

Abstract
We investigate the impact of antagonistic pleiotropy on the most widely used methods of estimation of the average coefficient of dominance of deleterious mutations from segregating populations. A proportion of the deleterious mutations affecting a given studied fitness component are assumed to have an advantageous effect on another one, generating overdominance on global fitness. Using diffusion approximations and transition matrix methods, we obtain the distribution of gene frequencies for nonpleiotropic and pleiotropic mutations in populations at the mutation-selection-drift balance. From these distributions we build homozygous and heterozygous chromosomes and assess the behavior of the estimators of dominance. A very small number of deleterious mutations with antagonistic pleiotropy produces substantial increases on the estimate of the average degree of dominance of mutations affecting the fitness component under study. For example, estimates are increased three- to fivefold when 2% of segregating loci are overdominant for fitness. In contrast, strengthening pleiotropy, where pleiotropic effects are assumed to be also deleterious, has little effect on the estimates of the average degree of dominance, supporting previous results. The antagonistic pleiotropy model considered, applied under mutational parameters described in the literature, produces patterns for the distribution of chromosomal viabilities, levels of genetic variance, and homozygous mutation load generally consistent with those observed empirically for viability in Drosophila melanogaster.
THE degree of dominance of genes controlling fitness components has a key role on many biological phenomena, such as the predictions on the evolution of selfing rates (e.g., Charlesworth and Charlesworth 1998, 1999), the evolution of sexual reproduction and genetic recombination (e.g., Chasnov 2000; Agrawal and Chasnov 2001; Otto 2003), the dominance hypothesis for Haldane's rule (Turelli and Orr 1995), and the maintenance of genetic variation and its changes under bottlenecks (e.g., Wang et al. 1998; Charlesworth and Hughes 1999; Rodríguez-Ramilo et al. 2004; Zhang et al. 2004). Unfortunately, the average coefficient of dominance of mutations (h) is generally difficult to estimate in terms of both statistical analysis and effort required. Although direct estimates of the average h can be obtained from newly arisen mutations on the basis of mutation-accumulation experiments where natural selection is partially avoided (see García-Dorado et al. 2004 for a recent review), indirect estimates are less costly and are often obtained. These can be inferred from the genetic structure of segregating populations assumed to be at the balance between mutation and directional selection.
The most widely used approach to obtain indirect estimates is based on the analysis of chromosomes extracted from natural populations both in homozygosis and in panmictic crosses (Mukai et al. 1972; Mukai and Yamaguchi 1974). Under the assumption of mutation-selection balance for non-fully recessive deleterious mutations, the variances and covariances of the heterozygous genotypic values (y) for a fitness component (e.g., viability) and the sum of their corresponding homozygous genotypic values (x) are
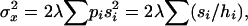 |
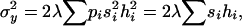 |
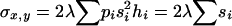 |
(Fernández et al. 2004), where si is the selection coefficient against mutation i in homozygosis, hi is the coefficient of dominance of the mutation, 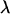 is the chromosome deleterious mutation rate,
is the chromosome deleterious mutation rate, 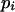 is the pervasiveness of the mutation (number of copies the mutation is expected to contribute before being lost; see García-Dorado et al. 2003), and the summation is over mutations. (The
is the pervasiveness of the mutation (number of copies the mutation is expected to contribute before being lost; see García-Dorado et al. 2003), and the summation is over mutations. (The 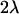 -factor was accidentally missed by Fernández et al. 2004, but this had no further consequences as the factor dropped in the derived expressions.) The regression by.x =
-factor was accidentally missed by Fernández et al. 2004, but this had no further consequences as the factor dropped in the derived expressions.) The regression by.x = 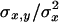 is the most extensively used method to estimate the average h from segregating populations (Mukai 1969; Mukai et al. 1972; Mukai and Yamaguchi 1974; Watanabe et al. 1976; Simmons and Crow 1977; Eanes et al. 1985; Hughes 1995; Johnston and Schoen 1995; Caballero et al. 1997; García-Dorado and Caballero 2000; Chavarrías et al. 2001). Thus, by.x estimates the harmonic mean of the average h for newly arisen mutations weighted by their homozygous effects,
is the most extensively used method to estimate the average h from segregating populations (Mukai 1969; Mukai et al. 1972; Mukai and Yamaguchi 1974; Watanabe et al. 1976; Simmons and Crow 1977; Eanes et al. 1985; Hughes 1995; Johnston and Schoen 1995; Caballero et al. 1997; García-Dorado and Caballero 2000; Chavarrías et al. 2001). Thus, by.x estimates the harmonic mean of the average h for newly arisen mutations weighted by their homozygous effects,
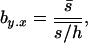 |
where the overbar indicates arithmetic mean. Simmons and Crow (1977) review estimates from this estimator, the most common values being ∼0.2, but with a large variation. Analogously, the inverse of the regression of x on y (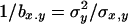 ) is assumed to estimate, under mutation-selection balance, the arithmetic mean of h for newly arisen mutations again weighted by their homozygous effects,
) is assumed to estimate, under mutation-selection balance, the arithmetic mean of h for newly arisen mutations again weighted by their homozygous effects,
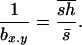 |
The empirical estimates from this estimator are generally very large (of the order of one or more; see, e.g., Mukai and Yamaguchi 1974; Watanabe et al. 1976).
Another simple indirect method of estimation of the average h involves estimates of the mean of a fitness component for outbred (WO) and inbred (WI) populations (Lynch and Walsh 1998, pp. 283–287). Assuming mutation-selection balance, the ratio R = (Wmax − WO)/2(Wmax − WI), where Wmax is the fitness of a genotype free of mutations, estimates the unweighted harmonic mean of dominance for newly arisen mutations,
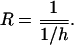 |
Empirical estimates for a range of vertebrates using data with different levels of inbreeding are ∼0.1 (Lynch and Walsh 1998). Because this estimator provides unweighted estimates of the average h it is less affected by some biasing factors. However, its reliability depends on the assumed value for the hypothetical genotype free of deleterious mutations. For absolute viability it is assumed that Wmax = 1. Since nongenetic sources of mortality imply Wmax < 1, the R estimate is usually expected to be biased upward (Lynch and Walsh 1998; García-Dorado et al. 1999; Fernández et al. 2004).
A more recently developed method (Deng and Lynch 1996), extending previous approaches from Morton et al. (1956) and Charlesworth et al. (1990), allows for the simultaneous estimation of mutation rates, average mutational effects, and average dominance coefficients, from experiments involving self-fertilization of naturally outbred populations or outcrossing in naturally self-fertilizing populations (Deng 1998b also extended the method to other forms of inbreeding). This method makes use of the expected mean fitness and genetic variance of fitness of infinite populations at mutation-selection balance. From Equations 3 and 4 in Deng and Lynch (1996) it can be shown that the mutation-selection balance prediction of the dominance estimator in this method is
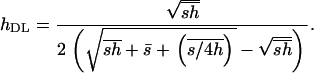 |
Assuming that h takes, even occasionally, values low enough so that 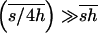 and
and 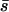 ,
,
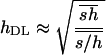 |
and estimates the geometric mean of the arithmetic and the harmonic average h weighted by s. For nonrecessive deleterious mutations, this is equivalent to 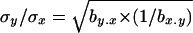 and, interestingly, it has the same prediction as the estimator used by Hughes (1995) and Charlesworth and Hughes (1999),
and, interestingly, it has the same prediction as the estimator used by Hughes (1995) and Charlesworth and Hughes (1999), 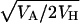 , where VA is the additive genetic variance and VH is the variance among homozygous genotypes.
, where VA is the additive genetic variance and VH is the variance among homozygous genotypes.
The analysis carried out in this article refers to the above estimators. Other methods are not considered, as they are less appropriate for fitness traits (Comstock and Robinson 1952), may involve the previous knowledge of other mutational parameters (Lynch et al. 1995; Deng et al. 2002; see also García-Dorado et al. 1999), or can be applied successfully only under simplistic mutational models (Kelly 2003).
Even if no substantial proportion of the standing genetic variability for the studied fitness component is maintained by mechanisms other than the balance between directional selection, mutation, and drift, indirect estimators of dominance generally produce biased estimates with respect to their mutation-selection balance expectations as a consequence of a number of factors (Fernández et al. 2004). A particularly important one is the finite size of populations as, for mutations with small heterozygous deleterious effect, the mutation-selection balance prediction for the pervasiveness is usually much larger than the actual one in finite populations. Other biasing factors are the correlation between s and h and the bottlenecking due to sampling. The counteracting sources of bias can be such that none of the estimators from segregating populations is very reliable.
Here we concentrate on the bias of indirect estimates caused by the existence of selectively maintained variability. In this respect, Mukai and Yamaguchi (1974) investigated the effect of overdominance for viability on the estimates of by.x and 1/bx.y. They showed that, in this instance, by.x becomes smaller, and 1/bx.y becomes larger than the corresponding values due only to partially recessive genes. The amount by which 1/bx.y is inflated is much larger than the amount by which by.x is reduced. In fact, the large empirical estimates obtained from 1/bx.y were suggested as an indication that genetic variability could be maintained by balancing selection (Mukai and Yamaguchi 1974; Watanabe et al. 1976; but see Fernández et al. 2004 for other likely sources of bias). The reduction in by.x by overdominance was also described by Deng (1998a) (see also Li et al. 1999), who investigated the usefulness of this regression (and an alternative method to be used when homozygous lines cannot be constructed) for discriminating between dominance and overdominance for the fitness component. The conclusion was that, despite the reductions in the estimate caused by overdominance, overwhelming contributions from overdominant loci should be necessary for the estimates of the average h to become negative and, therefore, indicative of overdominance.
Overdominance for the loci controlling life-history traits is decisively rejected by the data as a general model of maintenance of variation (Charlesworth and Hughes 1999). However, it is recognized that antagonistic pleiotropic effects on more than one fitness trait (Prout 1980; Rose 1982; Roff 1997) could give rise to overdominance for overall fitness, the so-called marginal overdominance (Falconer and Mackay 1996, p. 41). Antagonistic pleiotropy is considered as a central part of some evolutionary theories, such as the evolution of senescence, and as a plausible cause of maintenance of genetic variation for fitness components (e.g., Rose 1991; Stearns 1992; Charlesworth 1994; Roff 1997; Charlesworth and Hughes 1999; Merilä and Sheldon 1999), although some theoretical analyses suggest that the conditions under which antagonistic pleiotropy could work are very restrictive (Rose 1982; Curtsinger et al. 1994; Hedrick 1999).
Studies on individual mutations affecting fitness components (López and López-Fanjul 1993; Fernández and López-Fanjul 1996; Lynch and Walsh 1998, p. 345; Martorell et al. 1998) seem to support the view that pleiotropic effects of deleterious mutations are mostly in the same direction; i.e., detrimental mutations tend to have deleterious effects on several traits. This is what we are referring to in this article as strengthening pleiotropy. Several indirect arguments suggest that the overall effect on fitness of mutations affecting egg-to-adult viability in Drosophila melanogaster is about two to three times larger than their effect on viability alone (Mukai and Yamaguchi 1974; Mukai et al. 1974; Charlesworth and Hughes 1999), although these inferences have to be treated with caution because of the uncertainties on the assumptions involved in their calculations (Rodríguez-Ramilo et al. 2004). The effect of strengthening pleiotropy on the estimation of the average h from segregating populations has been shown to be weak (Fernández et al. 2004). However, a general scenario of strengthening pleiotropy is not incompatible with the possibility that a few particular deleterious mutations show antagonistic pleiotropic effects, generating some degree of overdominance for global fitness.
The main purpose of this article is to investigate the effect of a small proportion of mutations under balancing selection on the estimation of the average degree of dominance of deleterious mutations affecting a given fitness component in segregating populations at the mutation-selection-drift balance. To achieve this, we assume that all mutations are deleterious in their effect on the fitness component under study, but a small fraction of these mutations are under antagonistic pleiotropy (i.e., they have an advantageous effect on another fitness component), generating overdominance for their overall effects on fitness. A secondary objective is to investigate the impact of this model on the distribution of chromosomal mean viabilities and on the amount of genetic variance and homozygous mutation load, as well as to compare the patterns observed with empirical results obtained in D. melanogaster.
MUTATIONAL MODELS AND ESTIMATION PROCEDURE
Mutational models and parameters:
A mutational model was assumed in which the values for a fitness component of the wild homozygous, the heterozygous, and the mutant homozygous genotypes are 1, 1 − sh, and 1 − s, respectively. Because comparisons are made later with chromosome viabilities in D. melanogaster, we assume that the fitness trait under investigation is chromosome II viability, and mutational parameters refer to this widely studied trait.
Selection coefficients (s) for new mutations were sampled from a gamma distribution. Only positive values of s were assumed for the effects on viability; i.e., all mutations were assumed to be deleterious for this fitness component. However, some proportion of these mutations could also have a pleiotropic effect on other fitness components, and this effect could be also deleterious (strengthening pleiotropy) or advantageous (antagonistic pleiotropy). Because the effect on viability is always deleterious we maintain the term deleterious mutation even if it is not so on global fitness. Regarding viability deleterious mutations showing no antagonistic pleiotropy, we considered two mutation rates per chromosome and generation, λ = 0.006 and 0.04 (equivalent to 0.015 and 0.1 per haploid genome for D. melanogaster), with corresponding mean deleterious effects 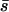 = 0.2 and 0.05, respectively. The shape parameters of the gamma distribution (β = 1 and 0.263, respectively) were chosen so that the whole-genome mutational variances explained by both models (0.0006) were the same and on the order of empirical estimates for nonlethal chromosomes (García-Dorado et al. 1999). These sets of mutational parameters are representative of most estimates reviewed by García-Dorado et al. (2004) and agree also with more recent estimates for viability in D. melanogaster by Charlesworth et al. (2004). Lethal mutations (sL = 1) were also included and assumed to occur at a rate λL = 0.006 per chromosome per generation with dominance coefficient hL = 0.02 (Simmons and Crow 1977). These parameters have been widely accepted, although higher values of λL from 0.010 to 0.015 have been recently reported (Fry et al. 1999; Charlesworth et al. 2004).
= 0.2 and 0.05, respectively. The shape parameters of the gamma distribution (β = 1 and 0.263, respectively) were chosen so that the whole-genome mutational variances explained by both models (0.0006) were the same and on the order of empirical estimates for nonlethal chromosomes (García-Dorado et al. 1999). These sets of mutational parameters are representative of most estimates reviewed by García-Dorado et al. (2004) and agree also with more recent estimates for viability in D. melanogaster by Charlesworth et al. (2004). Lethal mutations (sL = 1) were also included and assumed to occur at a rate λL = 0.006 per chromosome per generation with dominance coefficient hL = 0.02 (Simmons and Crow 1977). These parameters have been widely accepted, although higher values of λL from 0.010 to 0.015 have been recently reported (Fry et al. 1999; Charlesworth et al. 2004).
The dominance coefficient of mutations ranged between 0 and 1 (i.e., mutations over- and underdominant for viability itself were not considered). Dominance coefficients were assigned following the model proposed by Caballero and Keightley (1994). For this model, h-values are taken from a uniform distribution giving 0 < h < exp(−ks), where k is a constant allowing the mean dominance coefficient of newly arisen mutations, 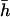 , to be the desired value. The average coefficients of dominance used were 0.2 and 0.4 for the two mutational rates, respectively. They were chosen because they explain, in combination with their respective mutation rates and average effects, the additive variance and inbreeding depression observed in a natural population of D. melanogaster (Rodríguez-Ramilo et al. 2004). The joint distribution obtained in this way for s and h implies a negative correlation between both variables, as suggested by empirical data (Greenberg and Crow 1960; Simmons and Crow 1977; Caballero and Keightley 1994) and biochemical arguments (Kacser and Burns 1981; Keightley 1996), and produces a variance of h-values of the order of that empirically observed by Mukai (1969) (see Fernández et al. 2004).
, to be the desired value. The average coefficients of dominance used were 0.2 and 0.4 for the two mutational rates, respectively. They were chosen because they explain, in combination with their respective mutation rates and average effects, the additive variance and inbreeding depression observed in a natural population of D. melanogaster (Rodríguez-Ramilo et al. 2004). The joint distribution obtained in this way for s and h implies a negative correlation between both variables, as suggested by empirical data (Greenberg and Crow 1960; Simmons and Crow 1977; Caballero and Keightley 1994) and biochemical arguments (Kacser and Burns 1981; Keightley 1996), and produces a variance of h-values of the order of that empirically observed by Mukai (1969) (see Fernández et al. 2004).
To investigate the effect of antagonistic pleiotropy we assumed a model where a given mutation has a deleterious effect on the fitness component under study (viability) and an advantageous one on another component. We usually considered a simple symmetrical model with equal effects for both traits,
Table 8.
| AA | Aa | aa | |
|---|---|---|---|
| Viability | 1 | 1 − sh | 1 − s |
| Other component | 1 − s | 1 − sh | 1 |
| Fitness | 1 − s′ | 1 | 1 − s′ |
where global fitness is obtained as the product of the individual components and is expressed, as usual, relative to the genotype with optimal value. Hence, the selective coefficient for fitness for each of the homozygotes is s′ = 1 − [(1 − s)/(1 − sh)2]. For h(1 − sh/2) ≤ 0.5 we get s′ > 0, so that overdominance for global fitness occurs at least when h < 0.5. In contrast, for h(1 − sh/2) > 0.5 there is underdominance for fitness. For the dominance model used, most mutations are recessive (h < 0.5) for the individual components, and pleiotropic mutations tend to be overdominant. The above model of dominance and equality of selection coefficients for pleiotropic effects is very favorable for the maintenance of polymorphisms (Hoekstra et al. 1985; Curtsinger et al. 1994; Hedrick 1999) and, therefore, will serve for illustration purposes. However, to evaluate the effect of relaxing the symmetry assumption we carried some runs where mutational effects and dominance values for viability and the other fitness component were drawn independently.
As it might be expected that antagonistic pleiotropic mutations are those with the smallest homozygous effects, mutations were sampled from the gamma distribution and antagonistic pleiotropic effects were assigned with a probability exp(−sα(1 − a)/a) and discarded otherwise. Here, α is the scale parameter of the gamma distribution of selection coefficients and a is a constant that takes values between 0 and 1 and gives the ratio of the average s-value for antagonistic pleiotropic mutations to that for nonpleiotropic ones.
The number (n) of antagonistic pleiotropic mutations assumed to be segregating in a chromosome ranged between 3 and 50 for the model of low mutation rate and between 10 and 300 for the model of larger mutation rate. The remaining mutations were assumed either to be nonpleiotropic (they affect fitness just through viability) or to show strengthening pleiotropy (the pleiotropic effect is also deleterious). In this case, the genotypic values for viability, other fitness components, and global fitness are
Table 9.
| AA | Aa | aa | |
|---|---|---|---|
| Viability | 1 | 1 − sh | 1 − s |
| Other component | 1 | 1 − sh | 1 − s |
| Fitness | 1 | 1 − s′h′ = (1 − sh)2 | 1 − s′ = (1 − s)2 |
where s′ = 2s − s2 and h′ = (2sh − s2h2)/(2s − s2). For small values of s, this model implies that the effect on fitness of a mutation affecting viability is about twice the viability effect (s′ 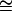 2s), in agreement with indirect calculations (e.g., Mukai and Yamaguchi 1974), and h′
2s), in agreement with indirect calculations (e.g., Mukai and Yamaguchi 1974), and h′ 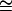 h.
h.
Distribution of mutant frequencies:
The expected distribution of mutant frequencies in a finite population at mutation-selection-drift balance was obtained by means of diffusion approximations in the case of nonpleiotropic or strengthening pleiotropic mutations and by means of transition matrices in the case of antagonistic pleiotropic mutations.
Diffusion approximations:
The procedure is described by Fernández et al. (2004). Briefly, we used Kimura's (1969) diffusion approximations under the infinite-sites model to obtain the stationary distribution of mutant frequencies. Thus, in a nonrecurrent mutation model for a randomly mating diploid population of N individuals and effective size Ne, the expected number of loci with i segregating mutant copies (1 ≤ i ≤ 2N − 1) and frequency q = i/2N is
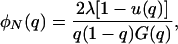 |
where G(q) = exp[2Nesq(q + 2h − 2hq)], and 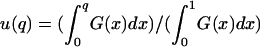 is the fixation probability of a mutant with initial frequency q. For strengthening pleiotropic mutations, the viability values of s and h in the expressions above were replaced by the corresponding fitness coefficients s′ and h′.
is the fixation probability of a mutant with initial frequency q. For strengthening pleiotropic mutations, the viability values of s and h in the expressions above were replaced by the corresponding fitness coefficients s′ and h′.
The expected number of loci with j copies in a random sample of M chromosomes taken from the population (φM(j)) was calculated using binomial sampling,
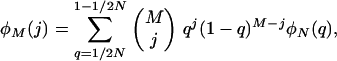 |
so that 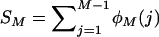 is the expected total number of segregating loci in the chromosome sample.
is the expected total number of segregating loci in the chromosome sample.
For each particular case and mutational model, a sufficiently large number (1000) of mutational s- and h-values were sampled to represent the whole distribution of effects, and their diffusion stationary frequencies were obtained. For each pair of mutational parameters (s, h), the expected distribution of gene copy numbers in a sample of M chromosomes, φM(j), and the expected total number of segregating loci in the sample, SM, were stored. Chromosomes in which a number of independent loci were segregating were then simulated. This number of loci was equal to the SM-value averaged over the 1000 sampled (s, h) pairs. The assignment of particular mutational values to each segregating locus was done by sampling with replacement from the available 1000 sampled (s, h) pairs with a probability proportional to their SM-value. The number j of copies of the mutant allele at each locus was sampled from a distribution proportional to the corresponding φM(j), and these j copies were randomly assigned to the simulated chromosomes.
Transition matrices:
To generate the distribution of frequencies for antagonistic pleiotropic mutations, a transition matrix A, of size (2N + 1) × (2N + 1), was constructed with terms
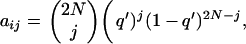 |
giving the probability that the number of copies of the mutant is j at a generation, given that it was i in the previous one, where q = i/2N(0 ≤ i ≤ 2N) and q′ = q + [pqs′(p − q)]/[1 − s′(p2 + q2)] is the expected frequency after selection for overdominant or underdominant genes. The state of the vector of frequencies at generation t is obtained from that at generation t − 1 as φN,t(j) = φN,t−1(j)A. To save computer time, the matrix and vectors considered a population of size 500, and selection coefficients of mutations were multiplied by a constant (N/500). Because the distribution of gene frequencies depends mainly on Ns, this procedure gives very accurate results (not shown).
For each (s, h) pair sampled for pleiotropic mutations, the number of copies was assumed to be zero at the initial generation, and 2Nλ mutations appeared each generation at frequency 1/2N with parameters s and h taken from the corresponding distribution. With overdominance for fitness, mutations tend to keep segregating in the population, so that the number of segregating loci can increase indefinitely. To solve this problem a maximum number of segregating overdominant loci was assumed. After this number was reached new mutations appeared only to restore those lost by drift. Note that this maximum number of segregating loci is arbitrary and irrelevant at this stage (100 loci were used), as the objective of the matrix approach is to provide the expected distribution of gene frequencies, from which the actual frequencies of pleiotropic segregating loci will be sampled. This distribution is independent of the number of loci used to compute it, provided this is large enough. The expected number of loci with j copies in a sample of M chromosomes (φM(j)) was calculated using binomial sampling, as explained above for the distributions obtained by diffusion approximations. For each sampled mutation the matrix iteration was continued until an equilibrium frequency distribution was reached. The criterion used to assure this was the following. Every 100 generations, the 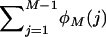 was calculated, and an equilibrium was assumed to occur when the absolute difference between two consecutive values of this parameter was <0.001. If this criterion was not achieved before 10,000 generations (in rare cases), the process was stopped at generation 10,000. Previous runs confirmed that this procedure is very reliable (not shown). Figure 1 shows the stationary distribution of frequencies in the sample of chromosomes for some examples of mutations with different values of s′, one of them being underdominant (s′ is negative) and the rest overdominant. The stationary distribution of frequencies depends on the equilibrium between drift and selection. For symmetric overdominant mutations the distribution is centered around a frequency of 0.5, and the larger the values of s, the smaller the dispersion.
was calculated, and an equilibrium was assumed to occur when the absolute difference between two consecutive values of this parameter was <0.001. If this criterion was not achieved before 10,000 generations (in rare cases), the process was stopped at generation 10,000. Previous runs confirmed that this procedure is very reliable (not shown). Figure 1 shows the stationary distribution of frequencies in the sample of chromosomes for some examples of mutations with different values of s′, one of them being underdominant (s′ is negative) and the rest overdominant. The stationary distribution of frequencies depends on the equilibrium between drift and selection. For symmetric overdominant mutations the distribution is centered around a frequency of 0.5, and the larger the values of s, the smaller the dispersion.
Figure 1.

Distribution of gene copy number (j) in a sample of 100 chromosomes, obtained by transition matrix, for several examples of underdominant (s′ < 0) and overdominant (s′ > 0) genes, where s′ is the fitness selection coefficient in favor of/against both homozygotes.
A number of mutational (s, h) values (1000) large enough to cover the distribution of effects were sampled, and their equilibrium distributions in the sample of chromosomes were obtained. A number (n, between 3 and 300) of these antagonistic pleiotropic mutations were randomly taken with replacement from the set of 1000 (s, h) pairs, with probability proportional to their SM-values. Frequencies for each of these n antagonistic mutations were sampled from the corresponding distribution, φM(j), and the j mutant copies were randomly assigned to the M chromosomes previously constructed with nonpleiotropic or strengthening pleiotropic mutations (obtained by diffusion approximations). All mutations, pleiotropic or not, were assumed to be unlinked.
For each case and mutational model, the sampling of M chromosomes was repeated 100 times and the estimates were averaged. The results given below refer to samples of M = 100 chromosomes taken, generally, from a population of size N = Ne = 104, but other population sizes N = 102–105 were also investigated. The whole procedure was then repeated 10 times [using 10 different samples of 1000 (s, h) values each] to obtain mean estimates and standard errors. Homozygous and heterozygous viabilities were calculated for all combinations of chromosomes [M homozygotes and M(M − 1)/2 heterozygotes], assuming a between-locus multiplicative fitness model.
Viability distribution for chromosomes and homozygous mutation loads:
Constructed chromosomes were classified in different categories, as a function of their mean viability in homozygosis. Thus, lethal chromosomes were those with homozygous viability <10% of the mean of the heterozygous viability, semilethal chromosomes were those with viability between 10 and 60%, and quasinormal chromosomes were those with viability >60%. The homozygous mutation load for quasinormal chromosomes was obtained as ln(WO/WI(QN)) (Greenberg and Crow 1960), where WO and WI are the heterozygous (outbred, O) and homozygous (inbred, I) mean viabilities, respectively, and the parentheses in the subscript indicate the fraction of homozygotes considered (quasinormals, QN, in this case). Analogously, for semilethal chromosomes the load was obtained as ln(WI(QN)/WI(NL)), where NL indicates all nonlethal chromosomes. The sum of the two loads above is the homozygous detrimental load (D) and, finally, the lethal load is obtained by subtracting this latter load from the total load, ln(WO/WI(T)), where T accounts for all chromosomes.
The additive and dominance genetic variances contributed to the original population by mutation i at frequency qi were calculated as 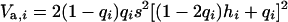 and
and 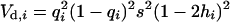 , respectively (Mukai et al. 1974). For nonpleiotropic and strengthening pleiotropic mutations, the total contributed additive (VA) and dominance (VD) variances were calculated as
, respectively (Mukai et al. 1974). For nonpleiotropic and strengthening pleiotropic mutations, the total contributed additive (VA) and dominance (VD) variances were calculated as 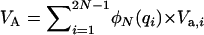 and
and 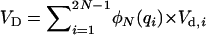 and averaged over mutations. The contribution of each antagonistic pleiotropic mutation was calculated in a similar way, using its equilibrium distribution frequency and summing up over all segregating mutations.
and averaged over mutations. The contribution of each antagonistic pleiotropic mutation was calculated in a similar way, using its equilibrium distribution frequency and summing up over all segregating mutations.
Estimation of the average dominance:
Estimates of the average coefficient of dominance from regressions involving heterozygous (y) and the sum of the corresponding homozygous (x) genotypic viabilities (by.x and 1/bx.y) used log-scaled viabilities to linearize multiplicative effects. Estimates were obtained for quasinormal chromosomes, as the main interest is usually on the average dominance coefficient of mildly deleterious mutations (Simmons and Crow 1977). Calculations for the R estimate, R = (1 − WO)/2(1 − WI), used the homozygous (WO) and heterozygous (WI) mean viabilities of nonlethal chromosomes.
Deng and Lynch (1996) estimates of dominance were obtained for untransformed viability, assuming that the outcrossing base population can be subject to self-fertilization. From each of the heterozygotes for nonlethal chromosomes, 40 selfed (S) progeny were obtained. The mean, 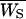 , and variance,
, and variance, 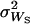 , of selfed offspring were computed. Using these estimates as well as the mean,
, of selfed offspring were computed. Using these estimates as well as the mean, 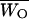 , and variance,
, and variance, 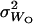 , of outcrossed parents, the following functions could be computed (Deng and Lynch 1996, Equations 2),
, of outcrossed parents, the following functions could be computed (Deng and Lynch 1996, Equations 2),
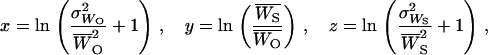 |
and the estimator of the average coefficient of dominance is
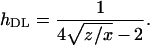 |
Accordingly, estimates of the mutation rate and mean selection coefficients were obtained as 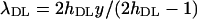 and
and 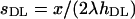 .
.
The above estimators were derived assuming constant selective and dominance effects and, therefore, are biased when mutational effects are variable. To correct for this, Deng and Lynch (1996) suggested that, if the coefficient of variation of dominance coefficients, 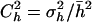 , and the coefficient of covariation between s and h,
, and the coefficient of covariation between s and h, 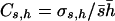 , were known, the estimators should be corrected as
, were known, the estimators should be corrected as
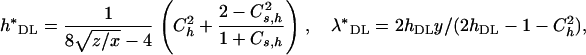 |
and
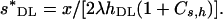 |
The possibility of knowing 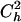 and
and 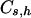 is, however, very unlikely.
is, however, very unlikely.
To check the procedure we made runs with some of the parameters used by Deng and Lynch (1996). For their case for outcrossing populations in their Table 2, using 2λ = 1.5, an exponential distribution of mutational effects with mean 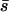 = 0.05, and dominance values h = exp(−13s)/2 for an average
= 0.05, and dominance values h = exp(−13s)/2 for an average 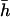 = 0.3, we obtained estimates of
= 0.3, we obtained estimates of 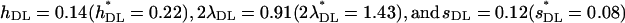 , which are very similar to those shown in their Table 2.
, which are very similar to those shown in their Table 2.
TABLE 2.
Estimates of the average coefficient of dominance for segregating mutations and other parameters for a model of many mutations of low average effect
| Estimates of average h
|
% chromosomes
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| n | Mean effect | by.x | R | 1/bx.y | hDL | QN | SL | D | VA | VD | VA/D |
| 0 (0, 0) | — | 0.07 | 0.15 | 0.28 | 0.09 | 88 | 12 | 0.169 | 0.0009 | 0.0005 | 0.005 |
| 50 (25, 25) | 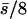 |
0.11 | 0.22 | 0.42 | 0.14 | 88 | 12 | 0.187 | 0.0017 | 0.0006 | 0.010 |
| 100 (50, 50) | 0.14 | 0.26 | 0.51 | 0.17 | 86 | 14 | 0.210 | 0.0025 | 0.0008 | 0.012 | |
| 150 (74, 76) | 0.18 | 0.29 | 0.56 | 0.20 | 85 | 15 | 0.233 | 0.0033 | 0.0009 | 0.014 | |
| 300 (149, 151) | 0.27 | 0.35 | 0.64 | 0.26 | 82 | 18 | 0.300 | 0.0057 | 0.0013 | 0.019 | |
| 10 (5, 5) | 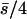 |
0.10 | 0.18 | 0.39 | 0.13 | 88 | 12 | 0.179 | 0.0021 | 0.0011 | 0.012 |
| 50 (23, 27) | 0.21 | 0.27 | 0.58 | 0.23 | 85 | 15 | 0.226 | 0.0051 | 0.0017 | 0.023 | |
| 100 (47, 53) | 0.29 | 0.32 | 0.67 | 0.30 | 81 | 19 | 0.283 | 0.0092 | 0.0025 | 0.032 | |
| 150 (70, 80) | 0.34 | 0.35 | 0.71 | 0.34 | 75 | 25 | 0.332 | 0.0129 | 0.0032 | 0.039 | |
| 10 (4, 6) |
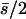
|
0.16 | 0.21 | 0.51 | 0.20 | 87 | 13 | 0.190 | 0.0046 | 0.0019 | 0.024 |
| 50 (21, 29) | 0.32 | 0.32 | 0.78 | 0.38 | 73 | 27 | 0.318 | 0.0182 | 0.0061 | 0.057 | |
| 100 (42, 58) | 0.35 | 0.37 | 0.95 | 0.45 | 51 | 49 | 0.486 | 0.0384 | 0.0115 | 0.079 | |
| 150 (63, 87) | 0.35 | 0.40 | 1.21 | 0.48 | 34 | 66 | 0.647 | 0.0552 | 0.0168 | 0.085 | |
Nonlethal mutational parameters: λ = 0.04, 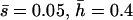 .
.
Other parameters and definitions are as in Table 1. The number of segregating nonpleiotropic mutations in the chromosome sample was 2396 ± 45. Ranges of SE: by.x, 0.002–0.005; R, 0.001–0.004; 1/bx.y, 0.003–0.028; hDL, 0.003–0.008; QN, 0.59–1.35; SL, 0.76–1.40; D, 0.005–0.010; VA, 0.0000–0.0006; and VD, 0.0000–0.0003.
Finally, to compare the sampling variance for the different estimators of dominance, some runs were done assuming the same total number of evaluated individuals in each case. Thus, estimates of by.x, R, and 1/bx.y were obtained as usual from 100 homozygotes (using 100 sampled chromosomes) and all combinations of heterozygotes (4950), whereas those for hDL were obtained from 50 heterozygous parents (again 100 sampled chromosomes) and 100 selfed progeny per parent.
RESULTS
Tables 1 and 2 show the average estimates of the coefficient of dominance obtained with four estimators (by.x, R, 1/bx.y, and hDL), for two mutational models, respectively. The first row in both tables refers to the case without pleiotropic mutations. Let us first consider the estimates under this situation. Note that most estimates are far from the arithmetic mean of h-values (i.e., 0.2 and 0.4 for Tables 1 and 2, respectively). This was already observed by Fernández et al. (2004), who also checked the diffusion results by simulations. The reason is that estimates provided by the different estimators do not give, in general, the arithmetic mean of dominance coefficients, but harmonic or arithmetic means weighted in different ways by the selection coefficients (see Introduction). In addition, the mutation-selection balance estimates are usually biased because of the finite size of the population, the correlation between selective and dominance coefficients, and other issues (Fernández et al. 2004). In the unlikely case that the coefficient of variation of dominance coefficients, 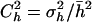 , and the coefficient of covariation between s and h,
, and the coefficient of covariation between s and h, 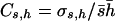 , were known (see above), Deng and Lynch (1996) estimates (hDL) can be roughly corrected to give the unweighted arithmetic average for the dominance coefficient of new mutations,
, were known (see above), Deng and Lynch (1996) estimates (hDL) can be roughly corrected to give the unweighted arithmetic average for the dominance coefficient of new mutations, 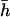 . The above necessary parameters calculated from 106 sampled new mutations were
. The above necessary parameters calculated from 106 sampled new mutations were 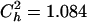 and
and 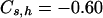 for the model in Table 1, and
for the model in Table 1, and 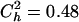 and
and 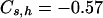 for the model in Table 2. The corrected estimates are
for the model in Table 2. The corrected estimates are 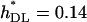 and 0.21 for Tables 1 and 2, respectively, still quite below the true arithmetic mean values (
and 0.21 for Tables 1 and 2, respectively, still quite below the true arithmetic mean values (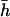 = 0.2 and 0.4, respectively). Accordingly, the regression by.x can be corrected in the also unlikely case that one could know the covariance between s2 and h,
= 0.2 and 0.4, respectively). Accordingly, the regression by.x can be corrected in the also unlikely case that one could know the covariance between s2 and h, 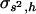 , and the mean of s2,
, and the mean of s2, 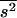 . Thus, from Caballero et al. (1997, Equation 3) the correction should be
. Thus, from Caballero et al. (1997, Equation 3) the correction should be 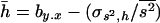 . The necessary parameters were
. The necessary parameters were 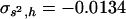 and
and 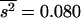 for the model in Table 1, and
for the model in Table 1, and 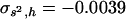 and
and 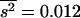 for the model in Table 2. The corrected estimates are 0.21 and 0.40 for Tables 1 and 2, respectively, in good agreement with the true arithmetic mean values.
for the model in Table 2. The corrected estimates are 0.21 and 0.40 for Tables 1 and 2, respectively, in good agreement with the true arithmetic mean values.
TABLE 1.
Estimates of the average coefficient of dominance for segregating mutations and other parameters for a model of few mutations of large average effect
| Estimates of average h
|
% chromosomes
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| n | Mean effect | by.x | R | 1/bx.y | hDL | QN | SL | D | VA | VD | VA/D |
| 0 (0, 0) | — | 0.04 | 0.04 | 0.18 | 0.06 | 88 | 12 | 0.114 | 0.0002 | 0.0008 | 0.002 |
| 10 (5, 5) | 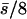 |
0.11 | 0.16 | 0.48 | 0.12 | 87 | 13 | 0.133 | 0.0015 | 0.0010 | 0.011 |
| 15 (7, 8) | 0.14 | 0.20 | 0.54 | 0.15 | 87 | 13 | 0.143 | 0.0022 | 0.0012 | 0.015 | |
| 30 (14, 16) | 0.23 | 0.28 | 0.61 | 0.21 | 86 | 14 | 0.179 | 0.0043 | 0.0015 | 0.024 | |
| 50 (23, 27) | 0.31 | 0.32 | 0.65 | 0.26 | 84 | 16 | 0.222 | 0.0069 | 0.0020 | 0.031 | |
| 3 (1, 2) | 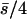 |
0.11 | 0.11 | 0.42 | 0.11 | 87 | 13 | 0.129 | 0.0022 | 0.0013 | 0.017 |
| 5 (2, 3) | 0.16 | 0.16 | 0.54 | 0.16 | 86 | 14 | 0.146 | 0.0033 | 0.0016 | 0.023 | |
| 10 (3, 7) | 0.25 | 0.23 | 0.65 | 0.24 | 85 | 15 | 0.180 | 0.0063 | 0.0023 | 0.035 | |
| 15 (5, 10) | 0.31 | 0.28 | 0.68 | 0.29 | 83 | 17 | 0.217 | 0.0094 | 0.0027 | 0.043 | |
| 3 (1, 2) |
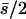
|
0.19 | 0.15 | 0.57 | 0.19 | 85 | 15 | 0.157 | 0.0082 | 0.0036 | 0.052 |
| 5 (1, 4) | 0.28 | 0.21 | 0.70 | 0.27 | 82 | 18 | 0.196 | 0.0143 | 0.0059 | 0.073 | |
| 10 (2, 8) | 0.35 | 0.28 | 0.83 | 0.38 | 71 | 29 | 0.299 | 0.0272 | 0.0105 | 0.091 | |
| 15 (3, 12) | 0.38 | 0.30 | 0.80 | 0.41 | 60 | 40 | 0.391 | 0.0402 | 0.0151 | 0.103 | |
Nonlethal mutational parameters: λ = 0.006, 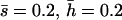 .
.
Lethal parameters: λL = 0.006, sL = 1, hL = 0.02. Population size: N = 10,000.
n, number of antagonistic pleiotropic mutations segregating per chromosome (the number of segregating nonpleiotropic mutations in the chromosome sample was 93 ± 1). In parentheses, number of underdominant and overdominant mutations, respectively. Mean effect, mean viability effect of antagonistic pleiotropic segregating mutations.
by.x, 1/bx.y, regression estimates using the heterozygous (y) viabilities and the sum of the homozygous viabilities (x) of quasinormal chromosomes.
R, estimate of the ratio of genetic loads between outbred and inbred populations.
hDL, estimate of h from Deng and Lynch (1996).
QN, SL, proportion of quasinormal and semilethal chromosomes, respectively.
D, VA, VD, detrimental homozygous load and additive and dominance genetic variances, respectively, for nonlethal chromosomes. Ranges of SE: by.x, 0.001–0.005; R, 0.001–0.003; 1/bx.y, 0.002–0.009; hDL, 0.001–0.006; QN, 0.37–0.64; SL, 0.32–0.63; D, 0.002–0.004; VA, 0.0000–0.0010; and VD, 0.0000–0.0004.
The effect of antagonistic pleiotropic mutations overdominant for fitness (n > 0) is to inflate the estimates of the average h. Note that, because of this inflation, estimates can sometimes be closer to 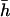 than estimates without antagonistic pleiotropic mutations. However, this is simply an artifact. The magnitude of the increase in the estimates of the average h increases with the number of antagonistic pleiotropic mutations and their average effect, but even a very small number of these loci are able to produce substantial biases. For example, three antagonistic pleiotropic mutations (n = 3; two of them overdominant) with an average effect half that for nonpleiotropic mutations increase the estimates three- to fivefold (Table 1). The average number of nonpleiotropic (partially recessive) mutations segregating in the chromosome sample is ∼93 in this case, so the proportion of antagonistic pleiotropic genes segregating in the chromosome is ∼3%. For a model with larger mutation rates (Table 2) the results are similar, but the number of antagonistic pleiotropic mutations has to be larger in absolute value to show an appreciable effect as, in this case, the number of segregating nonpleiotropic genes in the chromosome sample is ∼2400. In relative terms, however, the increase is similar to that produced with the previous mutational model. For example, 100 (4%) antagonistic pleiotropic mutations (2% overdominant), with an average effect half that for nonpleiotropic mutations, produce again a three- to fivefold increase of the estimates (Table 2). The cause for this increase is the large number of segregating copies of each allele overdominant for fitness, as inferences about the average h obtained from all estimators assume low frequencies for viability deleterious mutations (see discussion). Similar increases occur when all genes, except those showing antagonistic pleiotropy, show strengthening pleiotropy instead of being nonpleiotropic (Table 3). The large increases of by.x and hDL arising from overdominance for global fitness contrast with the decreases or small increases observed when overdominance occurs for viability itself (Mukai and Yamaguchi 1974; Deng 1998a; Li et al. 1999).
than estimates without antagonistic pleiotropic mutations. However, this is simply an artifact. The magnitude of the increase in the estimates of the average h increases with the number of antagonistic pleiotropic mutations and their average effect, but even a very small number of these loci are able to produce substantial biases. For example, three antagonistic pleiotropic mutations (n = 3; two of them overdominant) with an average effect half that for nonpleiotropic mutations increase the estimates three- to fivefold (Table 1). The average number of nonpleiotropic (partially recessive) mutations segregating in the chromosome sample is ∼93 in this case, so the proportion of antagonistic pleiotropic genes segregating in the chromosome is ∼3%. For a model with larger mutation rates (Table 2) the results are similar, but the number of antagonistic pleiotropic mutations has to be larger in absolute value to show an appreciable effect as, in this case, the number of segregating nonpleiotropic genes in the chromosome sample is ∼2400. In relative terms, however, the increase is similar to that produced with the previous mutational model. For example, 100 (4%) antagonistic pleiotropic mutations (2% overdominant), with an average effect half that for nonpleiotropic mutations, produce again a three- to fivefold increase of the estimates (Table 2). The cause for this increase is the large number of segregating copies of each allele overdominant for fitness, as inferences about the average h obtained from all estimators assume low frequencies for viability deleterious mutations (see discussion). Similar increases occur when all genes, except those showing antagonistic pleiotropy, show strengthening pleiotropy instead of being nonpleiotropic (Table 3). The large increases of by.x and hDL arising from overdominance for global fitness contrast with the decreases or small increases observed when overdominance occurs for viability itself (Mukai and Yamaguchi 1974; Deng 1998a; Li et al. 1999).
TABLE 3.
Estimates of the average coefficient of dominance for segregating mutations and other population parameters for a model where nonantagonistic mutations show strengthening pleiotropy
| Estimates of average h
|
% chromosomes
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| n | by.x | R | 1/bx.y | hDL | QN | SL | D | VA | VD | VA/D |
| A. | ||||||||||
| 0 (0, 0) | 0.04 | 0.03 | 0.17 | 0.05 | 92 | 8 | 0.069 | 0.0006 | 0.0006 | 0.009 |
| 3 (1, 2) | 0.23 | 0.17 | 0.58 | 0.21 | 90 | 10 | 0.109 | 0.0082 | 0.0033 | 0.075 |
| 5 (1, 4) | 0.32 | 0.23 | 0.68 | 0.30 | 87 | 13 | 0.151 | 0.0139 | 0.0053 | 0.092 |
| 10 (2, 8) | 0.38 | 0.30 | 0.80 | 0.41 | 77 | 23 | 0.249 | 0.0271 | 0.0102 | 0.109 |
| 15 (3, 12) | 0.37 | 0.33 | 0.91 | 0.46 | 65 | 35 | 0.349 | 0.0401 | 0.0147 | 0.115 |
| B. | ||||||||||
| 0 (0, 0) | 0.06 | 0.13 | 0.25 | 0.08 | 93 | 7 | 0.100 | 0.0016 | 0.0005 | 0.016 |
| 10 (4, 6) | 0.19 | 0.21 | 0.51 | 0.22 | 92 | 8 | 0.130 | 0.0053 | 0.0016 | 0.041 |
| 50 (21, 29) | 0.36 | 0.33 | 0.74 | 0.42 | 80 | 20 | 0.260 | 0.0195 | 0.0058 | 0.075 |
| 100 (42, 58) | 0.37 | 0.38 | 0.90 | 0.48 | 60 | 40 | 0.416 | 0.0364 | 0.0107 | 0.087 |
| 150 (63, 87) | 0.37 | 0.41 | 1.08 | 0.50 | 40 | 60 | 0.585 | 0.0554 | 0.0162 | 0.095 |
Nonlethal mutational parameters: (A) λ = 0.006, 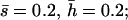 (B) λ = 0.04,
(B) λ = 0.04, 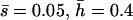 . The number of segregating strengthening pleiotropic mutations in the chromosome sample was 70 ± 1 for model A and 2102 ± 22 for model B. The mean selection coefficient of antagonistic pleiotropic mutations is
. The number of segregating strengthening pleiotropic mutations in the chromosome sample was 70 ± 1 for model A and 2102 ± 22 for model B. The mean selection coefficient of antagonistic pleiotropic mutations is 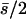 . Other parameters and definitions are as in Table 1. Ranges of SE: by.x, 0.001–0.005; R, 0.001–0.005; 1/bx.y, 0.002–0.011; hDL, 0.001–0.008; QN, 0.26–0.79; SL, 0.31–0.69; D, 0.002–0.006; VA 0.0000–0.0010; and VD, 0.0000–0.0004.
. Other parameters and definitions are as in Table 1. Ranges of SE: by.x, 0.001–0.005; R, 0.001–0.005; 1/bx.y, 0.002–0.011; hDL, 0.001–0.008; QN, 0.26–0.79; SL, 0.31–0.69; D, 0.002–0.006; VA 0.0000–0.0010; and VD, 0.0000–0.0004.
A compound estimate of the average h can also be obtained from 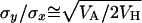 (Hughes 1995; Charlesworth and Hughes 1999), with a mutation-selection balance prediction identical to that from
(Hughes 1995; Charlesworth and Hughes 1999), with a mutation-selection balance prediction identical to that from 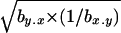 and similar to that from the estimator hDL. Values of
and similar to that from the estimator hDL. Values of 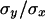 for quasinormal chromosomes were computed under the model in Table 1 for the case where pleiotropic mutations have an average effect
for quasinormal chromosomes were computed under the model in Table 1 for the case where pleiotropic mutations have an average effect 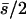 with n = 0, 5, and 15 antagonistic pleiotropic mutations. The values obtained were 0.10, 0.52, and 0.70, respectively, within the range of values observed for the estimators by.x and 1/bx.y.
with n = 0, 5, and 15 antagonistic pleiotropic mutations. The values obtained were 0.10, 0.52, and 0.70, respectively, within the range of values observed for the estimators by.x and 1/bx.y.
Tables 1–3 give results for a population of size N = 104 and a symmetric model of pleiotropy. Some of the cases in Table 1 are extended to different population sizes (Table 4A) and to an asymmetric model of pleiotropy (Table 4B). Results from both a range of population sizes and an asymmetric pleiotropic model are qualitatively similar to those shown previously, leading to identical conclusions.
TABLE 4.
Estimates of the average coefficient of dominance for segregating mutations and other parameters for a model of few mutations of large average effect, under different population sizes (A) and an asymmetric model of pleiotropy (B)
| Estimates of average h
|
% chromosomes
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| N | n | by.x | R | 1/bx.y | hDL | QN | SL | D | VA | VD | VA/D |
| A. | |||||||||||
| 102 | 0 | 0.15 | 0.15 | 0.33 | 0.18 | 98 | 2 | 0.018 | 0.0003 | 0.0005 | 0.017 |
| 15 | 0.39 | 0.34 | 0.62 | 0.44 | 96 | 4 | 0.095 | 0.0074 | 0.0023 | 0.078 | |
| 103 | 0 | 0.07 | 0.07 | 0.25 | 0.10 | 94 | 6 | 0.056 | 0.0003 | 0.0007 | 0.005 |
| 15 | 0.35 | 0.31 | 0.65 | 0.37 | 91 | 9 | 0.150 | 0.0090 | 0.0029 | 0.060 | |
| 105 | 0 | 0.03 | 0.02 | 0.16 | 0.03 | 77 | 23 | 0.206 | 0.0002 | 0.0013 | 0.001 |
| 15 | 0.27 | 0.22 | 0.72 | 0.20 | 73 | 27 | 0.296 | 0.0089 | 0.0034 | 0.030 | |
| B. | |||||||||||
| 3 | 0.08 | 0.15 | 0.40 | 0.10 | 86 | 14 | 0.132 | 0.0014 | 0.0010 | 0.011 | |
| 104 | 5 | 0.11 | 0.22 | 0.51 | 0.13 | 86 | 14 | 0.139 | 0.0021 | 0.0012 | 0.015 |
| 10 | 0.18 | 0.31 | 0.62 | 0.19 | 85 | 15 | 0.157 | 0.0041 | 0.0015 | 0.026 | |
| 15 | 0.24 | 0.35 | 0.69 | 0.24 | 83 | 17 | 0.178 | 0.0060 | 0.0018 | 0.034 | |
Nonlethal mutational parameters: λ = 0.006, 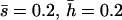 . N, population size. n, number of antagonistic pleiotropic mutations segregating per chromosome. The mean selection coefficient of antagonistic pleiotropic mutations is
. N, population size. n, number of antagonistic pleiotropic mutations segregating per chromosome. The mean selection coefficient of antagonistic pleiotropic mutations is 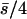 . Ranges of SE: by.x, 0.001–0.004; R, 0.000–0.003; 1/bx.y, 0.002–0.008; hDL, 0.001–0.008; QN, 0.18–0.81; SL, 0.11–0.39; D, 0.001–0.009; VA, 0.0000–0.0002; and VD, 0.0000–0.0001. Other parameters and definitions are as in Table 1.
. Ranges of SE: by.x, 0.001–0.004; R, 0.000–0.003; 1/bx.y, 0.002–0.008; hDL, 0.001–0.008; QN, 0.18–0.81; SL, 0.11–0.39; D, 0.001–0.009; VA, 0.0000–0.0002; and VD, 0.0000–0.0001. Other parameters and definitions are as in Table 1.
The sampling variance for the different estimators of dominance is shown in Table 5 for some of the cases in Table 1. The R-estimator shows the lowest sampling variance and 1/bx.y the highest one. Intermediate and similar sampling variances are shown by by.x and hDL.
TABLE 5.
Sampling variance of the estimates of the average coefficient of dominance for segregating mutations
| Variance (× 103) of the estimates of average h
|
||||
|---|---|---|---|---|
| n | by.x | R | 1/bx.y | hDL |
| A. | ||||
| 0 | 0.16 ± 0.01 | 0.07 ± 0.02 | 2.75 ± 0.21 | 0.22 ± 0.01 |
| 10 | 9.17 ± 0.24 | 1.66 ± 0.08 | 16.07 ± 1.29 | 7.98 ± 0.44 |
| B. | ||||
| 0 | 0.24 ± 0.01 | 0.17 ± 0.01 | 3.72 ± 0.22 | 0.25 ± 0.01 |
| 10 | 5.09 ± 0.31 | 0.61 ± 0.03 | 13.12 ± 1.07 | 6.59 ± 0.43 |
Mutational parameters: (A) λ = 0.006, 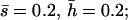 (B) λ = 0.04,
(B) λ = 0.04, 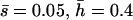 . n, number of antagonistic pleiotropic mutations segregating per chromosome. The mean selection coefficient of antagonistic pleiotropic mutations is
. n, number of antagonistic pleiotropic mutations segregating per chromosome. The mean selection coefficient of antagonistic pleiotropic mutations is 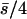 . Other parameters and definitions are as in Table 1.
. Other parameters and definitions are as in Table 1.
The method of Deng and Lynch (1996) allows us to estimate the average selection coefficient and the mutational rate of deleterious mutations using the previously estimated average coefficient of dominance. These estimates are presented in Table 6 for some cases corresponding to Tables 1 and 2. Uncorrected estimates (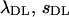 ) have some bias even in the case with no pleiotropic genes (see also Li et al. 1999 and Li and Deng 2000). This bias may become huge with the presence of pleiotropic mutations, although counteracting biases can make the estimates closer to the true values. Correction through mutational information leads estimates (
) have some bias even in the case with no pleiotropic genes (see also Li et al. 1999 and Li and Deng 2000). This bias may become huge with the presence of pleiotropic mutations, although counteracting biases can make the estimates closer to the true values. Correction through mutational information leads estimates (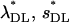 ) toward the true values on some occasions but not on others.
) toward the true values on some occasions but not on others.
TABLE 6.
Estimates of the per generation rate of mutations and the average selection coefficient of mutations by the method of Deng and Lynch (1996)
| n | 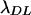 |
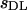 |
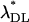 |
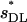 |
|
|---|---|---|---|---|---|
| A. | |||||
| True value: | 0.006 | 0.200 | 0.006 | 0.200 | |
| 0 | 0.009 | 0.409 | 0.012 | 0.307 | |
| 3 | 0.040 | 0.277 | 0.071 | 0.201 | |
| 10 | 0.188 | 0.150 | 0.265 | 0.092 | |
| B. | |||||
| True value: | 0.040 | 0.050 | 0.040 | 0.050 | |
| 0 | 0.022 | 0.205 | 0.038 | 0.131 | |
| 10 | 0.030 | 0.175 | 0.093 | 0.105 | |
| 100 | 0.394 | 0.067 | 0.537 | 0.014 |
Mutational parameters: (A) λ = 0.006, 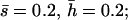 (B) λ = 0.04,
(B) λ = 0.04, 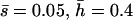 . n, number of antagonistic pleiotropic mutations segregating per chromosome. The mean selection coefficient of antagonistic pleiotropic mutations is
. n, number of antagonistic pleiotropic mutations segregating per chromosome. The mean selection coefficient of antagonistic pleiotropic mutations is 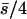 .
.
To assess the implications of segregating loci overdominant for fitness we analyzed the distribution of homozygous and heterozygous viabilities as well as the expressed homozygous mutation load. The proportion of lethal chromosomes was in the range 24–29% for all cases in Tables 1–4 (for N = 104–105), with a lethal mutation load in the range 0.27–0.34. These proportions are not affected by the models considered, as the assumed rate of lethal mutations was the same in all cases. They are of the same order as empirical estimates. Thus, the average proportion of lethal chromosomes II for Drosophila is between ∼20 and 40% (e.g., Mukai and Yamaguchi 1974; Mukai and Nagano 1983; Kusakabe and Mukai 1984), and the corresponding average lethal mutation load is 0.247 (with a range 0.13–0.50 from 17 studies) (Simmons and Crow 1977). Among the nonlethal chromosomes, the proportion of quasinormal and semilethal chromosomes is given in Tables 1–4. Tables 1–4 also show the homozygous mutation load due to nonlethal chromosomes (detrimental load) and the additive and dominance genetic variances contributed by nonlethal chromosomes. The presence of antagonistic pleiotropic (overdominant) mutations increases the fraction of semilethal chromosomes at the expense of quasinormal ones. For a sufficiently large number of overdominant mutations the proportion of semilethal chromosomes may exceed that of quasinormal ones (see the case with n = 150 segregating antagonistic pleiotropic mutations with average effect 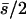 in Table 2). Segregating overdominant mutations increase therefore the detrimental load and the amount of additive and dominance genetic variance. However, the proportional increase in additive variance is much larger than that in the detrimental load, as indicated by their ratio. When pleiotropic genes show strengthening pleiotropy (Table 3), there is a slightly larger proportion of quasinormal chromosomes at the expense of semilethal ones, and the homozygous detrimental load is slightly smaller than in the case with nonpleiotropic genes (cf. Tables 1–3). Smaller population sizes also imply a larger proportion of quasinormal chromosomes and detrimental load (Table 4A). Asymmetrical models of pleiotropy produce slightly smaller detrimental loads and genetic variances than do symmetrical models (cf. Tables 1 and 4B).
in Table 2). Segregating overdominant mutations increase therefore the detrimental load and the amount of additive and dominance genetic variance. However, the proportional increase in additive variance is much larger than that in the detrimental load, as indicated by their ratio. When pleiotropic genes show strengthening pleiotropy (Table 3), there is a slightly larger proportion of quasinormal chromosomes at the expense of semilethal ones, and the homozygous detrimental load is slightly smaller than in the case with nonpleiotropic genes (cf. Tables 1–3). Smaller population sizes also imply a larger proportion of quasinormal chromosomes and detrimental load (Table 4A). Asymmetrical models of pleiotropy produce slightly smaller detrimental loads and genetic variances than do symmetrical models (cf. Tables 1 and 4B).
Figure 2 shows the distribution of chromosomal viabilities for some models in Tables 1 and 2. The top of each column shows the case with nonpleiotropic mutations (n = 0). In this case, the distribution of heterozygous chromosomes is closely centered around the mean, and the distribution of homozygotes has also a peak close to one. The inclusion of antagonistic pleiotropic mutations increases the variation of heterozygous and homozygous chromosome viabilities and moves the homozygous mean far away from the heterozygous one. The replacement of nonpleiotropic mutations by strengthening pleiotropic ones does not affect substantially the distribution of chromosomes (results not shown).
Figure 2.

Distribution of homozygous (continuous) and heterozygous (discontinuous) chromosome viabilities for different numbers (n) of antagonistic pleiotropic mutations segregating per chromosome (their average viability effects in parentheses). The two columns refer to predictions using mutational parameters λ = 0.006, 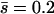 ,
, 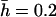 and λ = 0.04,
and λ = 0.04, 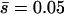 ,
, 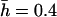 , respectively (λL = 0.006, sL = 1, and hL = 0.02 for lethals). Results were obtained by diffusion approximations for nonpleiotropic mutations and by transition matrix for antagonistic pleiotropic ones.
, respectively (λL = 0.006, sL = 1, and hL = 0.02 for lethals). Results were obtained by diffusion approximations for nonpleiotropic mutations and by transition matrix for antagonistic pleiotropic ones.
DISCUSSION
We have investigated the effect of antagonistic pleiotropy on indirect estimates of the average degree of dominance for a fitness component (say, viability). To achieve this, we have explored models with plausible mutational rates and effects, and we have included different proportions of antagonistic pleiotropic mutations that reduce viability but tend to be overdominant for fitness. We showed that even a very small proportion of such fitness-overdominant segregating mutations (down to 2%) can produce estimates of the average h inflated by up to fivefold. The reason is that the statistics studied (by.x, 1/bx.y, R, and hDL) can be interpreted only in terms of the corresponding average h-estimates (see the Introduction) on the assumption that segregating alleles reducing viability are always at low frequency, so that homozygotes for deleterious mutations can be neglected in a panmictic population. When this assumption does not hold, the above statistics give biased estimates. For example, from Equations 1–3 of Mukai and Yamaguchi (1974), which give variances of homozygous and heterozygous viabilities and their corresponding covariances, it can be shown that, if deleterious mutations are at frequency 0.5, we expect by.x = 0.5 irrespective of the value of h (see also Charlesworth and Hughes 1999), and the expected value for 1/bx.y ranges between 0.5 and 0.75 for different values of h. The high sensitivity of the estimators to a small fraction of pleiotropic mutations is due to the large proportion of genetic variance accounted for. Our results support the view that estimates of the average coefficient of dominance from segregating populations assuming a balance between directional selection and deleterious mutation are not very reliable and that direct estimates from mutation-accumulation experiments may be more appropriate (Fernández et al. 2004).
The approach followed in this work is similar to that from Mukai and Yamaguchi (1974), Deng (1998a), and Li et al. (1999), in that these authors also studied the effect, on the estimation of mutational parameters, of an increasing number of mutations under balancing selection in addition to deleterious mutations. The difference is that they assumed a model of pure overdominant mutations; i.e., mutations are overdominant for their effects on the trait itself under study. The result is a decrease or small increase of the estimates provided by by.x and hDL relative to the case where no overdominant mutations are included. In our model all mutations are deleterious for the trait under study, but a small proportion can be overdominant for overall fitness through an advantageous pleiotropic effect on another trait. The observed result is, in contrast, a huge increase of the estimates provided by by.x and hDL. Whereas the model of pure overdominance is, according to Charlesworth and Hughes (1999), decisively rejected by the data, marginal overdominance through antagonistic pleiotropy is a rather likely scenario (Charlesworth and Hughes 1999).
Mukai and Yamaguchi (1974) also considered the possibility that mutations affecting viability have a different effect on global fitness. They defined the ratio c from the degree of dominance for total fitness to that for viability, so that c > 1 would indicate that the overall heterozygous effect on fitness of a viability mutation is larger than the effect on viability itself. Indirect estimates of c suggesting values ∼2–3 have been obtained by Mukai and Yamaguchi (1974), Mukai et al. (1974), and Charlesworth and Hughes (1999). We have also considered a model of mutations with strengthening pleiotropy, roughly equivalent to a model where c = 2 for all segregating mutations affecting viability. As shown in Table 3, this model does not strongly affect the estimates of the average coefficient of dominance. Similar conclusions were reached by Fernández et al. (2004), who investigated the effect of strengthening pleiotropy on the estimation of the average h from segregating populations, assuming a model where the selection coefficients of mutations are increased by a factor between 1 and 3 in their global effects on fitness.
Our model of antagonistic pleiotropy usually assumes segregating loci that have symmetric effects on both fitness components, a given allele having opposite dominance relationships for each trait (termed reversal of dominance by Curtsinger et al. 1994). In other words, a given allele that is deleterious recessive for a fitness component will be advantageous dominant for the other. This model was chosen as it gives the most favorable conditions to maintain a polymorphism. Equal dominance relationships (i.e., a given allele is either recessive or dominant for both fitness components, termed parallel dominance by Curtsinger et al. 1994) substantially restrict the possibility of a stable polymorphism (Hoekstra et al. 1985; Curtsinger et al. 1994; Hedrick 1999). Branched multienzyme pathway analyses suggest that parallel dominance is the most general situation, as mutations affecting enzyme activity are likely to be recessive for all pleiotropic branches (Keightley and Kacser 1987). However, reversal of dominance may also occur under some particular situations (Keightley and Kacser 1987), and the phenomena illustrated in this article require only a very small fraction of mutations giving rise to balancing selection. In addition, we have also considered a more flexible and general model where mutational effects and their corresponding dominance coefficients are independently assigned to both fitness components. Thus, this model would include a range of dominance relationships. The results (Table 4B) are very similar to those obtained with the symmetrical reversal of dominance model (Table 1), suggesting that a variable relationship between the degrees of dominance of mutations for the pleiotropic traits can also maintain substantial polymorphism.
Conditions for the selective maintenance of stable polymorphisms by antagonistic pleiotropy are quite restrictive (Curtsinger et al. 1994; Hedrick 1999). However, the analyses leading to this conclusion consider only the maintenance of variation by balancing selection, ignoring the recurrent introduction of new alleles by mutation. A continual input of variation via mutation at loci subject to antagonistic pleiotropy, as assumed in our model, may additionally contribute to the maintenance of the polymorphism (Lynch and Walsh 1998, p. 656), but this is an issue out of the scope of this article.
The estimation method of Deng and Lynch (1996) allows for the estimation not only of the average dominance, but also of the average selection coefficient and the deleterious mutation rate. The biases incurred by the estimates of dominance with this method, hDL, are of the same order as those obtained by the other estimators (see Tables 1–5) and can be substantially lower than the arithmetic mean of h-values in the absence of pleiotropic effects. This may seem surprising as, apparently, hDL uses information from both the variance and the average viability in O and S populations. However, the averages are used to remove just the mean-variance dependence that arises because of the between-locus multiplicative nature assumed for fitness. This could be attained in a more simple way by assuming an additive model for multilocus effects or, alternatively, by using log-transformed data (M = ln(W)), for which the mutation-selection balance expectations for the basic parameters in Deng and Lynch (1996, Equations 1a–1d) become
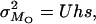 |
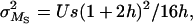 |
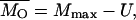 |
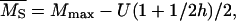 |
where U = 2λ. Therefore, parameters with the same expectation as the x, y, z used by Deng and Lynch (1996) reduce to
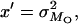 |
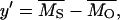 |
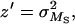 |
which can be used instead of x, y, z, in Deng and Lynch's (1996) estimation Equations 4a–4c, providing estimates for h, U, and s very close to those obtained for untransformed fitness (data not shown). This log-fitness version of the Deng and Lynch (1996) method reveals that hDL uses basically the ratio from the genetic variance between panmictic (outbred) individuals to that between individuals with some degree of inbreeding (selfed):
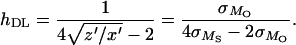 |
Because of this, its mutation-selection balance expectation approximates that for the estimator proposed by Charlesworth and Hughes (1999), which estimates h from the ratio from the genetic variance between panmictic individuals to that between homozygous ones, 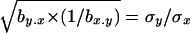 .
.
In addition to the bias incurred by hDL, substantial biases are also observed for the corresponding estimates of mutation rates and mean selective coefficients depending on the mutational model (Table 6). The presence of overdominant loci implies an increase in the estimates of hDL and λDL and a decrease in those of sDL (Table 1–6). A similar increase in the estimates of λDL and a decrease in those of sDL are observed under pure overdominance for the trait under study (Li et al. 1999).
T. Mukai and co-workers carried out analyses of viability in a number of natural populations of D. melanogaster. A summary and discussion of the results regarding the maintenance of variation has been provided by Mukai (1985, 1988). The analysis of variation suggested that there is a cline in the amount of additive variance from northern to southern populations, so that southern ones usually show higher additive variances than northern ones. Table 7 and Figure 3 show empirical estimates of the average coefficient of dominance and other parameters for chromosome II of D. melanogaster from the Aomori (Kusakabe and Mukai 1984), Raleigh (Mukai and Yamaguchi 1974), and Orlando (Mukai and Nagano 1983) populations, which are representatives of populations in the north-to-south cline. The northernmost population (Aomori) showed the lowest additive variance among all populations analyzed by Mukai (1985, 1988). The southernmost Orlando population was that with the highest additive variance. The Raleigh population had both intermediate latitude and variance. As shown in Table 7 and Figure 3, the proportion of detrimental chromosomes increased at the expense of the quasinormal ones toward the southern latitudes, and there was an increase in the homozygous detrimental load, but this was proportionately lower than the increase in additive variance. An increase in dominance variance was less apparent as this shows a large standard error for the Orlando population.
TABLE 7.
Empirical estimates of the average coefficient of dominance for segregating mutations (regression estimates using the average homozygous, x, and heterozygous, y, viabilities of chromosome II of Drosophila melanogaster) and other population parameters
| Population | by.x | 1/bx.y | QN | SL | D | VA | VD | VA/D |
|---|---|---|---|---|---|---|---|---|
| Aomoriab | 0.18 ± 0.06 | 0.67 ± 0.23 | 85 | 15 | 0.243 | 0.0027 ± 0.0010 | 0.0001 ± 0.0003 | 0.011 |
| Raleighac | 0.29 ± 0.07 | 1.01 ± 0.23 | 76 | 24 | 0.334 | 0.0096 ± 0.0025 | 0.0012 ± 0.0005 | 0.029 |
| Orlandoad | 0.48 ± 0.21 | 0.67 ± 0.28 | 64 | 36 | 0.403 | 0.0202 ± 0.0038 | 0.0006 ± 0.0057 | 0.056 |
Parameters and definitions are as in Table 1.
Mukai (1985).
Kusakabe and Mukai (1984).
Mukai and Yamaguchi (1974).
Mukai and Nagano (1983).
Figure 3.

Distribution of homozygous (continuous) and heterozygous (discontinuous) chromosome viabilities from empirical studies in three natural populations of Drosophila melanogaster: (A) Aomori (Kusakabe and Mukai 1984), (B) Raleigh (Mukai and Yamaguchi 1974), and (C) Orlando (Mukai and Nagano 1983).
It is also clear from Table 7 that the regression estimate by.x increased toward the south whereas the estimate 1/bx.y always showed large values. Another useful parameter to be compared among these populations is the variance among genotypic values for homozygous chromosomes, VH, because the ratio VA/VH is expected to be low under a mutation-selection balance model and large under balancing selection (Charlesworth and Hughes 1999; Kelly 1999). In fact, as stated above, the value of 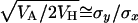 is an estimator of the average h with a mutation-selection balance expectation analogous to that of
is an estimator of the average h with a mutation-selection balance expectation analogous to that of 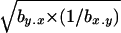 and hDL. Using the results from quasinormal chromosomes from Kusakabe and Mukai (1984), Mukai and Yamaguchi (1974), and Mukai and Nagano (1983), the estimates of
and hDL. Using the results from quasinormal chromosomes from Kusakabe and Mukai (1984), Mukai and Yamaguchi (1974), and Mukai and Nagano (1983), the estimates of 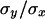 were 0.34, 0.66, and 0.57 for the Aomori, Raleigh, and Orlando populations, respectively. Another estimate for the Aomori population (0.17) can be obtained from Takano et al. (1987) (see also Charlesworth and Hughes 1999) to give an average of 0.25 for this population.
were 0.34, 0.66, and 0.57 for the Aomori, Raleigh, and Orlando populations, respectively. Another estimate for the Aomori population (0.17) can be obtained from Takano et al. (1987) (see also Charlesworth and Hughes 1999) to give an average of 0.25 for this population.
Mukai (1988) suggested that the variation in the northernmost populations could be explained by mutation-selection balance alone, whereas the excess in additive variance observed in the southernmost populations could be due to genotype-environment interaction (see also Santos 1997). This was suggested to be operating if the selection coefficients of mutations for viability take negative or positive values, depending on different environmental conditions, while the fitness of heterozygotes remains intermediate between the homozygotes but close to that of the fitter homozygote. In this situation, the maximum number of loci involved in diversifying selection would not be great (<200). Mukai and Yamaguchi (1974) also suggested the possible existence of a small number of overdominant loci affecting viability in the Raleigh population, responsible for a homozygous detrimental load of ∼0.1.
It should be noted that the effective population sizes estimated for the three populations discussed above were quite different: ∼2600 for the Aomori population, ∼20,000 for the Raleigh population, and effectively infinite for the Orlando population (Kusakabe and Mukai 1984). These estimates were obtained from the rate of allelism among lethal chromosomes and, therefore, they can be subject to large estimation errors. However, the results may indeed suggest different population sizes. If this is the case, it is possible that some of the patterns observed between populations for the north-south cline (Table 7) are due to this difference in population size. The larger the effective population size, the larger the expected proportion of semilethal chromosomes at the expense of quasinormal ones and the larger the homozygous load (Fernández et al. 2004), agreeing with the clinal results in Table 7. For example, the proportions of quasinormal chromosomes for the case without pleiotropic mutations (n = 0) in Tables 1 and 4 are 98, 94, 88, and 77% for N = 102, 103, 104, and 105, respectively, and the corresponding detrimental loads are 0.018, 0.056, 0.114, and 0.206, respectively. In contrast, small effective population sizes would usually imply inflated values for the average h-estimates, compared to their expectation assuming mutation-selection balance at an effectively infinite population (Fernández et al. 2004), whereas the observed results in the cline point toward the opposite direction. For example, for the same instances as above, the estimates of by.x are 0.15, 0.07, 0.04, and 0.03 for N = 102, 103, 104, and 105, respectively. Therefore, it may be quite feasible that the differences in variation, detrimental loads, and chromosome viability distributions are due to some form of balancing selection operating at a few loci, rather than to differences in effective population size.
The results of this article suggest the possibility that the enhanced variances in the southernmost populations studied by Mukai and co-workers could be caused by some deleterious mutations affecting viability and having an advantageous pleiotropic effect on other fitness components. The inclusion of a small number of such antagonistic pleiotropic mutations induces changes in the parameters studied, compatible with the main observations. Thus, in Tables 1–4 and Figure 2 it can be seen that pleiotropic mutations decrease the proportion of quasinormal chromosomes at the expense of semilethal ones, increase the homozygous detrimental load and the additive variance (the latter in a larger proportion than the former), and inflate the estimates of the regressions by.x and 1/bx.y. All these results are observed in the north-to-south cline in natural populations (Table 7 and Figure 3). Nevertheless, the increase in dominance variance is not so apparent in the empirical data (Table 7) (Mukai 1985, 1988). Theoretical arguments suggest that antagonistic pleiotropy should produce a dominance variance about half as large as the additive variance (Curtsinger et al. 1994). In agreement with this prediction, we obtain a dominance variance between one-third and one-half of the additive variance in the presence of antagonistic pleiotropic mutations (Tables 1–4). An extensive review including many wild and domestic species shows that life-history traits have levels of dominance variance that on average may be as high or even higher than those of additive variance (Crnokrak and Roff 1995), giving some indirect support for the antagonistic pleiotropy model or other models of balancing selection where recessive mutations are maintained at high frequencies. Whether or not this is a general scenario for viability in Drosophila, our results indicate that a few segregating loci maintained at large frequencies may importantly inflate indirect estimates of the average degree of dominance of deleterious mutations for fitness components, producing large average h-values in agreement with common empirical observations.
That antagonistic pleiotropy selection is likely to be a force occurring in nature has been deduced by different analyses (see review by Charlesworth and Hughes 1999), but quantifying the proportion of variation due to directional and balancing selection may be, however, a difficult task not yet resolved.
Acknowledgments
We thank Peter Keightley for a useful discussion on the dominance of pleiotropic multienzyme pathways. This article improved greatly with the suggestions from two anonymous referees. This work was supported by grants from Universidade de Vigo, Xunta de Galicia, and Ministerio de Ciencia y Tecnología and Fondos Feder (BMC 2002-00476, BMC2003-03022). B. Fernández was supported by a Xunta de Galicia predoctoral fellowship.
References
- Agrawal, A. F., and J. R. Chasnov, 2001. Recessive mutations and the maintenance of sex in structured populations. Genetics 158: 913–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero, A., and P. D. Keightley, 1994. A pleiotropic nonadditive model of variation in quantitative traits. Genetics 138: 883–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero, A., P. D. Keightley and M. Turelli, 1997. Average dominance for polygenes: drawbacks of regression estimates. Genetics 147: 1487–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth, B., 1994. Evolution in Age-Structured Populations. Cambridge University Press, Cambridge, UK.
- Charlesworth, B., and D. Charlesworth, 1998. Some evolutionary consequences of deleterious mutations. Genetica 102/103: 3–19. [PubMed] [Google Scholar]
- Charlesworth, B., and D. Charlesworth, 1999. The genetic basis of inbreeding depression. Genet. Res. 74: 329–340. [DOI] [PubMed] [Google Scholar]
- Charlesworth, B., and K. A. Hughes, 1999. The quantitative genetics of life history traits, pp. 369–392 in Evolutionary Genetics From Molecules to Morphology, edited by R. S. Singh and C. B. Krimbas. Cambridge University Press, Cambridge, UK.
- Charlesworth, B., D. Charlesworth and M. T. Morgan, 1990. Genetic loads and estimates of mutation rates in highly inbred plant populations. Nature 347: 380–382. [Google Scholar]
- Charlesworth, B., H. Borthwick, C. Bartolomé and P. Pignatelli, 2004. Estimates of the genomic mutation rate for detrimental alleles in Drosophila melanogaster. Genetics 167: 815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chasnov, J. R., 2000. Mutation-selection balance, dominance and the maintenance of sex. Genetics 156: 1419–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavarrías, D., C. López-Fanjul and A. García-Dorado, 2001. The rate of mutation and the homozygous and heterozygous mutational effects for competitive viability: a long-term experiment with Drosophila melanogaster. Genetics 158: 681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comstock, R. E., and H. F. Robinson, 1952. Estimation of average dominance of genes, pp. 494–516 in Heterosis, edited by J. W. Gowen. Iowa State College Press, Ames, IA.
- Crnokrak, P., and D. A. Roff, 1995. Dominance variance: associations with selection and fitness. Heredity 75: 530–540. [Google Scholar]
- Curtsinger, J. W., P. M. Service and T. Prout, 1994. Antagonistic pleiotropy, reversal of dominance, and genetic polymorphism. Am. Nat. 144: 210–228. [Google Scholar]
- Deng, H.-W., 1998. a Estimating within-locus nonadditive coefficient and discriminating dominance vs. overdominance as the genetic cause of heterosis. Genetics 148: 2003–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, H.-W., 1998. b Characterization of deleterious mutations in outcrossing populations. Genetics 150: 945–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, H.-W., and M. Lynch, 1996. Estimation of genomic mutation parameters in natural populations. Genetics 144: 349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, H.-W., G. Gao and J.-L. Li, 2002. Estimation of deleterious genomic mutation parameters in natural populations by accounting for variable mutation effects across loci. Genetics 162: 1487–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eanes, W. F., J. Hey and D. Houle, 1985. Homozygous and hemizygous viability variation on X chromosome of Drosophila melanogaster. Genetics 111: 831–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falconer, D. S., and T. F. C. Mackay, 1996. An Introduction to Quantitative Genetics, Ed. 4. Longman, Harlow, UK.
- Fernández, B., A. García-Dorado and A. Caballero, 2004. Analysis of the estimators of the average coefficient of dominance of deleterious mutations. Genetics 168: 1053–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández, J., and C. López-Fanjul, 1996. Spontaneous mutational variances and covariances for fitness-related traits in Drosophila melanogaster. Genetics 143: 829–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry, J. D., P. D. Keightley, S. L. Heinsohn and S. V. Nuzhdin, 1999. New estimates of the rates and effects of mildly deleterious mutation in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 96: 574–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Dorado, A., and A. Caballero, 2000. On the average coefficient of dominance of deleterious spontaneous mutations. Genetics 155: 1991–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Dorado, A., C. López-Fanjul and A. Caballero, 1999. Properties of spontaneous mutations affecting quantitative traits. Genet. Res. 74: 341–350. [DOI] [PubMed] [Google Scholar]
- García-Dorado, A., A. Caballero and J. F. Crow, 2003. On the persistence and pervasiveness of a new mutation. Evolution 57: 2644–2646. [DOI] [PubMed] [Google Scholar]
- García-Dorado, A., C. López-Fanjul and A. Caballero, 2004. Rates and effects of deleterious mutations and their evolutionary consequences, pp. 20–32 in Evolution of Molecules and Ecosystems, edited by A. Moya and E. Font. Oxford University Press, London/NewYork/Oxford.
- Greenberg, R., and J. F. Crow, 1960. A comparison of the effect of lethal and detrimental chromosomes from Drosophila melanogaster. Genetics 45: 1153–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick, P. W., 1999. Antagonistic pleiotropy and genetic polymorphism: a perspective. Heredity 82: 126–133. [Google Scholar]
- Hoekstra, R. F., R Bijlsma and A. J. Dolman, 1985. Polymorphism from environmental heterogeneity: models are only robust if the heterozygote is close in fitness to the favoured homozygote in each environment. Genet. Res. 45: 299–314. [DOI] [PubMed] [Google Scholar]
- Hughes, K. A., 1995. The inbreeding decline and average dominance of genes affecting male life-history characters in Drosophila melanogaster. Genet. Res. 65: 41–52. [DOI] [PubMed] [Google Scholar]
- Johnston, M. O., and D. J. Schoen, 1995. Mutation rates and dominance levels of genes affecting total fitness in two angiosperm species. Science 267: 226–229. [DOI] [PubMed] [Google Scholar]
- Kacser, H., and J. A. Burns, 1981. The molecular basis of dominance. Genetics 97: 639–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley, P. D., 1996. A metabolic basis for dominance and recessivity. Genetics 143: 621–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley, P. D., and H. Kacser, 1987. Dominance, pleiotropy, and metabolic structure. Genetics 117: 319–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly, J. K., 1999. An experimental method for evaluating the contribution of deleterious mutations to quantitative trait variation. Genet. Res. 73: 263–273. [DOI] [PubMed] [Google Scholar]
- Kelly, J. K., 2003. Deleterious mutations and the genetic variance of male fitness components in Mimulus guttatus. Genetics 164: 1071–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, M., 1969. The number of heterozygous nucleotide sites maintained in a finite population due to steady flux of mutations. Genetics 61: 893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusakabe, S., and T. Mukai, 1984. The genetic structure of natural populations of Drosophila melanogaster. XVII. A population carrying genetic variability explicable by the classical hypothesis. Genetics 108: 393–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J.-L., and H.-W. Deng, 2000. Estimation of parameters of deleterious mutations in partial selfing or partial outcrossing populations and in nonequilibrium populations. Genetics 154: 1893–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J.-L., J. Li and H.-W. Deng, 1999. The effect of overdominance on characterizing deleterious mutations in large natural populations. Genetics 151: 895–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López, M. A., and C. López-Fanjul, 1993. Spontaneous mutation for a quantitative trait in Drosophila melanogaster. II. Distribution of mutant effects on the trait and fitness. Genet. Res. 61: 117–126. [DOI] [PubMed] [Google Scholar]
- Lynch, M., and B. Walsh, 1998. Genetics and Analysis of Quantitative Traits. Sinauer, Sunderland, MA.
- Lynch, M., J. Conery and R. Burger, 1995. Mutation accumulation and the extinction of small populations. Am. Nat. 146: 489–518. [Google Scholar]
- Martorell, C., M. A. Toro and A. Gallego, 1998. Spontaneous mutation for life-history traits in Drosophila melanogaster. Genetica 102/103: 315–324. [PubMed]
- Merilä, J., and B. C. Sheldon, 1999. Genetic architecture of fitness and nonfitness traits: empirical patterns and development of ideas. Heredity 83: 103–109. [DOI] [PubMed] [Google Scholar]
- Morton, N. E., J. F. Crow and H. J. Muller, 1956. An estimate of the mutational damage in man from data on consanguineous marriages. Proc. Natl. Acad. Sci. USA 42: 855–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai, T., 1969. The genetic structure of natural populations of Drosophila melanogaster. VIII. Natural selection on the degree of dominance of viability polygenes. Genetics 63: 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai, T., 1985. Experimental verification of the neutral theory, pp. 125–145 in Population Genetics and Molecular Evolution, edited by T. Ohta and K. Aoki. Japan Scientific Society Press/Springer-Verlag, Tokyo/Berlin.
- Mukai, T., 1988. Genotype-environment interaction in relation to the maintenance of genetic variability in populations of Drosophila melanogaster, pp. 21–31 in Proceedings of the Second International Conference on Quantitative Genetics, Chap. 3, edited by B. S. Weir, E. J. Eisen, M. M. Goodman and G. Namkoong. Sinauer, Sunderland, MA.
- Mukai, T., and S. Nagano, 1983. The genetic structure of natural populations of Drosophila melanogaster. XVI. Excess of additive genetic variance of viability. Genetics 105: 115–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai, T., and O. Yamaguchi, 1974. The genetic structure of natural populations of Drosophila melanogaster. XI. Genetic variability in a large local population. Genetics 76: 339–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai, T., S. I. Chigusa, L. E. Mettler and J. F. Crow, 1972. Mutation rate and dominance of genes affecting viability in Drosophila melanogaster. Genetics 72: 333–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai, T., R. A. Cardellino, T. K. Watanabe and J. F. Crow, 1974. The genetic variance for viability and its components in a local population of Drosophila melanogaster. Genetics 78: 1195–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto, S. P., 2003. The advantages of segregation and the evolution of sex. Genetics 164: 1099–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prout, M., 1980. Some relationships between density dependent selection and density dependent population growth. Evol. Biol. 13: 1–68. [Google Scholar]
- Rodríguez-Ramilo, S. T., A. Pérez-Figueroa, B. Fernández, J. Fernández and A. Caballero, 2004. Mutation-selection balance accounting for genetic variation for viability in Drosophila melanogaster as deduced from an inbreeding and artificial selection experiment. J. Evol. Biol. 17: 528–541. [DOI] [PubMed] [Google Scholar]
- Roff, D. A., 1997. Evolutionary Quantitative Genetics. Chapman & Hall, New York.
- Rose, M. R., 1982. Antagonistic pleiotropy, dominance, and genetic variation. Heredity 48: 63–78. [Google Scholar]
- Rose, M. R., 1991. The Evolutionary Biology of Aging. Oxford University Press, Oxford.
- Santos, M., 1997. On the contribution of deleterious alleles to fitness variance in natural populations of Drosophila. Genet. Res. 70: 105–115. [DOI] [PubMed] [Google Scholar]
- Simmons, M. J., and J. F. Crow, 1977. Mutations affecting fitness in Drosophila populations. Annu. Rev. Genet. 11: 49–78. [DOI] [PubMed] [Google Scholar]
- Stearns, S. C., 1992. The Evolution of Life Histories. Oxford University Press, Oxford.
- Takano, T., S. Kusakabe and T. Mukai, 1987. The genetic structure of natural populations of Drosophila melanogaster. XX. Comparison of genotype-environment interaction in viability between a northern and a southern population. Genetics 117: 245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turelli, M., and A. Orr, 1995. The dominance theory of Haldane's rule. Genetics 140: 389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J., A. Caballero, P. D. Keightley and W. G. Hill, 1998. Bottleneck effect on genetic variance: a theoretical investigation on the role of dominance. Genetics 150: 435–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe, T. K., O. Yamaguchi and T. Mukai, 1976. The genetic variability of third chromosomes in a local population of Drosophila melanogaster. Genetics 82: 63–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X.-S., J. Wang and W. G. Hill, 2004. Influence of dominance, leptokurtosis and pleiotropy of deleterious mutations on quantitative genetic variation at mutation-selection balance. Genetics 166: 597–610. [DOI] [PMC free article] [PubMed] [Google Scholar]