

Abstract
Signals emanating from the receptor for interleukin-1 (IL-1), lipopolysaccharide (LPS) or osteoclast differentiation factor/receptor activator of NFκB ligand (ODF/RANKL) stimulate transcription factors AP-1 through mitogen-activated protein kinase (MAPK) activation and NFκB through IκB kinase (IKK) activation. These kinases are thought to be activated by tumor necrosis factor receptor-associated factor 6 (TRAF6). However, molecular mechanisms by which TRAF6 activates various downstream kinases remain to be elucidated. We identified functional domains of TRAF6 under physiological conditions established by appropriate expression of TRAF6 mutants in TRAF6-deficient cells. In IL-1 and LPS signaling pathways, the RING finger and first zinc finger domains are not required for NFκB activation but are required for full activation of MAPK. However, IL-1 and LPS signals utilize distinct regions within the zinc finger domains of TRAF6 to activate NFκB. Furthermore, the RING finger domain is not required for differentiation of splenocytes to multinuclear osteoclasts, but is essential for osteoclast maturation. Thus, TRAF6 plays essential roles in both the differentiation and maturation of osteoclasts by activating various kinases via its multiple domains.
Keywords: MAPK/NFκB/osteoclastogenesis/RANK/TRAF
Introduction
The tumor necrosis factor receptor (TNFR)-associated factor (TRAF) family proteins are cytoplasmic adapter proteins that mediate cytokine signaling (Arch et al., 1998). Among the receptors that recruit TRAF proteins are members of the TNFR superfamily and the Toll/interleukin-1 receptor (IL-1R) family. To date, six members of this family have been described. The TRAF domain is located in the C-terminal region of all members of the TRAF family and is composed of two subregions (Cheng et al., 1995): TRAF-C, which is the C-terminal half of the TRAF domain, is highly conserved among all members, and TRAF-N, which is the N-terminal half of the TRAF domain, is predicted to form a coiled-coil structure. Crystallographic studies of the TRAF domain of TRAF2 suggest that this domain forms a mushroom-shaped trimer, which is likely to interact with the trimer form of CD40 or TNFRII in response to ligand stimulation (McWhirter et al., 1999; Park et al., 1999). With the exception of TRAF1, all TRAFs contain an N-terminal RING finger domain and a stretch of predicted zinc fingers. Whereas TRAF2, TRAF5 and TRAF6 activate transcription factors NFκB through IκB kinase (IKK) activation and AP-1 through activation of mitogen-activated protein kinases (MAPKs) including Jun N-terminal kinase (JNK), p38 and extracellular signal-regulated kinase (ERK), neither TRAF3 nor TRAF4 activates these pathways (Arch et al., 1998). Recently, signal-dependent interaction of MAP kinase kinase kinase (MAP3K) with TRAFs was demonstrated. TRAF6 interacts with transforming growth factor β (TGFβ)-activated kinase 1 (TAK1) via TAK1 binding protein 2 (TAB2) to activate NFκB-inducing kinase (NIK) and JNK (Ninomiya et al., 1999; Takaesu et al., 2000). TRAF2 interacts with apoptosis signal-regulating kinase-1 (ASK1) (Nishitoh et al., 1998) and MEKK1 (Baud et al., 1999). Thus, TRAF2, TRAF6 and probably TRAF5 exert their function by activating downstream MAP3Ks or MAP4Ks.
TRAF6 has the most divergent TRAF-C and is the only TRAF that is involved in the signal from members of the Toll/IL-1R family by interacting with the IL-1R-associated kinase (IRAK) (Cao et al., 1996; Ishida et al., 1996a) and that from p75 neutrophin receptor (Khursigara et al., 1999). Furthermore, TRAF2, TRAF3 and TRAF5 bind to the membrane-distal domain in the cytoplasmic tail of CD40 and receptor activator of NFκB (RANK), whereas TRAF6 interacts with the membrane-proximal domain (Galibert et al., 1998; Wong et al., 1998; Tsukamoto et al., 1999). We have previously shown that TRAF6-deficient (TRAF6–/–) mice exhibit severe osteopetrosis and are defective in osteoclast formation due to defective signaling from RANK upon binding of osteoclast differentiation factor/RANK ligand (ODF/RANKL) (also known as OPGL and TRANCE) (Naito et al., 1999). Furthermore, TRAF6–/– mice are defective in normal B-cell differentiation, lymph node organogenesis and IL-1 signaling. Thus, TRAF6 plays pivotal roles in immune and inflammatory systems in vivo. Although TRAF6-mediated signal transduction is indispensable, the molecular mechanism by which TRAF6 exerts its biological effects remains unknown. Moreover, results of earlier studies designed to investigate the role of TRAF zinc-binding regions, which are required for TRAF signal transduction, were often contradictory, most likely because TRAF proteins were grossly overexpressed in cell lines that endogenously express TRAF (Cao et al., 1996; Takeuchi et al., 1996; Dadgostar and Cheng, 1998; Baud et al., 1999). In this study, we sought to identify critical regions within TRAF6 that are involved in activation of NFκB and MAPK pathways, and to define the essential roles of TRAF6 in osteoclastogenesis by expressing mutant TRAF6 transgenes at endogenous wild-type levels in TRAF6–/– cells.
Results
TRAF6 is essential for activation of NFκB, JNK and p38 by IL-1 and lipopolysaccharide (LPS) signaling
TRAF6–/– mice display osteopetrosis with defects in bone remodeling due to impaired ODF/RANKL–RANK signaling (Lomaga et al., 1999; Naito et al., 1999). However, we scarcely observed tartrate-resistant acid phosphatase-positive (TRAP+) osteoclast-like cells (OCLs) in bone tissue, whereas normal numbers of TRAP+ OCLs that lacked contact with bone surfaces were observed by Lomaga et al. (1999). Furthermore, our in vitro culture experiments revealed that osteoclast progenitors derived from TRAF6–/– mice are unable to differentiate into OCLs in response to ODF/RANKL. This discrepancy could be due to differences in targeting constructs. However, other unexpected genetic defects besides TRAF6 deficiency are possible. Thus, whether TRAF6 is essential for NFκB and MAPK activation via IL-1 and LPS signaling was first tested. Two independent mouse embryonic fibroblast (MEF) cell lines were generated from wild-type and TRAF6–/– mice (Figure 1B). MEF cells were stimulated with either IL-1 or LPS, and activation of NFκB, JNK and p38 was analyzed. Activation of NFκB, JNK and p38 in response to IL-1 and LPS was abrogated in TRAF6–/– MEF cells (Figures 2 and 3). In contrast, activation of NFκB and MAPK pathways by tumor necrosis factor α (TNFα) or sorbitol in TRAF6–/– MEF cells was similar to that in wild-type MEF cells (data not shown). To confirm that failure of MEF cells to respond to IL-1 and LPS was due solely to lack of TRAF6 expression, TRAF6 cDNA was introduced into TRAF6–/– MEF cells by a retrovirus vector carrying a puromycin resistance gene. The amount of TRAF6 expressed from the introduced cDNA was comparable to that of endogenous TRAF6 in wild-type MEF cells by incubating TRAF6–/– MEF cells with diluted virus solution to avoid multiple infection of virus (Figure 1B). Complementation of TRAF6–/– MEF cells with TRAF6 restored activation of NFκB, JNK and p38 in response to IL-1 and LPS treatment (Figures 2 and 3). These results indicate that TRAF6 is essential for IL-1 and LPS signaling linked to NFκB and MAPK activation.
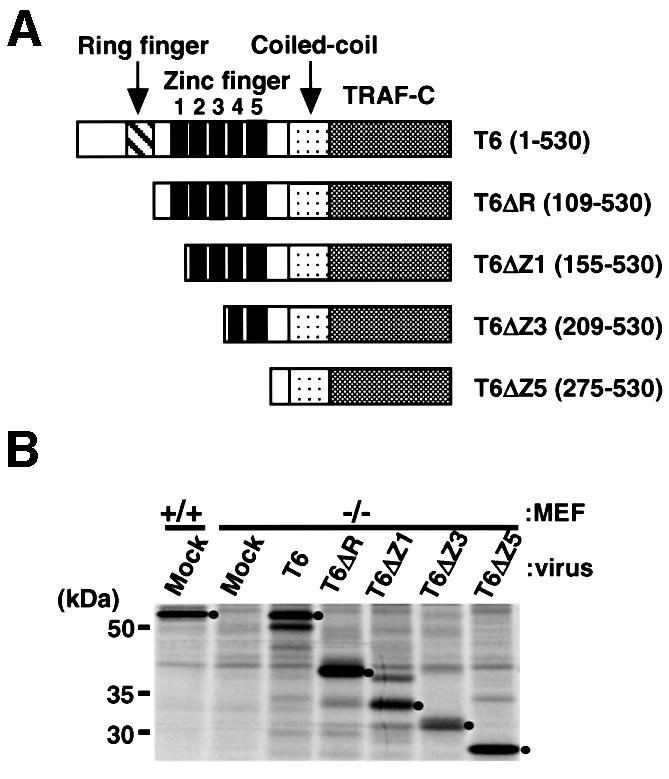
Fig. 1. Establishment of TRAF6–/– embryo-derived fibroblasts stably expressing physiological levels of exogenous wild-type or mutant TRAF6. (A) Structure of wild-type and N-terminally truncated TRAF6 mutants. Numbers in parentheses indicate amino acids that comprise wild-type and mutant TRAF6. (B) Expression level of wild-type and mutant TRAF6 in MEF cells. Cells were metabolically labeled with [35S]methionine, and wild-type and mutant TRAF6 were immunoprecipitated with anti-TRAF6 antibody. The number of methionine residues in each protein: T6 (18), T6ΔR (15), T6ΔZ1 (14), T6ΔZ3 (13) and T6ΔZ5 (10). Dots denote the positions of protein bands that correspond to the indicated form of TRAF6 (shown above each lane).

Fig. 2. The RING finger and first zinc finger of TRAF6 are not required for NFκB activation in response to IL-1 and LPS in MEF cells. MEF cells were derived from wild-type (+/+) or TRAF6–/– (–/–) mice and infected with recombinant retrovirus harboring either wild-type or mutant TRAF6 transgenes. (A) DNA binding activity of nuclear NFκB. Cells were either unstimulated (–) or stimulated (+) with IL-1 (20 ng/ml) for 30 min or LPS (200 ng/ml) for 60 min. Nuclear extracts were prepared and EMSAs were performed. The quantity of nuclear extracts used was confirmed by the DNA binding activity of Sp1. (B) NFκB-induced expression of endogenous IκBα gene. Cells were either unstimulated (–) or stimulated (+) with IL-1 (20 ng/ml) for 60 min or LPS (200 ng/ml) for 90 min. Expression of endogenous IκBα gene was determined by northern blotting. The fold increase in IκBα mRNA in stimulated cells compared with unstimulated cells was determined by phosphoimaging and normalized to the level of total RNA loaded into each lane as determined by ethidium bromide (EtBr) staining.

Fig. 3. Both the RING finger and the zinc finger domain are required for full activation of JNK and p38 in MEF cells. MEF cells were derived from wild-type (+/+) or TRAF6–/– (–/–) mice. (A) Activation of JNK. Cells were either unstimulated (–) or stimulated (+) with IL-1 (20 ng/ml) for 15 min or LPS (500 ng/ml) for 90 min. After stimulation, cells were assayed for endogenous JNK activity by immunocomplex kinase assay with GST–c-Jun (1–89) as a substrate (GST–c-Jun). The amount of JNK in the immunocomplex is also shown (JNK). The fold increase in endogenous JNK activity in stimulated cells compared with untreated cells was determined by phosphoimaging and normalized to the level of JNK in the immunocomplex, which was determined by immunoblotting using ImageQuant (Molecular Dynamics). (B) Activation of p38 MAPK. After stimulation as described in (A), cells were lysed with sample buffer. p38 MAPK (p38) and phosphorylated p38 (p-p38) were visualized by immunoblotting with anti-p38 and anti-p-p38 antibodies. The fold increase in the amount of p-p38 in stimulated cells compared with unstimulated cells was determined and normalized to the amount of p38 as measured by ImageQuant.
TRAF6 does not associate with TRAF2 or TRAF5 in response to IL-1
Since both TRAF2 and TRAF5 activate NFκB and MAPK (Rothe et al., 1994; Ishida et al., 1996b; Nakano et al., 1996), and since the TRAF family of proteins can associate to form homomeric or heteromeric complexes (Arch et al., 1998), TRAF6 is also likely to form complexes with TRAF2 or TRAF5. Thus, complex formation was analyzed under two different conditions where TRAF6 activates NFκB and MAPK pathways. TRAF6 did not associate with TRAF2 or TRAF5 when they were overexpressed in 293T cells, although TRAF6 forms a homomeric complex (Figure 4A). Furthermore, endogenous TRAF6 did not co-precipitate with TRAF2 or TRAF5 irrespective of IL-1 stimulation of MEF cells (Figure 4B). In contrast, stimulation-dependent association of TRAF6 with IRAK was reproduced (Cao et al., 1996). Thus, wild-type and mutant TRAF6 described below are not likely to associate with TRAF2 or TRAF5 in MEF cells, even upon stimulation.
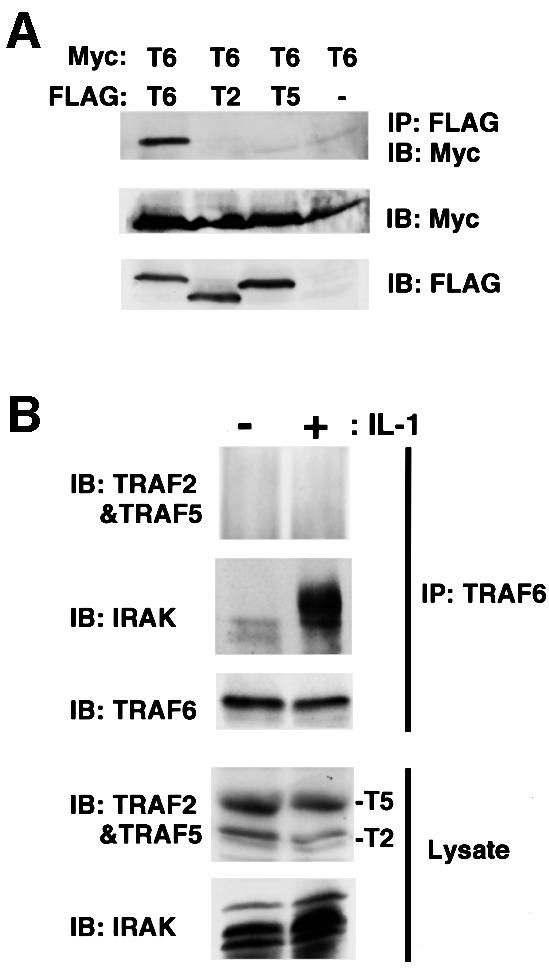
Fig. 4. TRAF6 does not associate with TRAF2 or TRAF5 in MEF cells. (A) TRAF6 does not heterodimerize with TRAF2 or TRAF5 when overexpressed in 293T cells. Myc-TRAF6 expression plasmid was co-transfected with control expression plasmid or expression plasmid encoding Flag-TRAF2, Flag-TRAF5 or Flag-TRAF6 into 293T cells. After 36 h, cell lysates were prepared and incubated with anti-FLAG monoclonal antibody. Co-precipitating Myc-TRAF6 was detected by immunoblot analysis with anti-Myc monoclonal antibody (top). Amounts of Myc-TRAF6 in total lysates (middle) and FLAG-TRAFs in immunocomplexes (bottom) are shown. (B) Endogenous TRAF6 does not associate with TRAF2 or TRAF5 upon IL-1 stimulation. MEF cells derived from wild-type mice were either unstimulated (–) or stimulated with IL-1 (20 ng/ml) for 15 min (+). Cell lysates were prepared and incubated with anti-TRAF6 antibody. Amounts of TRAF2, TRAF5, TRAF6 and IRAK in the immunoprecipitates were determined by immunoblot analysis with antibodies recognizing each protein (upper three panels). Amounts of endogenous TRAF2, TRAF5 and IRAK in the lysates were determined by immunoblotting (lower two panels).
IL-1 and LPS signals utilize distinct regions within the zinc finger domain of TRAF6 to activate NFκB pathways
Because IL-1- and LPS-dependent activation of NFκB and MAPK were restored by complementation of TRAF6–/– MEF cells with TRAF6, we attempted to delineate the domains in TRAF6 required for NFκB and MAPK activation. Previous studies designed to determine functional domains of TRAF6 were performed by overexpression of TRAF6 mutants programmed by transient transfection in cell lines having endogenous TRAF6 (Cao et al., 1996; Baud et al., 1999). Thus, the ability of TRAF6 mutants to transmit a physiological signal has never been appropriately addressed. Since the TRAF domain is required for binding to upstream molecules such as CD40 or IRAK (Cao et al., 1996; Ishida et al., 1996a), a series of N-terminal deletion mutants (Figure 1A) was generated, and their cDNAs were inserted into retrovirus vector. To avoid multiple integration of the mutant TRAF6 transgene, TRAF6–/– MEF cells were incubated with adequately diluted solutions of recombinant virus and subsequently selected for puromycin resistance. Expression levels of exogenously introduced TRAF6 mutants were comparable to that of endogenous TRAF6 in wild-type MEF cells (Figure 1B). The ability of TRAF6 mutants to activate NFκB in response to IL-1 was first determined by electrophoretic mobility shift assays (EMSAs) (Figure 2A). T6ΔR, T6ΔZ1 and T6ΔZ3 were capable of activating the NFκB pathway to a level almost comparable to that observed with wild-type TRAF6. Thus, these results indicate that the RING finger and first three zinc fingers were not required for NFκB activation in response to IL-1. T6ΔZ5, which does not have any zinc-binding region, still had residual yet detectable activity. These results were supported by the results of another set of experiments in which IL-1-induced expression of the IκBα gene was analyzed (Figure 2B). The IκBα gene is under the transcriptional control of NFκB (Sun et al., 1993). Consistent with the EMSA results, full induction of the IκBα gene was observed in MEF cells expressing wild-type TRAF6, T6ΔR, T6ΔZ1 and T6ΔZ3. However, expression of T6ΔZ5 did not result in IκBα gene induction.
LPS signal transduction was also analyzed (Figure 2A and B). Both EMSAs and IκBα northern blot analysis revealed that NFκB was fully activated in cells expressing T6ΔZ1. Thus, the RING finger and the first zinc finger are not required for LPS-induced NFκB activation. Interestingly, unlike IL-1 signaling, further deletion of the second and third zinc fingers completely abolished the ability of TRAF6 to mediate the NFκB signaling pathway. Taken together, results indicate that the RING finger and first zinc finger of TRAF6 are not required for full activation of the NFκB pathway by both IL-1 and LPS. Interestingly, the second and third zinc fingers were essential for NFκB activation by LPS but not required for that by IL-1. In contrast, NFκB activation by IL-1 required the fourth and fifth zinc fingers.
RING finger and zinc finger domains of TRAF6 are required for full activation of MAPKs
Domains of TRAF6 required for JNK or p38 activation were mapped. Wild-type MEF cells or TRAF6–/– MEF cells expressing full-length TRAF6 or one of the TRAF6 mutants described above were stimulated by either IL-1 or LPS. JNK activity was determined by in vitro kinase assay (Figure 3A), and p38 activation was estimated by immunoblotting phosphorylated p38 (Figure 3B). Deletion of the RING finger resulted in ∼40% reduction of IL-1-induced JNK and p38 activation. Further deletion of zinc fingers led to the gradual reduction of JNK and p38 activation, and the entire deletion of all zinc-binding domains abolished the ability of TRAF6 to activate JNK and p38 almost completely. Almost identical results were obtained when JNK and p38 activation were induced by LPS. Taken together, these results indicate that the zinc-binding motifs including the RING finger as well as all of the zinc fingers are required for full activation of JNK and p38. Thus, the RING finger and first zinc finger of TRAF6 are solely required for full activation of MAPK but not essential for activation of NFκB.
The RING finger domain of TRAF6 is essential for IL-1-induced activation of TAK1
TAK1, a member of MAP3K, is activated by association with TRAF6 via TAB2 in an IL-1 signal-dependent manner, and TAK1 in turn activates NIK, which is likely to activate the IKK complex (Ninomiya et al., 1999; Takaesu et al., 2000). TAK1 is capable of activating NFκB and JNK in the presence of TAB1. Thus, experiments designed to identify the domain of TRAF6 required for TAK1 activation by IL-1 were carried out. Activation of TAK1 coincides with phosphorylation of TAK1 and TAB1, which results in the mobility shift of these two proteins on SDS–polyacrylamide gels (Ninomiya et al., 1999). MEF cells were treated with IL-1, and lysates were prepared and assayed for TAB1 mobility shift (Figure 5). No activation of TAK1 was observed in TRAF6–/– MEF cells treated with IL-1, whereas TAK1 activation was observed in IL-1-treated TRAF6–/– MEF cells complemented with TRAF6. Thus, TRAF6 is essential for IL-1-induced activation of TAK1. Interestingly, in TRAF6–/– MEF cells expressing T6ΔR, no activation of TAK1 by IL-1 was detected, whereas IL-1 activated JNK by 60% in these cells (Figure 3A). These results indicate that TRAF6 activates other JNK-activating kinases via its zinc finger domain. Furthermore, since the RING finger is not required for NFκB activation, the contribution of TAK1 to NFκB activation via an IL-1 pathway is negligible in MEF cells. Therefore, involvement of TAK1 in NFκB activation is likely to be cell-type specific.

Fig. 5. The RING finger of TRAF6 is essential for TAK1 activation in response to IL-1. Various MEF cells were either unstimulated (–) or stimulated (+) with IL-1 (20 ng/ml) for 15 min. Cell lysates were analyzed by immunoblotting with anti-TAB1 antibody. Phosphorylated TAB1 (p-TAB1) shows less mobility than TAB1.
The RING finger domain of TRAF6 is essential for osteoclast maturation but is not required for osteoclast differentiation
Formation of functional osteoclasts requires differentiation of monocyte–macrophage lineage precursor cells (colony-forming unit monocyte–macrophage, CFU-M) to TRAP+ multinuclear cells and subsequent maturation, where cells polarize and form actin rings and ruffled border membranes for bone resorption (Teitelbaum, 2000). We have previously shown that TRAF6–/– mice exhibit severe osteopetrosis due to the lack of osteoclast formation (Naito et al., 1999). Furthermore, in vitro culture experiments revealed that osteoclast precursor cells derived from TRAF6–/– mice are unable to differentiate into osteoclasts in response to ODF/RANKL and macrophage colony-stimulating factor (M-CSF). However, Lomaga et al. (1999) demonstrated that the TRAF6–/– mice are osteopetrotic and have abundant dysfunctional osteoclasts. These observations prompted us to think that the RANK–TRAF6 pathway could be required for both differentiation and maturation of osteoclasts. Since the numbers of osteoclast progenitor cells in the spleens of wild-type and TRAF6–/– mice are comparable (Naito et al., 1999), we first checked various cytokine-signaling pathways in splenocytes pre-cultured for 3 days in the presence of M-CSF. Degradation of IκBα and phosphorylation of JNK and p38 in response to IL-1 or soluble ODF/RANKL (sODF/RANKL) were observed only in wild-type progenitor cells but not in TRAF6–/– progenitors (Figure 6). These results indicate that ODF/RANKL-induced RANK signaling is impaired in TRAF6–/– progenitor cells.

Fig. 6. TRAF6 is essential for ODF/RANKL-induced activation of NFκB and MAPK in osteoclast progenitors. Spleen cells derived from wild-type (+/+) or TRAF6–/– (–/–) mice were cultured for 3 days in the presence of M-CSF. After serum starvation for 30 min, cells were either unstimulated (None) or stimulated with IL-1 (10 ng/ml) or sODF/RANKL (100 ng/ml) for 20 min. Cell lysates were prepared and immunoblot analysis was performed with antibodies reactive with IκBα, JNK, p38, phosphorylated JNK (p-JNK) or phosphorylated p38 (p-p38).
To address the role of TRAF6 in the differentiation and maturation of osteoclasts, splenocytes from wild-type or TRAF6–/– mice were either mock infected or infected with retrovirus carrying wild-type TRAF6 or TRAF6 mutant in the presence of M-CSF. The infected cells were further cultured in the presence of both M-CSF and sODF/RANKL, and the number of OCLs identified as multinuclear TRAP+ cells was counted (Figure 7). Under this condition, splenocytes from wild-type mice efficiently differentiated into OCLs, ∼90% of which formed apparent actin rings. Non-infected or mock-infected splenocytes did not differentiate into OCLs. Expression of wild-type TRAF6 in TRAF6–/– splenocytes resulted in the appearance of OCLs, ∼50% of which formed actin rings (Figure 7A, c and d). Complementation of TRAF6–/– splenocytes with T6ΔR or T6ΔZ1 resulted in the appearance of significant numbers of OCLs, whereas complementation of these cells with T6ΔZ3 or T6ΔZ5 did not lead to formation of OCLs (Figure 7C). Interestingly, none of the OCLs generated by the expression of either T6ΔR or T6ΔZ1 was able to form the actin ring (Figure 7A, e and f and data not shown), although these OCLs expressed typical marker genes required for osteoclasts (Figure 7B). These results indicate that the RING finger domain is not required for differentiation of progenitor cells to TRAP+ OCLs, but is required for formation of the actin ring.

Fig. 7. The RING finger and first zinc finger of TRAF6 are not required for the formation of multinuclear TRAP+ cells. (A) Microscopic observation of TRAP+ cells and actin ring formation. Spleen cells from wild-type mice (a) and TRAF6–/– mice (b–f). TRAF6–/– cells were mock-infected (b) or infected with retrovirus encoding wild-type TRAF6 (c and d), or T6ΔR (e and f). After infection, cells were cultured in the presence of M-CSF (10 ng/ml) and sODF/RANKL (100 ng/ml). Then, cells were fixed and stained for TRAP (a, b, c and e) and F-actin (d and f) to visualize actin rings. (B) Expression of osteoclast marker genes. MMP-9 (nucleotides 111–864), cathepsin K (CATK) (nucleotides 1–505), calcitonin receptor (CTR) (nucleotides 1311–1765), TRAP (nucleotides 586–1082) and GAPDH (nucleotides 319–1047) were amplified by RT–PCR. (C) Quantitative analysis of the formation of OCLs. TRAP+ cells containing more than three nuclei were counted as OCLs.
We next analyzed the bone-resorbing activity of these OCLs. Splenocytes from TRAF6–/– mice infected with retrovirus harboring wild-type or mutant TRAF6 transgene were co-cultured with osteoblasts from wild-type mice in the presence of 1α,25-dihydroxyvitamin D3 and prostaglandin E2 for 7 days. The number of OCLs was counted, and the area of resorption pits was measured. Formation of OCLs was comparable to the results shown in Figure 7C, where recombinant M-CSF and sODF/RANKL were used instead of osteoblasts (data not shown). Complementation of TRAF6–/– splenocytes with wild-type TRAF6 restored resorption activity, as was shown by the generation of OCLs, which formed a number of resorption pits (Figure 8A). However, complementation of TRAF6–/– splenocytes with T6ΔR did not restore OCL resorption activity. Quantitation of resorption, expressed as the resorbed area per single OCL, revealed that OCLs derived from TRAF6–/– splenocytes expressing T6ΔR or T6ΔZ1 have impaired bone resorbing ability (Figure 8B). Therefore, the RING finger of TRAF6 is essential for the maturation of osteoclasts, but is not required for generating multinuclear TRAP+ OCLs. Because OCLs were absent from cultures of ODF/RANKL-treated TRAF6–/– splenocytes (Naito et al., 1999), it is clear that TRAF6 is required for both differentiation and maturation of osteoclasts.

Fig. 8. The RING finger of TRAF6 is essential for osteoclast maturation. (A) Microscopic view of resorption pits. Cells were cultured with normal osteoblasts on dentine slices in the presence of prostaglandin E2 (1 µM) and 1α,25-dihydroxyvitamin D3 (10 nM). Spleen cells from TRAF6–/– mice were mock infected (left) or infected with retrovirus encoding wild-type TRAF6 (center) and T6ΔR (right). (B) Quantitative analysis of the resorbed area. Resorption pits on dentine slices were visualized by staining with 0.5% toluidine blue, and the total pit area on dentine slices was measured. OCLs formed under identical conditions were counted, and the resorption area per single OCL was calculated.
Discussion
Identification of functional domains of TRAF6 under physiological conditions
In this paper, we have identified the signal-transmitting domains of TRAF6 by measuring the activity of N-terminally truncated mutants of TRAF6 under conditions where the two critical problems in previous reports can be overcome, as follows. First, the activity of mutants was analyzed in the absence of endogenous TRAF6 using TRAF6–/– MEF cells. Therefore, possible interaction of TRAF6 mutants and endogenous TRAF6 can be ruled out. Moreover, interaction of TRAF6 with TRAF2 or TRAF5, which also activates MAPK and NFκB, was not detected in MEF cells irrespective of the extracellular stimulation or the overexpression experiments in 293T cells (Figure 4). Both TRAF2- and TRAF5-deficient mice are likely to have intact ODF/RANKL signal transduction pathways, because they do not have osteopetrosis (Yeh et al., 1997; Nakano et al., 1999). Therefore, it is unlikely that NFκB activation exerted by expression of mutants T6ΔR, T6ΔZ1 and T6ΔZ3 was due to association of these mutants with an undetectable amount of TRAF2 or TRAF5 in the TRAF6–/– MEF cells. Secondly, the expression level of the TRAF6 mutants was adjusted to that of endogenous TRAF6 in wild-type MEF cells (Figure 1B). Thus, the system presented here is likely to be the most appropriate so far to identify functional domains of TRAF6.
In our assay system, neither IL-1 nor LPS activates NFκB or JNK in the absence of TRAF6, and complementation of TRAF6–/– MEF with wild-type TRAF6 protein restores all of these pathways (Figures 2 and 3). These results clearly indicate that TRAF6 is an essential component of both IL-1 and LPS signal transduction pathways linked to NFκB and MAPK activation.
The zinc-binding domain of TRAF6 consists of subdomains that interact with molecules linked to the distinct downstream pathways
Functional domains within the zinc-binding domain of TRAF6 identified in this paper are summarized in Figure 9. The NFκB activation domains are located in a relatively small segment within the zinc finger domain, while JNK activation domains were scattered throughout the zinc-binding domain. These results revealed that TRAF6 activates various downstream kinases, probably MAP3Ks or MAP4Ks, through its distinct subdomains present in the zinc-binding domain. In fact, the RING finger is essential for activating TAK1 (Figure 5), which is capable of activating both NFκB and MAPK pathways by its overexpression with TAB1 (Ninomiya et al., 1999). However, the T6ΔR mutant, which lacks the RING finger domain, still activates NFκB and MAPK significantly. These results clearly indicate that other unidentified kinases or adapter proteins are likely to be activated by the zinc finger domain of TRAF6. One candidate is MEKK1, which is activated by TNFα and associates with TRAF2 (Baud et al., 1999). Evolutionarily conserved signaling intermediate in Toll/IL-1 pathways (ECSIT), which bridges TRAF6 to MEKK1 and enhances MEKK1 processing, could also be a candidate, since ECSIT has been shown to bind to the TRAF domain of TRAF6 (Kopp et al., 1999). Interestingly, residual yet detectable NFκB activation was induced without a zinc-binding region in response to IL-1 (Figure 2A). Although this activation did not lead to IκBα induction (Figure 2B), there could be physiological target genes. Taken together, our results strongly suggest that there are subdomains in the zinc-binding domain of TRAF6 that lead to distinct post-receptor pathways.

Fig. 9. Functional domains of TRAF6. Functional domains in the zinc-binding region of TRAF6 identified in this study are summarized. Domains within the primary structure of TRAF6 (long rectangular box, top) are represented as smaller rectangular boxes having different patterns and are labeled accordingly. The underlined domains are required for the functions indicated to the right. A dotted line indicates weak NFκB activation domain in response to IL-1.
TLR4, a receptor for LPS (Poltorak et al., 1998), and IL-1R are thought to share the same intermediates, including MyD88, IRAK and TRAF6, for transmitting signals (Adachi et al., 1998; Kawai et al., 1999). Interestingly, distinct regions of TRAF6 are responsible for NFκB activation by IL-1 and LPS: IL-1 requires the fourth and fifth zinc fingers, whereas LPS requires the second and third (Figures 2 and 9). Although the molecular mechanisms underlying this discrepancy are not clear, results suggest that the downstream signaling components of TRAF6 for IL-1 signal transduction are different from those for LPS signaling. It is also possible that the upstream molecules that mediate TRAF6 activation by IL-1R occupation may be different from those of the LPS signaling pathways, even though these different pathways may share common molecules such as IRAK. This possibility is supported by studies showing that LPS is able to induce NFκB activation in cells derived from MyD88-deficient mice (Kawai et al., 1999), while IL-1- and IL-18-mediated NFκB activation are abrogated in these mice (Adachi et al., 1998).
Several groups reported functional domains within the zinc-binding region of TRAFs, which were identified by overexpression of TRAF mutants programmed by transient transfection. In domain mapping studies of TRAF2 and TRAF5, several discrepancies in the identification of various functional domains are apparent. However, TRAF2 and TRAF5 always required the RING finger domain for NFκB activation (Takeuchi et al., 1996; Dadgostar and Cheng, 1998; Baud et al., 1999). It was reported that NFκB activation by overexpression of TRAF6 lacking the RING finger was ∼60% of that by wild-type TRAF6, and that further deletion of the first two zinc fingers of TRAF6 resulted in the complete abrogation of NFκB activation (Cao et al., 1996). Although this suggests that TRAF6 could transmit signals in a different way from TRAF2 and TRAF5, the RING finger of all these TRAFs plays an important role in NFκB activation. However, our results show that deletion of the N-terminal region up to the first zinc finger scarcely affects the ability of TRAF6 to mediate IL-1- or LPS-induced NFκB activation. Furthermore, T6ΔZ3, which further lacks the second and third zinc fingers, is still capable of mediating IL-1-dependent NFκB activation (Figures 2 and 9). To explain discrepancies between our findings and those of others, we propose a model in which signal-dependent structural change or oligomerization of TRAF6 for signaling could require stabilization by both its zinc-binding region and an unidentified stabilizer protein. T6ΔR, when expressed at physiological levels, is likely to form putative, stabilizer-mediated oligomers that are sufficient for NFκB activation; however, these putative T6ΔR oligomers may be unstable or structurally insufficient for activation of molecules linked to MAPK, such as TAK1, due to lack of the RING finger. Overexpression of TRAF6 or T6ΔR could titrate out the stabilizer. However, the majority of TRAF6 could form stable oligomers without the stabilizer via its RING finger to activate both NFκB and MAPK, since the concentration of TRAF6 is significantly higher than the physiological condition. T6ΔR oligomers, which could form when overexpressed, are likely to be unstable because they lack the RING finger domain; thus, these molecules may only partially activate NFκB. The stabilizer is likely to be IRAK or the cytoplasmic tail of membrane receptors, including CD40, RANK and p75 NGFR. IL-1-induced TAK1 activation requires TAB2-mediated linkage of TAK1 to TRAF6 (Takaesu et al., 2000). Since the interaction of TAB2 with TRAF6 requires the entire structure of TRAF6, TAB2 is not likely to interact with unstable TRAF oligomers. It has been reported recently that the RING finger of TRAF6 is required when TRAF6 functions as an E3 ubiquitin protein ligase together with TRAF6-regulated IKK activator 1 (TRIKA1) to activate NFκB pathways (Deng et al., 2000). The discrepancy between this report and our results remains to be elucidated. However, there could be ubiquitylation-dependent and -independent IKK activation pathways downstream of TRAF6.
Role of TRAF6 in osteoclastogenesis: identification of the TRAF6 RING finger domain as a critical motif for osteoclast maturation
We have previously shown severe osteopetrosis in TRAF6–/– mice due to blockade of the ODF/RANKL–RANK pathway (Naito et al., 1999). The cytoplasmic tail of RANK, a receptor of ODF/RANKL, was shown to bind to TRAF6 through its membrane-proximal TRAF-binding domain (Galibert et al., 1998; Wong et al., 1998). Consistent with these findings, both ODF/RANKL- and RANK-deficient mice were characterized by profound osteopetrosis resulting from a block in osteoclast differentiation (Dougall et al., 1999; Kong et al., 1999). Here we show that expression of wild-type TRAF6 in TRAF6–/– osteoclast progenitor cells resulted in the appearance of mature osteoclasts, which were able to resorb bone (Figures 7 and 8). Results confirmed that TRAF6 is essential for differentiation of osteoclasts and ruled out other possible genetic defects that affect osteoclast differentiation in our TRAF6–/– cells. With this system, we provide evidence indicating that the RING finger of TRAF6 is responsible for the maturation of osteoclasts, whereas the second and third zinc fingers play essential roles in the differentiation of OCLs (Figure 9). Thus, we are the first to show that TRAF6 plays essential roles in both the differentiation and maturation of osteoclasts. Our findings strongly suggest that linkage between RANK and TRAF6 is also required for both steps of osteoclastogenesis.
Since the RING finger is required for the full activation of JNK and p38 but is not required for NFκB activation in MEF cells (Figures 2 and 3), the level of MAPK activation is likely to be critical for maturation of osteoclasts. Thus, the requirement of the RING finger for TAK1 activation suggests that TAK1-mediated JNK or p38 activation is responsible for osteoclast maturation. Osteoclasts in mice lacking c-fos are unable to differentiate (Grigoriadis et al., 1994). Moreover, expression of the Fra-1 transgene in c-fos–/– mice relieves osteopetrosis, and treatment of CFU-M with ODF/RANKL induces Fra-1 expression in a c-Fos-dependent manner (Matsuo et al., 2000). Thus, ODF/RANKL-induced AP-1 activation is likely to be linked to Fra-1 induction, which may be required for both differentiation and maturation of osteoclasts. On this basis, the partial activation of AP-1, which is a consequence of reduced JNK and p38 activation by T6ΔR and T6ΔZ1, is likely to induce differentiation but not maturation of osteoclasts. Therefore, the level of AP-1 activation required for osteoclast differentiation is likely to be lower than that for osteoclast maturation. NFκB is also essential for osteoclastogenesis because mice lacking NFκB subunits p50 and p52 do not form OCLs from CFU-M and consequently have osteopetrosis (Franzoso et al., 1997; Iotsova et al., 1997). Thus, the extent of AP-1 and NFκB activation mediated by the RANK–TRAF6 linkage is likely to control osteoclastogenesis.
The β3 integrin subunit and c-Src are two signal transduction molecules required for maturation of osteoclasts; mice lacking one of them have dysfunctional osteoclasts, leading to osteopetrosis (Lowe et al., 1993; McHugh et al., 2000). β3 integrin is a component of the αvβ3 integrin complex, which is a major functional adhesion receptor on osteoclasts. αvβ3 integrin is thought to transmit signals that are linked to the formation of actin rings and ruffled border membranes via Src-dependent tyrosine phosphorylation of PYK2 and c-Cbl (Tanaka et al., 1996; Duong et al., 1998). Interestingly, TRAF6 was recently shown to associate with and activate c-Src upon stimulation of RANK (Wong et al., 1999). Although the TRAF domain of TRAF6 was demonstrated to be sufficient for interaction with c-Src, the RING finger domain may be required for linkage of c-Src with downstream intermediates such as PYK2 and c-Cbl. However, expression of kinase-deficient Src mutant rescues osteopetrosis of c-src–/– mice almost completely (Schwartzberg et al., 1997). The relationship between TRAF6-mediated signal transduction and this kinase-independent function of c-Src remains to be elucidated. Further investigation is needed to broaden our understanding of the regulation of osteoclastogenesis by TRAF6-mediated signaling pathways.
Materials and methods
Plasmids and antibodies
To construct retrovirus vectors carrying a cDNA for wild-type or mutant TRAF6, each cDNA was inserted into pMX-puro vector (Kitamura, 1998). N-terminally truncated mutants of TRAF6 were generated by introducing an initiation methionine codon into the appropriate positions of TRAF6 cDNA by site-directed mutagenesis. The following mutants were used: T6ΔR (amino acids 109–530), T6ΔZ1 (amino acids 155–530), T6ΔZ3 (amino acids 209–530) and T6ΔZ5 (amino acids 275–530). Anti-TRAF6 (C-20), anti-TRAF2, anti-TRAF5, anti-Myc, anti-JNK and anti-p38 antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), anti-FLAG antibody from Sigma Chemicals (St Louis, MO), anti-IRAK and anti-IκBα antibodies from Transduction Laboratories (Lexington, KY), and anti-phospho-p38 and anti-phospho-JNK from NEB, Inc. (Beverly, MA).
Introduction of wild type and TRAF6 mutants into MEF cells by retrovirus vector
Two MEF cell lines each from 14.5-day-old TRAF6–/– and wild-type embryos were established. Similar results were obtained from both cell lines. Packaging cell line BOSC23 (2 × 106) was transfected with 2 µg of the retrovirus vector plasmid. Virus stocks were prepared by collecting the media from the cultures 48 h after transfection. MEF cells (3 × 105) were incubated with 2 ml of virus stock for 4 h in the presence of polybrene (1 µg/ml). Forty-eight hours after infection, the medium was changed to Dulbecco’s modified Eagle’s medium (DMEM)/10% fetal calf serum (FCS) containing 0.5 µg/ml puromycin. Puromycin-resistant cell pools were used for further experiments.
EMSAs
A double-stranded oligonucleotide containing a κB site from the mouse κ-light chain enhancer was used for NFκB and that containing a GC box was used for Sp1. Equal amounts of extract (5 µg protein) were incubated with 32P-labeled probe and the DNA–protein complex was analyzed on a 4% polyacrylamide gel.
Immunoprecipitation and western blotting
MEF cells were either left untreated or treated with IL-1 (20 ng/ml) or LPS (200 ng/ml for NFκB and 500 ng/ml for JNK and p38) for the period indicated. Cells were lysed in 500 µl of TNE buffer (50 mM Tris–HCl pH 8.0, 1% NP-40, 1 mM EDTA, 150 mM NaCl, 0.5 mM dithiothreitol) and centrifuged to remove cellular debris. The resulting supernatant was subjected to immunoprecipitation by adding 1 µg of the appropriate antibody and 20 µl of protein G–Sepharose (Amersham Pharmacia, UK). Mouse spleen cells (5 × 106 cells) from wild-type or TRAF6–/– mice were cultured in α-MEM/10% FCS for 3 days on 35 mm dishes in the presence of M-CSF (10 ng/ml). After serum starvation for 30 min, cells were stimulated with IL-1 (10 ng/ml) or sODF/RANKL (100 ng/ml) for 20 min, and whole-cell lysates were prepared. The immunoprecipitates or whole-cell lysates were separated by SDS–PAGE and transferred to PVDF membrane (Millipore Corp., Bedford, MA). The membranes were immunoblotted with specific antibodies, visualized with the appropriate horseradish peroxidase-conjugated secondary antibody using the ECL western blotting system (Amersham Pharmacia). For metabolic labeling of MEF cells, 2 × 106 cells were first incubated in methionine-free DMEM for 2 h and the medium was then changed to 5 ml of fresh methionine-free DMEM containing 250 µCi of [35S]methionine. After 20 h of labeling, cells were washed twice and lysed in TNE buffer. Wild-type and mutant TRAF6 were immunoprecipitated with anti-TRAF6 antibody that recognizes all of the TRAF6 mutants.
In vitro kinase assay of JNK
Endogenous JNK was immunoprecipitated with anti-JNK antibody. Immunoprecipitates were incubated with 2 µg of glutathione S-transferase (GST)–c-JUN (1–89) fusion protein (New England Biolabs Inc.) in 20 µl of kinase buffer [20 mM HEPES pH 7.5, 20 mM MgCl2, 5 µCi of [γ-32P]ATP (3000 Ci/mmol)] at 30°C for 30 min.
Northern analysis
Twenty micrograms of total RNA were electrophoresed in 1% agarose gel containing formaldehyde and transferred to nylon membrane (NEN Inc., Boston, MA). The filter was incubated with 32P-labeled DNA probe overnight at 42°C, and then washed in 0.5× SSC/0.2% (w/v) SDS for 30 min at 65°C. The entire coding region of IκBα cDNA was used as a probe.
In vitro differentiation and activation of osteoclasts
Spleen cells (1 × 106 cells) were cultured in α-MEM/10% FCS with M-CSF (10 ng/ml) overnight in 24-well plates. Cells were subsequently cultured in virus stock medium containing polybrene (4 µg/ml) for 4 h at 37°C. Infected cells were cultured for 2 days in the presence of M-CSF (10 ng/ml), and for an additional 4 days in the presence of both M-CSF (10 ng/ml) and sODF/RANKL (100 ng/ml). Adherent cells were then fixed with 10% formaldehyde, treated with ethanol–acetone (50:50), and stained for TRAP (Naito et al., 1999). TRAP+ multinucleated cells containing more than three nuclei were counted as osteoclasts. For visualizing actin rings, fixed cells were stained with rhodamine-conjugated phalloidin (Molecular Probes, Eugene, OR). For gene expression analysis, total RNA was isolated and subjected to RT–PCR using a One Step RNA PCR Kit (TAKARA, Tokyo) to amplify the DNA fragments of MMP-9, cathepsin K (CATK), calcitonin receptor (CTR), TRAP and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The PCR products were separated by electrophoresis on 1% agarose gels and visualized by ethidium bromide staining. For pit formation assays, osteoblasts from calvariae of wild-type newborn mice were cultured on dentine slices in α-MEM/10% FCS overnight in 48-well plates (two slices/well). Spleen cells were then co-cultured with these osteoblasts in the presence of 10 nM 1α,25-dihydroxyvitamin D3 and 10 µM prostaglandin E2 overnight prior to infection. Cells were then subjected to retroviral infection overnight. After the incubation for an additional week, cells were removed from dentine slices by adding 1 M NH4OH and resorption pits were visualized by 0.5% toluidine blue staining. The total pit areas were measured using an image analysis system (System Supply, Nagano, Japan). TRAP+ cells were counted in an identical set of cultures.
Acknowledgments
Acknowledgements
We thank Drs T.Kitamura and H.Hayashi for their helpful discussion and various materials. This work was supported by a grant-in-aid for Scientific Research on Priority Areas from the Ministry of Education, Science, Sports and Culture of Japan, a grant for AIDS Research from the Japan Health Science Foundation, a grant from the Takeda Science Foundation, and the Special Coordination Funds of the Science and Technology Agency of Japan.
References
- Adachi O., Kawai,T., Takeda,K., Matsumoto,M., Tsutsui,H., Sakagami,M., Nakanishi,K. and Akira,S. (1998) Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity, 9, 143–150. [DOI] [PubMed] [Google Scholar]
- Arch R.H., Gedrich,R.W. and Thompson,C.B. (1998) Tumor necrosis factor receptor-associated factors (TRAFs)—a family of adapter proteins that regulates life and death. Genes Dev., 12, 2821–2830. [DOI] [PubMed] [Google Scholar]
- Baud V., Liu,Z.G., Bennett,B., Suzuki,N., Xia,Y. and Karin,M. (1999) Signaling by proinflammatory cytokines: oligomerization of TRAF2 and TRAF6 is sufficient for JNK and IKK activation and target gene induction via an amino-terminal effector domain. Genes Dev., 13, 1297–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z., Xiong,J., Takeuchi,M., Kurama,T. and Goeddel,D.V. (1996) TRAF6 is a signal transducer for interleukin-1. Nature, 383, 443–446. [DOI] [PubMed] [Google Scholar]
- Cheng G., Cleary,A.M., Ye,Z.S., Hong,D.I., Lederman,S. and Baltimore,D. (1995) Involvement of CRAF1, a relative of TRAF, in CD40 signaling. Science, 267, 1494–1498. [DOI] [PubMed] [Google Scholar]
- Dadgostar H. and Cheng,G. (1998) An intact zinc ring finger is required for tumor necrosis factor receptor-associated factor-mediated NF-κB activation but is dispensable for c-Jun N-terminal kinase signaling. J. Biol. Chem., 273, 24775–24780. [DOI] [PubMed] [Google Scholar]
- Deng L., Wang,C., Spencer,E., Yang,L., Braun,A., You,J., Slaughter,C., Pickart,C. and Chen,Z.J. (2000) Activation of the IκB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell, 103, 351–361. [DOI] [PubMed] [Google Scholar]
- Dougall W.C. et al. (1999) RANK is essential for osteoclast and lymph node development. Genes Dev., 13, 2412–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong L.T., Lakkakorpi,P.T., Nakamura,I., Machwate,M., Nagy,R.M. and Rodan,G.A. (1998) PYK2 in osteoclasts is an adhesion kinase, localized in the sealing zone, activated by ligation of αvβ3 integrin and phosphorylated by src kinase. J. Clin. Invest., 102, 881–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzoso G. et al. (1997) Requirement for NF-κB in osteoclast and B-cell development. Genes Dev., 11, 3482–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galibert L., Tometsko,M.E., Anderson,D.M., Cosman,D. and Dougall,W.C. (1998) The involvement of multiple tumor necrosis factor receptor (TNFR)-associated factors in the signaling mechanisms of receptor activator of NF-κB, a member of the TNFR superfamily. J. Biol. Chem., 273, 34120–34127. [DOI] [PubMed] [Google Scholar]
- Grigoriadis A.E., Wang,Z.Q., Cecchini,M.G., Hofstetter,W., Felix,R., Fleisch,H.A. and Wagner,E.F. (1994) c-Fos: a key regulator of osteoclast–macrophage lineage determination and bone remodeling. Science, 266, 443–448. [DOI] [PubMed] [Google Scholar]
- Iotsova V., Caamano,J., Loy,J., Yang,Y., Lewin,A. and Bravo,R. (1997) Osteopetrosis in mice lacking NF-κB1 and NF-κB2. Nature Med., 3, 1285–1289. [DOI] [PubMed] [Google Scholar]
- Ishida T. et al. (1996a) Identification of TRAF6, a novel tumor necrosis factor receptor-associated factor protein that mediates signaling from an amino-terminal domain of the CD40 cytoplasmic region. J. Biol. Chem., 271, 28745–28748. [DOI] [PubMed] [Google Scholar]
- Ishida T.K., Tojo,T., Aoki,T., Kobayashi,N., Ohishi,T., Watanabe,T., Yamamoto,T. and Inoue,J. (1996b) TRAF5, a novel tumor necrosis factor receptor-associated factor family protein, mediates CD40 signaling. Proc. Natl Acad. Sci. USA, 93, 9437–9442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T., Adachi,O., Ogawa,T., Takeda,K. and Akira,S. (1999) Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity, 11, 115–122. [DOI] [PubMed] [Google Scholar]
- Khursigara G., Orlinick,J.R. and Chao,M.V. (1999) Association of the p75 neurotrophin receptor with TRAF6. J. Biol. Chem., 274, 2597–2600. [DOI] [PubMed] [Google Scholar]
- Kitamura T. (1998) New experimental approaches in retrovirus-mediated expression screening. Int. J. Hematol., 67, 351–359. [DOI] [PubMed] [Google Scholar]
- Kong Y.Y. et al. (1999) OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature, 397, 315–323. [DOI] [PubMed] [Google Scholar]
- Kopp E., Medzhitov,R., Carothers,J., Xiao,C., Douglas,I., Janeway,C.A. and Ghosh,S. (1999) ECSIT is an evolutionarily conserved intermediate in the Toll/IL-1 signal transduction pathway. Genes Dev., 13, 2059–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomaga M.A. et al. (1999) TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40 and LPS signaling. Genes Dev., 13, 1015–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe C., Yoneda,T., Boyce,B.F., Chen,H., Mundy,G.R. and Soriano,P. (1993) Osteopetrosis in Src-deficient mice is due to an autonomous defect of osteoclasts. Proc. Natl Acad. Sci. USA, 90, 4485–4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo K., Owens,J.M., Tonko,M., Elliott,C., Chambers,T.J. and Wagner,E.F. (2000) Fosl1 is a transcriptional target of c-Fos during osteoclast differentiation. Nature Genet., 24, 184–187. [DOI] [PubMed] [Google Scholar]
- McHugh K.P. et al. (2000) Mice lacking β3 integrins are osteosclerotic because of dysfunctional osteoclasts. J. Clin. Invest., 105, 433–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhirter S.M., Pullen,S.S., Holton,J.M., Crute,J.J., Kehry,M.R. and Alber,T. (1999) Crystallographic analysis of CD40 recognition and signaling by human TRAF2. Proc. Natl Acad. Sci. USA, 96, 8408–8413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito A. et al. (1999) Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice. Genes Cells, 4, 353–362. [DOI] [PubMed] [Google Scholar]
- Nakano H., Oshima,H., Chung,W., Williams-Abbott,L., Ware,C.F., Yagita,H. and Okumura,K. (1996) TRAF5, an activator of NF-κB and putative signal transducer for the lymphotoxin-β receptor. J. Biol. Chem., 271, 14661–14664. [DOI] [PubMed] [Google Scholar]
- Nakano H. et al. (1999) Targeted disruption of Traf5 gene causes defects in CD40- and CD27-mediated lymphocyte activation. Proc. Natl Acad. Sci. USA, 96, 9803–9808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninomiya T.J., Kishimoto,K., Hiyama,A., Inoue,J., Cao,Z. and Matsumoto,K. (1999) The kinase TAK1 can activate the NIK-IκB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature, 398, 252–256. [DOI] [PubMed] [Google Scholar]
- Nishitoh H., Saitoh,M., Mochida,Y., Takeda,K., Nakano,H., Rothe,M., Miyazono,K. and Ichijo,H. (1998) ASK1 is essential for JNK/SAPK activation by TRAF2. Mol. Cell, 2, 389–395. [DOI] [PubMed] [Google Scholar]
- Park Y.C., Burkitt,V., Villa,A.R., Tong,L. and Wu,H. (1999) Structural basis for self-association and receptor recognition of human TRAF2. Nature, 398, 533–538. [DOI] [PubMed] [Google Scholar]
- Poltorak A. et al. (1998) Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science, 282, 2085–2088. [DOI] [PubMed] [Google Scholar]
- Rothe M., Wong,S.C., Henzel,W.J. and Goeddel,D.V. (1994) A novel family of putative signal transducers associated with the cytoplasmic domain of the 75 kDa tumor necrosis factor receptor. Cell, 78, 681–692. [DOI] [PubMed] [Google Scholar]
- Schwartzberg P.L., Xing,L., Hoffmann,O., Lowell,C.A., Garrett,L., Boyce,B.F. and Varmus,H.E. (1997) Rescue of osteoclast function by transgenic expression of kinase-deficient Src in src–/– mutant mice. Genes Dev., 11, 2835–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S.C., Ganchi,P.A., Ballard,D.W. and Greene,W.C. (1993) NF-κB controls expression of inhibitor IκBα: evidence for an inducible autoregulatory pathway. Science, 259, 1912–1915. [DOI] [PubMed] [Google Scholar]
- Takaesu G., Kishida,S., Hiyama,A., Yamaguchi,K., Shibuya,H., Irie,K., Ninomiya-Tsuji,J. and Matsumoto,K. (2000) TAB2, a novel adaptor protein, mediates activation of TAK1 MAPKKK by linking TAK1 to TRAF6 in the IL-1 signal transduction pathway. Mol. Cell, 5, 649–658. [DOI] [PubMed] [Google Scholar]
- Takeuchi M., Rothe,M. and Goeddel,D.V. (1996) Anatomy of TRAF2. Distinct domains for NF-κB activation and association with tumor necrosis factor signaling proteins. J. Biol. Chem., 271, 19935–19942. [DOI] [PubMed] [Google Scholar]
- Tanaka S., Amling,M., Neff,L., Peyman,A., Uhlmann,E., Levy,J.B. and Baron,R. (1996) c-Cbl is downstream of c-Src in a signalling pathway necessary for bone resorption. Nature, 383, 528–531. [DOI] [PubMed] [Google Scholar]
- Teitelbaum S.L. (2000) Bone resorption by osteoclasts. Science, 289, 1504–1508. [DOI] [PubMed] [Google Scholar]
- Tsukamoto N., Kobayashi,N., Azuma,S., Yamamoto,T. and Inoue,J. (1999) Two differently regulated NFκB activation pathways triggered by the cytoplasmic tail of CD40. Proc. Natl Acad. Sci. USA, 96, 1234–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong B.R., Josien,R., Lee,S.Y., Vologodskaia,M., Steinman,R.M. and Choi,Y. (1998) The TRAF family of signal transducers mediates NF-κB activation by the TRANCE receptor. J. Biol. Chem., 273, 28355–28359. [DOI] [PubMed] [Google Scholar]
- Wong B.R., Besser,D., Kim,N., Arron,J.R., Vologodskaia,M., Hanafusa,H. and Choi,Y. (1999) TRANCE, a TNF family member, activates Akt/PKB through a signaling complex involving TRAF6 and c-Src. Mol. Cell, 4, 1041–1049. [DOI] [PubMed] [Google Scholar]
- Yeh W.C. et al. (1997) Early lethality, functional NF-κB activation and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity, 7, 715–725. [DOI] [PubMed] [Google Scholar]