

Abstract
The interaction between integrin lymphocyte function-associated antigen-1 (LFA-1) and its ligand intercellular adhesion molecule-1 (ICAM-1) is critical in immunological and inflammatory reactions but, like other adhesive interactions, is of low affinity. Here, multiple rational design methods were used to engineer ICAM-1 mutants with enhanced affinity for LFA-1. Five amino acid substitutions 1) enhance the hydrophobicity and packing of residues surrounding Glu-34 of ICAM-1, which coordinates to a Mg2+ in the LFA-1 I domain, and 2) alter associations at the edges of the binding interface. The affinity of the most improved ICAM-1 mutant for intermediate- and high-affinity LFA-1 I domains was increased by 19-fold and 22-fold, respectively, relative to wild type. Moreover, potency was similarly enhanced for inhibition of LFA-1-dependent ligand binding and cell adhesion. Thus, rational design can be used to engineer novel adhesion molecules with high monomeric affinity; furthermore, the ICAM-1 mutant holds promise for targeting LFA-1-ICAM-1 interaction for biological studies and therapeutic purposes.
Intercellular adhesion molecule-1 (ICAM-1)3 is a cell surface ligand for lymphocyte function-associated antigen-1 (LFA-1), a member of the integrin family of adhesion receptors (1, 2). The interaction of LFA-1 and ICAM-1 is critical to many immunological reactions, including T lymphocyte antigen-specific responses and leukocyte accumulation in inflamed tissues (3). Although the extracellular domains of LFA-1 are composed of the large and complex αL and β2 subunits, the ligand binding site is located exclusively in the inserted (I) domain of αL (4). Many antagonists of this interaction, including monoclonal antibodies to the I domain of LFA-1 and small molecules, have been developed to treat autoimmune diseases and prevent immune rejection in organ transplantation (5, 6). Whereas all available small molecule antagonists for LFA-1 are allosteric inhibitors, many inhibitory antibodies directly block ligand binding to the I domain (5, 7). We set out to explore a new class of competitive antagonists that mimic the native ligand, ICAM-1. Engineered high affinity ICAM-1 could serve as a biotherapeutic or a lead molecule in the development of competitive, small molecule agents for treatment of autoimmune diseases.
Allostery of LFA-1 on the cell surface, regulated physiologically by inflammatory stimuli and signal transduction through the LFA-1 α and β subunit transmembrane domains, alters affinity for ICAM-1 (5). In the isolated I domain, mutationally introduced disulfide bonds have been used to stabilize the open conformation with high affinity, the intermediate conformation with intermediate affinity, or the closed conformation with low affinity for ICAM-1 (8, 9). A recent crystal structure of the αL I domain in complex with ICAM-1 has revealed the binding interface between the αL I domain and ICAM-1 at 3.3-Å resolution (8). The I domain directly coordinates Glu-34 of ICAM-1 through a Mg2+, and a reorientation of Glu-241 of the I domain creates a critical salt bridge to Lys-39 of ICAM-1 (8). Further comparison of liganded and unliganded structures for both high affinity (HA) and intermediate affinity (IA) αL I domains reveals a shape-shifting pathway for integrin regulation by which allosteric signals convert the closed conformation to intermediate or open conformations. Binding of the IA I domain to ICAM-1 stabilizes the same open conformation as seen with the HA I domain (8). The affinity of the HA αL I domain for wild-type ICAM-1 is low (KD = 185 ± 12 nM) (9) compared with many other protein-protein interactions. Enhancement of this affinity is essential for therapeutic applications or for accurate measurement of physiologically induced increase in affinity of LFA-1 on the cell surface.
Recent advances in computational protein design algorithms (10–12) have markedly improved capabilities for generating novel proteins with optimized properties, including enhanced stability (13), altered substrate specificity (10), improved binding affinity (14, 15), and optimized pharmacokinetics (16). We have taken multiple structure-based approaches to design ICAM-1 variants with enhanced affinity for αLβ2. Moreover, we have measured the kinetics and affinity of I domains stabilized in different conformations for high affinity ICAM-1 mutants and investigated the inhibitory effects of our most improved variant on binding of ICAM-1 to cell surface αLβ2 and αLβ2-dependent adhesion.
EXPERIMENTAL PROCEDURES
Computational Design
The crystal structure of the αL I domain in complex with ICAM-1 (PDB code 1MQ8) was used as the starting template for computational calculations (8). One of the variant libraries was designed using combined output from Protein Design Automation® (PDA) (13, 17, 18) and Sequence Prediction Algorithm™ (SPA) (19) calculations. For PDA calculations, the conformations of amino acids at variable positions were represented as a set of backbone-independent side-chain rotamers derived from the rotamer library of Dunbrack and Cohen (20). The energies of all possible combinations of the considered amino acids at the chosen variable positions were calculated using a force field containing terms describing van der Waals, solvation, electrostatic, and hydrogen bond interactions. The optimal (ground state) sequence was determined using a Dead End Elimination algorithm, and a Monte Carlo algorithm was used to evaluate the energies of similar sequences around the predicted ground state. SPA calculations utilize a genetic algorithm to screen for low energy sequences, with energies being calculated during each round of “evolution” for those sequences being sampled. The conformations of amino acids were represented as a set of side-chain rotamers derived from a backbone-independent rotamer library using a flexible rotamer model (21). SPA calculations generated a list of 300 sequences that were subsequently clustered computationally into groups of similar sequences using a nearest neighbor single linkage hierarchical clustering algorithm. Parameters and other details for PDA and SPA calculations are described elsewhere (13, 17–19) and in unpublished results.
For these sets of calculations, critical contact residues Glu-34, Lys-39, Asn-68, and Gln-73 were fixed in both sequence and conformation. Calculations were carried out to evaluate single and combinatorial substitutions at variable residues Lys-29, Leu-30, Pro-38, Glu-41, Met-64, Tyr-66, and Thr-75. All residues in contact with these residues were floated, that is the amino acid conformation but not the amino acid identity was allowed to vary to allow for conformational adjustments. Final experimental substitutions, shown in Table 1, were chosen based on their predicted energies relative to wild type and their occupancy, that is the number times the substitution occurred in the set of 1000 Monte Carlo or 300 genetic algorithm sequences.
TABLE 1.
Mutants designed using PDA and SPA calculations
| Position | 29 | 30 | 38 | 41 | 64 | 66 | 75 | mAb bindinga | αLI domain tetramer bindingb | %OccPDAc | %OccSPAc |
|---|---|---|---|---|---|---|---|---|---|---|---|
| % of WT | % | ||||||||||
| WT | K | L | P | E | M | Y | T | 100 | 100 | ||
| %OccPDAc | 19 | 2 | 2 | 7 | 20 | 12 | 5 | ||||
| %OccSPAc | 0 | 0 | 92 | 0 | 0 | 0 | 9 | ||||
| 1 | K | 76 ± 15 | 0 ± 5 | 1 | 51 | ||||||
| 2 | Q | 88 ± 8 | 19 ± 10 | 0 | 0 | ||||||
| 3 | L | 80 ± 6 | 154 ± 12 | 73 | 32 | ||||||
| 4 | V | 80 ± 5 | 242 ± 12 | 3 | 0 | ||||||
| 5 | I | 92 ± 7 | 187 ± 20 | 17 | 2 | ||||||
| 6 | F | 75 ± 5 | 114 ± 8 | 0 | 0 | ||||||
| 7 | D | 93 ± 6 | 55 ± 3 | 5 | 0 | ||||||
| 8 | K | 74 ± 10 | 116 ± 10 | 5 | 0 | ||||||
| 9 | Q | 75 ± 5 | 115 ± 5 | 8 | 49 | ||||||
| 10 | W | A | 1 ± 2 | 1 ± 2 | 0, 0 | 54, 54 (54) | |||||
| 11 | A | L | W | 0 ± 1 | 3 ± 7 | 0, 73, 0 | 9, 32, 5 (2) | ||||
| 12 | A | W | I | 0 ± 1 | 312 ± 9 | 0, 0, 17 | 9, 5, 2 (1) | ||||
| 13 | F | V | −1 ± 1 | 317 ± 9 | 0, 0 | 31, 0 (0) | |||||
ICAM-1 expression was determined with CBR IC1/11 mAb, which recognizes a conformation-dependent epitope in domain 3 of ICAM-1. % mAb binding = (mutant mAb MFI − background mAb MFI)/(wild-type mAb MFI − background mAb MFI) × 100.
% tetramer binding = (mutant tetramer MFI − background tetramer MFI)/(wild-type tetramer MFI − background tetramer MFI) × 100.
% occupancies from PDA and SPA calculations are provided for the WT (under the WT residue) and variants (across from each variant). % occupancy represents the % occurrence of the given amino acid in the set of 1000 output sequences from the Monte Carlo (PDA) or 300 output sequences from the Genetic Algorithm (SPA). For double and triple variants (10–13), the % occupancy is presented for each substitution consecutively separated by a comma, with the % occupancy of the combined variant provided in parentheses.
Design calculations using Rosetta differed in the all-atom energy function (22, 23), as well as in the amino acid side-chain sampling and optimization protocol. As above, two sets of design calculations were carried out to identify substitutions predicted to stabilize the ICAM-1-I-domain interface. In the first round, only single amino acid substitutions at positions Lys-29, Leu-30, Gly-32, Pro-36, Pro-38, Lys-39, Glu-41, Gln-62, Met-64, Tyr-66, Asn-68, Gly-72, Gln-73, Thr-75, and Lys-77 were modeled. Residue choices evaluated at each position were: Lys-29: Arg, Asp, Glu, and Ser; Leu-30: Asp, Glu, Arg, Lys, Tyr, Trp, and Phe; Gly-32: Ala and Ser; Pro-36: all amino acids; Pro-38: all amino acids; Lys-39: Arg; Glu-41: Arg, Lys, Asp, Ser, and Asn; Gln-62: Arg, Lys, Asp, Glu, Ser, and Asn; Met-64: Leu, Ile, Phe, Tyr, and Val; Tyr-66: Phe and His; Asn-68: Thr, Tyr, Arg, Asp, and Glu; Gly-72: Lys, Arg, Asp, Glu, Asn, Ser, Gln, and Pro; Gln-73: Thr, Asn, Ser, Arg, Asp, and Glu; Thr-75: all residues; and Lys-77: all residues. In a second round, 11 interface residues (Lys-29, Leu-30, Pro-36, Pro-38, Lys-39, Met-64, Tyr-66, Asn-68, Gln-73, Thr-75, and Lys-77) were designed (allowed to change to all 20 naturally occurring amino acids, including the native amino acid type but excluding cysteine) simultaneously. In each case, amino acid side chains contacting the substituted amino acid side chains were repacked (allowing all rotamers of the native amino acid type). Sequences and conformations with low energies were selected using a Monte-Carlo simulated annealing procedure as described previously (24). All resulting protein complex models (in the simultaneous design runs, several hundred models with similar energies were generated) were rescored by computing a predicted binding energy as described (22). Final sequences were selected for the lowest binding energy as described in the main text and are shown in Table 2.
TABLE 2.
Mutants designed with Rosetta
| Position | 30 | 36 | 38 | 39 | 64 | 68 | 75 | 77 | mAb bindinga | αLI domain tetramerbindinga | Predicted binding energy (relative to WT) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| % of WT | kcal/mol | ||||||||||
| WT | L | P | P | K | M | N | T | K | 100 | 100 | 0 |
| 1 | K | 75 ± 10 | 75 ± 9 | 31.0 | |||||||
| 2 | Q | 50 ± 12 | 35 ± 6 | 31.4 | |||||||
| 3 | R | 50 ± 15 | 50 ± 15 | 31.6 | |||||||
| 4 | F | 105 ± 8 | 138 ± 12 | 30.5 | |||||||
| 5 | Y | 90 ± 6 | 135 ± 10 | 30.5 | |||||||
| 6 | E | 100 ± 5 | 120 ± 8 | 31.5 | |||||||
| 7 | Y | 86 ± 12 | 30 ± 16 | 31.2 | |||||||
| 8 | W | 55 ± 9 | 110 ± 15 | 30.6 | |||||||
| 9 | R | 150 ± 10 | 210 ± 30 | 31.3 | |||||||
| 10 | F | 95 ± 6 | 120 ± 6 | 30.5 | |||||||
| 11 | Y | 90 ± 7 | 115 ± 8 | 30.5 | |||||||
| 12 | I | 92 ± 7 | 187 ± 20 | 30.7 | |||||||
| 13 | Y | E | W | R | F | I | F | 80 ± 12 | 10 ± 10 | 33.4 | |
See Table 1 for details.
PARE (predicting association rate enhancement) mutations were kindly provided by Dr. Gideon Schreiber and Yossi Kuttner (Weizmann Institute of Science, Rehovot, Israel) to alter electrostatic interactions outside of the binding interface to enhance kon without affecting koff (12, 25).
Construction and Expression of Mutant Libraries
cDNA of human ICAM-1 cloned in vector pAprM8 was used as the template. QuikChange (Stratagene) was used to generate single or multiple substitution mutations in the ICAM-1 D1 domain. Mutations were confirmed by DNA sequencing. Transient transfection of 293T cells was as described previously (26).
Preparation of I Domain Tetramer
A BirA enzyme recognition tag (LGGIFEAMKMELRD) was fused through a Gly-Gly-Gly-Ser linker to the N terminus of the soluble HA mutant αL I domain (residues Gly-128 to Tyr-307, mutant K287C/K294C) (9, 27–29). The cDNA was cloned into the NdeI and BamHI sites of the pET-20b vector. Protein was expressed in Escherichia coli BL21(DE3) (Novagen). The transformed bacteria were cultured in rich media (20 g/liter Tryptone, 10 g/liter yeast extract, 5 g/liter NaCl, 20 ml/liter glycerol, 50 mm K2HPO4, 10 mM MgCl2, 10 g/liter glucose, 100 mg/liter ampicillin). Expression was induced by addition of isopropyl 1-thio-β-d-galactopyranoside (Invitrogen) to a final concentration of 1 mm when the A600 of cultures was 0.6–1.0. The I domain was refolded and purified as described (9) with some modifications in the step of refolding. Briefly, frozen cells were resuspended in 20 mm Tris (pH 8.0), 150 mm NaCl, with 1 mg/ml lysozyme at 37 °C for 15 min and then disrupted by ultrasonication. Inclusion bodies were harvested by centrifugation. After extensive washing with washing buffer (20 mm Tris (pH 8.0), 23% (w/v) sucrose, 0.5% (v/v) Triton X-100, 1 mm EDTA), the pellet was solubilized by adding 6 m guanidine HCl, 50 mm Tris (pH 8.0), 1 mm dithiothreitol. The I domain was rapidly diluted in a redox buffer (20 mm Tris (pH 8.0), 100 mm NaCl, 5% glycerol, 5 mm cysteamine/2.5 mm cystamine) to a final concentration of 25 μg/ml and then incubated at 4 °C with slow stirring. Refolding was performed for 4–5 days until no free thiol group was detected with 5,5′-dithio-bis-(2-nitrobenzoic acid) (Pierce). The I domain was precipitated with ammonium sulfate and purified by Superdex S-200 gel-filtration in phosphate-buffered saline. Biotin was ligated to the tag with the BirA ligase (Avidity, Denver, CO). Typically, I domain at a concentration of 1–2 mg/ml was incubated with BirA (15–20 μg/ml) at room temperature overnight in a buffer containing 20 mm Tris (pH 8.0), 100 mm NaCl, 10 mm magnesium acetate, 50 mm Bicine (pH 8.3), 10 mm ATP, and 50 μm biotin. The unbound biotin was removed by passing the sample through a Superdex S-200 column (Amersham Biosciences). The biotinylated I domain was mixed with streptavidin-FITC (BIOSOURCE International) or streptavidin (BIO-SOURCE International) at a molar ratio of 8:1 for 2 h at 21 °C. The mixture was then subjected to Superdex S-200 chromatography in TBS (20 mm Tris·HCl, pH 8.0, 150 mm NaCl) to separate the tetrameric complex peak from free I domain.
Immunofluorescent Flow Cytometry
Plasmids encoding full-length ICAM-1 were transfected into 293T cells using calcium phosphate precipitation (26). The mAbs RR1/1, CBR IC1/11, CBR IC1/12, and CA-7 were used to stain transfected cells as described (30). For soluble I domain tetramer binding, the ICAM-1-transfected cells were washed once with Hanks’ Balanced Salt solution (Invitrogen), 10 mm EDTA, and twice with 20 mm Hepes (pH 7.4), 150 mm NaCl (HBS)/5.5% glucose/1% bovine serum albumin. Cells were incubated with streptavidin-I domain tetramer (2 μg/ml)/HBS/5.5% glucose/1% bovine serum albumin in the presence of 1 mm Mg2+ at room temperature for 1 h, and washed three times with HBS/1 mm Mg2+. FITC-goat anti-streptavidin (10 μg/ml, Vector Laboratories) and Cy3-labeled CBR IC1/11 (10 μg/ml), a nonblocking antibody to LFA-1 mapped to domain 3 of ICAM-1 (31), were added and incubated with the cells on ice for 15 min. After washing three times with HBS/1 mm Mg2+, cells were subjected to fluorescence flow cytometry. Mean FITC fluorescence intensity (MFI) of Cy3-positive cells was determined. In all experiments, background CBR IC1/11 binding to mock transfected cells and background tetramer binding to ICAM-1-transfected cells in the presence of 5 mm EDTA were subtracted to obtain specific ICAM-1 expression and ligand binding, respectively: % binding = (mutant MFI − background MFI)/(wild-type MFI − background MFI) × 100.
Preparation of Soluble ICAM-1
cDNA of the extracellular domain of wild-type or mutant ICAM-1 was fused to pEF-Fc vector (32, 33) with a thrombin cleavage site (LVPRGS) between ICAM-1 and human IgG Fc portions. Culture supernatants from GnTI− HEK293S cells (34, 35) were collected 5–7 days after transient transfection. ICAM-1 Fc fusion proteins were purified from supernatants on a protein G column (Invitrogen). Contaminating IgGs were removed by gel filtration on a Superdex 200 column (Amersham Biosciences) in TBS (20 mm Tris·HCl, pH 8.0, 150 mm NaCl). ICAM-1 Fc eluted earlier than IgGs. After thrombin cleavage (1 mg of protein/20 units of thrombin, Amersham Biosciences) at room temperature overnight, soluble ICAM-1 D1–5 was further purified by gel filtration to remove the Fc portion and trace amounts of thrombin. Typical yield was 5 mg/liter.
Antibody Binding Assay
Antibody-binding assay for soluble ICAM-1-Fc was performed as described (36) except using goat anti-human IgG (I-3391, Sigma) as the capture antibody.
Surface Plasmon Resonance
ICAM surfaces or a control surface were prepared by injecting ICAMs (20 μg/ml) in 10 mm sodium acetate buffer (pH 4.0) or buffer only (control) over the flow cells activated with N-ethyl-N′-(3-dimethylaminopropyl)carbodiimide hydrochloride/N-hydroxysuccinimide and then blocking these surfaces with ethanolamine. To prepare I domain surfaces, biotinylated HA I domain (20 μg/ml) or 5 μm biotin as control in 10 mm sodium acetate buffer (pH 4.0) was directly captured on a streptavidin-conjugated sensor chip (Biacore) using 10 mm HEPES (pH 7.4), 150 mm NaCl (HBS-N, Biacore) as running buffer. I domains or ICAMs were infused in 20 mm Tris (pH 8.0), 150 mm NaCl, 1 mm MgCl2, and regeneration was done in 20 mM Tris (pH 8.0), 0.3 m NaCl, 20 mm EDTA. The KD value was calculated by curve fitting with Langmuir 1:1 binding model or Scatchard analysis if the binding reached steady-state. koff was derived from curve fitting on the dissociation phases. kon was calculated as koff/KD.
Soluble ICAM-1 Binding
Binding of soluble, multimeric ICAM-1-IgA-Fc/FITC-anti-IgA to K562 cells expressing wild-type LFA-1 was as described previously (37). For competition assay, the Hi3-ICAM-1 mutant or wild-type ICAM-1 D1–D5 was mixed with ICAM-1-IgA-Fc (5 μg/ml)/FITC-anti-IgA (25 μg/ml) at a series of concentrations and then incubated with the cells at room temperature for 1 h.
Cell Adhesion to Immobilized ICAM-1
Interleukin-15 cultured peripheral blood lymphocytes were prepared and maintained as described (38). Adhesion of peripheral blood lymphocytes in the presence of phorbol 12-myristate 13-acetate (1 μm) to wild-type ICAM-1 was performed in the presence of the indicated concentration of ICAM-1 D1–D5 as described (36). Cell adhesion in the presence of ICAM-1-blocking mAb was <5% of input cells.
RESULTS
Design of High Affinity ICAM-1 Mutants
We used multiple rational design methods to design ICAM-1 mutants with increased affinity for LFA-1. The 3.3-Å crystal structure of αL I domain in the complex with ICAM-1 (8) was used as a starting template (Fig. 1A).
FIGURE 1. The interface between the αL I domain and ICAM-1.
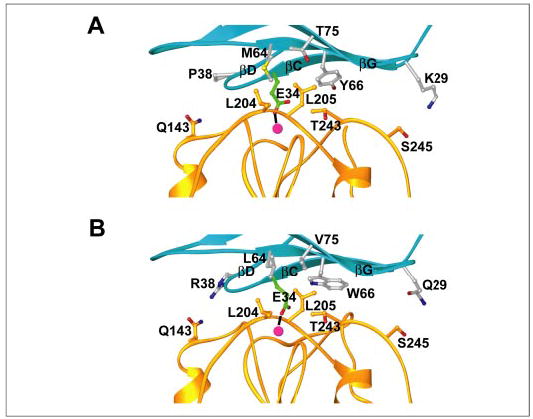
A, the αL I domain in complex with wild-type ICAM-1 (PDB number 1MQ8). B, the complex with the Hi3 ICAM-1 mutant. Model was made with Model Mutant in Genemine (Molecular Applications Group). Ribbons of ICAM-1 D1 and the αL I domain are shown in cyan and gold, respectively. Side chains of residues of ICAM-1 that were mutated in Hi3 are shown in silver, and Glu-34 is shown in green. Nitrogen and oxygen atoms are in blue and red, respectively. The Mg2+ ion is shown as a magenta sphere. Interacting residues on the I domain are shown in gold. This figure was prepared with RIBBONS (47).
One set of variants was designed using Protein Design Automation (PDA) (13, 17, 18) and Sequence Prediction Algorithm (SPA) (19) calculations to optimize the free energy of the ICAM-1·LFA-1 complex. To preserve key contacts at the ICAM-1/LFA-1 interface, Glu-34, Lys-39, Asn-68, and Gln-73 of ICAM-1 were fixed as wild-type in the calculations (8). Glu-34 coordinates the Mg2+ ion of the αL I domain, and Lys-39 forms salt bridges with Glu-241 and His-264 of the I domain that appear to be indirectly important for metal chelation. ICAM-1 Asn-68 and Gln-73 form a well ordered H-bonding network with the LFA-1 Thr-243 backbone carbonyl and the side chain of Asn-207. All other residues at or near the binding interface were allowed to mutate, including Lys-29, Leu-30, Pro-38, Glu-41, Met-64, Tyr-66, and Thr-75. The rotamers of all residues in contact with mutated residues were allowed to vary for conformational adjustments. Experimental variants (Table 1) were chosen based on occupancy in PDA and/or SPA calculations (see “Experimental Procedures”) and visual inspection of output model structures. In a number of cases, similar residues were substituted for functional or biophysical reasons: Gln was used instead of Glu (%Occ SPA =8) at position 64 due to the lack of a visible interacting residue on LFA-1 yet in proximity to the negative and critical Glu-34 on ICAM-1; Val was included at position 75 because of similarity to the isosteric Thr (wild type) and high occupancy of Ile (PDA and SPA); Phe was included at position 75 because of a preference for hydrophobics in the calculations; Val was used at position 66 instead of the isosteric Thr (all 31% Phe-30 predictions occurred with Thr-66), because of the otherwise complete preference for hydrophobics despite the fact that the predicted γ-hydroxyl H-bond was intramolecular.
Predictions made by Rosetta (10, 23, 24) are summarized in Table 2. In this case, all residues forming contacts in the ICAM-1/LFA-1 interface except the metal coordinating Glu-34 residue were redesigned, allowing either single substitutions, or simultaneous design of the entire interface (see “Experimental Procedures”). Substitutions were selected based on a predicted increase in binding affinity of the protein·protein complex (22). All single substitutions with a predicted interface stabilization of more than 0.6 kcal/mol lower in energy than wild type (see “Experimental Procedures” for residue choices at each position) were tested experimentally, except for positions 64 and 77, where the mutations with the largest effect were predicted to stabilize the interface by 0.5 kcal/mol (Table 2).
We also designed mutations based on inspection of the complex structure (Table 3). Furthermore, charged substitutions were also predicted that aimed to enhance electrostatic attraction and increase kon by mutating residues outside of the interface (Table 3) (12, 25).
TABLE 3. Mutants from expert design (variants 1–10) and PARE (variants 11–12).
Expert design was based on visualization of the structure. To alter electrostatic interactions outside of the binding interface to enhance kon without affecting koff , PARE (predicting association rate enhancement) was used.
| Position | 30 | 38 | 41 | 59 | 64 | 66 | 75 | mAb bindinga | αLI domain tetramer bindinga |
|---|---|---|---|---|---|---|---|---|---|
| % of WT | |||||||||
| WT | L | P | E | E | M | Y | T | 100 | 100 |
| 1 | L | 80 ± 6 | 154 ± 12 | ||||||
| 2 | F | 80 ± 6 | 180 ± 10 | ||||||
| 3 | W | 61 ± 12 | 245 ± 15 | ||||||
| 4 | I | 92 ± 7 | 187 ± 20 | ||||||
| 5 | V | 80 ± 5 | 242 ± 12 | ||||||
| 6 | F | 93 ± 6 | 25 ± 7 | ||||||
| 7 | Y | 86 ± 12 | 30 ± 16 | ||||||
| 8 | W | 55 ± 9 | 110 ± 15 | ||||||
| 9 | E | 90 ± 6 | 120 ± 5 | ||||||
| 10 | R | 90 ± 6 | 120 ± 6 | ||||||
| 11 | K | 85 ± 5 | 116 ± 10 | ||||||
| 12 | K | 90 ± 6 | 105 ± 9 | ||||||
See Table 1 for details.
Experimental Screen of Mutant Library
To establish an assay appropriate for screening a large number of ICAM-1 mutants expressed on the surface of 293T cell transfectants, we prepared a soluble multimeric HA αL I domain as described under “Experimental Procedures,” because the monomeric I domain binding was too weak for robust detection of ICAM-1 transfectants using flow cytometry (Fig. S1A). The N-terminal biotin tag did not affect the monomeric affinity of the HA αL I domain mutant for ICAM-1 as measured by SPR (Fig. S1B). Monomeric and tetrameric I domain bound in the presence of Mg2+ to immobilized ICAM-1, and binding was abolished by EDTA (Fig. S1, B and C). This specific, Mg2+-dependent binding is as previously described for binding of the αL I domain to ICAM-1 (9), in agreement with the central location of the MIDAS in the binding site (Fig. 1A).
Two color immunofluorescent flow cytometry of mutant ICAM-1 transfectants was used to simultaneously measure binding to I domain tetramer using FITC-anti-streptavidin and to measure ICAM-1 surface expression using Cy3-labeled mAb CBR IC1/11 to domain 3 of ICAM-1 (31) (Fig. S1D). All of the single mutants were expressed at levels within 50–150% of wild-type levels (Tables 1–3), whereas simultaneous mutation of two or three residues (Table 1) or seven residues (Table 2) was much less successful. Six single ICAM-1 mutants showed enhanced I domain tetramer binding of 150–250% relative to wild-type ICAM-1. These mutants were M64L (Tables 1 and 3), T75V (Tables 1 and 3), T75I (Tables 1–3), P38R (Table 2), and Y66F and Y66W (Table 3). Another predicted mutant, K29Q, showed binding relative to wild type of 115% (Table 1). The monomeric affinity of a representative mutant was tested by SPR. The affinity of the T75V mutant for the HA αL I domain was increased 2.7-fold (Table 4), in good agreement with the results of the tetramer binding assay.
TABLE 4.
Affinity of ICAM-1 mutants measured with SPR
| Immobilized ligand | Analyte | kon | koff | KDa | Increase in affinity |
|---|---|---|---|---|---|
| M−1 s−1 × 10−4 | s−1× 102 | nM | |||
| HA I domain | WT-ICAM-1 | 6.0 ± 0.75 | 2.50 ± 0.18 | 277 ± 40 | 1 |
| HA I domain | T75V-ICAM-1 | 10.5 ± 0.61 | 1.60 ± 0.30 | 102 ± 27 | 2.7 |
| HA I domain | Hi1-ICAM-1 | 10.7 ± 1.00 | 0.75 ± 0.035 | 47 ± 2.2 | 5.9 |
| WT-ICAM-1 | HA I domain | 7.2 ± 0.50 | 1.88 ± 0.27 | 263 ± 59 | 1 |
| Hi1-ICAM-1 | HA I domain | 9.1 ± 1.50 | 0.39 ± 0.09 | 43 ± 2.0 | 6.1 |
| Hi2-ICAM-1 | HA I domain | 11.1 ± 2.00 | 0.21 ± 0.06 | 19 ± 3.0 | 13.8 |
| Hi3-ICAM-1 | HA I domain | 16.7 ± 1.60 | 0.20 ± 0.05 | 12 ± 3.0 | 21.9 |
| WT-ICAM-1 | IA I domain | 11.6 ± 1.05 | 42.0 ± 5.0 | 3600 ± 500 | 1 |
| Hi1-ICAM-1 | IA I domain | 19.8 ± 0.60 | 8.1 ± 0.3 | 410 ± 60 | 8.8 |
| Hi2-ICAM-1 | IA I domain | 21.7 ± 3.60 | 6.0 ± 0.9 | 276 ± 25 | 13.0 |
| Hi3-ICAM-1 | IA I domain | 31.9 ± 2.60 | 6.0 ± 0.8 | 188 ± 30 | 19.1 |
The KD value was determined from Scatchard plots of the steady-state equilibrium response levels when ICAMs were immobilized on the chip. When the I domain was immobilized, the KD value was calculated from curve fitting using 1:1 Langmuir model because steady-state equilibrium was not reached. koff was derived from curve fitting to the post-injection part of the sensorgrams. kon was calculated as koff/KD. The values are mean ± S.D. for three separate experiments.
Combining Single Mutations for Higher Affinity
The locations of affinity-enhancing single mutations were examined with molecular graphics. Met-64, Tyr-66, and Thr-75 are located at the center of the binding interface and form van der Waals contacts with one another. Pro-38 is on the CD loop, and is essentially structurally uncoupled to Met-64, Tyr-66, and Thr-75 (Fig. 1A). Lys-29 is also uncoupled from Met-64, Tyr-66, and Thr-75, at the opposite side of the interface in the BC loop (Fig. 1A). The three residues in contact with one another were introduced simultaneously, and the non-interacting residues were subsequently introduced one by one (Table 5). The ICAM-1 mutant of the three contacting residues M64L/Y66W/T75V (Hi1) was enhanced in affinity for the HA αL I domain by 6-fold (Table 4 and Fig. 2). Similar results were obtained with either the αL I domain or the ICAMs immobilized on the SPR chip (Table 4). The decrease in koff was greater than the increase in kon. Addition of the P38R mutation in the Hi2 mutant (Table 5) resulted in a further decrease in koff (Table 4 and Fig. 2). The total increase in affinity compared with wild-type ICAM-1 was 14-fold. Further addition of the E41K mutation markedly lowered expression (not shown). By contrast, addition of the K29Q mutation in Hi3 (Table 5) had no deleterious effect on expression and showed a 22-fold higher affinity for the HA I domain than wild-type ICAM-1 (Table 4 and Fig. 2). Binding of the Hi1, Hi2, and Hi3 ICAM-1 mutants to an IA I domain mutant revealed 9-, 13-, and 19-fold affinity increases, respectively (Table 4). These increases are very similar to those observed with the HA I domain.
TABLE 5.
Hi ICAM-1 mutants
|
Position |
|||||
|---|---|---|---|---|---|
| 29 | 38 | 64 | 66 | 75 | |
| WT | K | P | M | Y | T |
| Hi1 | L | W | V | ||
| Hi2 | R | L | W | V | |
| Hi3 | Q | R | L | W | V |
FIGURE 2. Affinity measurements using SPR.
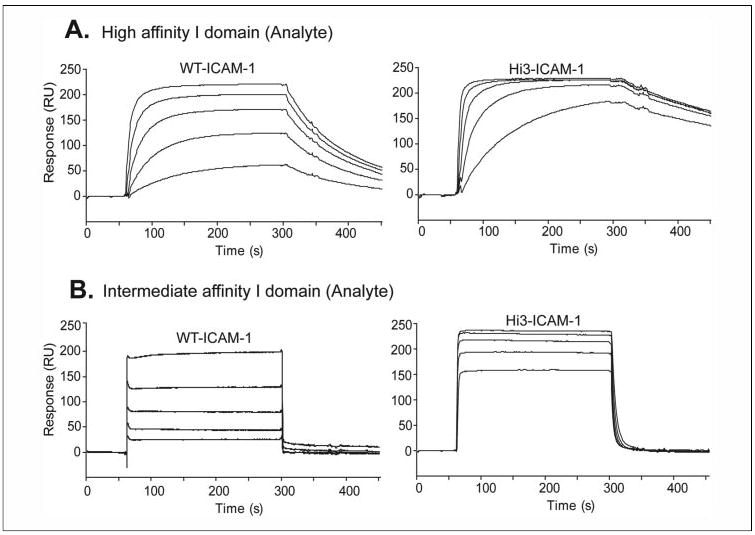
A, wild-type ICAM-1 or Hi3 ICAM-1 was immobilized on the sensor chip. HA αL I domain was injected at 250, 111, 49, 22, and 10 nM in the curves shown top to bottom. B, IA αL I domain was injected at 5, 2.2, 1, 0.45, and 0.19 μM in the curves shown top to bottom.
Structural Integrity and Inhibitory Effect of Hi3-ICAM-1
Consistent with the increased expression on the cell surface of the P38R mutant (Table 2), addition of this mutation in the Hi2-ICAM-1 and Hi3-ICAM-1 mutants increased secretion relative to Hi1-ICAM-1 (Fig. 3A). Hi3-ICAM-1 reacted with a panel of mAbs to ICAM-1 that do not block binding to integrin αLβ2. CBR IC1/11, CBR IC1/12, and CA-7, which recognize epitopes in D3, D4, and D5 of ICAM-1, reacted with Hi3 as well as wild-type ICAM-1 (Fig. 3B), suggesting that the structural integrity of the ICAM-1 mutant was not disturbed by these mutations. By contrast, binding of function-blocking mAb RR1/1 to Hi3-ICAM-1, but not to Hi2-ICAM-1 and Hi1-ICAM-1, was significantly decreased by 70% (Fig. 3B). This finding suggests that Lys-29 is part of the RR1/1 epitope, in agreement with earlier results (39).
FIGURE 3. Characterization of Hi1-, Hi2-, and Hi3-ICAM-1_Fc mutants, and inhibition of soluble ligand binding and adhesion by Hi3-ICAM-1 D1–D5.

A, supernatants from 293S cells transfected with the same amount of the indicated constructs were captured by goat anti-human IgG and detected in enzyme-linked immunosorbent assay with mAb CBR IC1/11. B, purified ICAM-1_Fc preparations (1 μg/ml) were captured as described in A and subjected to enzyme-linked immunosorbent assay with mAbs RR1/1, CBR IC1/11, CBR IC1/12, and CA-7. Results are presented as the percentage of binding to wild-type ICAM-1_Fc. C, inhibition by soluble, monomeric ectodomain fragments of Hi3 or wild-type ICAM-1 of binding of soluble, multim-eric ICAM-1/IgA-Fc to αLβ2 on K562 cells in the presence of 1 mm Mn2+ D, inhibition by soluble Hi3 or wild-type ICAM-1 D1–D5 of binding of phorbol 12-myristate 13-acetate-stimulated peripheral blood lymphocytes to immobilized wild-type ICAM-1 in the presence of 5 mm Mg2+ Results are mean ± S.D. of multiple experiments (n = 2– 4)
To examine antagonism, we tested inhibition by Hi3-ICAM-1 D1–D5 of binding of soluble, multimeric ICAM-1/IgA-Fc to αLβ2-expressing stable K562 cells in the presence of 1 mm Mn2+ (Fig. 3C). Soluble Hi3-ICAM-1 D1–D5 inhibited this interaction with an IC50 value of 0.2 μm. Inhibition by wild-type ICAM-1 D1–D5 was much weaker, with an IC50 value of 10 μM (Fig. 3C). Furthermore, Hi3-ICAM-1 D1–D5 antagonized adhesion of peripheral blood lymphocytes activated by phorbol 12-myristate 13-acetate to immobilized ICAM-1 more effectively than wild-type ICAM-1 D1–D5 (Fig. 3D). The IC50 values in this assay were 0.05 μm and 15 μm for Hi3 and wild-type ICAM-1, respectively.
DISCUSSION
In this study, a combined methodology of structure-based rational design and empirical testing has resulted in a substantial increase in affinity of an adhesion molecule. The use of computational structure-based methods was directed toward two goals: directly optimizing the ICAM-1-LFA-1 interaction and ensuring that mutations resulted in stable, properly folded proteins. The ability of computational methods to enrich the hit rate of variant libraries (40) was particularly important in this study because of the need for mammalian expression and the low throughput nature of the screen. Also critical was an efficient yet sensitive screen that allowed us to distinguish true positives. Designed libraries were reduced to a desired size according to our experimental capacity, and the subsequent I domain tetramer binding screen allowed us to distinguish which of these mutations improved binding. Combining successful single amino acid substitutions yielded the Hi1, Hi2, and Hi3 series of ICAM-1 mutants with substantially enhanced affinity for the αL I domain. This serial approach to generation of multiple variants was more successful than their design de novo, which typically resulted in much lower levels of expression relative to wild-type ICAM-1 (Tables 1 and 2). The success of our predictions, despite the lack of atomic resolution (3.3 Å) of the complex structure, suggests that our computational methods are robust with respect to structural quality. Furthermore, the distinct PDA and Rosetta computational methods each predicted some successful substitutions not found by the other, which when combined yielded increased affinity. The particular receptor-ligand pair studied here was not well suited for prediction of association rate enhancement, because no electrostatic hotspots susceptible to substantial rate enhancement at physiologic ionic strength were found. The successfully predicted stabilizing mutations in Tables 1 and 2 are almost exclusively non-polar or aromatic, suggesting that packing interactions are predicted more accurately than electrostatic or polar interactions. It is further emphasized that experimental screening of large combinatorial libraries was not required, an advantage of our engineering approach over display methodologies, which are not well developed for mammalian expression systems.
Our kinetic measurements together with the 3.3-Å structure of the interface of the αL I domain with ICAM-1 (8) and the more recently determined 1.65-Å structure of the closely related interface with ICAM-3 (41) provide insight into the effects of the mutations. The affinity of Hi1-ICAM-1 (M64L, Y66W, and T75V), relative to wild type, was increased by 7.4 ± 1.3-fold (average for the HA and IA I domains plus or minus the difference from the mean) (Table 4). The mutations primarily affected koff (5.0 ± 0.2-fold decrease) rather than kon (1.5 ± 0.2-fold increase) (Table 4). These results suggest that the mutations contribute directly to the protein-protein interaction interface rather than promoting initial contact (42–44). Leu-64, Trp-66, and Val-75 are predicted to pack favorably against the αL I domain (Fig. 1B), providing lower free energy and driving tighter binding than Met-64, Tyr-66, and Thr-75 in wild-type ICAM-1 (Fig. 1A). These substitutions increase the hydrophobicity of the region surrounding the metal coordinating residue, Glu-34, suggesting that the affinity increase is due to the strengthened nonpolar environment at the center of the interface. The recent 1.65-Å structure of the HA αL I domain in complex with ICAM-3 (41) shows less hydrophobicity surrounding the metal-coordinating residue Glu in ICAM-3 than in ICAM-1. Comparison of the ICAM-1-αL and ICAM-3-αL complex structures and the ICAM-2 structure shows a rank order of hydrophobicity in this interface, with ICAM-1 > ICAM-2 > ICAM-3. The affinity parameters for HA αL I domain binding to ICAM-1, ICAM-2, and ICAM-3 follow exactly the same order, with KD = 173 nM, 605 nM, and 4320 nM, respectively (9).
Pro-38 and Lys-29 are located in the CD and BC loops in the periphery of the interface (Fig. 1A). Whereas the complex structure shows no interaction of Pro-38 in wild-type ICAM-1 with the I domain, the much longer and polar side chain of Arg-38 may be able to interact with αL Gln-143 through hydrogen bonding (Fig. 1B). Intriguingly, the mutation increases cell surface expression, which generally correlates with greater stability (Table 2). In agreement with this, Pro-38 has non-optimal Phi-Psi angles that place it in the generously allowed region in a Ramachandran plot (not shown). However, an effect of backbone conformation alone on affinity in the P38R mutation is not supported by the failure of the P38D and P38E substitutions to increase affinity (Tables 1–3). The affinity enhancement by P38R in Hi2 compared with Hi1 stemmed primarily from the decrease in koff (1.6 ± 0.26-fold) rather than the increase in kon (1.16 ± 0.06-fold) (Table 4). By contrast, the K29Q substitution in Hi3 compared with Hi2 enhanced solely kon (1.5 ± 0.02-fold) and not koff (1.0 ± 0.02-fold). The shorter side chain of the glutamine was predicted by PDA to interact better with Ser-245 of the αL I domain; the mechanism by which this may benefit association rate is unclear (Fig. 1B).
The affinity increase of our ICAM-1 mutants was not biased by the particular αL I domain mutants that were used in measurements. Thus, the affinity of Hi3 for HA and IA I domain mutants was increased proportionally by 21.9-fold and 19.1-fold, respectively. These data suggest that the same conformation of the IA and HA mutant αL I domain binds to ICAM-1 and that the HA and IA mutations just determine the population of the states, i.e. at any given time a much higher percentage of the HA mutant molecules than the IA mutant molecules populate the open, ligand binding conformation. The results agree with the findings that the IA mutant has the same open conformation as the HA αL I domain mutant when bound to ICAM-1 (8).
Our high affinity ICAM-1 mutant could be potentially useful for many biological and therapeutic applications. For example, the Hi3 ICAM-1 mutant bound to HA and IA αL I domain mutants with KD values of 12 nm and 188 nm, respectively. This places the hypothetical physiologic intermediate affinity state of integrin αLβ2 on cell surfaces (8, 9, 45) within the range of affinities for Hi3-ICAM-1 that would be accessible by saturation binding assays with whole cells. Thus, we may be able to differentiate high and intermediate affinity states of αLβ2 and measure dynamic conformational transitions of αLβ2 on cell surfaces. Furthermore, our high affinity ICAM-1 mutant is a potential biotherapeutic agent and could serve as a lead molecule to develop novel antagonists that can bind to αLβ2 like ICAM-1. Inhibition of the binding of soluble ICAM-1/IgA-Fc to αLβ2 on the cell surface and cell adhesion through ICAM-1 to αLβ2 by the Hi3-ICAM-1 mutant revealed significant potency. There has been much interest in the rational development of αLβ2 antagonists based on the structure of ICAM-1; i.e. competitive antagonists. However, each of the two classes of antagonists developed to date is allosteric and binds to sites distinct from the ICAM-1 binding site (5). A new class of direct I domain antagonists remains to be developed.
Overall, our approach results in rapid discovery of novel mutants with targeted properties, allowing incorporation of information from structural and experimental data, and accessing broad sequence diversity. Because the affinity of cell surface adhesion receptors is generally low compared with the affinity of receptors for soluble ligands (46), this approach should be applicable to the design of other high affinity adhesive molecules for biological and therapeutic studies.
Supplementary Material
Acknowledgments
We thank Dr. Gideon Schreiber and Yossi Kuttner for calculations with the program PARE and Dr. Philip J. Reeves for the HEK293S GnTI− cell line.
Footnotes
This work was supported by National Institutes of Health Grant CA31798.
The on-line version of this article (available at http://www.jbc.org) contains Fig. S1.
The abbreviations used are: ICAM-1, intercellular adhesion molecule-1; LFA-1, lymphocyte function-associated antigen-1; PDA, protein design automation; SPA, sequence prediction algorithm; SPR, surface plasmon resonance; PARE, predicting association rate enhancement; MFI, mean fluorescence intensity; HA, high affinity; IA, intermediate affinity; Bicine, N,N-bis(2-hydroxyethyl)glycine; FITC, fluorescein isothiocyanate; mAb, monoclonal antibody; HBS, Hanks’ balanced salt solution.
References
- 1.Springer TA. Nature. 1990;346:425–433. doi: 10.1038/346425a0. [DOI] [PubMed] [Google Scholar]
- 2.Springer TA. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 3.Dustin, M. L., and Springer, T. A. (1999) in Guidebook to the Extracellular Matrix and Adhesion Proteins (Kreis, T., and Vale, R., eds) 2nd. Ed., pp. 216–220, Sambrook and Tooze, New York
- 4.Shimaoka M, Takagi J, Springer TA. Annu Rev Biophys Biomol Struct. 2002;31:485–516. doi: 10.1146/annurev.biophys.31.101101.140922. [DOI] [PubMed] [Google Scholar]
- 5.Shimaoka M, Springer TA. Nat Rev Drug Disc. 2003;2:703–716. doi: 10.1038/nrd1174. [DOI] [PubMed] [Google Scholar]
- 6.Curley GP, Blum H, Humphries MJ. Cell Mol Life Sci. 1999;56:427–441. doi: 10.1007/s000180050443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang C, Zang Q, Takagi J, Springer TA. J Biol Chem. 2000;275:21514–21524. doi: 10.1074/jbc.M002286200. [DOI] [PubMed] [Google Scholar]
- 8.Shimaoka M, Xiao T, Liu JH, Yang Y, Dong Y, Jun CD, McCormack A, Zhang R, Joachimiak A, Takagi J, Wang Jh, Springer TA. Cell. 2003;112:99–111. doi: 10.1016/s0092-8674(02)01257-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shimaoka M, Lu C, Palframan R, von Andrian UH, Takagi J, Springer TA. Proc Natl Acad Sci U S A. 2001;98:6009–6014. doi: 10.1073/pnas.101130498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kortemme T, Baker D. Curr Opin Chem Biol. 2004;8:91–97. doi: 10.1016/j.cbpa.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 11.Lazar GA, Marshall SA, Plecs JJ, Mayo SL, Desjarlais JR. Curr Opin Struct Biol. 2003;13:513–518. doi: 10.1016/s0959-440x(03)00104-0. [DOI] [PubMed] [Google Scholar]
- 12.Selzer T, Albeck S, Schreiber G. Nat Struct Biol. 2000;7:537–541. doi: 10.1038/76744. [DOI] [PubMed] [Google Scholar]
- 13.Luo P, Hayes RJ, Chan C, Stark DM, Hwang MY, Jacinto JM, Juvvadi P, Chung HS, Kundu A, Ary ML, Dahiyat BI. Protein Sci. 2002;11:1218–1226. doi: 10.1110/ps.4580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shimaoka M, Shifman JM, Jing H, Takagi J, Mayo SL, Springer TA. Nat Struct Biol. 2000;7:674–678. doi: 10.1038/77978. [DOI] [PubMed] [Google Scholar]
- 15.Martin L, Stricher F, Misse D, Sironi F, Pugniere M, Barthe P, Prado-Gotor R, Freulon I, Magne X, Roumestand C, Menez A, Lusso P, Veas F, Vita C. Nat Biotechnol. 2003;21:71–76. doi: 10.1038/nbt768. [DOI] [PubMed] [Google Scholar]
- 16.Sarkar CA, Lowenhaupt K, Horan T, Boone TC, Tidor B, Lauffenburger DA. Nat Biotechnol. 2002;20:908–913. doi: 10.1038/nbt725. [DOI] [PubMed] [Google Scholar]
- 17.Dahiyat BI, Mayo SL. Protein Sci. 1996;5:895–903. doi: 10.1002/pro.5560050511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dahiyat BI, Mayo SL. Science. 1997;278:82–87. doi: 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]
- 19.Raha K, Wollacott AM, Italia MJ, Desjarlais JR. Protein Sci. 2000;9:1106–1119. doi: 10.1110/ps.9.6.1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dunbrack RL, Jr, Cohen FE. Protein Sci. 1997;6:1661–1681. doi: 10.1002/pro.5560060807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mendes J, Baptista AM, Carrondo MA, Soares CM. Proteins. 1999;37:530–543. doi: 10.1002/(sici)1097-0134(19991201)37:4<530::aid-prot4>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 22.Kortemme T, Baker D. Proc Natl Acad Sci U S A. 2002;99:14116–14121. doi: 10.1073/pnas.202485799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kortemme T, Morozov AV, Baker D. J Mol Biol. 2003;326:1239–1259. doi: 10.1016/s0022-2836(03)00021-4. [DOI] [PubMed] [Google Scholar]
- 24.Kuhlman B, Baker D. Proc Natl Acad Sci U S A. 2000;97:10383–10388. doi: 10.1073/pnas.97.19.10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Selzer T, Schreiber G. J Mol Biol. 1999;287:409–419. doi: 10.1006/jmbi.1999.2615. [DOI] [PubMed] [Google Scholar]
- 26.Lu C, Springer TA. J Immunol. 1997;159:268–278. [PubMed] [Google Scholar]
- 27.Altman JD, Moss PAH, Goulder JR, Barouch DH, McHeyzer-Williams MG, Bell JI, McMichael AJ, Davis MM. Science. 1996;274:94–97. doi: 10.1126/science.274.5284.94. [DOI] [PubMed] [Google Scholar]
- 28.Crawford F, Kozono H, White J, Marrack P, Kappler J. Immunity. 1998;8:675–682. doi: 10.1016/s1074-7613(00)80572-5. [DOI] [PubMed] [Google Scholar]
- 29.Schatz PJ. Biotechnology. 1993;11:1138–1143. doi: 10.1038/nbt1093-1138. [DOI] [PubMed] [Google Scholar]
- 30.Lu C, Oxvig C, Springer TA. J Biol Chem. 1998;273:15138–15147. doi: 10.1074/jbc.273.24.15138. [DOI] [PubMed] [Google Scholar]
- 31.Parkos CA, Colgan SP, Diamond MS, Nusrat A, Liang TW, Springer TA, Madara JL. Mol Med. 1996;2:489–505. [PMC free article] [PubMed] [Google Scholar]
- 32.Tian L, Yoshihara Y, Mizuno T, Mori K, Gahmberg CG. J Immunol. 1997;158:928–936. [PubMed] [Google Scholar]
- 33.Tamada A, Yoshihara Y, Mori K. Neuroscience. 1998;240:163–166. doi: 10.1016/s0304-3940(97)00951-8. [DOI] [PubMed] [Google Scholar]
- 34.Reeves PJ, Callewaert N, Contreras R, Khorana HG. Proc Natl Acad Sci U S A. 2002;99:13419–13424. doi: 10.1073/pnas.212519299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reeves PJ, Kim JM, Khorana HG. Proc Natl Acad Sci U S A. 2002;99:13413–13418. doi: 10.1073/pnas.212519199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jun CD, Shimaoka M, Carman CV, Takagi J, Springer TA. Proc Natl Acad Sci U S A. 2001;98:6830–6835. doi: 10.1073/pnas.121186998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shimaoka M, Salas A, Yang W, Weitz-Schmidt G, Springer TA. Immunity. 2003;19:391–402. doi: 10.1016/s1074-7613(03)00238-3. [DOI] [PubMed] [Google Scholar]
- 38.Kim M, Carman CV, Yang W, Salas A, Springer TA. J Cell Biol. 2004;167:1241–1253. doi: 10.1083/jcb.200404160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Staunton DE, Dustin ML, Erickson HP, Springer TA. Cell. 1990;61:243–254. doi: 10.1016/0092-8674(90)90805-o. [DOI] [PubMed] [Google Scholar]
- 40.Marshall SA, Lazar GA, Chirino AJ, Desjarlais JR. Drug Discov Today. 2003;8:212–221. doi: 10.1016/s1359-6446(03)02610-2. [DOI] [PubMed] [Google Scholar]
- 41.Song G, Yang Y, Liu Jh, Casasnovas J, Shimaoka M, Springer TA, Wang Jh. Proc Natl Acad Sci U S A. 2005;102:3366–3371. doi: 10.1073/pnas.0500200102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu LC, Tuot DS, Lyons DS, Garcia KC, Davis MM. Nature. 2002;418:552–556. doi: 10.1038/nature00920. [DOI] [PubMed] [Google Scholar]
- 43.Clackson T, Wells JA. Science. 1995;267:383–386. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 44.Clackson T, Ultsch MH, Wells JA, de Vos AM. J Mol Biol. 1998;277:1111–1128. doi: 10.1006/jmbi.1998.1669. [DOI] [PubMed] [Google Scholar]
- 45.Kim M, Carman CV, Springer TA. Science. 2003;301:1720–1725. doi: 10.1126/science.1084174. [DOI] [PubMed] [Google Scholar]
- 46.Wang J. Trends Biochem Sci. 2002;27:122–126. doi: 10.1016/s0968-0004(01)02038-2. [DOI] [PubMed] [Google Scholar]
- 47.Carson M. Methods Enzymol. 1997;277:493–505. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.