reorganization (Bunnell et al., 1996), we explored a role for Itk in regulating the T-cell actin cytoskeleton. Jurkat T cells transiently expressing GFP or GFP–Itk fusion proteins were sorted, stimulated with anti-CD3 antibody and then stained with biotin-labeled phalloidin and streptavidin–phycoerythrin (PE). CD3 stimulation of Jurkat T cells expressing either GFP or GFP–Itk resulted in a 5- to 6-fold increase in actin polymerization (Figure 9). In contrast, CD3 stimulation of Jurkat T cells expressing GFP–KN/Itk or GFP–PH Itk resulted in only a 2.5-fold increase in actin polymerization. Thus, these results suggest that Itk plays an important role in regulating changes in the actin cytoskeleton upon CD3 stimulation that are critical to integrin function.

Fig. 9. CD3-induced actin polymerization is dependent on Itk. Jurkat T cells transiently transfected with vectors encoding either GFP, GFP–Itk (GFP–WT Itk), GFP–KN/Itk or GFP–PH Itk were sorted to isolate a homogeneous population of GFP+ T cells, and then left unstimulated (open curves) or CD3-stimulated for 10 min at 37°C (closed curves). Cells were stained with biotin-labeled phalloidin and PE-conjugated streptavidin, and analyzed by flow cytometry. Numbers in parentheses in each set of curves indicate the fold increase in mean fluorescence intensity of phalloidin staining upon CD3 stimulation.
Discussion
The results in this study demonstrate a novel function for the Tec family tyrosine kinase Itk in the regulation of β1 integrin-mediated adhesion by the CD3–TCR complex that is dependent on activation of both PI 3-K and the src kinase Lck. We propose that coordination of signaling between these kinases is critical for activation-dependent redistribution of Itk to an appropriate compartment in the T-cell membrane, where it becomes accessible to Lck, a kinase that regulates Itk tyrosine kinase activity (August et al., 1997; Heyeck et al., 1997). Previous studies have demonstrated a critical role for PI 3-K in the regulation of β1 integrin function by CD3–TCR (Woods et al., 2000) as well as several other cell surface receptors (Shimizu and Hunt, 1996; Zell et al., 1996; Chan et al., 1997; Kivens et al., 1998; Kinashi et al., 1999). Several lines of evidence argue that a prominent function of PI 3-K in integrin regulation is membrane recruitment of Itk mediated via binding of the PH domain of Itk to PI(3,4,5)-P3 produced upon PI 3-K activation by CD3–TCR: (i) CD3-mediated increases in membrane recruitment of Itk are blocked by PI 3-K inhibitors or by deletion of the PH domain of Itk; (ii) Itk binding to PI (3,4,5)-P3 is dependent on the Itk PH domain; (iii) the PH domain of Itk is sufficient to bind PI(3,4,5)-P3 and to inhibit CD3-induced increases in β1 integrin function; and (iv) constitutive membrane targeting of Itk with a farnesylation sequence can synergize with CD4 stimulation, which activates Lck, to enhance β1 integrin-mediated adhesion in the absence of CD3–TCR stimulation. Our results are consistent with previous studies in COS cells indicating a role for PI 3-K in membrane recruitment of Itk (August et al., 1997; Heyeck et al., 1997), although CD3-dependent membrane recruitment of Itk via PI 3-K in Jurkat T cells has not been consistently observed in earlier reports (Lu et al., 1998; Ching et al., 1999; Shan and Wange, 1999; Bunnell et al., 2000). The functional significance of the PH domain of Itk in CD3-dependent regulation of β1 integrin function further highlights the critical role that lipid binding to PH domains of Tec family tyrosine kinases plays in Tec family kinase function, as first vividly illustrated by the analysis of PH domain mutations in Btk that result in B-cell immunodeficiency (Sideras and Smith, 1995). Although the Tec family tyrosine kinase Etk is also expressed in Jurkat T cells (data not shown), expression of kinase-inactive Etk or the Etk PH domain did not inhibit CD3-induced increases in T-cell adhesion to FN. This suggests a specific role for Itk in regulating β1 integrin function in T cells.
The src family tyrosine kinase Lck is one of several molecules that are preferentially localized in DIGs (Xavier et al., 1998; Ilangumaran et al., 1999; Langlet et al., 2000), specialized regions of the plasma membrane that provide an important scaffold for the assembly of functional signaling complexes. CD3–TCR stimulation results in enhanced localization of several signaling molecules in DIGs, including ZAP-70 (Xavier et al., 1998; Salojin et al., 2000), LAT (Zhang et al., 1998; Lin et al., 1999), the p85 subunit of PI 3-K (Xavier et al., 1998) and now Itk. CD3-dependent recruitment of Itk to DIGs places Itk in proximity to Lck, which has been shown to enhance Itk tyrosine kinase activity via tyrosine phosphorylation of Itk (Gibson et al., 1996; August et al., 1997; Heyeck et al., 1997). Indeed, we observed that CD3 stimulation induced dramatic increases in tyrosine phosphorylation of DIG-localized Itk. In addition, expression of kinase-inactive Itk or the PH domain of Itk specifically inhibited CD3-induced tyrosine phosphorylation of endogenous Itk found in DIGs, as well as CD3-mediated increases in Itk found in DIGs. Thus, PI 3-K functions specifically to promote the co-localization of Itk with its upstream regulatory tyrosine kinase. Such redistribution of key signaling molecules into and out of DIGs following CD3–TCR stimulation represents a powerful mode of regulating signal transduction via careful assembly of kinases with molecules that regulate their enzymatic activity.
Although previous studies have suggested a role for src family kinases in regulating both PI 3-K and Itk in COS cells (August et al., 1997), our results suggest that Lck and PI 3-K play distinct and complementary roles in CD3–TCR regulation of β1 integrin-mediated adhesion. Stimulation of the CD4 co-receptor, which efficiently activates Lck (Turner et al., 1990; Baldari et al., 1995), by itself was insufficient to induce increases in β1 integrin functional activity. CD4 signaling resulted in increased β1 integrin functional activity only in the presence of constitutively active PI 3-K or membrane-targeted Itk. Analysis of Lck-deficient Jurkat T cells illustrated that the ability of CD4 signaling to enhance β1 integrin functional activity in conjunction with active PI 3-K was dependent on Lck expression. Thus, these results support the model that PI 3-K serves to play a critical role in targeting of Itk to DIGs, while Lck plays a role in regulating Itk once it has been recruited to DIGs.
The inability of CD3 stimulation to enhance β1 integrin-mediated adhesion of Lck-deficient J.CaM1 T cells indicates that Lck is required for β1 integrin activation by CD3–TCR. In addition to its proposed role in regulating Itk tyrosine kinase activity (August et al., 1997), Lck probably also plays a central role in regulating β1 integrin function via its central role in recruiting and regulating ZAP-70 tyrosine kinase activity (Iwashima et al., 1994; Chan et al., 1995). Recent studies using peripheral T cells and ZAP-70-deficient Jurkat T cells indicate that β1 integrin activation induced by CD3–TCR stimulation requires ZAP-70 tyrosine kinase activity (Epler et al., 2000). ZAP-70, like Lck, may participate in CD3–TCR-mediated activation of β1 integrins via regulation of Itk, since activation of Itk is dependent on ZAP-70 function in Jurkat T cells (Shan and Wange, 1999). The exact role of ZAP-70 in regulating Itk activity is currently unclear, although ZAP-70 cannot phosphorylate the Itk kinase domain directly (Shan and Wange, 1999). One possibility that must be considered is that ZAP-70 might regulate PI 3-K-dependent localization of Itk to DIGs, since the Syk tyrosine kinase regulates PI 3-K activity in B cells (Beitz et al., 1999; Pogue et al., 2000). However, the reported lack of effect of loss of ZAP-70 expression on CD3-dependent localization of Itk to DIGs (Shan and Wange, 1999) is inconsistent with this hypothesis. Unlike our results, this prior study did not observe significant relocalization of Itk to DIGs upon CD3 stimulation of wild-type Jurkat T cells. The reasons for this discrepancy are unclear, but may relate to differences in the cell lines employed, such as the use of specific Jurkat T-cell subclones or Jurkat T cells expressing SV40 large T antigen. Since CD3 stimulation also results in the association of Itk with the ZAP-70 substrate LAT (linker for activation of T cells) (Ching et al., 2000), ZAP-70 might serve to participate in Itk-dependent regulation of β1 integrin function via effects on Itk–LAT association.
The membrane localization of the Tec family tyrosine kinase Btk is also regulated by PI 3-K (Salim et al., 1996; Li et al., 1997). Although Btk is not found at appreciable levels in resting B cells, there is basal membrane and DIG localization of Itk in unstimulated Jurkat T cells. Recent studies have demonstrated that this basal membrane localization of Itk is due to lack of expression in Jurkat T cells of the PTEN phosphatase, which dephosphorylates the PI 3-K lipid product PI(3,4,5)-P3 (Shan et al., 2000). However, our analysis of PTEN+ human T-cell blasts and PTEN– Jurkat T cells indicated that CD3 stimulation of both cell types induced wortmannin-sensitive increases in the localization of Itk to DIGs. Furthermore, Itk found in DIGs in unstimulated Jurkat T cells was not appreciably tyrosine phosphorylated. In addition, we observed that expression of kinase-inactive Itk was equally effective in inhibiting CD3-dependent increases in β1 integrin function in PTEN– Jurkat T cells and in PTEN+ human peripheral T-cell blasts. These results suggest that PTEN levels may regulate basal levels of Itk at the T-cell membrane, but that CD3 stimulation can still enhance Itk membrane localization and Itk kinase activity. In prior studies, we have noted that unstimulated Jurkat T cells generally exhibit higher levels of basal β1 integrin- mediated adhesion than unstimulated peripheral T cells (Mobley et al., 1994). It is possible that PTEN might regulate basal β1 integrin functional activity by regulating the constitutive amount of D-3-phosphorylated lipids found at the membrane of unstimulated T cells. We have been unable to test directly the function of PTEN in β1 integrin function, since expression of PTEN in our Jurkat T-cell line results in death of the transient transfectants (data not shown), similar to what has been observed by other groups (Wang et al., 2000).
Changes in β1 integrin conformation that result in enhanced ligand binding affinity have been proposed as one mechanism by which activation increases β1 integrin-mediated adhesion (Bazzoni and Hemler, 1998). However, CD3 stimulation of Jurkat T cells does not lead to enhanced binding of soluble FN or increased expression of antibody epitopes used as markers for β1 integrins with increased affinity (Woods et al., 2000). Alternatively, changes in the cytoskeleton have been proposed to regulate integrin microclustering, thereby altering the avidity of integrin-mediated adhesion (Kucik et al., 1996; van Kooyk et al., 1999; Calderwood et al., 2000). The ability of kinase-inactive Itk and the Itk PH domain to inhibit CD3-induced increases in actin polymerization suggests that Itk regulates β1 integrin functional activity via regulation of the T-cell actin cytoskeleton. The association of Itk with cytoskeletal regulatory proteins, such as WASP (Bunnell et al., 1996), and a proposed role for Btk in regulating Rho-family GTPase activity in B cells, are consistent with this hypothesis (Nore et al., 2000). Since stimulation of the CD28 co-receptor also activates β1 integrin function (Zell et al., 1996) as well as Itk tyrosine kinase activity (August et al., 1994), Itk may play a similar role in regulating β1 integrin function by other cell surface receptors in addition to the T-cell receptor.
In summary, we have defined a novel function for the Tec family tyrosine kinase Itk in regulating β1 integrin functional activity induced by CD3–TCR stimulation. Itk-dependent regulation of β1 integrin functional activity involves PI 3-K-dependent localization of Itk to DIGs and Lck-dependent regulation of Itk kinase activity. It will be important in future studies to determine how other signaling proteins found in DIGs might participate in the regulation of β1 integrin function, how Itk impacts on T-cell cytoskeletal reorganization, and whether other Tec family tyrosine kinases serve a similar regulatory function.
Materials and methods
Antibodies and reagents
The CD3-specific monoclonal antibody (mAb) OKT3 and the CD4-specific mAb OKT4 were purchased from the American Type Culture Collection (ATCC; Manassas, VA). The anti-Itk, anti-Lck, anti-Erk1 and anti-CD45 antibodies were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). The anti-hemagglutinin (HA) antibody 3F10 was purchased from Boehringer-Mannheim (Indianapolis, IN) and the anti-HA mAb 16B12 was purchased from BabCo (Berkeley, CA). The anti-PTEN polyclonal antibody was purchased from Upstate Biotechnology (Lake Placid, NY). The inhibitory anti-β1 integrin mAb AIIB2 was obtained from the Developmental Studies Hybridoma Bank (Iowa City, IA). FN was provided by Dr J.McCarthy (University of Minnesota, Minneapolis, MN). Horseradish peroxidase (HRP)-, FITC- and biotin-conjugated forms of cholera toxin B subunit were purchased from Sigma Chemical Co. (St Louis, MO). Stock solutions of wortmannin (Sigma), LY294,002 (Alexis Corporation, San Diego, CA) and PMA (LC Services Corp., Woburn, MA) were dissolved in dimethyl sulfoxide (DMSO) and stored at –70°C.
Cell culture and stimulation conditions
The JE64-6A Jurkat T-cell line (Mobley et al., 1994) is a subclone of the Jurkat E6-1 cell line available from the ATCC (Manassas, VA). The Lck-deficient J.CaM1 cell line was also obtained from the ATCC. Both cell lines were maintained in RPMI 1640 medium supplemented with 10% fetal calf serum (FCS) (Atlanta Biologicals, Norcross, GA), l-glutamine and penicillin/streptomycin. The Lck+ stable transfectant of J.CaM1 was kindly provided by Dr David Straus (University of Chicago, Chicago, IL) and was cultured as previously described (Denny et al., 2000). Human peripheral blood T-cell blasts were prepared by stimulating human peripheral blood lymphocytes with PHA for 4 days as previously described (Epler et al., 2000). For CD3–TCR stimulation, T cells were incubated on ice with the anti-CD3 mAb OKT3 at 1 µg/ml per 106 cells. The cells were washed 3× in ice-cold phosphate-buffered saline (PBS), and incubated for 30 min on ice with goat anti-mouse IgG at 0.5 µg/ml per 106 cells. For the confocal microscopy experiments in Figure 4B, rat anti-mouse IgG was used at 0.5 µg/ml per 106 cells. The cells were then rapidly warmed to 37°C for the indicated period of time.
Cellular fractionation
Cellular fractionation was adapted from previously described methods for isolating cytosolic, membrane and DIG fractions (Xavier et al., 1998). Briefly, 50 × 106 Jurkat cells or 100 × 106 PHA T-cell blasts were resuspended in 1 ml of hypotonic buffer [10 mM Tris pH 8.0, 1 mM MgCl2, 1 mM NaOVO4, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 µg/ml aprotinin, 1 µg/ml leupeptin] and sonicated for 1 min. Thirty microliters of 5 M NaCl were added and the lysate was spun down at 200 g for 10 min. The supernatant containing cytosolic and membrane proteins was collected and 3 ml of isotonic buffer (0.6 M NaCl, 1 mM NaOVO4, 1 mM PMSF, 1 µg/ml aprotinin, 1 µg/ml leupeptin) were added. The lysate was then spun at 100 000 g for 45 min. The supernatant containing cytosolic proteins was collected and 0.3 ml of cytosolic adjusting buffer (1% Triton, 1% SDS, 1% sodium deoxycholate, 1 mM NaOVO4, 1 mM PMSF, 1 µg/ml aprotinin, 1 µg/ml leupeptin) were added. The pellet containing membrane and DIG proteins was resuspended in 1 ml of MBS (25 mM MES, 150 mM NaCl pH 6.5, 2 mM EDTA, 0.5% Triton X-100, 1 mM NaOVO4, 1 mM PMSF, 1 µg/ml aprotinin, 1 µg/ml leupeptin) and sonicated for 1 min. The lysates were then gently mixed with 1 ml of 80% sucrose in MBS, which was then overlayed with 35% sucrose in MBS and 5% sucrose in MBS. The sucrose gradient was spun at 200 000 g for 16 h at 4°C. DIG-associated proteins were collected from the 5/35% interphase. Membrane proteins were collected from the bottom 1 ml of the sucrose gradient. For anti-PTEN immunoblotting, Jurkat T cells and human T-cell blasts were lysed in lysis buffer (1% Triton X-100, 1% sodium deoxycholate, 158 mM NaCl, 10 mM Tris–HCl, 5 mM EDTA, 2 mM sodium vanadate, 20 µg/ml leupeptin, 20 µg/ml aprotinin, 2 mM PMSF) as previously described (Epler et al., 2000).
Western blotting
Western blotting was performed as previously described (Hunter and Shimizu, 1997). The anti-Itk, anti-Lck, anti-ERK1, anti-CD45 and anti-PTEN antibodies were used at a 1:500 dilution in PBS/5% milk. Cholera toxin B subunit–HRP was used at a 1:200 dilution in PBS/5% milk. Secondary reagents were HRP-conjugated donkey anti-goat IgG (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA), donkey anti-rabbit IgG (Amersham Pharmacia Biotech, Inc., Piscataway, NJ), goat anti-mouse IgG (Caltag Inc., South San Francisco, CA) and rat anti-mouse IgG (Jackson). Membranes were developed using ECL (Pierce, Rockford, IL).
Plasmid constructs
The HA-tagged wild-type Itk and ΔPH Itk cDNA clones were inserted into the XhoI–EcoRI sites of the pEGFP-C3 vector (Clontech, Palo Alto, CA) and into the BamHI–EcoRI sites of the pGEX-4T-1 GST fusion protein vector (Amersham Pharmacia Biotech). The kinase-inactive (K391R) Itk construct was first subcloned into the pMEXNeo vector, which added an in-frame HA tag at the 5′ end, and then directionally cloned into the GFP and GST vectors as described above. Each plasmid was sequenced to confirm that the inserts were inserted into the appropriate reading frame. The stop codon of the pEGFP-F vector coding for farnesylated GFP (Clontech) was mutated to an alanine and then the HA-ΔPH Itk cDNA was inserted in-frame at the 3′ end of the eGFP coding sequence. The kinase-inactive K391R mutation was created using standard site-directed mutagenesis techniques (Stratagene). The GFP–Itk fusion protein constructs used in this study are shown in Figure 3. The Etk cDNA clones (Qiu et al., 1998) were subcloned into the KpnI–ApaI site of the pEGFP-C1 fusion protein vector (Clontech). The constitutively active PI 3-K p110 cDNA (ACT.p110) was purchased from Upstate Biotechnology (Lake Placid, NY).
Transient transfections
Transient transfections were performed as previously described using a BTX square wave electroporator (BTX, San Diego, CA) set at 240 V with a pulse length of 25 ms (Romzek et al., 1998). After electroporation, cells were incubated for 10 min at room temperature before resuspension at 1 × 106 cells/ml in RPMI 1640 medium supplemented with 10% FCS, l-glutamine and penicillin/streptomycin. Cells were harvested after 14–18 h for use in western blotting or in adhesion assays.
Itk and GFP fusion protein immunoblotting
Cells were transiently transfected as described above. The total number of GFP+ cells was determined by flow cytometry for western blotting with anti-HA. In the anti-phosphotyrosine experiment shown in Figure 4D, transfected cells were sorted by flow cytometry to obtain a homogeneous population of GFP+ cells using a Becton Dickinson FACSVantage. Equal numbers of GFP+ cells from each transfectant were either unstimulated or CD3-stimulated as described above. Cytosolic fractions, membrane fractions and DIGs were prepared as described above. Immuno precipitations were performed using anti-HA (3F10)-coated protein A–Sepharose beads or anti-Itk-coated protein G beads as previously described (Hunter and Shimizu, 1997). The samples were separated on a 7.5% SDS–PAGE mini-gel, transferred to PVDF, and immunoblotted with anti-HA mAb (16B12) or anti-pTyr (4G10) followed by HRP-conjugated goat anti-mouse IgG. Detection was by ECL.
Confocal microscopy
Jurkat T cells, either untransfected or transiently transfected with GFP–Itk constructs, were left unstimulated or CD3 stimulated, and then fixed in 4% paraformaldehyde for 30 min at room temperature. The GFP–Itk-transfected cells were added to poly-l-lysine-coated slides and allowed to settle. Untransfected cells were washed in 1× Perm Buffer [PBS containing 0.5% saponin (Sigma), 2% FCS and 0.2% sodium azide] followed by a 1× wash in Superperm Buffer (3 parts Perm Buffer + 1 part FCS). The cells were incubated in Perm buffer containing anti-Itk at 2 µg/106 cells for 30 min at room temperature. Following a 2× wash in Perm Buffer, the cells were incubated for an additional 30 min at room temperature with biotin-labeled rabbit anti-goat IgG (Southern Biotechnology, Birmingham, AL) in Perm Buffer. The cells were subsequently stained for 30 min with Streptavidin–APC (BD PharMingen, San Diego, CA) in Perm Buffer. The cells were then added to poly-l-lysine-coated slides and allowed to settle. After the untransfected cells had settled, the slides were blocked with 2% FCS in PBS, then stained with FITC-conjugated cholera toxin B subunit at 8 µg/ml for 30 min. Slides were analyzed on a Bio-Rad MRC 1024 confocal microscope.
Phospholipid extraction
Jurkat cells were stimulated in the presence or absence of 50 µM LY294,002 with the anti-CD3 mAb OKT3 and goat anti-mouse IgG for 10 min at 37°C as described above in the presence of 100 µM ATP, 10 µCi 32P-ATP, 12% TransPort reagent (Life Technologies, Gaithersburg, MD). The cells were first incubated on ice for 60 min then rapidly warmed to 37°C for 5 min. Lipids were extracted, spotted onto TLC plates with the lipid standards PI, PI(4)-P, PI(4,5)-P2 (Sigma), PI(3)-P, PI(3,4)-P2 and PI(3,4,5)-P3 (Matreya, Inc., Street Pleasant Gap, PA) and developed as previously described (L.M.O’Rourke et al., 1998). The 32P incorporated into the phosphatidylinositols was quantitated on a phosphoimager. The percentage of individual phosphoinositides was determined by dividing the volume (intensity as determined by the phosphoimager) of each individual spot by the total volume (intensity of all phospholipid spots as determined by the phosphoimager) and multiplying by 100. Extracted lipids and lipid standards were developed in an iodine tank and compared.
FAT western blotting
Binding of GST–Itk fusion proteins to purified phospholipids was performed as previously described (Stevenson et al., 1998). Briefly, PI(3)-P, PI(4)-P, PI(4,5)-P2, PI(3,4)-P2 and PI(3,4,5)-P3 were spotted onto nitrocellulose membranes and dried. The membrane was blocked with 3% fatty acid-free bovine serum albumin, then GST, GST–wt Itk, GST–PH Itk and GST–ΔPH Itk fusion proteins were incubated with the membrane. Binding of the GST fusion proteins was detected by chemiluminescence following incubation with goat anti-GST mAb conjugated to HRP. Lipids were developed in an iodine tank and compared to ensure equal loading.
Adhesion assays
The adhesion of transiently transfected T cells to FN (0.3 µg/well) was analyzed as previously described (Kivens and Shimizu, 1998; Kivens et al., 1998). For PMA stimulation, T cells were added to wells containing 10 ng/ml PMA. For CD3 and CD4 stimulation, cells pre-coated with OKT3 mAb (anti-CD3) or OKT4 mAb (anti-CD4) were added to wells containing 1 µg/ml goat anti-mouse IgG. Plates were then rapidly warmed to 37°C for 10 min and washed to remove non-adherent cells. An aliquot of each cell sample representing the same volume used in each well for the adhesion assay was prepared for flow cytometric analysis for verification of the cell numbers added per well. Adherent cells were removed with PBS/0.1% EDTA and collected. Cells from six replicate wells were pooled into a tube, pelleted and resuspended in 200 µl of PBS/5% FCS supplemented with 50 µl of PKH26 reference microbeads (Sigma) and 25 µl of propidium iodide (Sigma). Each sample was then analyzed on a Becton-Dickinson FACScan as previously described (Kivens and Shimizu, 1998; Kivens et al., 1998). For each sample analyzed, the total number of reference microbeads acquired was divided by the bead density to obtain the total volume of sample acquired. Post-acquisition gating was used to define GFP-negative and -positive subpopulations. Within these two subpopulations, the total number of T cells in each sample was then determined by the following equation: [(T cells acquired)/(ml of sample acquired)](0.275 ml). Initial numbers of T cells added to each well at the start of the adhesion assay were calculated by the same procedure using the pre-adherent cell samples.
Actin polymerization
Jurkat T cells were transfected with GFP–Itk constructs and sorted to 95% purity as described above. Sorted cells were left unstimulated or stimulated by mAb cross-linking of CD3, then fixed in 4% paraformaldehyde containing 1 mg/ml lysophosphatidylcholine (Sigma) for 15 min at room temperature. Following fixation, biotin–XX– phalloidin (Molecular Probes, Eugene, OR) was added at 1 µM and the cells were incubated for 15 min at 37°C. Cells were then stained with streptavidin–PE (Southern Biotechnology Associates) at 1 µg/106 cells for 20 min at room temperature. Cells were analyzed on a Becton-Dickinson FACScan.
Acknowledgments
Acknowledgements
We thank Dr D.Straus for providing critical reagents and helpful comments, and J.Peller of the University of Minnesota Cancer Center Flow Cytometry Core Facility for technical assistance with cell sorting. This work was supported by NIH grant AI31126 (Y.S.) and by NIH postdoctoral fellowship grants AI09993 (M.L.W.) and AR09438 (W.J.K.). Y.S. is the Harry Kay Chair of Cancer Research at the University of Minnesota.
References
- August A., Gibson,S., Kawakami,Y., Kawakami,T., Mills,G.B. and Dupont,B. (1994) CD28 is associated with and induces the immediate tyrosine phosphorylation and activation of the Tec family kinase ITK/EMT in the human Jurkat leukemic T-cell line. Proc. Natl Acad. Sci. USA, 91, 9347–9351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- August A., Sadra,A., Dupont,B. and Hanafusa,H. (1997) Src-induced activation of inducible T cell kinase (ITK) requires phosphatidylinositol 3-kinase activity and the pleckstrin homology domain of inducible T cell kinase. Proc. Natl Acad. Sci. USA, 94, 11227–11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldari C.T., Milia,E., Di Somma,M.M., Baldoni,F., Valitutti,S. and Telford,J.L. (1995) Distinct signaling properties identify functionally different CD4 epitopes. Eur. J. Immunol., 25, 1843–1850. [DOI] [PubMed] [Google Scholar]
- Bazzoni G. and Hemler,M.E. (1998) Are changes in integrin affinity and conformation overemphasized. Trends Biochem. Sci., 23, 30–34. [DOI] [PubMed] [Google Scholar]
- Beitz L.O., Fruman,D.A., Kurosaki,T., Cantley,L.C. and Scharenberg, A.M. (1999) SYK is upstream of phosphoinositide 3-kinase in B cell receptor signaling. J. Biol. Chem., 274, 32662–32666. [DOI] [PubMed] [Google Scholar]
- Bunnell S.C., Henry,P.A., Kolluri,R., Kirchhausen,T., Rickles,R.J. and Berg,L.J. (1996) Identification of Itk/Tsk Src homology 3 domain ligands. J. Biol. Chem., 271, 25646–25656. [DOI] [PubMed] [Google Scholar]
- Bunnell S.C., Diehn,M., Yaffe,M.B., Findell,P.R., Cantley,L.C. and Berg,L.J. (2000) Biochemical interactions integrating Itk with the T cell receptor-initiated signaling cascade. J. Biol. Chem., 275, 2219–2230. [DOI] [PubMed] [Google Scholar]
- Calderwood D.A., Shattil,S.J. and Ginsberg,M.H. (2000) Integrins and actin filaments: reciprocal regulation of cell adhesion and signaling. J. Biol. Chem., 275, 22607–22610. [DOI] [PubMed] [Google Scholar]
- Caron E., Self,A.J. and Hall,A. (2000) The GTPase Rap1 controls functional activation of macrophage integrin αM β2 by LPS and other inflammatory mediators. Curr. Biol., 10, 974–978. [DOI] [PubMed] [Google Scholar]
- Chan A.C., Dalton,M., Johnson,R., Kong,G., Wang,T., Thoma,R. and Kurosaki,T. (1995) Activation of ZAP-70 kinase activity by phosphorylation of tyrosine 493 is required for lymphocyte antigen receptor function. EMBO J., 14, 2499–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan A.S.H., Mobley,J.L., Fields,G.B. and Shimizu,Y. (1997) CD7-mediated regulation of integrin adhesiveness on human T cells involves tyrosine phosphorylation-dependent activation of phos phatidylinositol 3-kinase. J. Immunol., 159, 934–942. [PubMed] [Google Scholar]
- Ching K.A., Kawakami,Y., Kawakami,T. and Tsoukas,C.D. (1999) Emt/Itk associates with activated TCR complexes: role of the pleckstrin homology domain. J. Immunol., 163, 6006–6013. [PubMed] [Google Scholar]
- Ching K.A., Grasis,J.A., Tailor,P., Kawakami,Y., Kawakami,T. and Tsoukas,C.D. (2000) TCR/CD3-induced activation and binding of Emt/Itk to linker of activated T cell complexes: requirement for the Src homology 2 domain. J. Immunol., 165, 256–262. [DOI] [PubMed] [Google Scholar]
- Denny M.F., Patai,B. and Straus,D.B. (2000) Differential T-cell antigen receptor signaling mediated by the Src family kinases Lck and Fyn. Mol. Cell. Biol., 20, 1426–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cristofano A. and Pandolfi,P.P. (2000) The multiple roles of PTEN in tumor suppression. Cell, 100, 387–390. [DOI] [PubMed] [Google Scholar]
- Dustin M.L. and Springer,T.A. (1989) T-cell receptor cross-linking transiently stimulates adhesiveness through LFA-1. Nature, 341, 619–624. [DOI] [PubMed] [Google Scholar]
- Epler J.A., Liu,R., Chung,H., Ottoson,N.C. and Shimizu,Y. (2000) Regulation of β1 integrin-mediated adhesion by T cell receptor signaling involves ZAP-70 but differs from signaling events that regulate transcriptional activity. J. Immunol., 165, 4941–4949. [DOI] [PubMed] [Google Scholar]
- Gibson S., August,A., Kawakami,Y., Kawakami,T., Dupont,B. and Mills,G.B. (1996) The EMT/ITK/TSK (EMT) tyrosine kinase is activated during TCR signaling—LCK is required for optimal activation of EMT. J. Immunol., 156, 2716–2722. [PubMed] [Google Scholar]
- Heyeck S.D., Wilcox,H.M., Bunnell,S.C. and Berg,L.J. (1997) Lck phosphorylates the activation loop tyrosine of the Itk kinase domain and activates Itk kinase activity. J. Biol. Chem., 272, 25401–25408. [DOI] [PubMed] [Google Scholar]
- Hughes P.E., Renshaw,M.W., Pfaff,M., Forsyth,J., Keivens,V.M., Schwartz,M.A. and Ginsberg,M.H. (1997) Suppression of integrin activation: a novel function of a Ras/Raf-initiated MAP kinase pathway. Cell, 88, 521–530. [DOI] [PubMed] [Google Scholar]
- Hunter A.J. and Shimizu,Y. (1997) α4β1 integrin-mediated tyrosine phosphorylation in human T cells: characterization of Crk- and Fyn-associated substrates (pp105, pp115 and human enhancer of filamentation-1) and integrin-dependent activation of p59fyn. J. Immunol., 159, 4806–4814. [PubMed] [Google Scholar]
- Ilangumaran S., Arni,S., Van Echten-Deckert,G., Borisch,B. and Hoessli,D.C. (1999) Microdomain-dependent regulation of Lck and Fyn protein-tyrosine kinases in T lymphocyte plasma membranes. Mol. Biol. Cell, 10, 891–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwashima M., Irving,B.A., Van Oers,N.S.C., Chan,A.C. and Weiss,A. (1994) Sequential interactions of the TCR with two distinct cytoplasmic tyrosine kinases. Science, 263, 1136–1139. [DOI] [PubMed] [Google Scholar]
- Janes P.W., Ley,S.C. and Magee,A.I. (1999) Aggregation of lipid rafts accompanies signaling via the T cell receptor. J. Cell Biol., 147, 447–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane L.P., Lin,J. and Weiss,A. (2000) Signal transduction by the TCR for antigen. Curr. Opin. Immunol., 12, 242–249. [DOI] [PubMed] [Google Scholar]
- Katagiri K., Hattori,M., Minato,N., Irie,S., Takatsu,K. and Kinashi,T. (2000) Rap1 is a potent activation signal for leukocyte function-associated antigen 1 distinct from protein kinase C and phosphatidylinositol-3-OH kinase. Mol. Cell. Biol., 20, 1956–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinashi T., Asaoka,T., Setoguchi,R. and Takatsu,K. (1999) Affinity modulation of very late antigen-5 through phosphatidylinositol 3-kinase in mast cells. J. Immunol., 162, 2850–2857. [PubMed] [Google Scholar]
- Kivens W.J. and Shimizu,Y. (1998) Tracking integrin-mediated adhesion using green fluorescent protein and flow cytometry. In Guan,J.L. (ed.), Signaling Through Cell Adhesion Molecules. CRC Press, Boca Raton, FL, pp. 219–234.
- Kivens W.J., Hunt,S.W.,III, Mobley,J.L., Zell,T., Dell,C.L., Bierer,B.E. and Shimizu,Y. (1998) Identification of a proline-rich sequence in the CD2 cytoplasmic domain critical for regulation of integrin-mediated adhesion and activation of phosphoinositide 3-kinase. Mol. Cell. Biol., 18, 5291–5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klarlund J.K., Guilherme,A., Holik,J.J., Virbasius,J.V., Chawla,A. and Czech,M.P. (1997) Signaling by phosphoinositide-3,4,5-trisphosphate through proteins containing pleckstrin and Sec7 homology domains. Science, 275, 1927–1930. [DOI] [PubMed] [Google Scholar]
- Kolanus W., Nagel,W., Schiller,B., Zeitlmann,L., Godar,S., Stockinger,H. and Seed,B. (1996) αLβ2 integrin/LFA-1 binding to ICAM-1 induced by cytohesin-1, a cytoplasmic regulatory molecule. Cell, 86, 233–242. [DOI] [PubMed] [Google Scholar]
- Kucik D.F., Dustin,M.L., Miller,J.M. and Brown,E.J. (1996) Adhesion-activating phorbol ester increases the mobility of leukocyte integrin LFA-1 in cultured lymphocytes. J. Clin. Invest., 97, 2139–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlet C., Bernard,A.-M., Drevot,P. and He,H.-T. (2000) Membrane rafts and signaling by the multichain immune recognition receptors. Curr. Opin. Immunol., 12, 250–255. [DOI] [PubMed] [Google Scholar]
- Lemmon M.A., Falasca,M., Ferguson,K.M. and Schlessinger,J. (1997) Regulatory recruitment of signalling molecules to the cell membrane by pleckstrin-homology domains. Trends Cell Biol., 7, 237–242. [DOI] [PubMed] [Google Scholar]
- Li Z., Wahl,M.I., Eguinoa,A., Stephens,L.R., Hawkins,P.T. and Witte,O.N. (1997) Phosphatidylinositol 3-kinase-γ activates Bruton’s tyrosine kinase in concert with Src family kinases. Proc. Natl Acad. Sci. USA, 94, 13820–13825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao X.C. and Littman,D.R. (1995) Altered T cell receptor signaling and disrupted T cell development in mice lacking Itk. Immunity, 3, 757–769. [DOI] [PubMed] [Google Scholar]
- Lin J., Weiss,A. and Finco,T.S. (1999) Localization of LAT in glycolipid-enriched microdomains is required for T cell activation. J. Biol. Chem., 274, 28861–28864. [DOI] [PubMed] [Google Scholar]
- Liu K.Q., Bunnell,S.C., Gurniak,C.B. and Berg,L.J. (1998) T cell receptor-initiated calcium release is uncoupled from capacitative calcium entry in Itk-deficient T cells. J. Exp. Med., 187, 1721–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.L., Cuevas,B., Gibson,S., Khan,H., Lapushin,R., Imboden,J. and Mills,G.B. (1998) Phosphatidylinositol 3-kinase is required for CD28 but not CD3 regulation of the TEC family tyrosine kinase EMT/ITK/TSK: functional and physical interaction of EMT with phosphatidylinositol 3-kinase. J. Immunol., 161, 5404–5412. [PubMed] [Google Scholar]
- Mobley J.L., Ennis,E. and Shimizu,Y. (1994) Differential activation-dependent regulation of integrin function in cultured human T-leukemic cell lines. Blood, 83, 1039–1050. [PubMed] [Google Scholar]
- Moran M. and Miceli,M.C. (1998) Engagement of GPI-linked CD48 contributes to TCR signals and cytoskeletal reorganization: a role for lipid rafts in T cell activation. Immunity, 9, 787–796. [DOI] [PubMed] [Google Scholar]
- Nagel W., Zeitlmann,L., Schilcher,P., Geiger,C., Kolanus,J. and Kolanus,W. (1998) Phosphoinositide 3-OH kinase activates the β2 integrin adhesion pathway and induces membrane recruitment of cytohesin-1. J. Biol. Chem., 273, 14853–14861. [DOI] [PubMed] [Google Scholar]
- Nore B.F. et al. (2000) Redistribution of Bruton’s tyrosine kinase by activation of phosphatidylinositol 3-kinase and Rho-family GTPases. Eur. J. Immunol., 30, 145–154. [DOI] [PubMed] [Google Scholar]
- O’Rourke A.M., Shao,H. and Kaye,J. (1998) Cutting edge: a role for p21ras/MAP kinase in TCR-mediated activation of LFA-1. J. Immunol., 161, 5800–5803. [PubMed] [Google Scholar]
- O’Rourke L.M., Tooze,R., Turner,M., Sandoval,D.M., Carter,R.H., Tybulewicz,V.L. and Fearon,D.T. (1998) CD19 as a membrane-anchored adaptor protein of B lymphocytes: costimulation of lipid and protein kinases by recruitment of Vav. Immunity, 8, 635–645. [DOI] [PubMed] [Google Scholar]
- Perez-Villar J.J. and Kanner,S.B. (1999) Regulated association between the tyrosine kinase Emt/Itk/Tsk and phospholipase-Cγ1 in human T lymphocytes. J. Immunol., 163, 6435–6441. [PubMed] [Google Scholar]
- Pogue S.L., Kurosaki,T., Bolen,J. and Herbst,R. (2000) B cell antigen receptor-induced activation of Akt promotes B cell survival and is dependent on Syk kinase. J. Immunol., 165, 1300–1306. [DOI] [PubMed] [Google Scholar]
- Qiu Y., Robinson,D., Pretlow,T.G. and Kung,H.J. (1998) Etk/Bmx, a tyrosine kinase with a pleckstrin-homology domain, is an effector of phosphatidylinositol 3′-kinase and is involved in interleukin 6-induced neuroendocrine differentiation of prostate cancer cells. Proc. Natl Acad. Sci. USA, 95, 3644–3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reedquist K.A., Ross,E., Koop,E.A., Wolthuis,R.M., Zwartkruis,F.J., van Kooyk,Y., Salmon,M., Buckley,C.D. and Bos,J.L. (2000) The small GTPase, Rap1, mediates CD31-induced integrin adhesion. J. Cell Biol., 148, 1151–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romzek N.C., Harris,E.S., Dell,C.L., Skronek,J., Hasse,E., Reynolds,P.J., Hunt,S.W.,III and Shimizu,Y. (1998) Use of a β1 integrin-deficient human T cell to identify β1 integrin cytoplasmic domain sequences critical for integrin function. Mol. Biol. Cell, 9, 2715–2727. [PMC free article] [PubMed] [Google Scholar]
- Salim K. et al. (1996) Distinct specificity in the recognition of phosphoinositides by the pleckstrin homology domains of dynamin and Bruton’s tyrosine kinase. EMBO J., 15, 6241–6250. [PMC free article] [PubMed] [Google Scholar]
- Salojin K.V., Zhang,J., Meagher,C. and Delovitch,T.L. (2000) ZAP-70 is essential for the T cell antigen receptor-induced plasma membrane targeting of SOS and Vav in T cells. J. Biol. Chem., 275, 5966–5975. [DOI] [PubMed] [Google Scholar]
- Schaeffer E.M. and Schwartzberg,P.L. (2000) Tec family kinases in lymphocyte signaling and function. Curr. Opin. Immunol., 12, 282–288. [DOI] [PubMed] [Google Scholar]
- Schaeffer E.M. et al. (1999) Requirement for Tec kinases Rlk and Itk in T cell receptor signaling and immunity. Science, 284, 638–641. [DOI] [PubMed] [Google Scholar]
- Shan X.C. and Wange,R.L. (1999) Itk/Emt/Tsk activation in response to CD3 cross-linking in Jurkat T cells requires ZAP-70 and Lat and is independent of membrane recruitment. J. Biol. Chem., 274, 29323–29330. [DOI] [PubMed] [Google Scholar]
- Shan X.C., Czar,M.J., Bunnell,S.C., Liu,P.H., Liu,Y.S., Schwartzberg, P.L. and Wange,R.L. (2000) Deficiency of PTEN in Jurkat T cells causes constitutive localization of Itk to the plasma membrane and hyperresponsiveness to CD3 stimulation. Mol. Cell. Biol., 20, 6945–6957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu Y. and Hunt,S.W.,III (1996) Regulating integrin-mediated adhesion: one more function for PI 3-kinase? Immunol. Today, 17, 565–573. [DOI] [PubMed] [Google Scholar]
- Shimizu Y., van Seventer,G.A., Horgan,K.J. and Shaw,S. (1990) Regulated expression and binding of three VLA (β1) integrin receptors on T cells. Nature, 345, 250–253. [DOI] [PubMed] [Google Scholar]
- Shimizu Y., Rose,D.M. and Ginsberg,M.H. (1999) Integrins and the immune response. Adv. Immunol., 72, 325–380. [DOI] [PubMed] [Google Scholar]
- Sideras P. and Smith,C.I. (1995) Molecular and cellular aspects of X-linked agammaglobulinemia. Adv. Immunol., 59, 135–223. [DOI] [PubMed] [Google Scholar]
- Stevenson J.M., Perera,I.Y. and Boss,W.F. (1998) A phosphatidyl inositol 4-kinase pleckstrin homology domain that binds phosphatidylinositol 4-monophosphate. J. Biol. Chem., 273, 22761–22767. [DOI] [PubMed] [Google Scholar]
- Stewart M.P., Cabañas,C. and Hogg,N. (1996) T cell adhesion to intercellular adhesion molecule-1 (ICAM-1) is controlled by cell spreading and the activation of integrin LFA-1. J. Immunol., 156, 1810–1817. [PubMed] [Google Scholar]
- Turner J.M., Brodsky,M.H., Irving,B.A., Levin,S.D., Perlmutter,R.M. and Littman,D.R. (1990) Interaction of the unique N-terminal region of tyrosine kinase p56lck with cytoplasmic domains of CD4 and CD8 is mediated by cysteine motifs. Cell, 60, 755–765. [DOI] [PubMed] [Google Scholar]
- van Kooyk Y., van de Wiel-van Kemenade,P., Weder,P., Kuijpers,T.W. and Figdor,C.G. (1989) Enhancement of LFA-1-mediated cell adhesion by triggering through CD2 or CD3 on T lymphocytes. Nature, 342, 811–813. [DOI] [PubMed] [Google Scholar]
- van Kooyk Y., Van Vliet,S.J. and Figdor,C.G. (1999) The actin cytoskeleton regulates LFA-1 ligand binding through avidity rather than affinity changes. J. Biol. Chem., 274, 26869–26877. [DOI] [PubMed] [Google Scholar]
- Wang X.D., Gjörloff-Wingren,A., Saxena,M., Pathan,N., Reed,J.C. and Mustelin,T. (2000) The tumor suppressor PTEN regulates T cell survival and antigen receptor signaling by acting as a phos phatidylinositol 3-phosphatase. J. Immunol., 164, 1934–1939. [DOI] [PubMed] [Google Scholar]
- Ward S.G., Ley,S.C., MacPhee,C. and Cantrell,D.A. (1992) Regulation of D-3 phosphoinositides during T cell activation via the T cell antigen receptor/CD3 complex and CD2 antigens. Eur. J. Immunol., 22, 45–49. [DOI] [PubMed] [Google Scholar]
- Woods M.L., Cabañas,C. and Shimizu,Y. (2000) Activation-dependent changes in soluble fibronectin binding and expression of β1 integrin activation epitopes in T cells: relationship to T cell adhesion and migration. Eur. J. Immunol., 30, 38–49. [DOI] [PubMed] [Google Scholar]
- Xavier R. and Seed,B. (1999) Membrane compartmentation and the response to antigen. Curr. Opin. Immunol., 11, 265–269. [DOI] [PubMed] [Google Scholar]
- Xavier R., Brennan,T., Li,Q.Q., McCormack,C. and Seed,B. (1998) Membrane compartmentation is required for efficient T cell activation. Immunity, 8, 723–732. [DOI] [PubMed] [Google Scholar]
- Zell T., Hunt,S.W.,III, Finkelstein,L.D. and Shimizu,Y. (1996) CD28-mediated upregulation of β1 integrin-mediated adhesion involves phosphatidylinositol 3-kinase. J. Immunol., 156, 883–886. [PubMed] [Google Scholar]
- Zhang W.G., Trible,R.P. and Samelson,L.E. (1998) LAT palmitoylation: its essential role in membrane microdomain targeting and tyrosine phosphorylation during T cell activation. Immunity, 9, 239–246. [DOI] [PubMed] [Google Scholar]
- Zhang Z.H., Vuori,K., Wang,H.G., Reed,J.C. and Ruoslahti,E. (1996) Integrin activation by R-ras. Cell, 85, 61–69. [DOI] [PubMed] [Google Scholar]